-
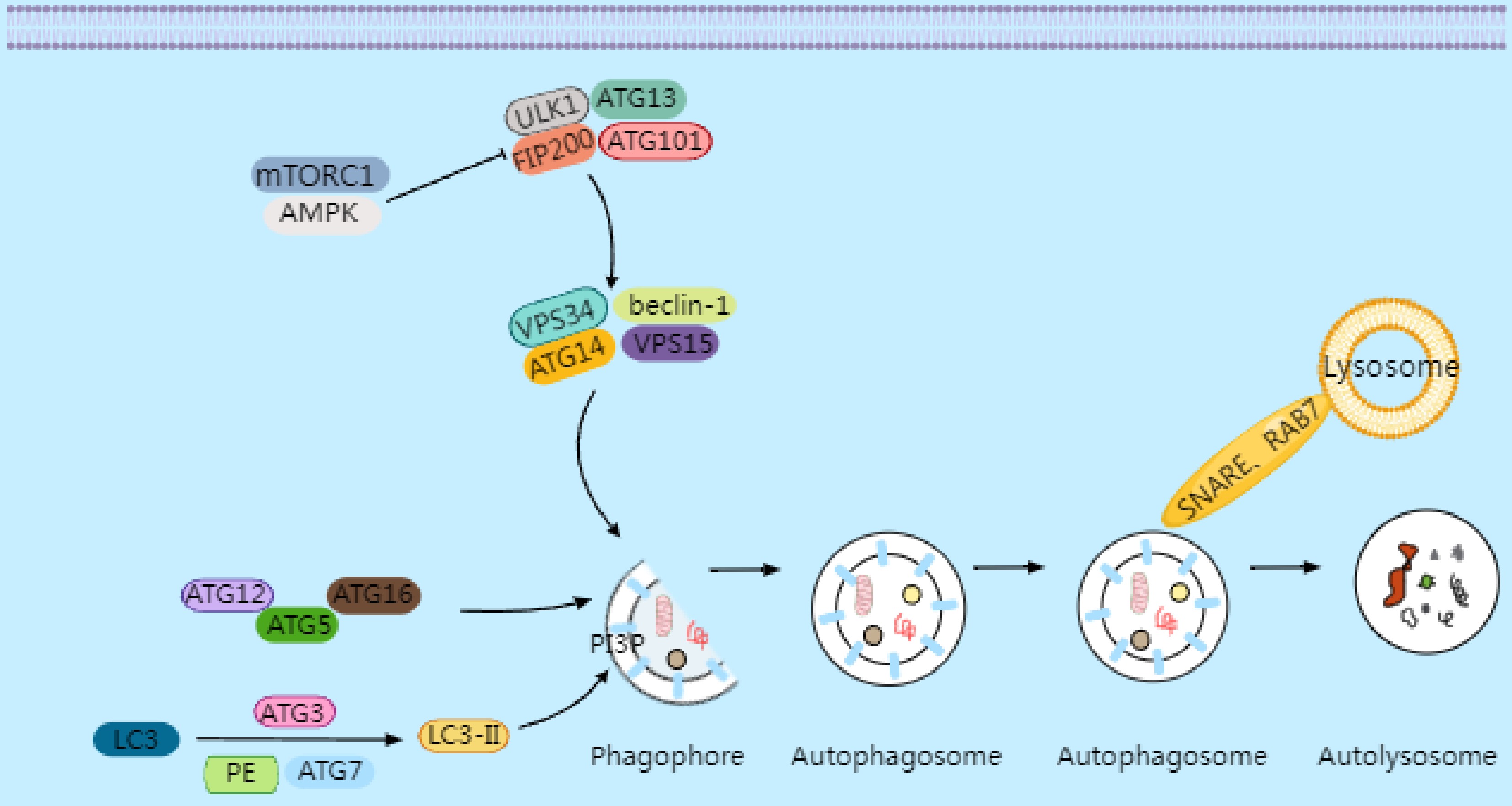
Figure 1.
Under conditions of cellular stress, nutrient deprivation, or inactivation of mammalian target of rapamycin complex 1 (mTORC1), the inhibition of the ULK1 complex is relieved, initiating the formation of the phagophore. Downstream from the ULK complex is the autophagy-specific VPS34 complex I, which catalyzes the production of phosphatidylinositol 3-phosphate (PI3P) on autophagic membranes, thereby promoting the expansion of autophagosome membranes. PI3P triggers the recruitment of autophagy's conjugation machinery, including the ATG16–ATG5-ATG12 complex, ATG3, and ATG7. These proteins facilitate the conjugation of microtubule-associated protein 1A/1B light chain 3 (LC3) to phosphatidylethanolamine (PE), converting it into LC3-II (LC3's lipidated form). LC3-II binds to newly formed autophagosome membranes and remains associated until the autophagosome fuses with lysosomes. The fusion process between autophagosomes and lysosomes requires Rab GTPase family members and soluble NSF attachment protein receptor (SNARE)proteins. Rab7 collaborates with SNARE proteins to drive membrane fusion between autophagosomes and lysosomes, forming autolysosomes. Following the degradation of the contents by hydrolases, small-molecule nutrients are released and recycled back to the cytoplasm.
-
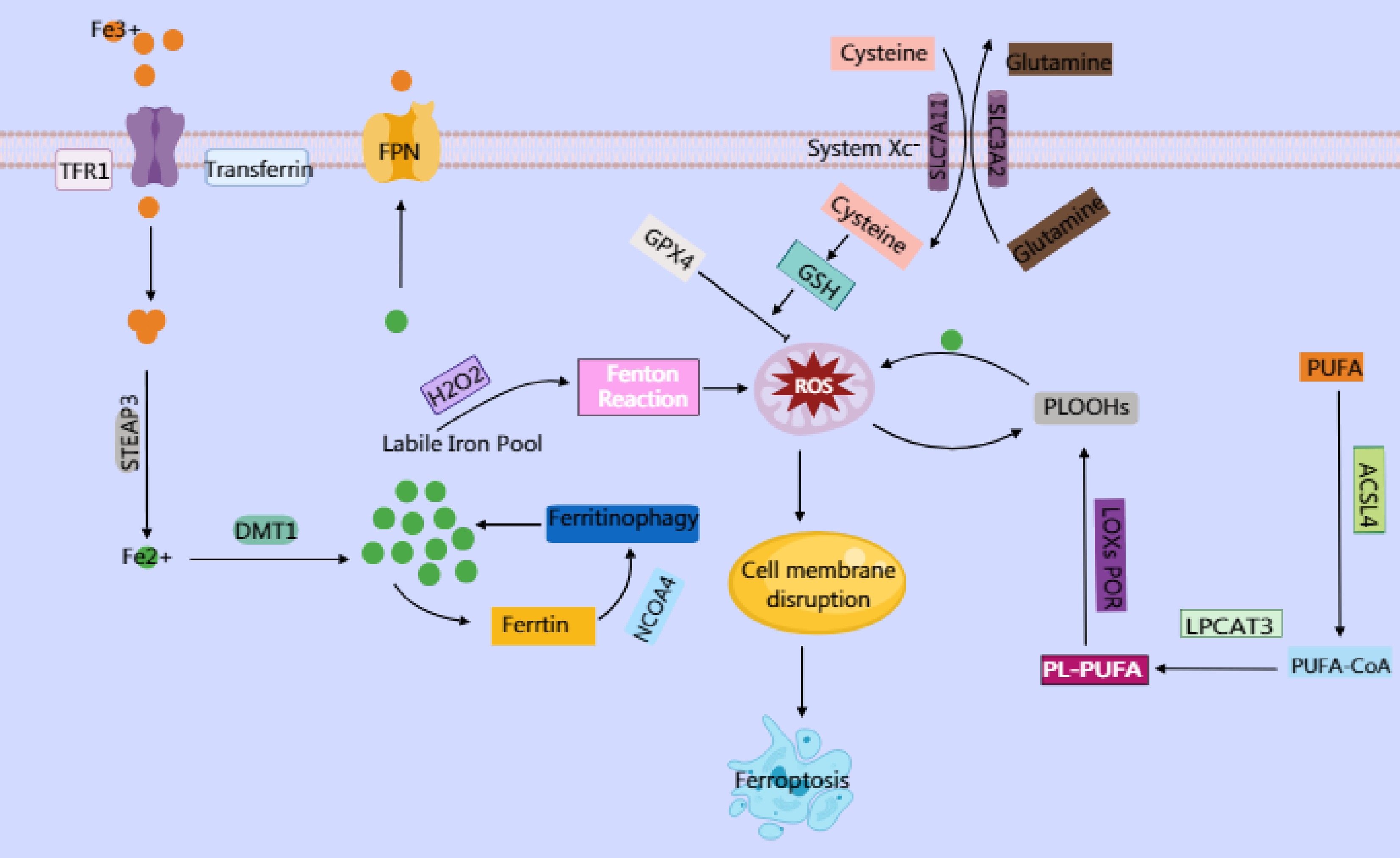
Figure 2.
Ferroptosis is an iron-dependent cell death characterized by lipid peroxidation. Elevated intracellular iron imported via transferrin and transferrin receptor (TFR1) is reduced by six-transmembrane epithelial antigen of prostate 3 (STEAP3) to its ferrous form (Fe2+), which is then transported by divalent metal transporter 1 (DMT1) into the labile iron pool. This labile iron contributes to the generation of reactive oxygen species (ROS), particularly hydroxyl radicals (·OH), through the Fenton reaction, where Fe2+ catalyzes the conversion of hydrogen peroxide (H2O2) into highly reactive ROS. In turn, ROS trigger oxidative damage, including the peroxidation of polyunsaturated phospholipids (PUFA-PLs), which serve as critical substrates for ferroptosis. PUFA-PLs are formed through the incorporation of polyunsaturated fatty acids (PUFAs) into membrane phospholipids by Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), followed by their oxidation with the involvement of lipoxygenases (LOXs) and cytochrome P450 oxidoreductase (POR). Under normal physiological conditions, the body utilizes GPX4 to reduce toxic lipid hydroperoxides (PLOOHs) into nontoxic lipid alcohols (PLOHs) by consuming GSH, thereby maintaining redox homeostasis. GPX4 mediates cystine uptake through the cystine–glutamate antiporter (System Xc−), a heterodimeric transmembrane transporter composed of SLC7A11 and SLC3A2. Intracellular cystine is reduced to cysteine, whereas Glu is exported. Cysteine serves as a critical precursor for synthesizing GSH, which is an essential cofactor for GPX4. However, when GPX4 function is impaired or GSH synthesis is disrupted, lipid peroxides accumulate.
-
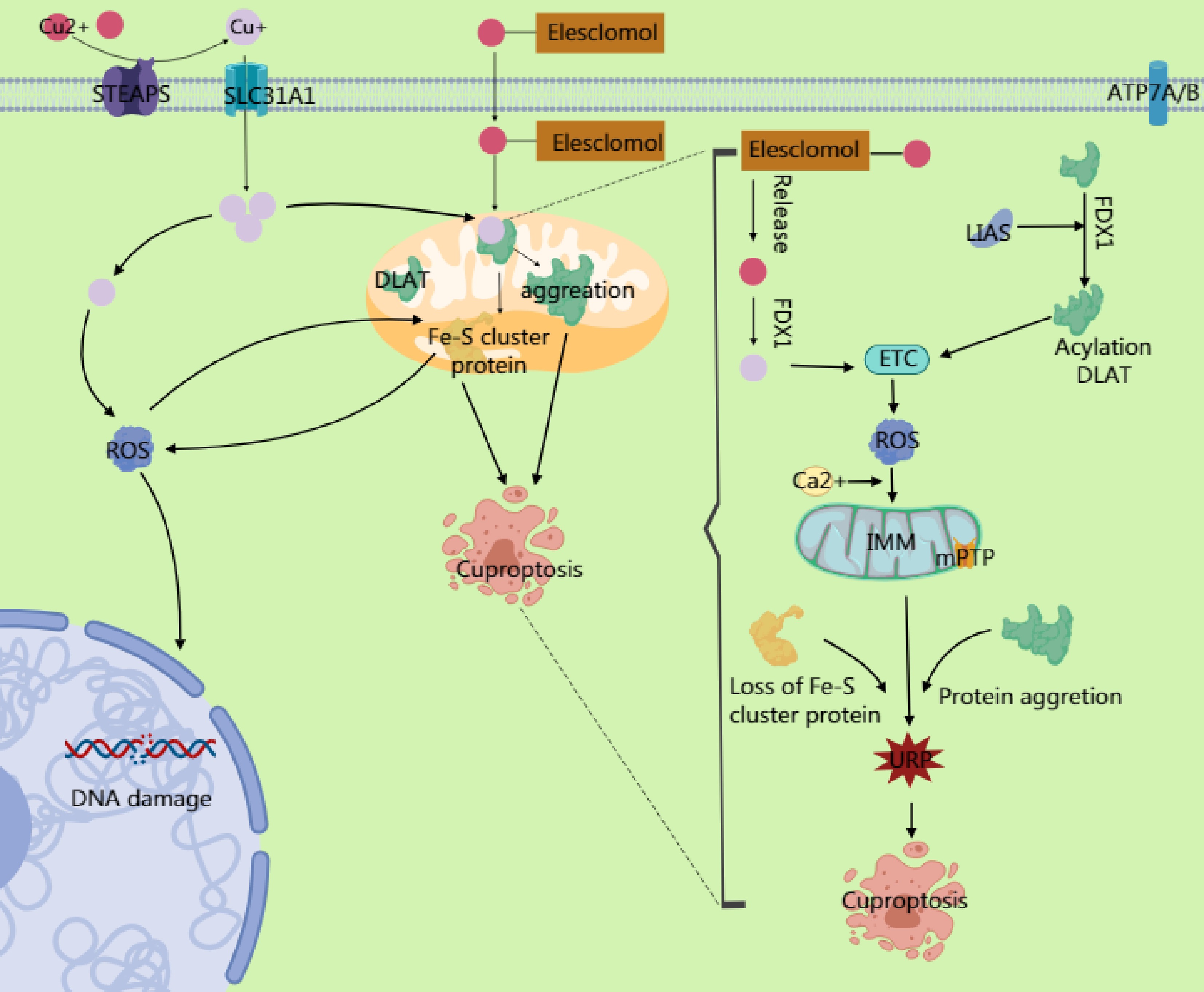
Figure 3.
Copper ions are reduced to Cu+ by six-transmembrane epithelial antigen of prostate 3 (STEAP) family proteins, and their dynamic balance is maintained through the transport proteins SLC31A1 and ATP7A/B. Excess Cu2+ can be converted to Cu+ via ferredoxin 1 (FDX1). The generated Cu+ induces reactive oxygen species (ROS) production through the Fenton reaction, leading to DNA damage and other effects. Simultaneously, it inhibits Fe-S cluster protein synthesis. Excess Cu2+ enters the mitochondrial matrix through transporters on the mitochondrial membrane, targeting enzymes modified with lipoic acid. Cu2+ binds to the lipoylation site of the TCA cycle enzyme dihydrolipoamide S-acetyltransferase (DLAT), causing the dsyfunction in the pyruvate dehydrogenase (PDH) complex and impairing electron transport chain (ETC) activity. This ETC impairment increases electron leakage and massive ROS generation. The ROS further oxidizes cysteine residues in Fe–S cluster proteins, forming a positive feedback loop that exacerbates Fe–S cluster degradation. The accumulated ROS from ETC dysfunction, combined with Ca2+ overload in the mitochondrial matrix (caused by copper-induced disruption of mitochondrial Ca2+ homeostasis), directly activates the components of the mitochondrial permeability transition pore (mPTP), prompting its opening. The mPTP, a high-conductance channel in the mitochondria, compromising the integrity of the inner mitochondrial membrane (IMM) and releasing proapoptotic factors. Additionally, the accumulation of mitochondrial ROS and degradation of the Fe–S cluster may activate endoplasmic reticulum stress through calcium signaling or ROS diffusion, triggering the unfolded protein response (UPR). This upregulates proapoptotic factors, ultimately driving the cell toward death.
-
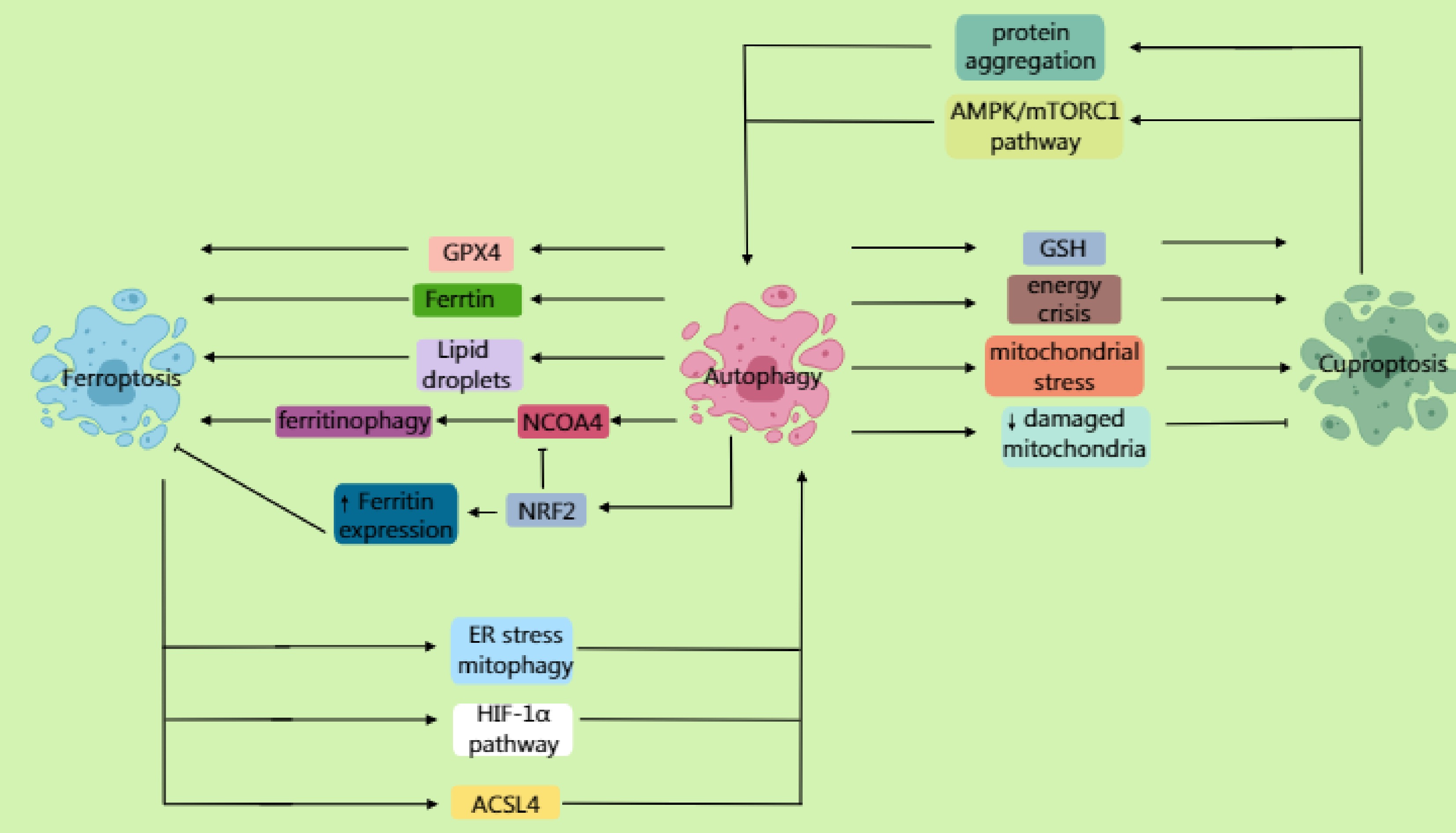
Figure 4.
Autophagy not only mediates the lysosomal degradation of ferritin through the receptor NCOA4, promoting the Fenton reaction to generate reactive oxygen species (ROS) and accelerate lipid peroxidation, but also degrades lipid droplets, releasing polyunsaturated fatty acids (PUFAs). Furthermore, by selectively degrading GPX4, it promotes the ferroptosis process. NRF2 antagonizes ferroptosis by upregulating the expression of ferritin to promote iron storage while inhibiting NCOA4-mediated ferritinophagy. Inhibition of the mTOR pathway activates autophagy, enhancing susceptibility to ferroptosis via the ferritinophagy or GPX4 degradation pathways. Ferroptosis can further induce autophagy; ferroptosis-dependent lipid ROS damage the endoplasmic reticulum (ER) and mitochondria, triggering ER stress and mitophagy. It also activates pathways such as HIF-1α, upregulating autophagy genes. ACSL4-mediated membrane phospholipid peroxidation products can directly activate autophagosome formation. In cuproptosis, autophagy may degrade important intracellular copper chelators (such as glutathione and metallothioneins), leading to elevated levels of free copper ions. Moreover, under specific conditions, excessive autophagy may exacerbate a cellular energy crisis and mitochondrial stress, sensitizing cells to cuproptosis. Additionally, autophagy (specifically mitophagy) clears the damaged mitochondria caused by copper overload, reducing cellular sensitivity to cuproptosis. Beyond promoting cell death, autophagy may delay cuproptosis by clearing damaged mitochondria or abnormally aggregated lipoylated proteins. Cuproptosis can similarly induce autophagy. Copper ions bind to mitochondrial tricarboxylic acid (TCA) cycle enzymes, inducing abnormal protein aggregation and activating mitophagy. Copper overload increases mitochondrial ROS, inducing autophagy through AMPK/mTORC1 imbalance.
-
Cell death
typeCombination strategy Mechanism of action Indication/target Clinical stage/
evidence levelKey findings Autophagy Autophagy inhibitors chloroquine/hydroxychloroquine (CQ/HCQ) + ICIs Blocks lysosomal acidification, reduces exosomal PD-L1 secretion, and enhances tumor sensitivity to PD-1/PD-L1 inhibitors Advanced gastroesophageal junction cancer, Epstein–Barr virus (EBV) gastric cancer Phase II trials (NCT04132505, NCT03755440) Combination significantly prolonged median progPFS vs monotherapy group. Rab27a/nSMase2 inhibitors + anti-vascular endothelial growth factor (VEGF) Inhibits exosomal PD-L1 packaging and release, blocks pre-metastatic niche formation Colorectal cancer metastasis models Preclinical studies Significantly reduced liver metastases when combined with bevacizumab. MAPK/ERK kinase (MEK) inhibitor (trametinib) + autophagy inhibitor Blocks autophagy-dependent metabolic reprogramming Colorectal cancer liver metastasis Preclinical studies Synergistically inhibited liver metastasis progression. Ferroptosis Ferroptosis inducers (erastin/RSL3) + PD-1 inhibitor CD8+ T-cell secreted IFN-γ inhibits SLC7A11, enhances ferroptosis sensitivity, creates a positive immune feedback loop Gastric cancer mouse model Preclinical studies Increased tumor-infiltrating T-cells (three- to five fold) and enhanced IFN-γ secretion. VPS34 inhibitor (SAR405) + Atezolizumab Blocks autophagosome formation to induce ferroptosis, synergistically enhances anti-PD-L1 efficacy Advanced colorectal cancer, pancreatic cancer Preclinical studies Significantly suppressed tumor growth; clinical trials needed to confirm mechanism. Ferroptosis nanoparticles + STING agonist Co-delivers ferroptosis inducer and ADU-S100, activates DCs and inhibits PD-L1 Colon cancer mouse model Preclinical studies Reversed immunosuppressive microenvironment, achieved significant tumor suppression. Ferroptosis induction + photothermal therapy (PTT) + anti-PD-1 PTT directly kills cells and releases tumor-associated antigens (TAAs), ferroptosis enhances immunogenic cell death (ICD), reverses T-cell exhaustion Gastrointestinal tumors (theoretical model) Preclinical studies Triple synergy provides a 'heat-sensitization + immune activation' strategy. Copper ionophore (Elesclomol) + immune checkpoint inhibitors (ICIs) Copper accumulation triggers lipoylated protein aggregation, inducing ICD; concurrently downregulates PD-L1 transcriptional activity Advanced gastric adenocarcinoma/gastroesophageal junction cancer Phase III Trial (GEMSTONE-303) Sugemalimab (anti-PD-L1) combined with antibody-dependent phagocytosis significantly improved survival. Cuproptosis Cuproptosis inducer + anti-V-domain Ig suppressor of T cell activation (VISTA) antibody Blocks VISTA-mediated immunosuppression by myeloid-derived suppressor cells (MDSCs), synergizes with cuproptosis to activate immune response Adenomatous polyposis coli (APC)-mutant colorectal cancer mouse model Preclinical studies Significantly suppressed tumor progression. Table 1.
Summary of clinical research on targeting cell death pathways in cancer immunotherapy.
Figures
(4)
Tables
(1)