-
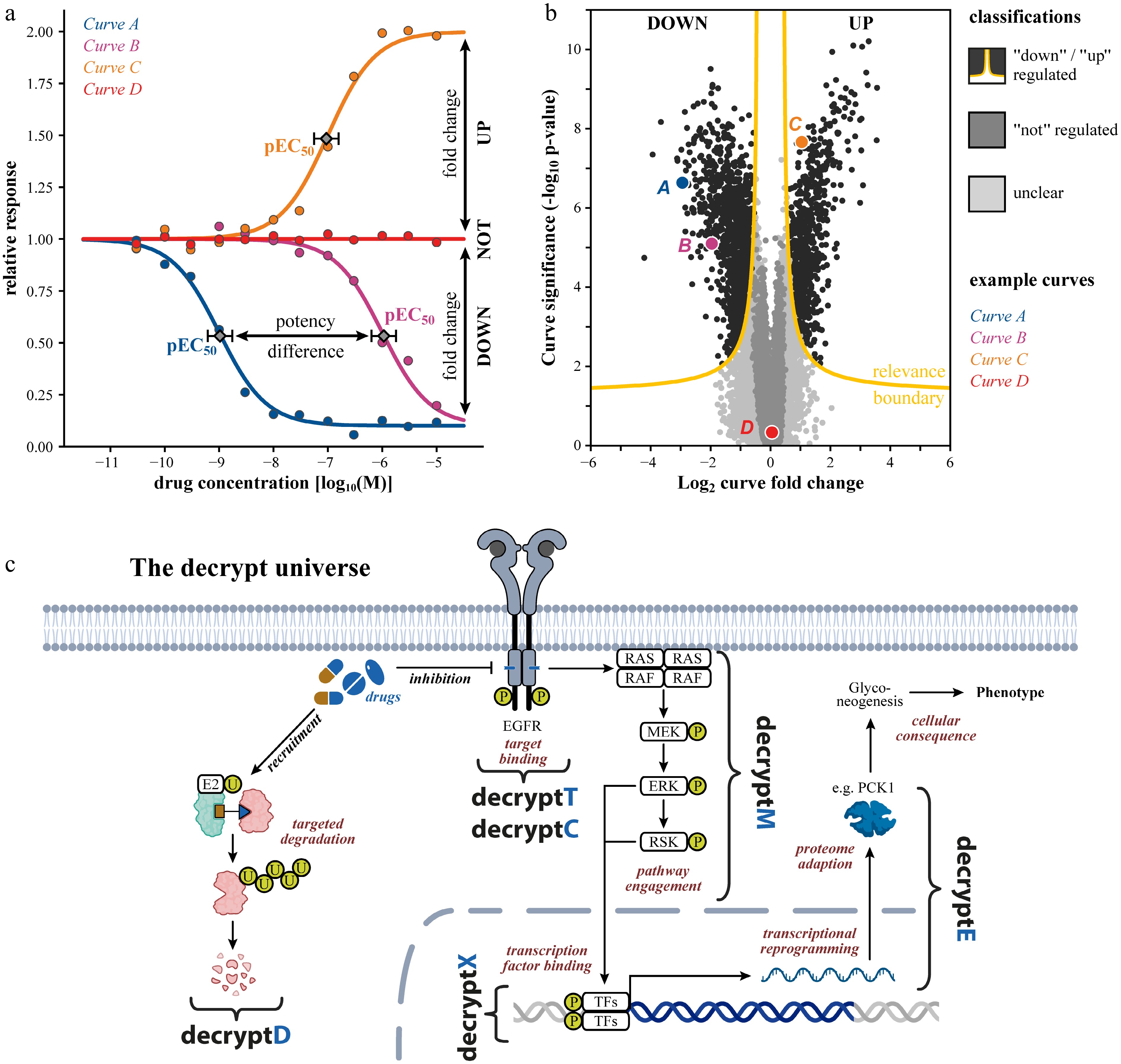
Figure 1.
Tracing drug mechanism of action by dose-dependent proteome- and PTM-wide measurements. (a) Schematic dose response curves illustrating extraction of pEC50: −log10 effective drug concentration to achieve 50% of the maximal effect as quantitative measures of target or pathway potency. (b) Example volcano plot of a typical omics dose-response dataset, where each data point is a dose-response curve. The relevance boundary (combination of fold change and alpha threshold) identifies down- and up-regulated responses with false discovery rate (FDR) control. (c) Schematic of how a drug's mechanism unfolds in a cell: target binding or degradation (decryptT/decryptC/decryptD), pathway engagement (decryptM/decryptX), proteome reprogramming (decryptE), and ultimately cellular consequences. TFs: transcription factors.
-
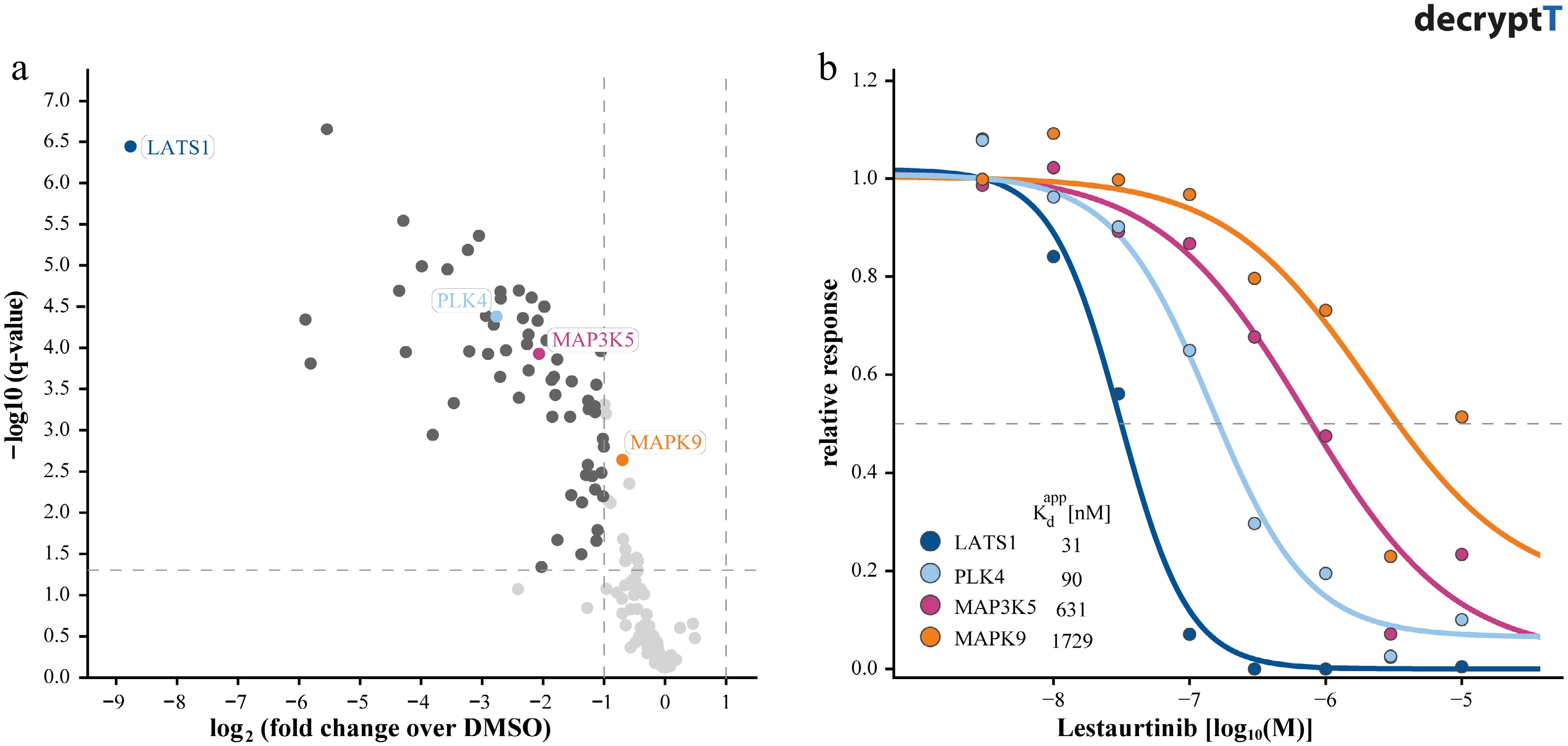
Figure 2.
Distinguishing targets of Lestaurtinib by dose-response competition affinity profiling. (a) Volcano plot showing kinases identified in replicate single-dose (1 µM) competition binding experiments using Kinobeads, illustrating limited selectivity resolution. (b) Corresponding dose-response curves (dose range, 0–30 µM) for selected kinases derived from Kinobead profiling, enabling quantification of apparent binding affinities (pKdapp: −log apparent dissociation constant).
-
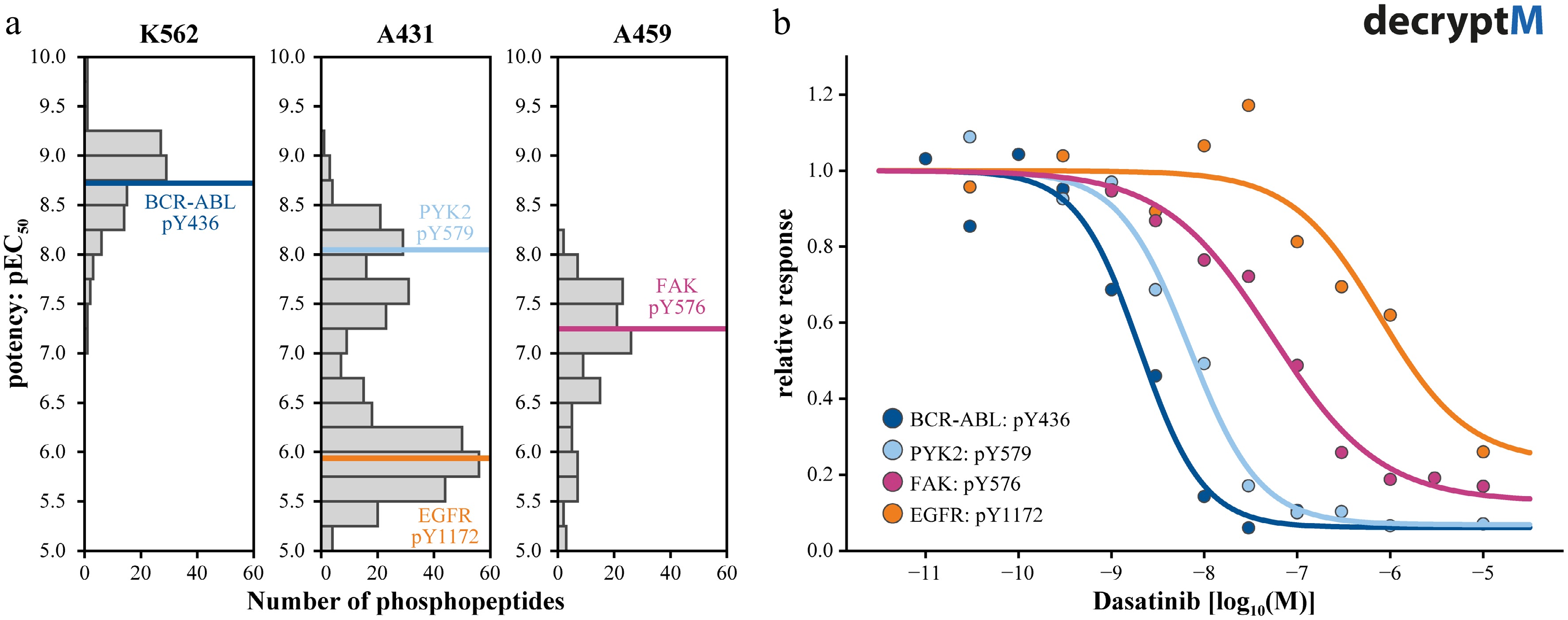
Figure 3.
Measuring pathway engagement by decryptM profiling. (a) Summary plots of the potencies by which phosphopeptides are regulated by Dasatinib showing that the polypharmacology of Dasatinib leads to different profiles in different cell lines. Phosphorylation sites on Datastinib target proteins are marked in color. (b) Dose-response curves of the four examples highlighted in panel (a), illustrating the different potencies with which Dasatinib inhibits its targets and, in turn, leading to different but matching potencies of kinase substrate phosphorylation.
-
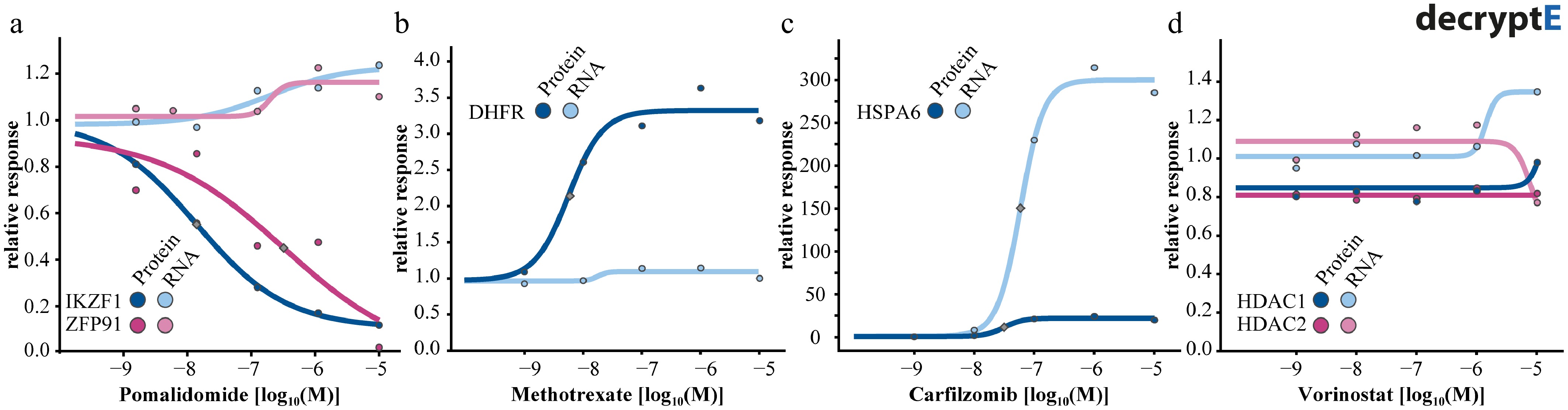
Figure 4.
Dose-response curves for proteins and their mRNA transcripts for (a) the degrader pomalidomide, (b) the DHFR inhibitor methotrexate, (c) the proteoasome inhibitor carfilzomib, and (d) the HDAC1-3 inhibitor vorinostat.
-
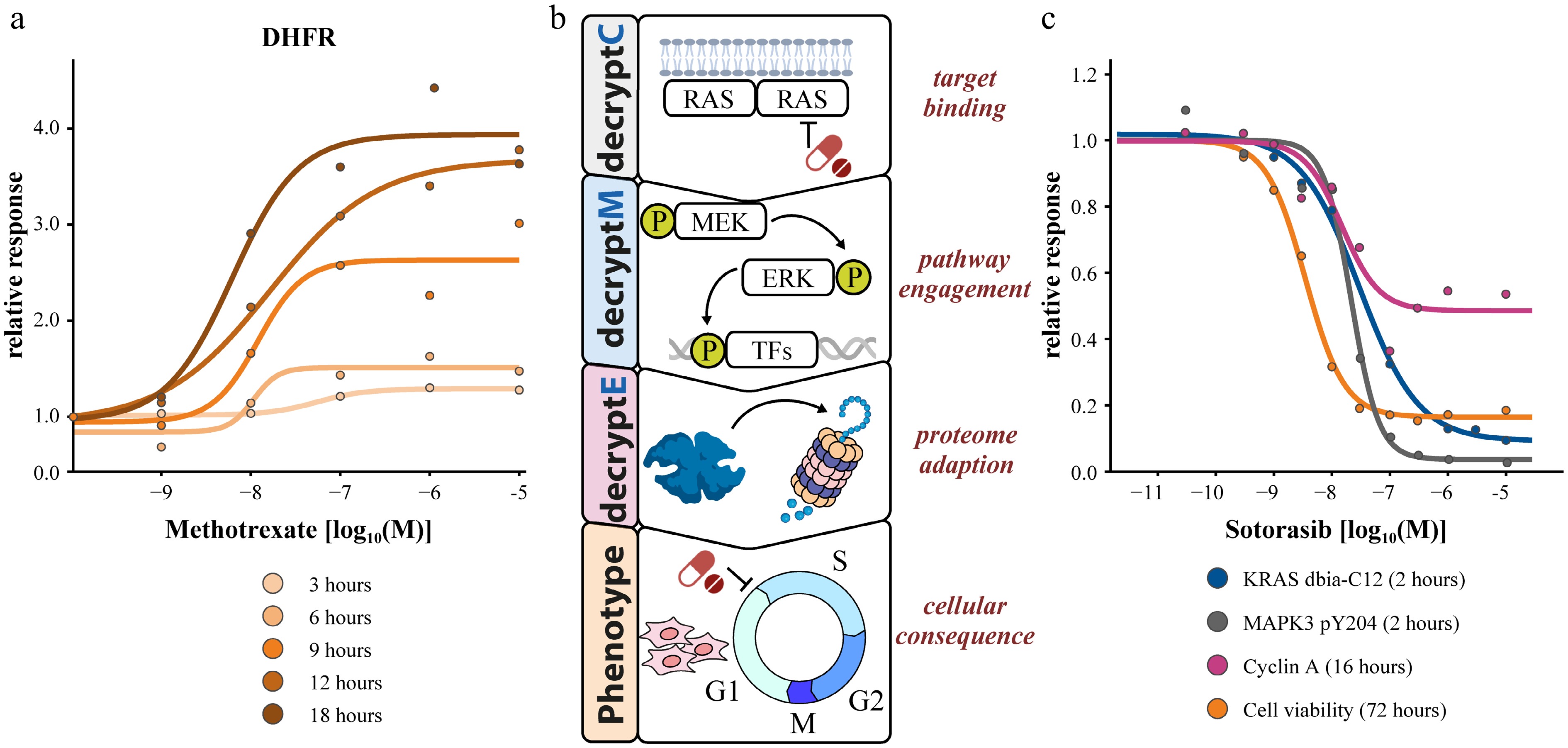
Figure 5.
Multi-dimensional dose-response measurements. (a) 2D time-dose coupling of DHFR abundance changes in response to methotrexate. (b) Multi-decrypt-dimension profiling of the KRAS inhibitor sotorasib revealing potency coherence at all levels of MoA elucidation.
-
Technique Examples What it's used for How DRC concept adds value Pros/cons and our view Ref. Target binding Affinity enrichment-based Immobilized probes/matrices (Kinobeads, HDAC beads, tailored resins) Capture drug-protein interactions in native proteome; Immobilized matrices enrich stable, high-affinity interactions; PAL uses UV-induced crosslinking to also
capture weak, transient interactions that could otherwise be lost during washes. Both enable identification of direct binders and selectivity profiling.Converts single pulldowns into quantitative DRC allow calculation of Kdapp, EC50; Filters out sticky or nonspecific binders; Reveals relative selectivity across isoforms, families, or complexes. Pros: Use of native lysates preserves PTMs, cofactors, complexes; broad family applicability (kinases, HDACs) or individual compounds; PAL extends to weak/transient binders; enrichment reduces proteome complexity.
Cons: Loses intact-cell context; Immobilization bias; PAL probe derivatization required; unselective photocrosslinking.
Recommendation: Use for quantitative target space mapping and selectivity profiling.[6,8,15,
29]Photoaffinity labeling (PAL) [12,13] Activity enrichment-based 1st-gen family-directed probes (e.g., serine hydrolases, DUBs, kinases) Covalent probes enable labeling of active enzymes or identification of drug targets. Competitive incubations reveal target engagement by blocking probe labeling. First-generation probes profile enzyme family activity, while second-generation approaches achieve proteome-wide reactivity and site-specific ligandability mapping. Single-dose competition assays can overestimate weak or nonspecific interactions, producing false-positives for ligandability. DRC enable quantitative extraction of IC50/Ki values, target occupancy, and relative selectivity across enzymes, nucleophile classes, or reactive cysteines, allowing rigorous ranking of cysteine reactivity and ligandability. Pros: Enables target deconvolution and covalent ligand discovery; isoTOP-ABPP provides site-specific, quantitative readouts. Some probes (e.g., kinase probes XO44) cell-permeable, allowing live cell profiling.
Cons: Probe chemistry and steric bias can influence labeling patterns; Bulky enrichment handles often limit cell permeability; requires well-designed electrophiles tailored to nucleophile classes.
Recommendation: Use for covalent drug profiling and site-specific ligandability mapping. The competition format is also valuable for confirming intracellular target engagement and deconvolution of covalent compounds.[44−46] 2nd-gen proteome-wide (e.g., DecryptC: cysteine-reactivity, isoTOP-ABPP, TRAP) [9−11,
47,48]Denaturation-based (thermal) 2D-TPP, ITDR,
conc-PISA, mTSAInfer drug-target engagement in living cells or lysates via changes in thermal stability and protein solubility after ligand binding. In living cells, PTMs and PPIs influence thermal stability; dose-response curves clarify dose-dependent stabilization, reduce false positives, and rank direct vs indirect targets. Pros: Applicable in intact cells; no compound modification required; Detects diverse binding modes, including allosteric interactions; Dose-response analysis enables ranking and hit prioritization beyond binary hit-calling.
Cons: 2D-TPP requires large sample numbers (dose x temperature); Condensed TPP/iTSA are higher throughput but less sensitive; Indirect effects complicate interpretation; Not all proteins show measurable thermal shifts (false negatives); PTMs and PPIs can cause apparent stability changes (false positives), though DRC mitigates this.
Recommendation: Use to confirm intracellular target engagement, ideally in combination with orthogonal approaches for higher confidence.[18−20,
23,29]Denaturation-based (chemical) iSPP, solvent-induced precipitation (ethanol, acetone), chaotropic/ kosmotropic agents (urea, guanidinium), pH shifts Used to probe protein stability, folding, and ligand binding, particularly useful for thermolabile proteins or systems where temperature-based denaturation is unsuitable. Dose-dependent stabilization reveals specific, saturable ligand binding, distinguishing true from indirect targets. It enables quantitative estimation of apparent affinity (EC50/
pEC50), supports target ranking by stabilization potency, and reduces false positives from indirect or nonspecific effects.Pros: Suitable for proteins and complexes sensitive to heat; reveals ligand-induced stabilization or destabilization; flexible across solvents, salts, and pH, compatible with DRC analysis.
Cons: Can have reduced proteome coverage for very insoluble or membrane-associated proteins; depends on chemical buffer composition.
Recommendation: Use as a complementary approach to thermal profiling when heat is unsuitable, particularly for ligand screening or dose-response target identification.[22−27] Conformational/Accessibility-based Lip-Quant, PELSA Detects ligand-induced structural changes via altered protease or chemical reactivity, supporting target validation and binding-site mapping. DRC analysis confirms ligand-induced protection, ranks stabilization, maps binding sites, and limits indirect false positives. Pros: Can provide site-specific binding information.
Cons: Not all compounds induce structural changes that alter the accessibility of the protein, resulting in false negatives.
Recommendation: Use for precise binding site mapping and target validation, ideally in combination with orthogonal methods to confirm hits and mitigate the risk of missed interactions.[32,33,
49]Pathway modulation/consequence of binding DecryptM Phosphorylation, acetylation, or ubiquitinylation Measures dose-dependent PTM responses to reveal pathway engagement and cellular mechanisms of drug on/off-target activity. Enables separation of direct and indirect effects, clusters PTM sites by shared potency (EC50) to reveal pathway wiring and compares pathway engagement across compounds or cell types. Pros: Provides systems-level resolution of downstream signaling; distinguishes multiple pathways engaged by a drug; links potency information to pharmacological effects; available across different PTMs.
Cons: interpretation can be complicated by parallel pathways or cell-type context.
Recommendation: Use to gain mechanistic insight beyond direct targets, connecting drug binding to functional signaling outcomes. Best applied over multiple doses and, ideally, across different cell models.[1,37−
40]Cellular consequence/adaptation/death DecryptE Drug-induced proteome abundance changes Measures dose-dependent proteome changes to reveal how drugs remodel proteostasis through direct and adaptive effects. Differentiates rapid abundance changes linked to direct target effects from slower, adaptive reprogramming; allows quantification of potency windows; distinguishes primary MoA from secondary downstream effects; and helps correlate molecular changes with phenotypic outcomes. Pros: Provides a global view of proteome remodeling with broad coverage and quantifiable EC50 values, uncovering unexpected drug effects beyond known targets.
Cons: Expression changes are often indirect and slow, complicating mechanistic interpretation; requires careful temporal design.
Recommendation: Use to map systems-level consequences of drug treatment, compare potency windows across pathways and phenotypes, and identify adaptive mechanisms, ideally in combination with faster-acting assays like phosphoproteomics for mechanistic resolution.[2] Phenotypic Cell viability, apoptosis, differentiation, cell cycle arrest, metabolic activity assays Measures the functional consequences of drug treatment on cellular phenotypes across a range of concentrations, providing insight into efficacy, toxicity, and therapeutic windows. Resolves the concentrations at which phenotypic changes arise relative to molecular target engagement, distinguishing direct drug effects from downstream or off-target consequences. Enables correlation of molecular EC50 values with phenotypic outcomes and informs safe, effective dosing ranges. Pros: Directly links molecular perturbations to cellular outcomes; enables ranking of compounds by potency and efficacy; provides translationally relevant data for therapeutic assessment.
Cons: Often integrate multiple direct and indirect mechanisms, complicating mechanistic interpretation; slower phenotypic responses may lag behind molecular changes; high variability may require multiple replicates and careful experimental design.
Recommendation: Use in combination with molecular profiling (e.g., decryptM or decryptE) to map drug potency, mechanism, and therapeutic window, and to validate that molecular engagement translates into functional cellular effects.[2,39,
42,50]Table 1.
Collection of experimental approaches probing each layer of drug mode of action including their advantages, limitations, and the added value of dose-response analysis.
Figures
(5)
Tables
(1)