-
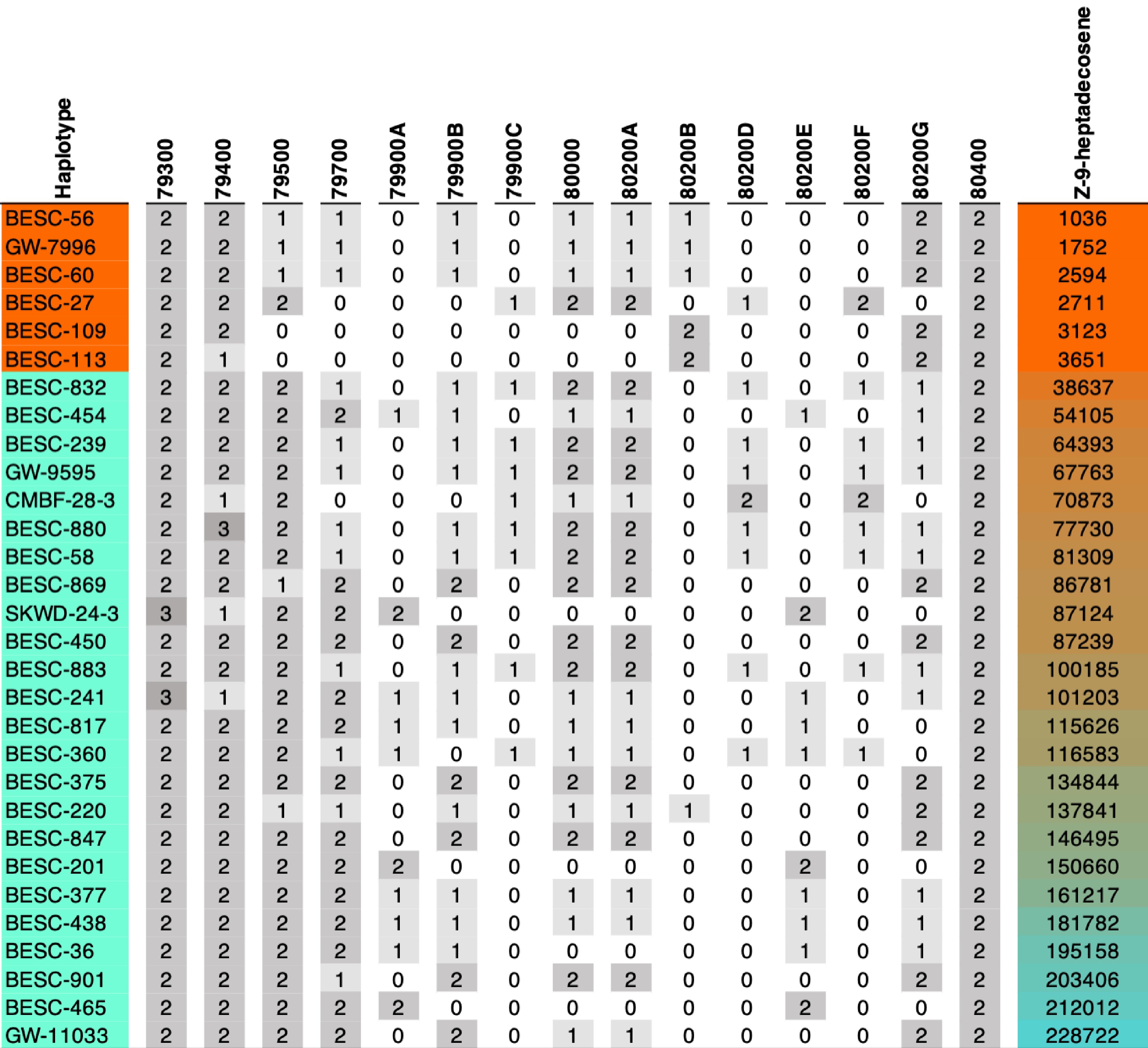
Figure 1.
KCS gene copy counts for each genotype and their alkene levels colored by mean Z-9-heptadecosene amount. The first six genotypes are alkene minus (AM) accessions. The gray heatmap indicates copy numbers for genes listed as columns. A more detailed breakdown by differing amino acids is also provided in the Supplementary Table S2a.
-
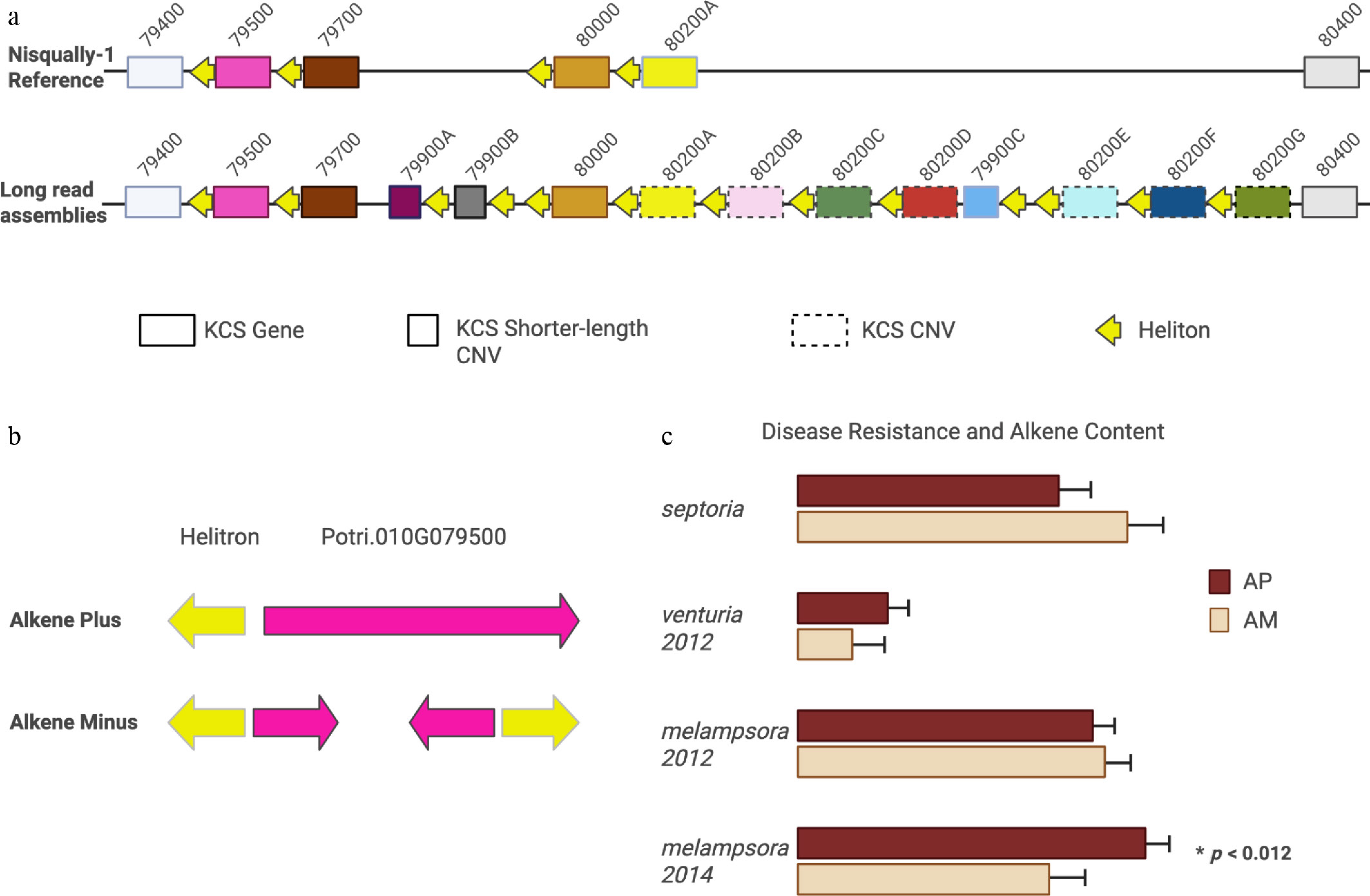
Figure 2.
Comparison of our results to published reports of the KCS locus. (a) KCS genes in the Nisqually-1 reference and the new 78 haploid assemblies. Boxes with solid edges are genes (same position across genomes), boxes with broken edges are copy number variants (different positions across genomes), and short boxes represent shorter-length KCS variants with intact open reading frames. Nomenclature corresponds to Nisqually-1 reference. Yellow arrows indicate Helitrons with archetypal CTAG palindrome. Note, each gene is paired with a Helitron. (b) Gene model for Potri.010G079500 in alkene-minus (AM) and alkene plus (AP) haplotypes. In AM individuals, the 5' end of Potri.010G079500 was inverted and placed on the reverse strand, deleting portions of the coding sequence, confirming the prediction of Gonzales-Vigil et al. (c) Differences in disease severity in AM and AP phenotypes. Contrary to Gonzales-Vigil et al.[3] we did not find differences in S. musiva (Septoria) severity but did find decreased severity of Melampsora in 2014 (p < 0.012) but not in 2012.
-
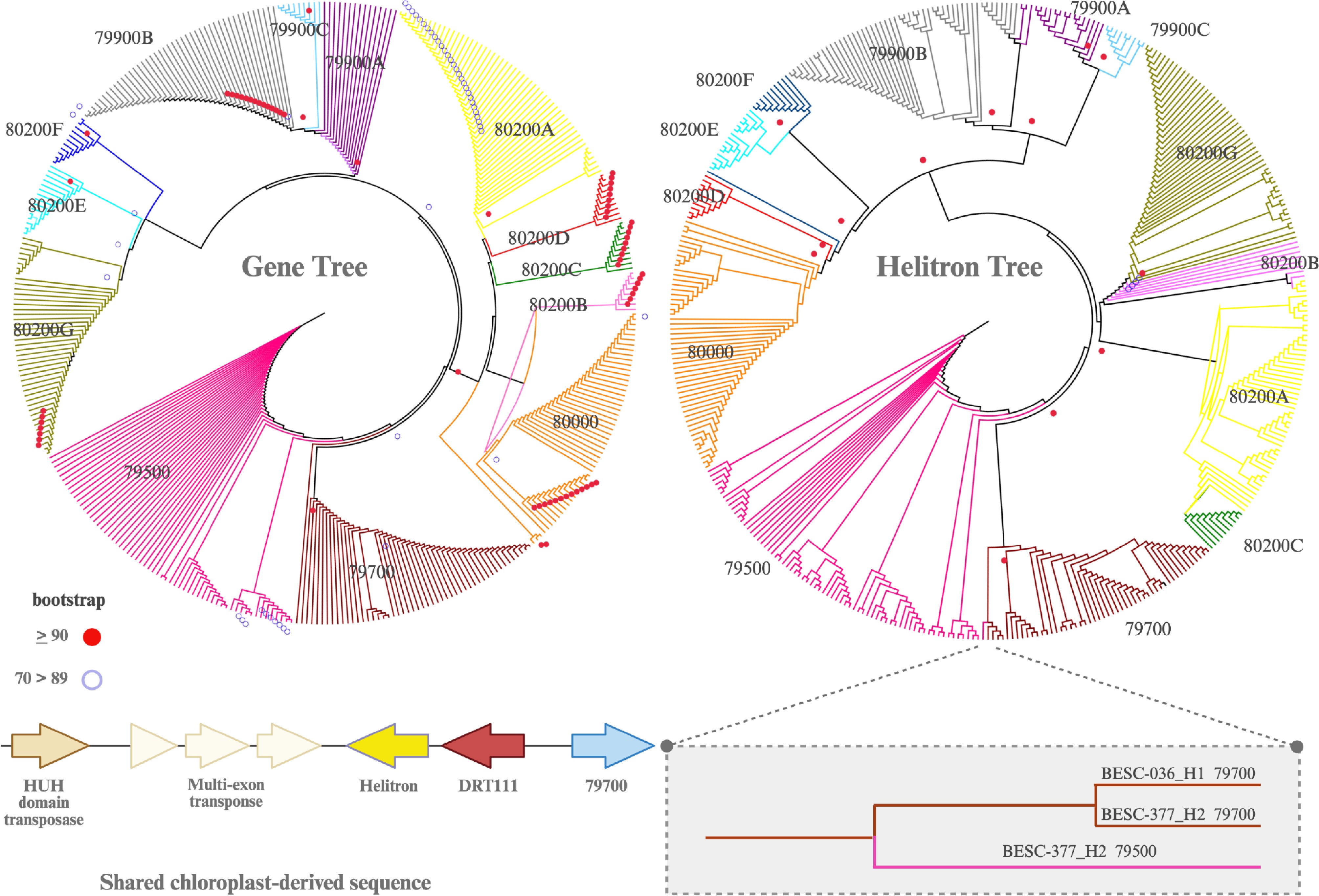
Figure 3.
Neighbor-joining (NJ) trees based on exons of the genes and copy number variants (left) and their paired Helitrons (right). Colors correspond to those in Fig. 1a. Helitron sequences are mostly conserved, and the topology is congruent with the exon-based tree, with one major discrepancy (inset box). The coding sequences of Potri.010G079500 and Potri.010G079700 in 'BESC-377' vary, even though their Helitrons are nearly identical. The sequences shared between Potri.010G079500 and Potri.010G079700 are unique in that they both harbor a chloroplast-derived gene (DRT111) and two transposases. This DRT111-transposase sequence is found in all haplotypes with a full-length Potri.010G079700 gene. In BESC-377_H2 it also appears upstream from Potri.010G079500, suggesting a shared evolutionary history. Bootstrap values for nodes greater than 90 and between 70 and 90 are shown.
-
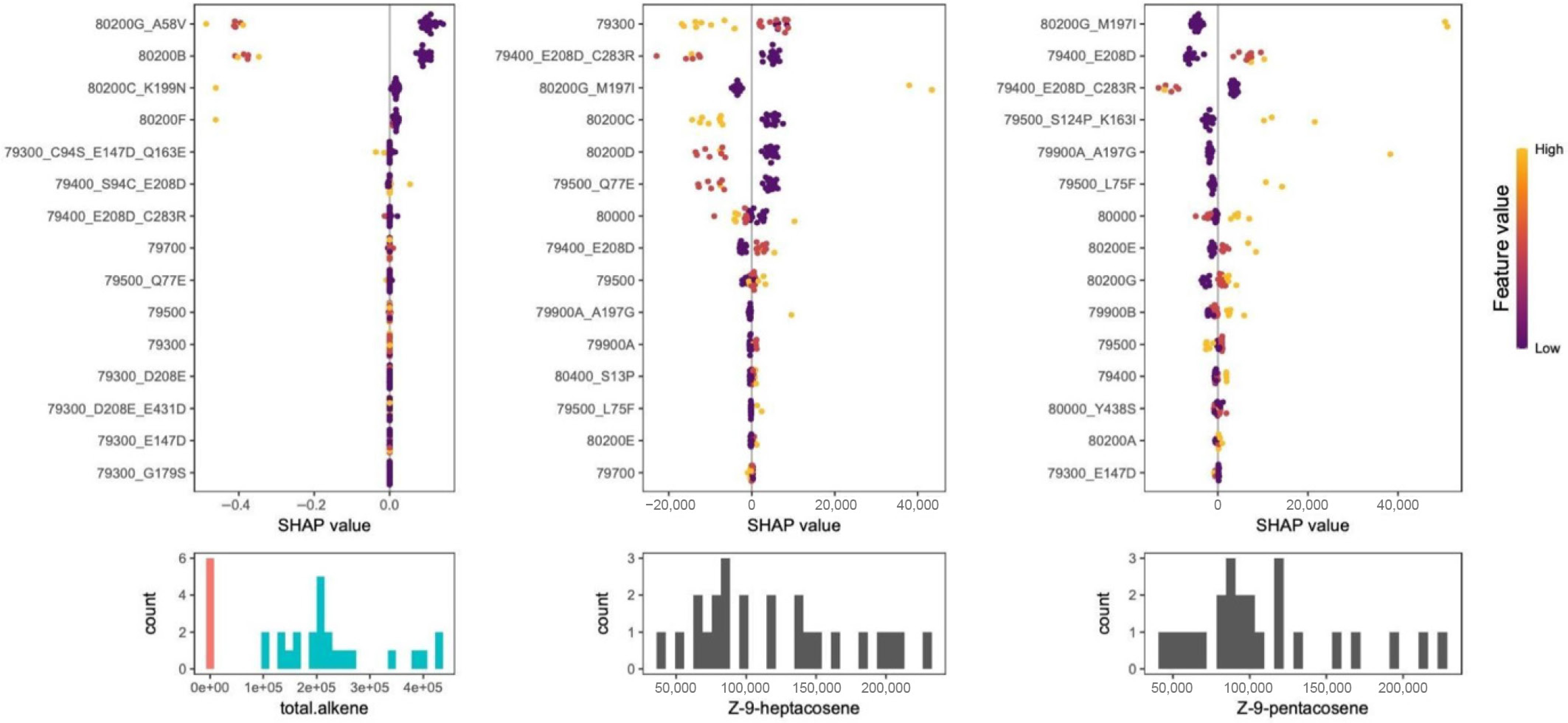
Figure 4.
Explainable AI analysis of iterative Random Forest (iRF) models for different alkene phenotypes. The y-axis represents the SHAP value of each individual gene or copy number variant. Each filled circle represents an individual and the color represents the feature values, which is the number (dosage) of that gene or CNV for that individual. Purple indicates 0 alleles and yellow is the maximum number of alleles for a gene variant (usually two but can be one). Histograms below each SHAP plot depict the phenotype's distribution and SHAP values represent the tendency for that feature-allele combination to drive the phenotype prediction for an individual towards either low (left side) or high (right side) values. The model to predict alkene deficient (AM, red bars) vs alkene plus (AP, blue bars) phenotypes indicate 80200B and 80200G_A58V are the most accurate predictors for predicting AM or AP class (left plot), where non-zero dosage (1-red or 2-yellow) strongly drives individuals towards the AM class. The models that predict heptacosene (center) and pentacosene (right) identify a variant of the type 2 KCS protein Potri.010G079400 as important (E208D) for both, but addition of the C283R mutation decreases these alkenes. The presence of Potri.010G080000 has opposing effects on the two alkene types and different variants of the Potri.010G079500 are important for predicting pentacosene levels but not heptacosene.
-
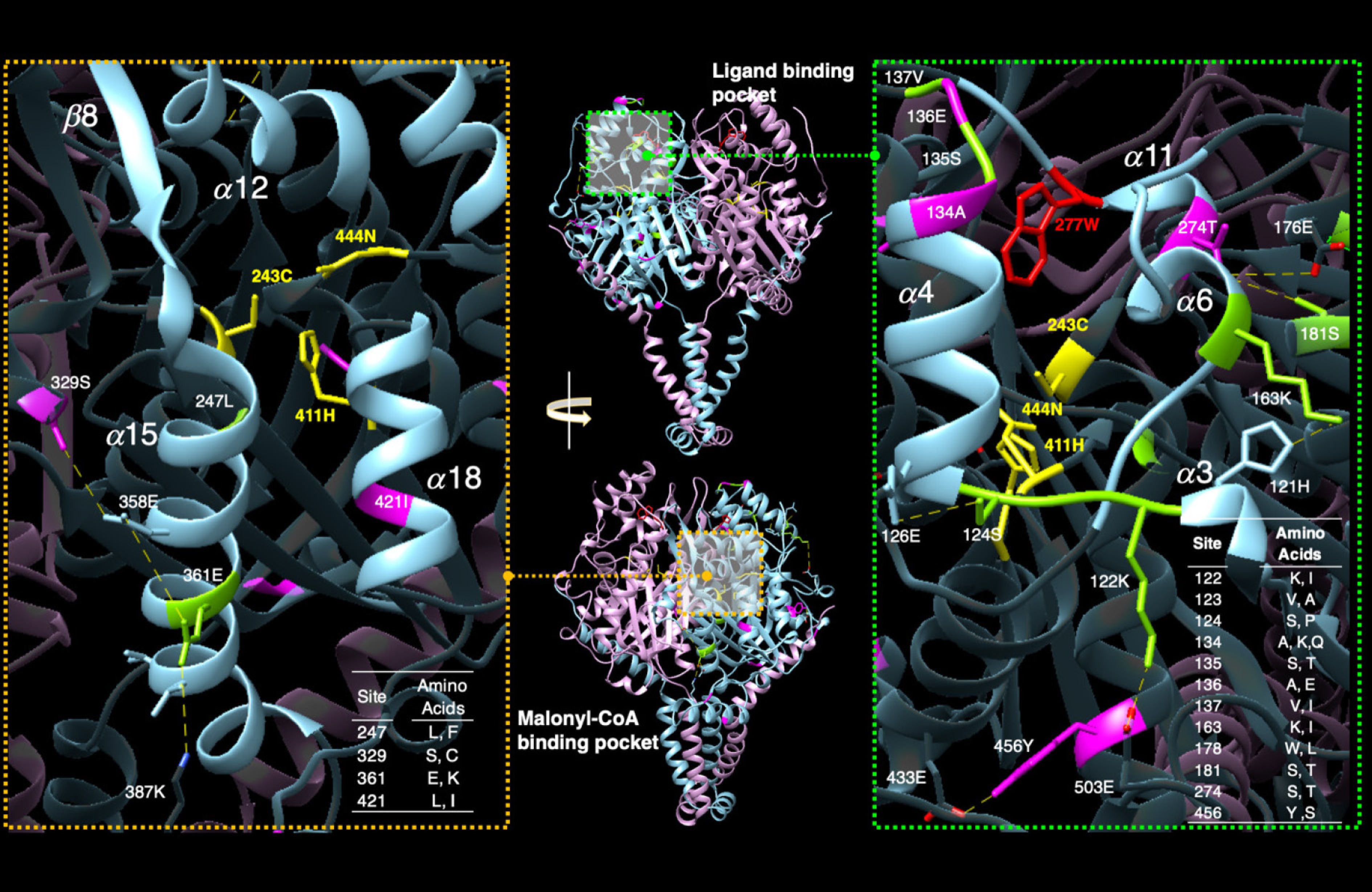
Figure 5.
Amino acid substitutions overlaid on the AlphaFold predicted structure for Potri.010G080200. Previous work identified the binding pockets for the ligand (top, center panel) and the two-carbon donor malonyl-CoA (bottom, center panel). Substitutions among KCS variants in general (green residues in left and right panel) and those specific to the Potri.010G080200 copies (pink residues left and right panels), cluster around the ligand binding pocket (magnified view with light blue coloring, right panel). The tryptophan at position 277 (as shown in red in the right panel) is necessary for function, active Cys-His-Asn triad (in yellow). Substitutions are also found in the malonyl-CoA binding pocket (magnified in the left panel). Changes are predicted to alter the hydrophobicity and shape of the region and likely affect ligand binding/activity (malonyl-CoA binding).
-
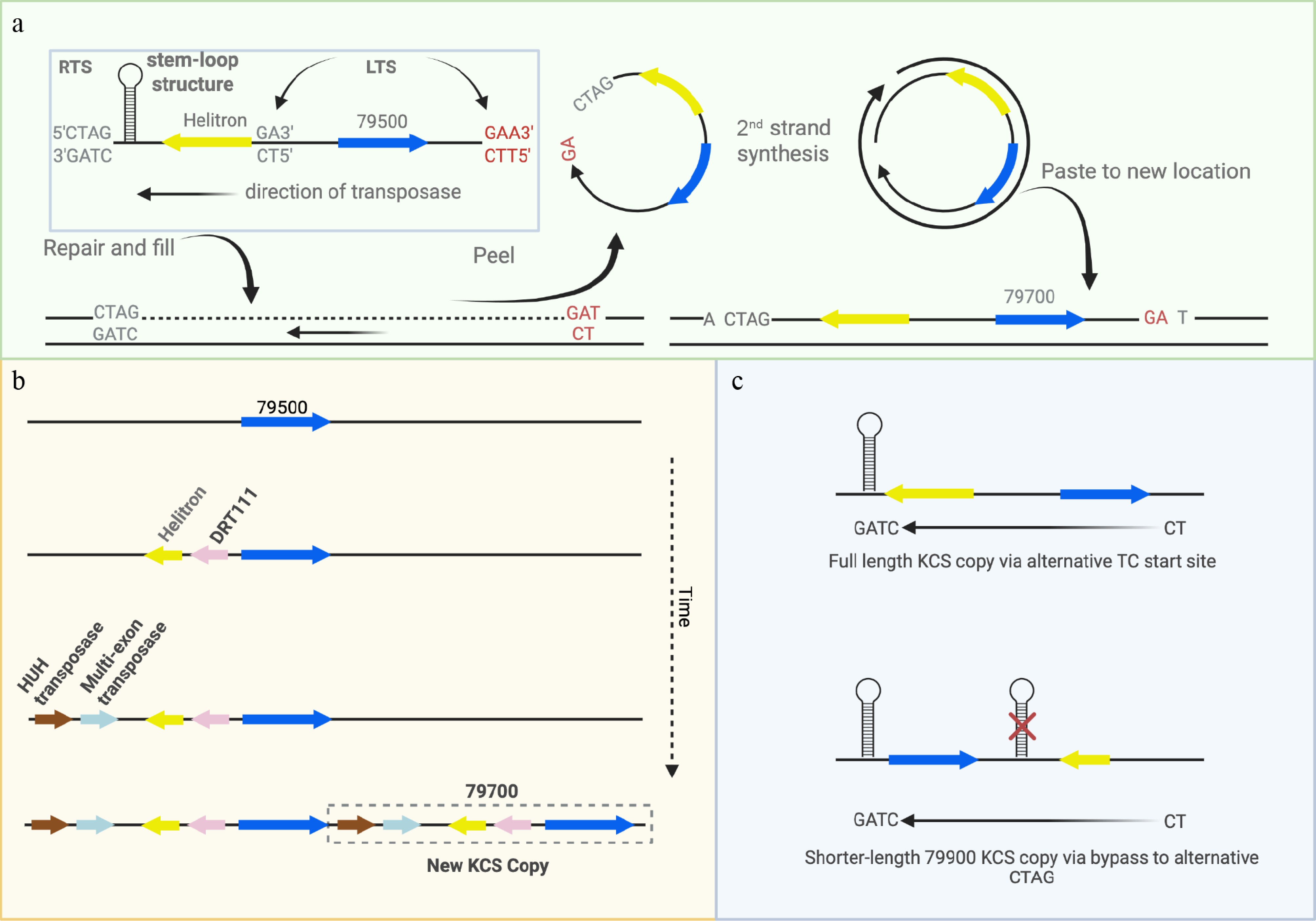
Figure 6.
Model of possible mechanism of Helitron-driven copy number variation. (a) Helitrons have a unique peel-and-paste method of transposition in which a rolling circle is generated from a "TC" left terminal sequence (LTS) and a "CTAG" right terminal sequence (RTS) and inserted between an AT target nucleotide pair, with no duplications at the end (note KCS Helitrons are on the negative strand so LTS and RTS are reversed). The single strand from which it was derived is restored, retaining the original gene. Nearly all Helitrons harbor a 'T' upstream from the 'CTAG' site, and an 'ATC' just downstream from its KCS paired gene (Supplementary Tables S7, S8a, S8b), as would be expected if they were inserted as Helitrons (potential alternative LTS shown in red). (b) Hypothetical evolutionary model of copy number generation via Helitrons. Given the shared Helitron between Potri.010G79500 and Potri.010G079700 in BESC-377_H2 and the presence of that Helitron in all Potri.010G79700 sequences, we propose that Potri.010G079500 represents the origin of KCS copies. Once the transposase machinery and the DRT111 gene were inserted proximal to the Helitron in Potri.010G79500 it was able to replicate itself and generate Potri.010G79700. (c) For the shorter-length KCS genes 79900A, 79900B and 79900C, the Helitron is downstream, rather than upstream and therefore a model consistent with Helitron replication would require a bypass of the original CTAG site.
Figures
(6)
Tables
(0)