









-
Glucosamine (2-amino-2-deoxy-d-glucose, GlcN), an important amino sugar, which is found in almost all organisms, including bacteria, fungi, and animals, with shrimp and crab shells being particularly rich in this compound[1]. GlcN has a wide range of applications in the biomedical, food, and cosmetic industries[2, 3]. This compound is believed to be a beneficial food supplement for the treatment of osteoarthritis by aiding cartilage growth and repair, and therefore GlcN has become a popular non-prescription nutritional supplement with a currently estimated market volume of
${\$}$ ${\$}$ -
All reagents and chemicals were used as received from commercial suppliers without further purification or modification. Silica gel TLC plates were obtained from Merck KG (Type 60 F254, Darmstadt, Germany). Nickel-nitrilotriacetic acid Sefinose Resin was purchased from BBI Life Sciences (Shanghai, China). NMR spectra were generated on a Bruker AV-400 instrument using the residual solvent signal as the internal standard. Chromatographic analyses were performed by a Shimadzu LCMS 2020 system (Shimadzu Corporation, Kyoto, Japan).
Synthesis of the fluorogenic glucosamine derivative
-
Glucosamine hydrochloride (539 mg, 2.5 mmol), 1,3-diphenyl-1,3-propanedione (DPPD, 560 mg, 2.5 mmol), sodium bicarbonate (42 mg, 0.5 mmol) and 5 mL of methanol (MeOH)/H2O (3:1) were combined and mixed in a 50 mL sealable glass tube. The tube was heated to 110 °C for 17 h. The reaction mixture was allowed to cool to room temperature and dried under reduced pressure. The solid residue was dissolved in MeOH and mixed with 1 g of silica, then dried again. The mixture was loaded onto the silica column, which was then eluted with MeOH/CHCl3 (5:95). All fractions were analyzed by thin-layer chromatography (TLC, 1:9, MeOH/CHCl3) at 254 nm and 362 nm, and fractions containing the fluorescent product (Rf = 0.7) were pooled together and lyophilized. The dry samples were re-dissolved in H2O and purified on reversed-phase C18 SPE cartridges (Supelco-5706, 500 mg bed weight). The C18 reversed-phase cartridges were preconditioned with acetonitrile (ACN) and equilibrated with water. The samples were applied to the cartridges followed by washing with water, and the cartridges were successively eluted with water (10:90), ACN/water (50:50), and ACN. Fractions were analyzed by TLC, and the fluorescent products (Rf = 0.7) were pooled together and lyophilized (yielding 8 mg).
1H NMR (400 MHz, MeOD): δ 2.16 (1H, dd, J 14, 2, H5´´(Z)), 2.28 (1H, dd, J 13.2, 6, H5´(E)), 2.44−2.56 (1H, m, H5´(Z)) 2.48 (2H, dd, J 13.6, 6, H5´´(E)), 3.63-3.80 (2H, m, H8´, H8´´), 4.17 (1H, app q, J 4.8, H7(Z)), 4.30 (1H, td, J 4.8, 2.4, H7(E)), 4.47 (1H, td, J 6, 4.8, H6(Z)), 4.54 (1H, dt, J 6, 2.4, H4(E)), 5.48 (1H, s, H4(Z)), 5.53 (1H, s, H4(E)), 7.60−7.54 (2H, m, H2), 7.68−7.62 (1H, m, H1), 7.88−7.84 (2H, m, H3).
13C NMR (100 MHz, MeOD): δ 203.64 (C8(Z)), 202.60 (C8(E)); 178.58 (C5(Z)), 177.91 (C5(E)); 134.20 (C1); 134.16 (C4); 130.21 (C2); 128.63 (C3); 97.25 (C7(E)), 96.42 (C7(Z)); 93.43 (C6(E)), 92.86 (C6(Z)); 90.99 (C11(E)), 89.66 (C11(Z)); 73.90 (C10(E)), 73.14 (C10(Z)); 63.91 (C12(E)), 63.20 (C12(Z)); 44.41 (C9(E)), 42.20 (C9(Z)).
HPLC analysis
-
Chromatographic analyses were carried out using a Shimadzu LCMS 2020 system (Shimadzu Corporation, Kyoto, Japan) consisting of an LC-30AD pump equipped with a low-pressure gradient mixing unit, a SIL-30AC autosampler, an RF-20Axs fluorescence detector (excitation 362 nm, emission 450 nm), and an ESI mass spectrometer. The analytes were separated on a reversed phase HPLC column (Phenomenex Hyperclone 5 μm ODS 120 Å, 250 × 4.6 mm). The mobile phases were NH4COOH (pH 4.5, 50 mM) in water and acetonitrile for solvents A and B, respectively. The flow rate was 1.5 mL/min. After injection of 5 μL of the sample, a linear gradient of 12%−45% B was applied from 0 to 3 min; B was then increased to 95% over 1 min and held at 95% for 2 min. B was then decreased to 12% over 1 min, and the column was equilibrated with the initial conditions for 3 min.
Plasmid construction, expression, and purification of GPDA
-
A putative glucosamine-6-phosphate deaminase gene from the bacterium Enterococcus canintestini (GenBank Accession Number WP_071864541.1) containing Nde I and Xho I restriction site overhangs was synthesized and ligated on the pET30a vector by Tsingke Biotechnology Company (Nanjing, China). The constructed expression vector was transformed into Escherichia coli BL21 (DE3) competent cells and plated on lysogeny broth (LB) agar containing 50 µg/mL kanamycin. A single colony was transferred into a 2 L Erlenmeyer shaking flask containing 400 mL of LB medium and shaken at 37 °C until the culture density reached an absorbance at λ = 600 nm of 0.5. The final concentration of 1 mM isopropyl 1-thio-β-D-galactopyranoside (IPTG) was added to start the recombinant protein expression. After 20 h of induction at 18 °C, the biomass of the fermentation culture was pelleted by centrifugation at 5,000 g for 20 min. The cell pellet was re-suspended in 10 mL of lysis buffer (100 mM NaCl, 50 mM Tris, 1% (w/v) Triton X-100, 1 mM PMSF, adjusted to pH 8.0) and sonicated for 20 min. The cell homogenate was cleared by centrifugation (20 min at 12,000 g) and then the supernatant was subject to nickel-nitrilotriacetic acid (Ni-NTA) column chromatography (5 mL of bed volume). Unspecifically bound proteins were washed off the Ni-NTA column with 150 mL of WB-solution (consisting of 50 mM NaCl, and 50 mM Tris, adjusted to pH 8.0 with HCl), and the target protein was eluted with 10 mL of EB-solution (WB-solution containing 500 mM imidazole). The purity and quantity of recombinant GPDA was estimated by SDS-PAGE, and samples were subsequently stored at −80 °C for further experiments.
GPDA mutant library design
-
Two different approaches were taken for the generation of mutant libraries. The first approach was to generate a randomized GPDA mutant library by error-prone PCR based on the method described by Lin-Goerke et al.[22] In brief, the pet30a-GPDA plasmid (10 ng/µl) was used as a template for amplification of the PCR product using the T7 forward/reverse primers (Supplemental Table S1). The 2×Taq Polymerase Master Mix (Vazyme, Nanjing, China) was supplemented with 0.8 mM of additional dCTP/dTTP, and 100 µM MnCl2, which resulted in an error rate of ~1 mutation/700 base pairs.
The second approach was to generate focused[23] GPDA mutant libraries through the simultaneous randomization of three conserved amino acids adjacent to the substrate binding pocket of the NagB/GlcN6P crystal complex from S. mutans (PDB accession 2RI1)[24]. These amino acids were then randomized with the QuickChange XL site-directed mutagenesis protocol (Stratagene) by replacing the three conserved amino acid codon triplets with an 'NDTNDTNDT' nucleotide sequence using primer pairs bearing this randomized sequence motif (Supplemental Table S1). The mutated plasmids were verified by DNA sequencing and transformed into Escherichia coli BL21 (DE3) competent cells for performing the in-solution and colony screening assays.
GPDA mutant screen
-
The screening assays for GPDA precipitation mutants were conducted with both liquid and membrane-based screening methods. For the liquid screening assay, a modified M9 minimal growth medium which was sterilized and prepared according to the description by Elbing & Brent[25] consisted of Na2HPO4•12H2O (17.1 g/L), KH2PO4 (3 g/L), NaCl (0.5 g/L), NH4Cl (1 g/L), MgSO4 (240 mg/L, heat-sterilized separately), CaCl2 (11.1 mg/L, heat-sterilized separately), isopropyl thio-β-galactoside (IPTG, 23.8 mg/L, filter-sterilized separately), and glucosamine (5 g/L, filter-sterilized separately). Five mL of minimum growth medium were transferred into glass vials and 5 µL of each E. coli Bl21 (DE3) mutant library were shaken for 12 h at 37 °C. Then, 20 µL of each culture (OD600 < 0.05) were transferred into fresh vials containing 5 mL of minimum medium and shaken for another 12 h at 37 °C. This procedure was repeated seven times, with passages 5−7 showing an obvious cell growth after the 12 h incubation (OD600 > 0.5). From each final incubation vial, 200 µL of 10−4-dilutions were transferred onto LB agar plates (9 cm diameter), which resulted after 16 h incubation at 37 °C in 400−600 colonies/plate. Three colonies from each library were picked for identifying the selected mutants by Sanger sequencing.
For the membrane-based GPDA precipitation screen, 200 µL of 10−4-dilutions of each mutant libraries were incubated on circular mixed cellulose ester (MCE) membranes (0.1 mm thickness, 0.45 μm pore size) located on LB agar plates (9 cm diameter). After 6 h incubation at 37 °C, the MCE membranes were transferred onto 9 cm petri dishes containing solid minimum medium (medium supplemented with 10 g/L agarose and 71.4 mg/L IPTG) and incubated for 16 h at 37 °C. The colonies were then visualized using a solidified MTT reaction mixture in 9 cm petri dishes (consisting of 5 mg MTT (3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium bromide) and 100 mg agarose in 10 mL of phosphate-buffered saline solution). Dark enlarged colonies were selected for mutational analysis by Sanger sequencing.
GPDA mutant variant characterization and protein modeling
-
Selected GPDA variants were expressed in recombinant form and purified as described above. The purified GPDA variants (typically 40 µL) were then incubated with 40 µL of a 100 mM GlcN solution, and the protein precipitation was monitored at 340 nm using the method described by Shmueli et al.[26] using a 384-well-microplate reader (Thermo Multiskan). The three GPDA mutant variants which showed the highest precipitation rate were then further tested for their precipitation behavior when exposed to glycine or a series of other carbohydrates (glucose, N-acetylglucosamine, mannose, galactose, fructose, 50 mM final concentration). In addition, protein precipitation was also analyzed at different GlcN concentrations (final GlcN concentration between 1 mM and 200 mM). The structural models of the apo-forms of the GPDA wild-type and mutant variants were generated using the AlphaFold tool of the ChimeraX software package (Version 1.5rc)[27]. GlcN was modeled into the active site of the GPDA variants using the molecular scaffold of glucosamine-6-phosphate shown in complex with PDB 2RI1[24] and replacing the phosphate moiety with a hydroxyl group in a similar manner as previously described by Hu et al.[28] GPDA variants were overlayed using the MatchMaker tool of the Chimera software package (Version 1.16)[29].
-
Our research group reported previously an efficient strategy for the labeling of mono- and oligosaccharides containing 2-hydroxyl groups using non-fluorescent 1,3-di(2-pyridyl)-1,3-propanedione as the derivatization reagent[30,31]. These works allowed the synthesis of a fluorescent 2-pyridylfuran derivative via Knoevenagel condensation between 1,3-di(2-pyridyl)-1,3-propanedione and the reducing end of a carbohydrate followed by intramolecular oxa-Michael cyclization. Applying similar reaction conditions as reported before, the treatment of GlcN with 1,3-diphenyl-1,3-propanedione (DPPD) in the presence of sodium hydrogen carbonate led to highly fluorogenic cis/trans isomers (compounds 1a and 1b, Fig. 1a). Thin-layer chromatography (TLC) revealed these compounds 1a and 1b as a single spot when the plate was irradiated with UV light (Fig. 1b). The synthesized compounds were also observed by HPLC analysis using a fluorometric detector (retention time: 4.2 min, Fig. 1c), confirming the fluorescent nature of the glucosamine derivative. As shown in Supplemental Fig. S1, HPLC-based analysis of the obtained compound demonstrated excitation and emission maxima at wavelengths of 362 nm and 450 nm, respectively. Interestingly, the excitation wavelength appears in the UV whereas the emission wavelength is inside the visible light, producing a violet/blue color. The unusually large difference between recorded excitation and emission wavelengths, 88 nm, makes this compound a fascinating small dye.
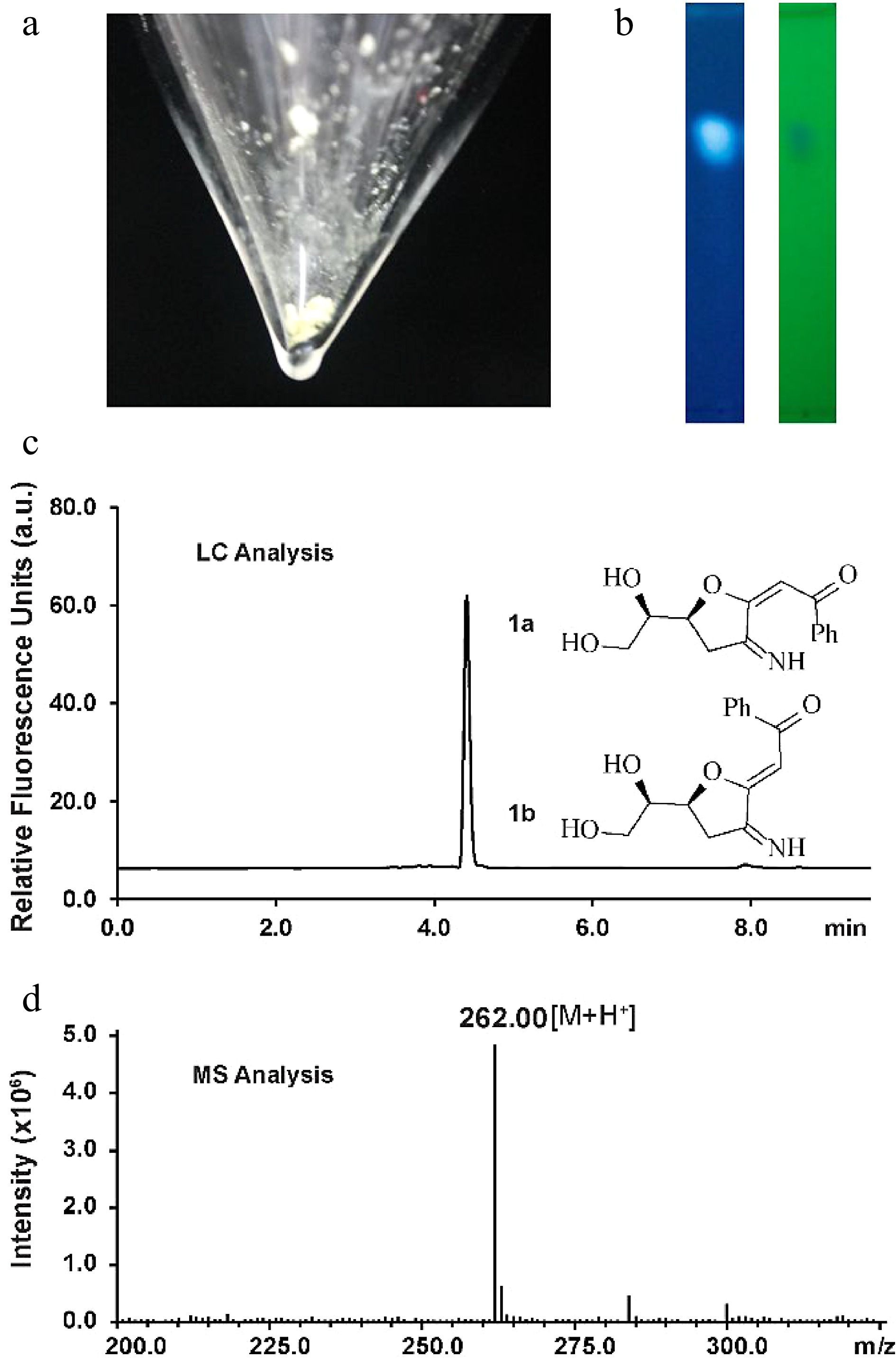
Figure 1.
Characterization of the fluorescent glucosamine derivative. (a) Isolated compound 1a and 1b mixture after silica-gel chromatography purification. (b) TLC analysis by irradiation with UV light (left: 362 nm, right: 254 nm). (c) HPLC chromatogram of compounds 1a and 1b (excitation 362 nm, emission 450 nm). (d) Positive ion electrospray-ionization (ESI) mass spectrum of compounds 1a and 1b.
Silica-gel chromatography purification allowed the isolation of the fluorescent compound as a light yellow solid (Fig. 1a). The structure of the new compound was elucidated by NMR (Supplemental Figs S2−S7). NMR analysis indicated that the isolated mixture was composed of compounds 1a(E) and 1b(Z) in a 2:1 ratio. The measured m/z value of glucosamine derivatives 1a and 1b ([M+H]+ = 262.0) compared well with the calculated mass ([M+H]+ = 262.1) (Fig. 1d). The signal corresponding to H4 of 1a appears at 5.53 ppm whereas the H4 proton in 1b generates a signal at 5.48 ppm, indicating that 1a is the (E) isomer judging from previously reported NMR data for enolates[32]. To elucidate the size of the ring (possible structures as shown in Supplemental Fig. S8), an NMR analysis based on the angles/coupling constants, Karplus equation, between vicinal ring protons was performed. A 7.2 Hz coupling constant (corresponding with an angle of about 150°−160°) was previously reported for vicinal trans protons in a similar unsaturated 7-membered ring whereas the signal corresponding to H7 appears in our spectrum as an apparent triplet of doublets with coupling constants of 4.8 and 2.4 Hz in the case of the 1a and as an apparent quartet a coupling constant of 4.8 Hz in the case of the 1b, indicating that we can discount this possibility[33]. This result is in agreement with the HMBC spectra wherein there are only correlations between both H6 and H7 and C7 and not between H8' and H8'' and C7. Coupling constants of trans vicinal protons in unsaturated 6-membered ring structures (unsaturated rings with two sp2 carbons should have similar conformations to our conjugated system), such as ᴅ-glucal or other unsaturated derivatives[34,35] are about 9−11 Hz (corresponding with an angle of almost 180°) which is not in agreement with the coupling constants observed for H7 suggesting that a structure incorporating a 6-membered ring can discounted. The signal corresponding to H6 appears as an apparent doublet of triplets with coupling constants of 6 and 2.4 Hz for 1a or as an apparent triplet of doublets with coupling constants of 6 Hz and 4.8 Hz in compound 1b. The trans vicinal protons of 2,3-dihydrofuran derivatives show a coupling constant of 6−8 Hz (corresponding with an angle of about 150°) whereas cis vicinal protons show a coupling constant of about 4.5 Hz (corresponding with an angle of 30°−40°) which is consistent with the results observed in our1H NMR spectrum for H6 indicating that 1a/1b is a 5-membered ring[36].
The synthesis of 1 can be explained through the mechanism shown in Fig. 2. After Knoevenagel condensation between DPPD and the reducing end of GlcN to produce 2, the 5-membered ring 3 is formed by intramolecular oxa-Michael cyclization. The irreversible, base-promoted decarboxylation of 3 then results in compound 4[37]. Then, compound 4 is oxidized by an unknown oxidant (presumably molecular oxygen in the alkaline environment) at the carbonyl group forming α,β-unsaturated carbonyl compounds 5a/5b. Then, the dehydratation of compounds 5a/5b results in the formation of the 1a/1b mixture. Finally, the labeling of GlcN with different propane-1,3-dione derivatives, such as 1,3-dipyridin-2-ylpropane-1,3-dione, dibenzoylmethane, and 1-benzoyl-3,3,3-trifluoroacetone, and the ester ethyl benzoylacetate, was also studied (Supplemental Fig. S9). All reactions were examined by TLC analysis. UV-active compounds with Rf values similar to that observed for 1a/1b could be detected when 1,3-dipyridin-2-ylpropane-1,3-dione, dibenzoylmethane and ethyl benzoylacetate were used as the labeling reagents whereas no UV-active compounds were detected after the labeling with 1-benzoyl-3,3,3-trifluoroacetone. The blue/violet color of the dot when the TLC plate was irradiated with a 362 nm light indicated the formation of a fluorescent derivative.
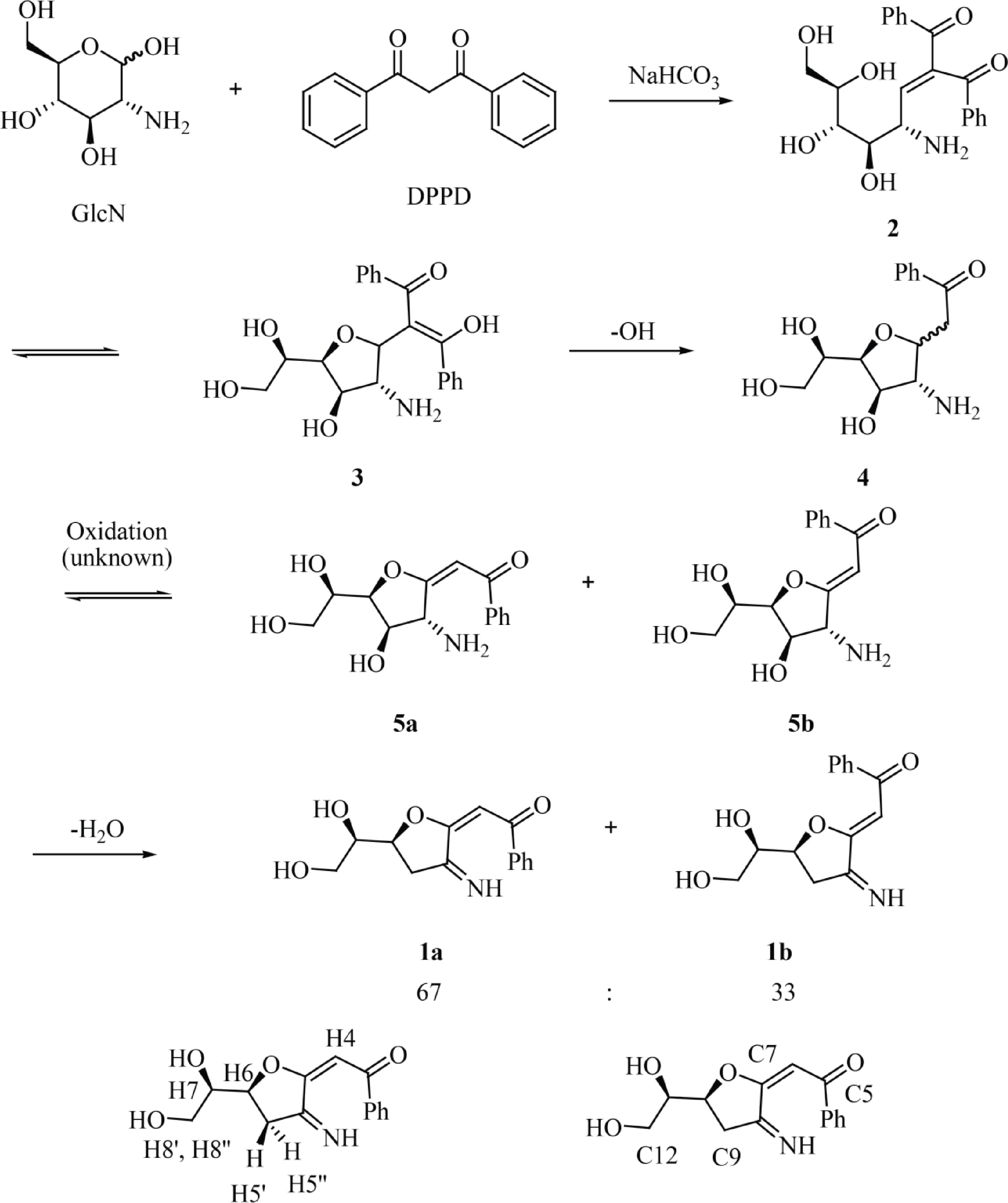
Figure 2.
Reaction of GlcN with DPPD to afford fluorescent glucosamine derivatives 1a and 1b with atom numbering corresponding to that used for NMR assignment.
Protein-mediated detection of GlcN by GPDA
-
Protein-mediated detection of carbohydrates was first described in the late 19th century, showing that protein extracts from plants were able to agglutinate red blood cells depending on their cell surface carbohydrates[38]. However, this phenomenon usually requires a multivalent binding mode of the same carbohydrate moieties to various individual binding sites of the protein units to trigger protein precipitation via the formation of an insoluble three-dimensional carbohydrate-protein network[39]. So far, sugar moieties which can precipitate lectins and are not organized in larger oligomeric branched structures such as natural glycoconjugates[40] or artificial glycodentrimers[41] were not yet reported. Therefore, lectins may not be suitable for the detection of monomeric carbohydrates. We observed that GPDA, which is an enzyme that catalyzes the conversion of glucosamine 6-phosphate into fructose 6-phosphate and ammonium, showed a peculiar precipitation behavior in the presence of the monosaccharide GlcN (Fig. 3a). The addition of other monosaccharides such as glucose, N-acetylglucosamine, mannose, galactose, or fructose did not result in any observable GPDA precipitation (Supplemental Fig. S10a). However, the GPDA precipitation required incubation times of several hours, and therefore we attempted to engineer the glucosamine 6 phosphate binding site of GPDA to increase its precipitation performance. This type of mutational approach was previously reported for changing the binding specificity of ACG, a carbohydrate binding protein specific for β-galactosides, which changed its carbohydrate binding profile to α1,3-linked GalNAc after the substitution of an asparagine with alanine at position 46 (Asn46Ala)[42]. Therefore, one random mutation library (generated by error-prone PCR, ~900 clones) and two focused libraries mutating amino acids 36G37S38T and 124G123I126G (generated by site directed mutagenesis using the primers listed in Supplemental Table S1, ~700 clones each). These three libraries were transformed into the BL21(DE3) expression host and GPDA mutants which had a faster growth rate were selected in eight passes by the liquid medium screening method described above. This resulted in the isolation of two individual GPDA variants with the amino acid substitutions 36F37R38I or 124N123Y126N. Applying the three mutant libraries to the membrane-based GPDA precipitation assay, which selected mutant variants with precipitation activity based on the size of the colonies (Supplemental Fig. S11), GPDA variants 36F37H38L, 36Y37Y38Y, 36V37V38V, 36I37V38D, and 46I (from the error-prone PCR derived library) were selected (Fig. 3b). To rule out that the increased growth performance of the mutant variants was due to spontaneous mutations in the BL21(DE3) expression host’s genomic DNA, the plasmids of all GPDA mutant variants were re-transformed into fresh BL21(DE3) cells.
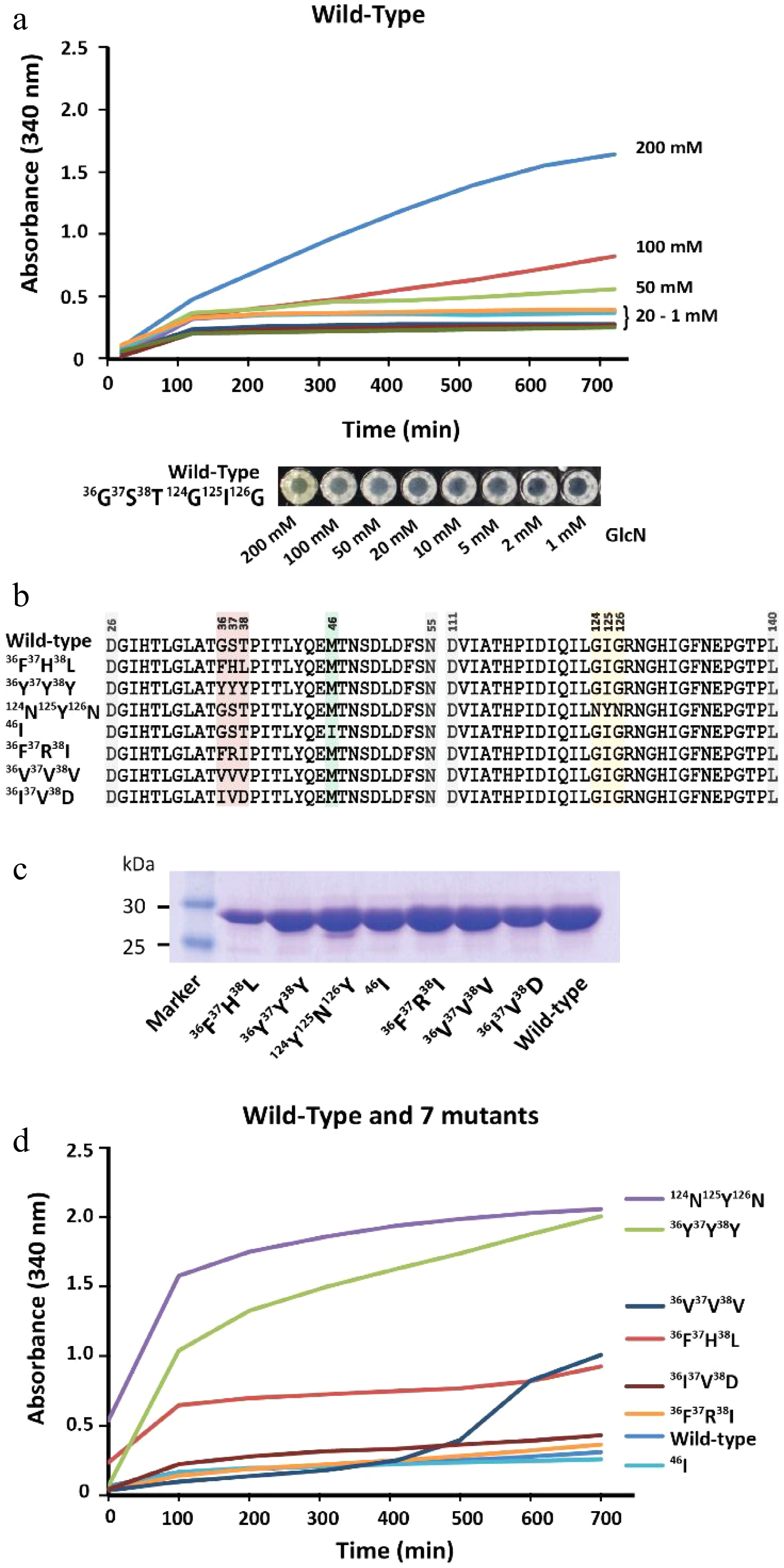
Figure 3.
Characterization of GPDA wild-type and mutant variants. (a) Precipitation behavior of wild-type GPDA in the presence of various concentrations of GlcN. (b) Protein sequence comparison of the selected GPDA mutant variants. (c) Comparison of protein yields of GPDA mutant and wild-type variants. (d) Precipitation behavior of the GPDA variants in the presence of 50 mM GlcN.
Generally, the GPDA mutant variants showed comparable expression levels to the wild-type variant (Fig. 3c), with the exception of GPDA variant 36F37H38L, which showed slightly reduced expression levels. The precipitation behavior of the purified GPDA variants in the presence of 50 mM GlcN showed that mutants 36F37H38L, 36Y37Y38Y, and 124N123Y126N precipitated significantly faster compared to the GPDA wild-type and other mutant variants (Fig. 3d). Therefore, these 36F37H38L, 36Y37Y38Y, and 124N123Y126N mutant variants were chosen for further characterization. Similar to the wild-type GPDA, the addition of other monosaccharides than GlcN or the addition of the amino acid glycine did not result in any protein precipitation (Supplemental Fig. S10b−d). The GPDA mutant variants and the wild-type showed comparable enzymatic activity for up to 24 h exposure at standard reaction conditions (pH 8.0, 37 °C). However, the three most active mutant variants were less active when the enzymes were pre-incubated for more than 3 h at 65 °C (Supplemental Fig. S12). Although these differences in heat stability between the GPDA wild-type and mutant variants have only little consequence at the ambient temperatures used for the precipitation reaction, it may still be a contributing factor to the enhanced overall precipitation performance of each mutant variant.
The precipitation efficacy of 36F37H38L, 36Y37Y38Y, and 124N123Y126N strongly depended on the concentration of GlcN, with no to little precipitation observed between 1 mM and 20 mM of GlcN (Fig. 4a). Strong precipitation was observed for samples containing 50, 100, and 200 mM of GlcN, and the limit of detection was determined to be between 10−20 mM of GlcN (Supplemental Fig. S13a−c). GPDA mutant 124N123Y126N showed the fastest response of all tested variants, and protein precipitation started almost immediately upon the addition of GlcN. The structural comparison of the GPDA wild-type and mutant variants using AlphaFold revealed that the amino acid substitutions of all mutants led to a reduced size of the glucosamine-6-phosphate binding site (Fig. 4b). We speculate that these substitutions may facilitate the accommodation of a sterically smaller GlcN. Despite observing a clear difference in the precipitation behavior between the GPDA wild-type and mutant variants in the presence of GlcN, this effect can currently not be pinned down to a single amino acid substitution. Therefore, we presume that stearic effects are the main cause for this functional changes. A comparison of the amino acid substitutions showed a significant increase of the overall size for all mutants (wild-type 36G37S38T = 263 Da vs. mutant 36F37H38L = 415 Da vs. mutant 36Y37Y38Y = 507 Da; wild-type 124G123I126G = 245 Da vs. mutant 124N123Y126N = 409 Da), which further substantiated the presumption of a sterically guided precipitation effect.
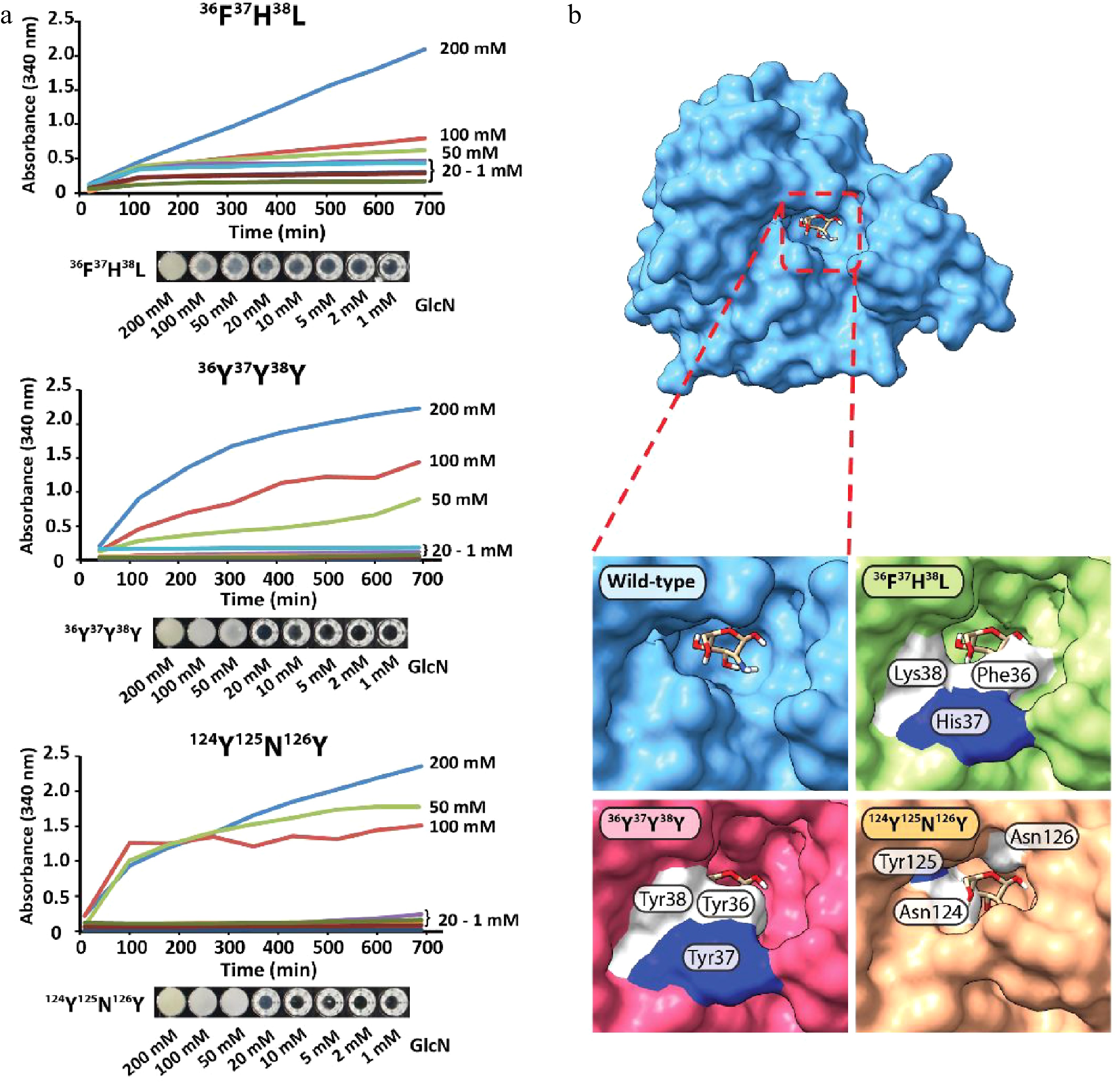
Figure 4.
(a) Protein precipitation behavior of GPDA mutant variants and (b) visualization of the GlcN binding site of the GPDA wild-type and mutant variants protein models generated by AlphaFold.
-
In summary, we presented two previously undescribed methods for the detection of GlcN in aqueous solutions. The first method describes a one-step synthetic strategy for the synthesis of a fluorescent glucosamine derivative. This derivative possesses very unusual fluorescent properties, showing a massive Stokes shift of 88 nm, and might therefore be useful in tracing GlcN. This GlcN derivatization can be performed in a single step reaction and does not require any sample workup prior to HPLC analysis, and although this method allows the derivatization of small quantities of GlcN, a fluorescence detector is required for the detection of the analyte. The second method presented is based on the precipitation behavior of the GPDA protein in the presence of GlcN, and the generation and screening of engineered GPDA mutants resulted in the selection of three variants with increased properties. Although the detection limit for GlcN of this method is still relatively high (10−20 mM of GlcN), further mutational screens of GPDA are likely to result in the discovery of more sensitive mutant variants. Furthermore, no derivatization of GlcN is required prior to the analysis, and the precipitation can be either monitored by the naked eye or photometrically, and therefore enabling the development of high-throughput screens. We envisage that these methods will be a useful addition to the currently available toolbox for GlcN detection.
This work was supported in parts by the National Natural Science Foundation of China (grant numbers 31471703, 31671854, and 31871754), and the 100 Foreign Talents Plan (grant number JSB2014012).
-
Josef Voglmeir is the Editorial Board members of Journal Food Materials Research. He was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of these Editorial Board members and their research groups.
- Supplemental Table S1 Oligonucleotide primers used in this study. The underlined nucleotides show where mutations were introduced.
- Supplemental Fig. S1 Excitation and emission spectra of the fluorescent glucosamine derivative 1a/1b monitored by HPLC.
- Supplemental Fig. S2 1H NMR spectrum of 67:33 1a/1b E/Z equilibrium.
- Supplemental Fig. S3 Amplified 1H NMR spectrum between 4.58-4.14 ppm and 2.56-2.10 ppm.
- Supplemental Fig. S4 13C NMR spectrum of 1a/1b cis/trans equilibrium.
- Supplemental Fig. S5 Two-dimensional COSY spectrum.
- Supplemental Fig. S6 Two-dimensional HSQC spectrum.
- Supplemental Fig. S7 Two-dimensional HMBC spectrum.
- Supplemental Fig. S8 Possible structures of 1a/1b depending on the ring size.
- Supplemental Fig. S9 TLC analysis of the reactions between GlcN and DPPD analogues such as 1,3-dipyridin-2-ylpropane-1,3-dione, dibenzoylmethane, ethyl benzoylacetate or 1-benzoyl-3,3,3-trifluoroacetone.
- Supplemental Fig. S10 Protein precipitation performance in the presence of GlcN, glucose, fructose, mannose, galactose, GlcNAc, and glycine (a) Wild-type; (b) Mutant variant 36F37H38L (c) Mutant variant 36Y37Y38Y; (d) Mutant variant 124Y125N126Y.
- Supplemental Fig. S11 Mutant variant library (mutations at amino acids 36-38) visualized on a solidified MTT reaction mixture. Large colonies were selected for sequencing.
- Supplemental Fig. S12 Heat stability test of GPDA wild-type and mutant variants. The activity test was conducted as follows: A reaction mixture (50 μL) containing 50 mM MES buffer (pH 6.5), 1.2 mM MgCl2, 4 mM GlcN-6-P, 4 μL of purified GPDA enzyme variant (pre-incubated for either 0, 3, 6, 9 or 12 h at 65 °C) was incubated for 80 minutes at 37 °C. 23 μL of this mixture were mixed with 57 μL of resorcinol reagent (0.18 g resorcinol dissolved in 100 mL of 4M HCl solution). After heating at 95 °C for 10 minutes, 65 μL of the mixture was transferred into a 384-well plate and the absorbance was measured at λ=505 nm.
- Supplemental Fig. S13 Concentration-depending absorbance values using purified GPDA enzyme variants (a) 36F37H38L, (b) 36Y37Y38Y, and (c) 124N123Y126N. The red arrow indicates the minimum detectable concentration. Error bars show the standard deviation from 3 independent experiments.
- Copyright: © 2022 by the author(s). Published by Maximum Academic Press on behalf of Nanjing Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Chen L, Laborda P, Cai Z, Hagan AK, Lu A, et al. 2022. Novel chemical- and protein-mediated methods for glucosamine detection. Food Materials Research 2:19 doi: 10.48130/FMR-2022-0019 

Novel chemical- and protein-mediated methods for glucosamine detection
- Received: 16 November 2022
- Accepted: 14 December 2022
- Published online: 30 December 2022
Abstract: We describe two novel approaches for the determination of glucosamine (GlcN). The first approach is based on the chemical derivatization of GlcN with the non-fluorophor 1,3-diphenyl-1,3-propanedione (DPPD), which results in a condensation product with interesting fluorescent properties. The obtained compound was isolated by silica-gel chromatography and its structure elucidated by NMR and mass spectrometry. The second approach is based on a previously undescribed sensitivity of the enzyme glucosamine-6-phosphate deaminase (GPDA) towards GlcN, which resulted in the precipitation of the enzyme. Using a rational enzyme engineering approach and both liquid-based and plate-based screening methods, mutational GPDA variants with significantly improved precipitation properties were identified and characterized. These novel glucosamine detection methods may be a useful addition to the repertoire of currently available glucosamine detection sensors.