









-
The chloroplast (cp) is involved in a variety of biological processes in the cells of plants, including photosynthesis, carbon fixation, and stress responses[1,2]. The genome of cp is smaller (75–250 kb) than the nuclear genome, and the genome sequences are more easily obtained via new sequencing technology; furthermore, the influence from homologous locations is lower[3]. The cp genome is a popular method used to identify differences among species, due to its short sequence length and fairly simple analysis. Its genome genes, ndhF, matK, and trnS-trnG, have been widely amplified for species recognition, barcoding, and phylogeny[4]. In angiosperms, a large single-copy (LSC, 80–90 kb), a small single-copy (SSC, 16–27 kb), and two copies of inverted repeats (IRa/b, 20–28 kb) make up the typical quadripartite structure of cp genomes[5,6]. Previous research verified that the gene sequence, gene information, and genome structure of the cp genomes were extremely stable in plants[7]. Moreover, the cp genome has some specific characteristics, such as uniparental inheritance, natural haploid, and a minimum number of recombination, which have assisted in understanding the phylogeny and evolution of numerous genera, such as Oncidiinae[8], Camellia[9], and Fritillaria[10].
C. illinoinensis, commonly known as pecan, belongs to the family Juglandaceae, which is located in Asia and North America's tropical and temperate zones[11]. In China, pecan is a well-known nut crop that has been grown extensively in recent years[12,13]. C. illinoinensis is a monoecious, dichogamous, and wind-pollinated species[14]. The timing of its pollination is crucial, as the stigma surface of the pistil only receives pollen during a relatively short period[15]. The pecan cultivar 'Pawnee' is protandrous, meaning that the pollen is shed before the pistil is receptive. It was introduced to China in 1998[16], and is the only early pollination tree; this leads to a serious pollination deficiency in the orchards of China[17]. The 'Xinxuan-4' is also protandrous, and was selected from an autochthonous individual tree growing in Nanjing Botanical Garden Men. Sun Yat-sen in the 1950s[18,19]. The maturation of its anthers in male flowers occurs two days earlier than that of the 'Pawnee', which could satisfy the early pollination of pecan trees. However, the genetic information of 'Xinxuan-4' remains exclusive.
Recently, the cp genomes of C. illinoinensis cv. Pawnee[20], C. illinoinensis cv. Wichita[21], C. illinoinensis cv. 87MX3-2.11, and C. illinoinensis cv. Lakota[22] were identified. The release of more cp genomes will help identify genetic variations, and offer new perspectives on the interspecific relationships among the Carya species. This research sequenced the 'Xinxuan-4' cp genome and the first comparative analysis of its sequence with other published Carya cp genomes.
Our main objectives in this study were to: (1) ascertain the cp genome's structure and composition; (2) carry out an analysis of codon preference; (3) detect repeats and microsatellite patterns; (4) determine highly divergent regions; (5) define the phylogenetic analysis. The research will unveil the maternal origin of the 'Xinxuan-4' by cp sequencing and will contribute to the future genetic breeding of pecan.
-
The pecan cultivar 'Xinxuan-4' is protandrous, with a very early-season pollen shed; it was planted in Nanjing Botanical Garden Men. Sun Yat-sen, Nanjing City, Jiangsu Province, China. The 'Xinxuan-4' should be a good pollenizer for the 'Mahan', 'Wichita', and 'Mohawk' varieties, which have plenty of flowers, red stigma, and small fruit (Fig. 1). Fresh leaves of 'Xinxuan-4' were obtained and rapidly stored at −80 °C. The adjusted CTAB protocol was adopted to extract DNA[23].

Figure 1.
Carya illinoinensis cv. Xinxuan-4. (a) Male flower. (b) Female flower. (c) Fruits on the tree. (d) Fruit without husk, scale bar = 2 cm.
Cp genome sequencing
-
The pecan cultivar 'Xinxuan-4' was used for the cp genome sequencing. After sequencing, the adapters of the raw data were removed and the low-quality reads were cleaned by fastp v20.0. The clean reads were obtained to assemble the 'Xinxuan-4' cp genome using SPAdes 3.11.0 software[24]. The assembly contigs were blasted to the Carya laciniosa cp genome, and the gaps were repaired using GapCloser 1.12 software.
Genome annotation
-
Prodigal v2.6.3 software was used to annotate the cp genome, and Hammer v3.1 b2 software was utilized to scan the tRNA genes. The rRNA genes were identified using Aragorn v1.2.38. Organellar Genome DRAW v1.3.1[25] was used to generate the map. The cp genomic sequence of the 'Xinxuan-4' was uploaded to GenBank with accession number PRJNA795859.
Analysis of microsatellites
-
A simple sequence repeat (SSR) marker is a kind of tandem repeat sequence made up of a dozen nucleotides, which has several repeat units (usually 1 to 6). CpSSR markers are SSR markers present in the genomes of cps. CpSSR analysis was performed using the Misa software[26]. The parameters used were as follows: mono-nucleotides repeated eight times; di-nucleotides repeated five times; trinucleotides repeated four times, tetra-, penta-, and hexa-nucleotides repeated three times.
Repeat sequence and synonymous codon usage analysis
-
REPuter software was employed to examine repeat structures, containing forward (F), reverse (R), complement (C), and palindromic (P) repeats[27]. The tandem repeats finder 4.07b was used to search for tandem repeats. Using CodonW1.4.4, the synonymous codon usage was characterized using relative synonymous codon usage (RSCU).
Cp comparison and phylogenetic analysis
-
The sequence information of five Carya genotypes, including C. illinoinensis cv. Xinxuan-4 (PRJNA795859), C. illinoinensis cv. Pawnee (MN9771241), C. illinoinensis cv. 87MX3-2.11 (MH909600), C. illinoinensis cv. Lakota (MH909599), and C. cathayensis (PE00820836)[28], were obtained from the Gene Bank for the comparative analysis. The cp genome of 'Xinxuan-4' was compared to those of four chosen Carya materials using the mVISTA program[29]. The nucleotide variability (Pi) in the whole cp genome was evaluated by DnaSp v6[30]. To analyze the cp genome difference between 'Xinxuan-4' and its close relatives, the relative rates of synonymous (Ks) and non-synonymous (Ka) substitution rates were determined using the Ka/Ks Calculator software[31]. The sequence data of 17 Juglandaceae species were retrieved from the NCBI to examine the evolutionary relations among these species. The PhyML program (v3.0)[32] was adopted to produce the phylogenetic tree.
-
The cp genome of 'Xinxuan-4' was 160,819 bp in length (Fig. 2), which was the same as that of 'Pawnee'[20] and 'Lakota' (Table 1)[22]. Carya species showed similar cp genome sizes (Fig. 3), and the cp genome of 'Wichita' was the shortest genome published thus far[21]. Similarly to most angiosperms, the cp genome presented a common quadripartite architecture consisting of LSC (90,022 bp), SSC (18,791bp), and IRa/b, (each 26,003 bp). Comparatively, the lengths of LSC, SSC, and IR of 'Xinxuan-4' were the same as that of 'Pawnee', whereas the 'Wichita' had the shortest LSC (89,799 bp), SSC (18,751bp), and IR (25,991bp). The GC content is an important indicator of affinity in various species. The 'Xinxuan-4' cp genome had a total GC content of 36.1%, which means it was exactly like other species in the genus Carya (35.8%–36.3%)[33−35]. Specifically, the LSC, SSC, and IR locations had GC contents of 33.74%, 29.89%, and 42.58%, respectively. In the IR locations, the high GC content may be attributed to the high GC content in four rRNA genes (rrn16, 56.41%; rrn23, 55.64%; rrn4.5, 47.97%; rrn5, 52.07%) in this region (Fig. 3)[36].

Figure 2.
Circular representation of the cp genome of C. illinoinensis cv. Xinxuan-4.
Table 1. Features of the cp genomes of C. illinoinensis cv. Xinxuan-4 and five related materials.
Species Xinxuan-4 Pawnee 87MX3-2.11 Lakota Wichita Cathayensis Genome size (bp) 160,819 160,819 160,545 160,819 160,532 160,825 LSC size (bp) 90,022 90,022 89,933 90,041 89,799 90,115 SSC size (bp) 18,791 18,791 18,576 18,790 18,751 18,760 IR size (bp) 26,003 26,003 26,018 25,994 25,991 25,975 Total genes 132 131 124 123 128 129 Protein-coding genes 87(8) 86(7) 84(6) 83(6) 83(6) 84 tRNAs 37(7) 37(8) 32(6) 32(6) 37(8) 37(7) rRNAs 8(4) 8(4) 8(4) 8(4) 8(4) 8(4) GC content (%) 36.1 36.1 36.2 36.1 36.2 36.1 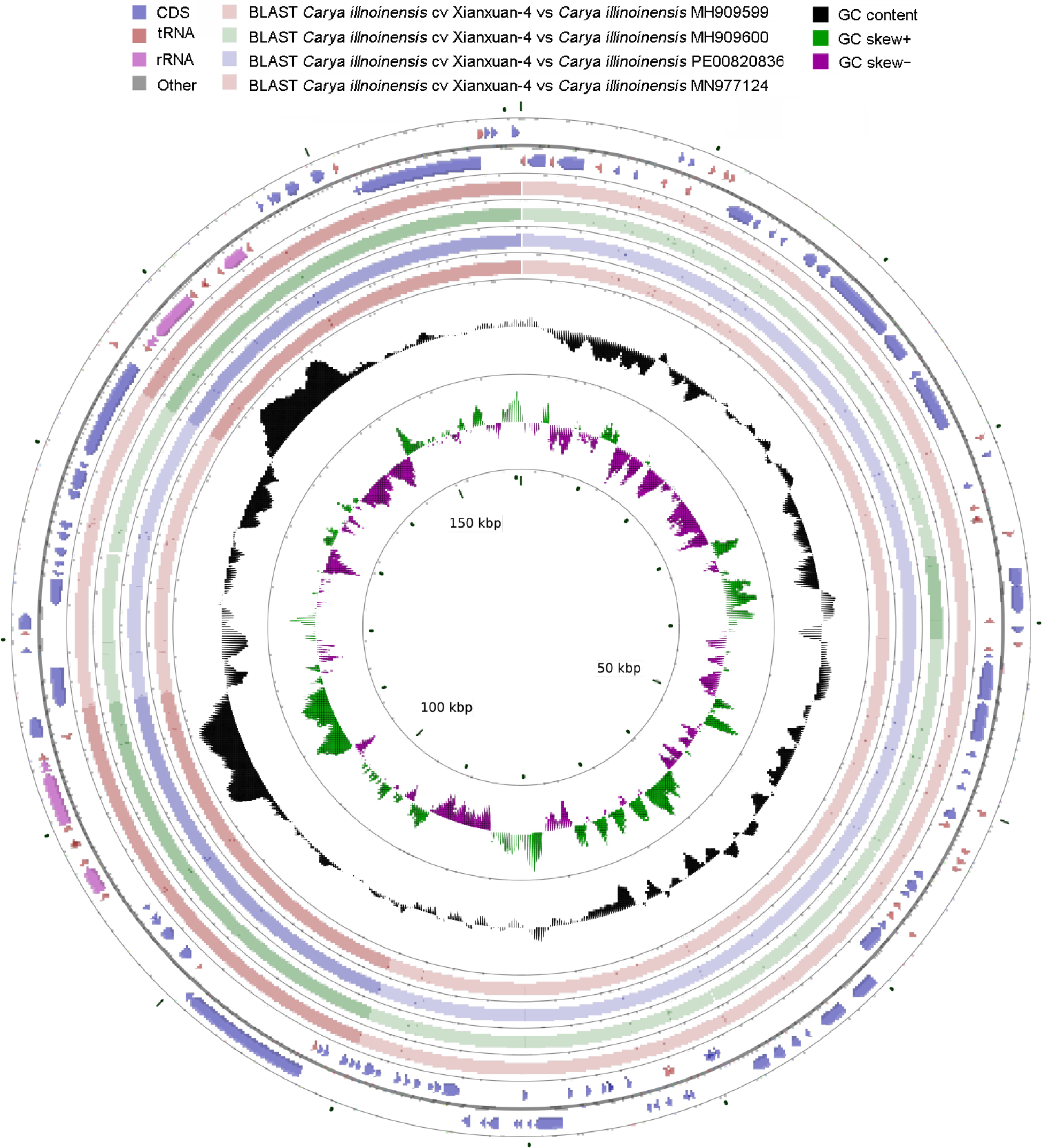
Figure 3.
GC content of the C. illinoinensis cv. Xinxuan-4 cp genome.
The cp genome of 'Xinxuan-4' contained 132 genes, comprising 87 protein-coding genes, 37 tRNA genes, and eight rRNA genes (Table 1). Nineteen duplicate genes were discovered: eight were protein-coding genes (rpl2, rpl23, rps7, rps12, yct1, ycf15, ycf2, and ndhB), seven were tRNA (trnA-UGC, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, and trnV-GAC), and four were rRNAs genes (rrn16, rrn23, rrn4.5, and rrn5) (Table 2). In the 'Xinxuan-4' cp genome, there were 18 intron-containing genes, of which 15 genes (nine protein-coding genes and six tRNA genes) contained one intron, and three genes (rps12, ycf3, and clpP1) possessed two introns (Table 2 & Supplemental Table S1). With a length of 2,559 bp, the intron of the trnK-UUU gene was the longest. The rps12 gene, consisting of one intron in the LSC location and the other two exons in the IR locations, was a trans-spliced gene[37,38].
Table 2. Annotated genes in the cp genome of C. illinoinensis cv. Xinxuan-4.
Category Gene group Photosynthesis Subunits of photosystem I psaA, psaB, psaC, psaI, psaJ Subunits of photosystem II psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ Subunits of NADH ndhAb, ndhBab, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK Cytochrome b/f complex petA, petBb, petDb, petG, petL, petN Subunits of ATP synthase atpA, atpB, atpE, atpFb, atpH, atpI Large subunit of rubisco rbcL Self-replication Large ribosomal subunit rpl14, rpl16b, rpl2ac, rpl20, rpl22, rpl23a, rpl32, rpl33, rpl36 Small ribosomal subunit rps11, rps12ab, rps14, rps15, rps16b, rps18, rps19, rps2, rps3, rps4, rps7a, rps8 Subunits of RNA polymerase rpoA, rpoB, rpoC1b , rpoC2 Ribosomal RNAs rrn16a, rrn23a, rrn4.5a, rrn5a Transfer RNAs trnA-UGCab, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCCb, trnG-UCC, trnH-GUG, trnI-CAUa, trnI-GAUab, trnK-UUUb, trnL-CAAa, trnL-UAAb, trnL-UAG, trnM-CAU, trnN-GUUa, trnP-UGG, trnQ-UUG, trnR-ACGa, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GACa, trnV-UACb, trnW-CCA, trnY-GUA, trnfM-CAU Other genes Maturase matK Protease clpPc Envelope membrane protein cemA Acetyl-CoA carboxylase accD c-type cytochrome synthesis gene ccsA Unknown function genes Conserved open reading frames ycf1a, ycf15a, ycf2a, ycf3c, ycf4 a Gene with two copies, b Gene with one intron, c Gene with two introns. Genes can be gained or eliminated in the cp genomes during the process of evolution[37,39]. Comparing the cp genome of Carya 'Pawnee', '87MX3-2.11', 'Lakota', and 'C. cathayensis' in GenBank, the gene rps12 was absent in 'Lakota', but existed in the other cp genomes, with one duplicate in the '87MX3-2.11' and two duplicates in the remaining species (Table 3 & Supplemental Table S2). Previous results verified that the 'Lakota' cp genome's rps12 gene was missing a reading frame[22]. All C. illinoinensis contained the genes of psbZ and ycf15, but could not be found in C. cathayensis. Gene rps16 existed in the other three Carya genotypes ('Xinxuan-4', 'Pawnee' and 'C. cathayensis') with one copy; however, it was absent from cp genomes of the 'Lakota' and '87MX3-2.11'. The rps16 gene encoded in the cp genome of most organisms was also not discovered in Populus alba[40]. The loss of this gene in the cp genomes was compensated by the rps16 gene from mitochondria[41].
Table 3. Different genes and gene copies in the cp genomes among five Carya materials.
Gene group Different gene Xinxuan4 Pawnee 87MX3-2.11 Lakota Cathayensis Protein-coding genes rps12 2 2 1 0 2 rps16 1 1 0 0 1 psbZ 1 1 1 1 0 lhbA 0 0 0 0 1 ycf15 2 2 2 2 0 tRNA genes trnA-UGC 2 2 2 2 0 trnG-UCC 1 0 0 0 0 trnI-GAU 2 2 0 0 0 trnL-UAA 1 1 0 0 1 trnV-UAC 1 1 0 0 1 rna-UGC 0 0 0 0 2 The most notable differences in Carya were in the tRNA genes, six tRNA genes (trnA-UGC, trnG-UCC, trnI-GAU, trnL-UAA, trnV-UAC, and rna-UGC) were found to be different (Table 3). All of the C. illinoinensis lacked duplicates of rna-UGC, but two duplicates were discovered in C. cathayensis. Both pecans varieties, '87MX3-2.11' and 'Lakota', lost copies of trnG-UCC, trnI-GAU, trnL-UAA, trnV-UAC, and rna-UGC, and a single duplicate of trnG-UCC was detected only in 'Xinxuan-4'. Approximately 110–130 individual genes were found in the majority of cp genomes, most of which were coding DNA sequence (CDS) or protein-coding genes; the rest of the genes were tRNA and rRNA genes[42]. The 'Xinxuan-4' and 'Pawnee' cp genomes included 132 and 131 genes, respectively, compared with 123 ('87MX3-2.11') or 128 ('Wichita') genes in the Carya. The protein-coding genes and rRNA genes remained stable among the five Carya genotypes (77–83 CDS and four rRNA genes). In general, the number of tRNA genes was the most variable among the annotations in the Carya cp genomes.
Codon preference analysis
-
Due to the degeneracy of codons, each amino acid is encoded by multiple codons (synonymous codons) in the organisms[43]. The utilization rate of genome codons differs greatly in various species; this inequality in the use of synonymous codons is known as RSCU. Natural selection in the organisms is believed to have generated the RSCU, which can be categorized into four models: no preference (RSCU < 1.0), low preference (1.0 < RSCU < 1.2), moderate preference (1.2 > RSCU < 1.3), and high preference (RSCU > 1.3)[44]. The RSCU of the 'Xinxuan-4' cp encoding sequence was calculated. The findings demonstrated that 26,643 codons encoded all of the genes and that 20 amino acids were encoded by 68 different kinds of codons. The most commonly utilized codons were ATT (isoleucine), AAA, (lysine), and GAA (glutamic acid), which were 1,159 (4.35%), 1,071 (4.02%), and 1,049 (3.94%) codons, respectively (Fig. 4 & Supplemental Table S3). In the 'Xinxuan-4' cp genome, 31 out of 68 codons had RSCU values > 1, of which 22 displayed a strong preference, six had a median preference and three showed a low preference. With the exception of TTG being G-ending, all of the codons in the 'Pawnee' cp genome showed a preference for an A/T ending[20]. In this study, we also detected that the codons in the 'Xinxuan-4' cp genome preferred A/ T ending. Similarly, A/T ending has been found in C. cathayensis and other angiosperms[45,46]. This preference of the codons may be due to the high conservation in the cp genes. In contrast, numerous codons ending in G or C showed RSCU values below 1, suggesting that these condons were less frequent in Carya cp genes. The specific species characteristics of synonymous codon usage could be utilized to study the regulation of gene expression, differentiation, and evolutionary processes of Carya in the future.

Figure 4.
Codon usage frequency of the C. illinoinensis cv. Xinxuan-4 cp genome.
Repeat sequence and CpSSR analysis
-
Repeat sequences are thought to be crucial for the rearrangement and recombination of the cp genome[47,48]. In the present study, a total of 59 long repeats, comprising 32 forward, 23 palindromic, three reverse, and one complementary repeat, were found in the cp genome (Fig. 5 & Supplemental Table S4). The lengths for the majority of the repeats varied from 30 to 90 bp. There were 46, 7, and 12 long repeats located in the LSC, SSC, and IR locations, respectively. Most of the repeats occurred in the intergenic spacers (IGS), and the remaining repeats were located in the coding regions associated with the protein-coding genes ycf3, ycf2, rrn4.5s ndhA, psaB, psaA, and tRNA genes trns-UGA, and trns-GCU. These conclusions were consistent with the findings for C. illinoinensis and C. cathayensis[20,28].

Figure 5.
Size and type of the long repeats located in the C. illinoinensis cv. Xinxuan-4 cp genome.
Short DNA sequences, or SSRs, exhibit high polymorphism in related species. CpSSR markers are a wonderful tool for studying interspecific evolution and identification, as well as intraspecific population genetic variation[49,50]. According to a prior study, 213 SSRs were examined in the cp genome of C. illinoinensis[20]; in the present study, a total of 278 SSRs were detected in the 'Xinxuan-4' cp genome using MISA. There were six different types of SSR, the most prominent of which were mononucleotide repeats (189, 67.98%), followed by tri-nucleotide (70, 25.18%) and di-nucleotide (15, 5.39%) repeats. The other three kinds of SSRs were less prevalent: tetra-nucleotide (2, 0.72%), pentanucleotide (1, 0.36%), and hexanucleotide (1, 0.36%) repeats (Fig. 6a). These results were also reported in C. illinoinensis [20] and C. cathayensis [28], in which mono- and tri-nucleotide type cpSSRs were found at high rates, while at low frequencies were di-, tetra-, penta-, and hexa-nucleotide type cpSSRs. Moreover, the majority of cpSSRs belonged to the A/T types (65.11%), while only eight C/G types (2.87%) cpSSRs were identified in the cp genome, indicating that short A or T repeats made up the largest number of cpSSRs in the 'Xinxuan-4' cp genome (Fig. 6b & Supplemental Table S5). These results confirmed the hypothesis of G or C repeats that were uncommon in cpSSRs, which consisted of short A or T repeats[28].

Figure 6.
SSRs distribution in the cp genomes of the C. illinoinensis cv. Xinxuan-4. (a) Type and number of SSRs. (b) Type and number of SSR repeats. (c) Number of SSRs in various location. (d) Number of SSRs in three locations.
In this study, we also evaluated the distribution of 278 cpSSRs in the cp genome, which were predicted to be 191 (68.71%), 45 (16.18%), and 42 (15.11%) in the LSC, SSC, and IR locations, respectively (Fig. 6c). In addition, 167, 66, and 45 SSRs existed in the intergenic regions, introns, and coding sequences, respectively (Fig. 6d). The noncoding regions in the cp genome of the 'Xinxuan-4' contained the majority of the cpSSRs, similar distribution preferences of cpSSRs have been observed in C. illinoinensis, C. cathayensis, and Avena sativa[20,28,51]. The majority of genes had mono- or tri-nucleotide SSRs, whereas only one protein-coding gene, ycf1, contained tetranucleotide SSR (Supplemental Table S5). Therefore, the specific SSR to the 'Xinxuan-4' in different gene regions could be used as a molecular marker to choose an appropriate cultivar for early pollination breeding material and the management of the pure line.
IR contraction and expansion
-
In the cp genomes of angiosperms, the expansion and contraction of the IR and SSC boundaries are frequently observed, giving rise to size differences among cp genomes[52,53]. To further explore the structural characteristics of the cp genome of 'Xinxuan-4', we examined the IR/SSC and IR/LSC junctions using four different Carya materials, namely 'Pawnee', '87MX3-2.11', 'Lakota', and C. cathayensis. The results are demonstrated in Fig. 7. Our results showed that the 'Xinxuan-4', 'Pawnee', and 'Lakota' had the same size of chloroplast genomes (160,819 bp), while C. cathayensis had the largest cp genomes (160,825 bp). All five materials had IR, SSC, and LSC regions of similar sizes, and their IRb boundaries all reached into the ycf1 gene, with lengths ranging from 1,093 bp (C. illinoinensis) to 1,109 bp (C. cathayensis). Correspondingly, the 'Xinxuan-4', 'Pawnee', and 'Lakota' varieties had the same IR sequence (26,003 bp), which was highly conserved. However, '87MX3-2.11' contained a slightly longer IR region (26,030 bp), and C. cathayensis possessed a smaller IR location (25,975 bp). It was suggested that the IRa/b region of the '87MX3-2.11' had experienced expansion and C. cathayensis underwent contraction during evolution. In angiosperm plastomes, the SSC/IR border is relatively conserved and mostly located within ycf1[54]. Similar expansions or contractions have been published in Jasminum nudiflorum Lindl[55] and Avena sativa[51].

Figure 7.
Comparing the LSC, SSC, and IR locations of five selected cp genomes in the Carya.
Comparative analysis of cp genome structure
-
Further investigation of the variations in the cp sequences was conducted with the five Carya genera using mVISTA, with 'Xinuan-4' as a reference. The findings showed that there were extremely high sequence similarities among the cp genome sequences of 'Xinuan-4', 'Pawnee', and 'Lakota', as demonstrated in Fig. 8. Compared to the encoding regions, the non-coding locations showed a comparatively higher level of divergence. Some notable divergences in the non-coding locations included the following: trnS-GCU–trnG-GCC, trnR-UCU–atpA, atpF–atpH, trnD-GUC–trnE-UUC, trnT-GGU–psbD, trnG-UCC–trnfM-CAU, ndhC-trnV-UAC, accD–pasI, rpl32–trnL-UAG, and ndhG–ndhI. Genes such as matK, rpoC2, and ycf1 were discovered to contain variation coding genes. These findings were consistent with those reported for the related family Juglandacea, the genus Quercus, in the Fagaceae family[56].
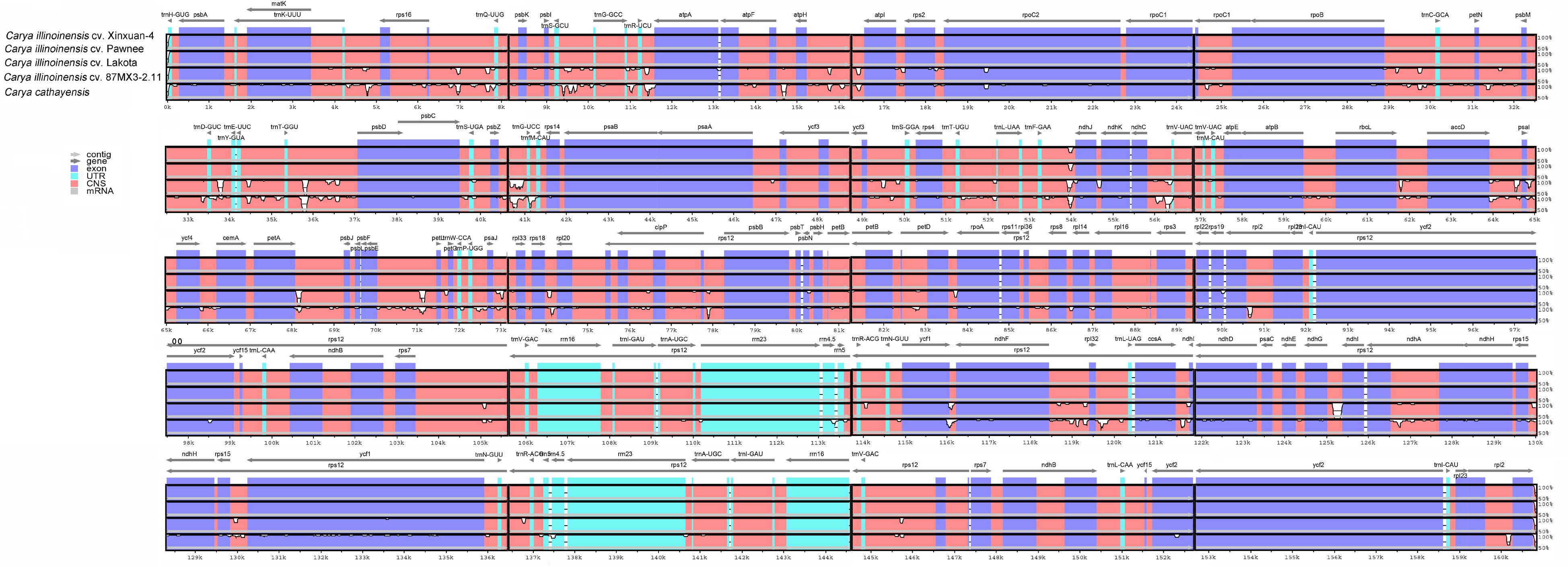
Figure 8.
Sequence identity plot comparing the cp genomes among Carya with C. illinoinensis cv. Xinxuan-4 set as a reference.
In order to test whether the cp genes of C. illinoinensis underwent selection, the Ka/Ks were computed to identify the variations among genes. The results showed that the Ka/Ks values of most genes were NA, and only 14 genes had the values (Supplemental Table S6). Most of the (seven of 12) genes had values below 1 in the cp genomes of C. illinoinensis, suggesting these kinds of genes were the target of purifying selection. The remaining five genes, which were petD, rpl16, rpoC2, rpoC1, and rps12, had Ka/Ks ratios that were generally greater than 1, meaning positive selection in comparison to the other C. illinoinensis species.
Moreover, the gene nucleotide variability value (pi) can offer prospective molecular markers for genetics in population applications and show variations in nucleic acid sequences of various species[51]. In this study, the Pi values of five Carya genotypes, including 'Xinxuan-4', 'Pawnee', '87MX3-2.11', 'Lakota', and C. cathayensis, are shown in Fig. 7. The figure demonstrated that the nucleotide diversity of the SSC and LSC locations was significantly higher than that of the IR locations (Fig. 9 & Supplemental Table S7). Gene nucleotide variability values of LSC. rps12, psbL, petD, and IR. trnV-GAC were higher genes. The remaining genes' values were less than 0.003, which suggested that the Carya species had a low nucleotide diversity.
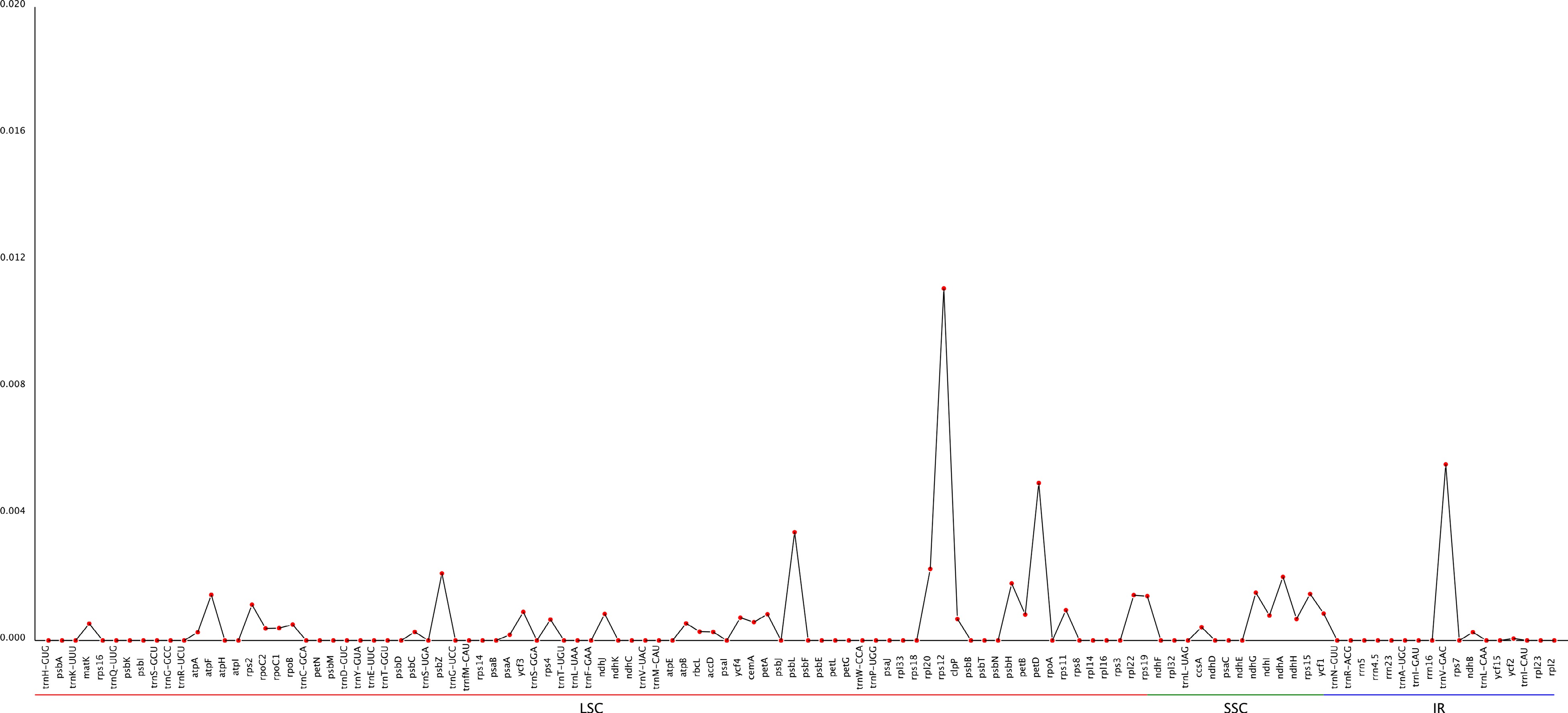
Figure 9.
Sliding window analysis of the cp genome for nucleotide diversity (pi) of three species in Carya.
Phylogenetic analysis
-
The encoding sequences from the cp genomes of 18 species were used to create a phylogenetic tree. As shown in Fig. 10, the phylogenetic tree indicated that the genera Carya and Juglandaceae were both monophyletic and that Carya proved more related to the group formed by the genus Juglandaceae, which was in line with earlier research[33,57]. Interestingly, C. cathayensis was grouped with C. sinensis instead of C. illinoinensis. Previous research demonstrated that C. cathayensis belonged to one of the typical species of the Asian sect. Sinocarya, while C. illinoinensis represented one of the typical species of the North American sect. Apocarya[20]. This could be the cause of the separated between the groups of C. illinoinensis and C. cathayensis. In addition, the 'Xinxuan-4' and 'Pawnee' varieties formed a single clade, suggesting that the 'Xinxuan-4' variety was more closely related to 'Pawnee', which inferred that 'Xinxuan-4' seedling may have come from North America.
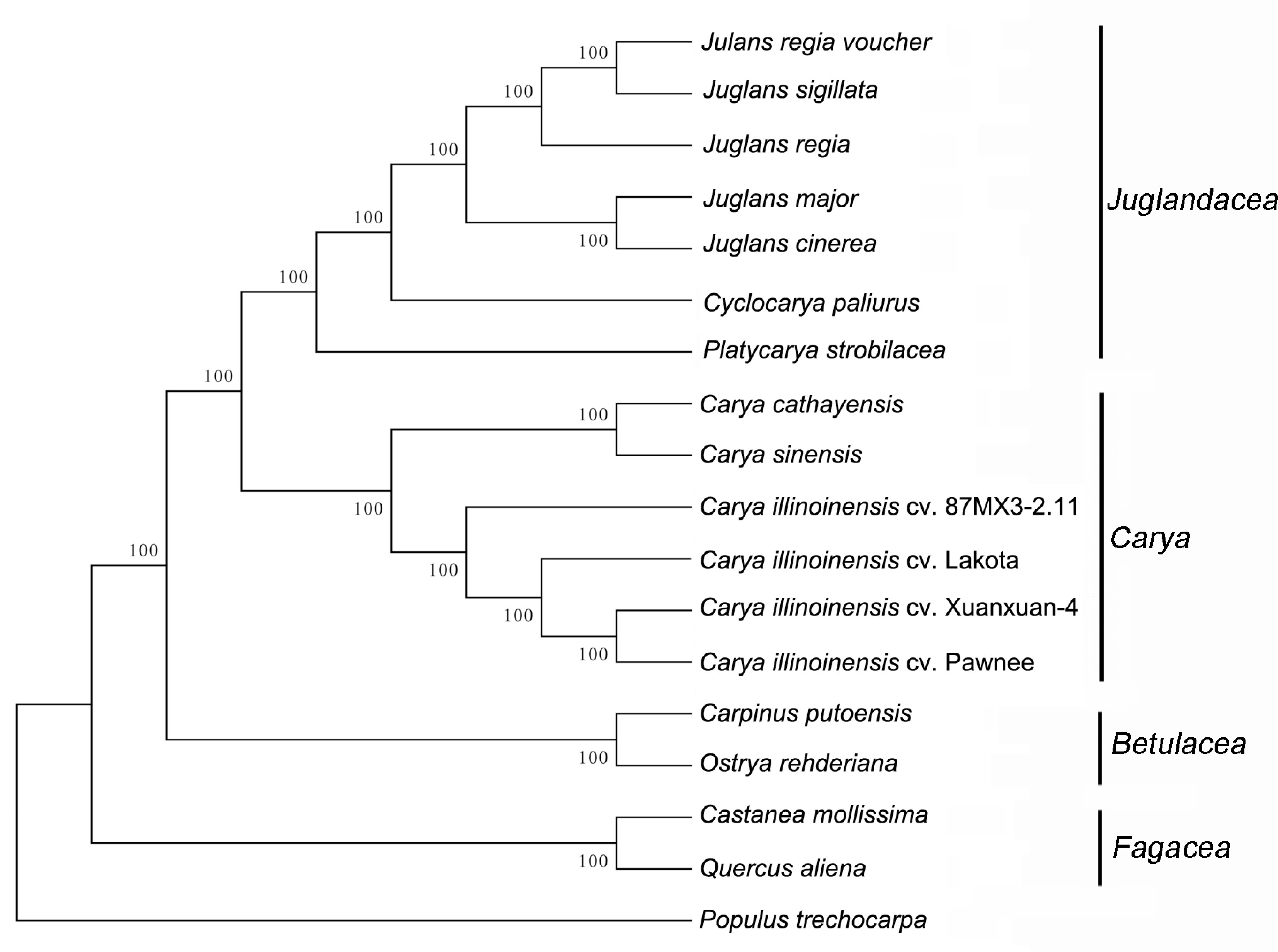
Figure 10.
Phylogenetic tree of 18 related species based on the whole cp genome.
-
In this study, we published the cp genome sequence of 'Xinxuan-4', which came from a seedling selection. The genome showed similar features to those of 'Pawnee'. The genome was estimated to contain 112 unique genes, comprising 79 protein coding genes, 29 tRNAs, and four rRNAs. We identified 278 cpSSRs and 59 long repeats that can be applied as prospective molecular markers for genetics of the population and evolutionary studies. The RSCU analysis showed that all of the genes were encoded by 26,643 codons, and 68 kinds of codons encoded 20 amino acids. We also detected that the codons in the genome preferred A/T endings. Phylogenetic analysis showed that the 'Xinxuan-4' variety was more closely related to 'Pawnee'. These results not only offer a useful database to recognize the maternal origin of the 'Xinxuan-4' cultivars, but also contribute to the analysis of the phylogenetic relationship and application of germplasm resources for Carya species.
-
The authors confirm contribution to the paper as follows: study conception and design: Chen Y; data collection: Zhang S, Chen Y; software, visualization: Wang W, Mo Z; draft manuscript preparation: Chen Y, Chen X; review and editing: Zhao Y, Zhu C. All authors reviewed the results and approved the final version of the manuscript.
-
The data that support the findings of this study are openly available in the GenBank of NCBI at
www.ncbi.nlm.nih.gov (accessed on 10 September 2022), reference number (PRJNA795859). This research was supported by the National Natural Science Foundation of China (32001344), the Natural Science Foundation of Jiangsu Province, China (BK20200290, BK20210166), Key Research and Development Plan of Jiangsu Province (BE2021406).
-
The authors declare that they have no conflict of interest.
- Supplemental Table S1 Summary of introns and exons of genes in chloroplast of C. illinoinensis cv. Xinxuan-4.
- Supplemental Table S2 Annotated genes in the chloroplast genome among five Carya.
- Supplemental Table S3 Codon usage of C. illinoinensis cv. xinxuan-4 Chloroplast genome.
- Supplemental Table S4 Long repeat sequences in the C. illinoinensis cv. xinxuan-4 chloroplast genome.
- Supplemental Table S5 Simple sequence repeats (SSR) in the C. illinoinensis cv. xinxuan-4 Chloroplast genome.
- Supplemental Table S6 The KaKs ratios of genes in the C. illinoinensis.
- Supplemental Table S7 Nucleotide variability values among the Carya.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Chen Y, Zhang S, Wang W, Chen X, Zhao Y, et al. 2024. Chloroplast genome sequencing of Carya Illinoinensis cv. Xinxuan-4, a new pecan pollinated cultivar. Fruit Research 4: e012 doi: 10.48130/frures-0024-0006 

Chloroplast genome sequencing of Carya Illinoinensis cv. Xinxuan-4, a new pecan pollinated cultivar
- Received: 27 June 2023
- Revised: 03 November 2023
- Accepted: 17 November 2023
- Published online: 07 March 2024
Abstract: Carya illinoinensis, is a highly valuable nut plant that is cultivated worldwide. As a precious pecan pollination resource, C. illinoinensis cv. Xinxuan-4 is protandrous, with a very early bud break in China. In this study, the chloroplast (cp) genome of 'Xinxuan-4' was sequenced and compared with closely related cultivars. The cp genome was found to be 160,819 bp in length, and it had a common quadripartite architecture with one large single copy (LSC; 90,022 bp), one small single copy (SSC; 18,791 bp), and two inverted repeats (IRs; 26,003 bp). The genome contained 132 genes, including 87 protein-coding genes, 37 tRNA genes, and eight rRNA genes, with a GC content of 36.1%. Furthermore, 278 simple sequence repeats and 59 long repeat sequences were identified, and the genome comparisons revealed that there was a greater divergence in the noncoding regions than in the coding regions. According to the gene selective pressure analysis, five genes (petD, rpl16, rps12, rpoC2, and rpoC1) were identified to be potentially under positive selection when contrasted with the other Carya genotypes. Phylogenetic analysis of the cp genome of 'Xinxuan-4' and 17 other species inferred that Carya is monophyletic and that the genetic relationship between 'Xinxuan-4' and 'Pawnee' is quite close from an evolutionary perspective. The currently characterized cp genome of 'Xinxuan-4' offers useful data for subsequent research on this pecan species.
-
Key words:
- Pecan /
- Chloroplast /
- Genome comparison /
- Phylogenetic analysis