









-
Hereditary transthyretin amyloidosis with a protein variant (ATTRv) is a rare autosomal dominant disease resulting from genetic mutations in the TTR gene, leading to abnormal extracellular deposition knonw as amyloidosis. This insoluble fibrillar protein accumulates in affected organs or tissues[1]. Among the various types of hereditary amyloidosis, ATTRv is the most common and best studied[2−4].
Transthyretin (TTR), also known as prealbumin, is a 127-amino-acid tetrameric protein mainly synthesized in the liver, choroid plexuses, retinal pigment epithelial cells, and pancreas[5,6]. It plays an essential role in the transport of thyroxine and vitamin A in vertebrates[4]. To date, more than 140 amyloidogenic TTR variants have been identified worldwide[2,7]. About 30 variants are associated with ocular involvement[8]. Mutations in TTR are a major cause of hereditary vitreous amyloidosis, with vitreous involvement occurring in 5.4%−35% of ATTRv patients[7,9].
Vitreous opacity (VO) may represent the first or even the sole clinical manifestation in patients with certain TTR mutations, such as Gly83Arg (c.307G > C) and Ile107Met (c.381T > G)[10,11]. Although the TTR Gly83Arg variant is rare worldwide, it is one of the most prevalent TTR variants in China and is associated with a characteristic phenotype, including VO, other ocular abnormalities, polyneuropathy, and cardiac amyloidosis[11−27]. Clinical manifestations show their relationship with neuro-ophthalmology. In this study, we conduct a comprehensive literature review of all reported ATTRG83R cases to provide a more complete understanding of the genotype–phenotype correlations.
Literature review of transthyretin Gly83Arg amyloidosis cases
-
Multiple search strategies of the title and abstract fields, including "hereditary transthyretin amyloidosis AND Gly83Arg", "transthyretin AND Gly83Arg", "ATTR G83R", "transthyretin AND p.Gly103Arg", "transthyretin AND p. G103R", "transthyretin AND c.307G > C", and "vitreous amyloidosis AND transthyretin AND Gly83Arg", were conducted on 1 August 2025 in PubMed (
http://pubmed.ncbi.nlm.nih.gov ). Since this genotype has only been reported in China until now, the same search terms were translated into Chinese and were applied to a Chinese literature database, CNKI (www.cnki.net ). The inclusion criteria were as follows: (1) studies reported cases with ATTR Gly83Arg; (2) papers written in English or in Chinese with an English abstract. The exclusion criteria were as follows: (1) studies with no ATTR Gly83Arg case presented; (2) review articles or basic medical research; (3) papers written in Chinese with no English abstract or in a language other than English or Chinese. The characteristics of family history, onset age, genetic findings, and clinical manifestations in these studies were acquired to make a summary table.After removing duplicate and previously reported cases, a total of 25 publications were identified (Fig. 1). Two were written entirely in Chinese, and one review article and six basic research articles that lacked clinical data, were excluded, meaning that 12 case series (16 articles) were included in the analysis. In total, 17 unrelated families and 80 individuals with ATTRG83R amyloidosis have been reported since the first description in 2008[28] (Table 1). Most studies (7/12, 58.3%) originated from southwestern China. The majority of patients (79/80, 98.8%) presented with blurred vision as the initial symptom, with VO as the first ophthalmic finding. Only one patient initially experienced upper limb numbness[24].
Figure 1.
Flow diagram of the literature search, exclusion, and inclusion.
Table 1. Family history features and clinical manifestations of all reported ATTRG83R amyloidosis cases.
Study
No.Author, year Number of
familiesFamily members
(Gly83Arg/all affected)Onset age (yrs),
mean (range)Clinical manifestation Region, ethnic
groupRef. VO Beyond VO Other systems 1 Xu 2025;
Xu 20131 18/25 47 (37−61) + RAA, NV, GLC, RD − Guizhou, China, NA [13,14]* 2 Chen 2025;
Su 2023;
Xie 20171 14/20 41 (29−52) + VH, RAA, NV, GLC PN Guizhou, China, Han [15−17] 3 Xiong 2024 1 3/10 NA + − PN China, NA [18] 4 Feng 2023 1 2/8 46 + RD − China, NA [19] 5 Shen 2022 1 4/9 35 (31−41) + VH, RAA − China, NA [20] 6 Li 2021 3 Family I: 3/6;
Family II: 1/5;
Family III: 2/335 (34−36);
42;
43 (42−43);
Total 39 (34−43)+ − PN, AN, heart China, Han [21] 7 Yu 2021 1 5/8 NA (32−43) + GLC − Guizhou, China, Han [22]* 8 Yin 2014 1 2/3 NA + − − Yunnan, China, NA [23] 9 Liu 2014 1 12/22 41 (37−47) + RAA, GLC PN, heart Guizhou, China, NA [24] 10 Zhang 2013 3 Family A: 5/11
Family B: 2/8
Family C: 2/734 (30−19);
43 (40−45);
40 (40−40);
Total 37 (30−45)+ − − Yunnan, China, Han [25] 11 Xie 2012 1 3/4 40s + − − Guizhou, China, Li [26]* 12 Chen 2011;
Chen 20082 Family A: 1/6;
Family B: 1/9NA (41−43) + RAA − China, NA [27,28]* Family members (Gly83Arg/all affected), the number of affected family members who were confirmed to carry the TTR Gly83Arg variant divided by the number of all symptomatic family members. Beyond VO, ocular manifestations other than vitreous opacity. NA, not available; RAA, retinal amyloid angiopathy; NV, neovascularization; GLC, glaucoma; RD, retinal detachment; VH, vitreous hemorrhage; PN, peripheral neuropathy; AN, autonomic neuropathy; heart, cardiac involvement; Han, Han ethnic group; Li, Li ethnic group. * The paper was published in Chinese with an English abstract. The mean onset age of most cases ranged across 30−47 years (range, 29 to 61 years). Beyond VO, frequent ocular complications included retinal amyloid angiopathy (RAA), secondary glaucoma (GLC), and tractional retinal detachment (RD). The reported sequelae of RAA encompassed retinal microangiomas, retinal vessel sheaths, retinal ischemia, vessel wall staining in fundus fluorescein angiography, retinal or iris neovascularization and vitreous hemorrhage (VH).
Although ATTRv is classified as a systemic amyloidosis[29], ATTRG83R typically manifests as ocular findings first (Table 1). Peripheral neuropathy (PN) was the most common extraocular manifestation. A comprehensive study combining 3 new and 41 previously reported cases was included[18]. Among 32 ATTRG83R amyloidosis patients who underwent a neurological examination, 53.1% (17/32) presented with PN, such as sensorimotor polyneuropathy (SMPN) and carpal tunnel syndrome (CTS) characterized by limb numbness, weakness, muscle atrophy, decreased sensation, and abnormal neuroelectrophysiological findings[18,21]. SMPN developed at 7.2 ± 5.5 years on average (range: 3−15 years) after the disease's onset[18]. No central nervous system involvement has been reported. Autonomic neuropathy (AN) occurred in 13.7% (6/44) of 44 patients reviewed, presenting as a positive upright tilt test result (syncope or orthostatic hypotension), diarrhea, constipation, dyspareunia, or dry mouth[18].
Of 28 patients who underwent a cardiac evaluation, only 3 (10.7%) exhibited cardiac abnormalities, including cardiac hypertrophy or atrioventricular blockage, substantially fewer than in cardiomyopathy-predominant variants such as ATTRV122I[30].
Genetic testing of asymptomatic family members in several reports revealed carriers without clinical manifestations (Studies 2−4 and 6−12). These individuals were presumed to be below the typical age of onset[14,20,24,25].
Visual prognosis after pars plana vitrectomy (PPV) was generally favorable, with a marked improvement in best-corrected visual acuity (BCVA) postoperatively[24]. However, VO recurrence was common several years after surgery[16,23,24]. A recent 15-year follow-up study of 14 ATTRG83R patients demonstrated VO recurrence in 94.4% of cases at approximately 8 years post-PPV[15]. Secondary glaucoma developed in 33%–36% of patients, often 2–20 years after surgery[25,26], and the mean onset age was 53−55 years (range: 43−65 years)[13,15]. Retinal photocoagulation was applied for nonperfusion areas in the retina to prevent neovascularization, and intravitreal anti-vascular endothelial growth factor (VEGF) was used to treat neovascular glaucoma[13]. Multiple treatments might be required to control intraocular pressure in GLC patients, including antiglaucoma medication or surgery, but the visual prognosis in glaucoma-affected eyes remained poor[15].
To date, no disease-related deaths have been reported, likely reflecting the low prevalence of cardiac amyloidosis in this genotype.
Critical role of TTR Gly83
-
A schematic of the TTR variant was drawn (Fig. 2a). Multiple sequence alignment revealed that glycine at Position 83 of TTR (Position 103 of the TTR precursor) is highly conserved across vertebrate species, suggesting its functional importance (Fig. 2b). Wild-type TTR is a tetramer comprising four identical subunits. Previous studies have revealed that glycine at Position 83 of the TTR protein is a part of the EF loop and Schellman helix C-capping motif, which helps stabilize the EF helix structure (Fig. 2c)[31,32]. Through a protein–protein interaction (PPI) network, retinol-binding protein 4 (RBP4) was found to have the closest relationship with TTR (combined score: 0.999) (Fig. 2d). Structural prediction of the missense mutation for TTR-RBP4's biological assembly (Protein Data Bank [PDB] ID: 1QAB) was performed, and structural damage of an impermissible phi/psi angle was detected by Missense3D (Fig. 2e).
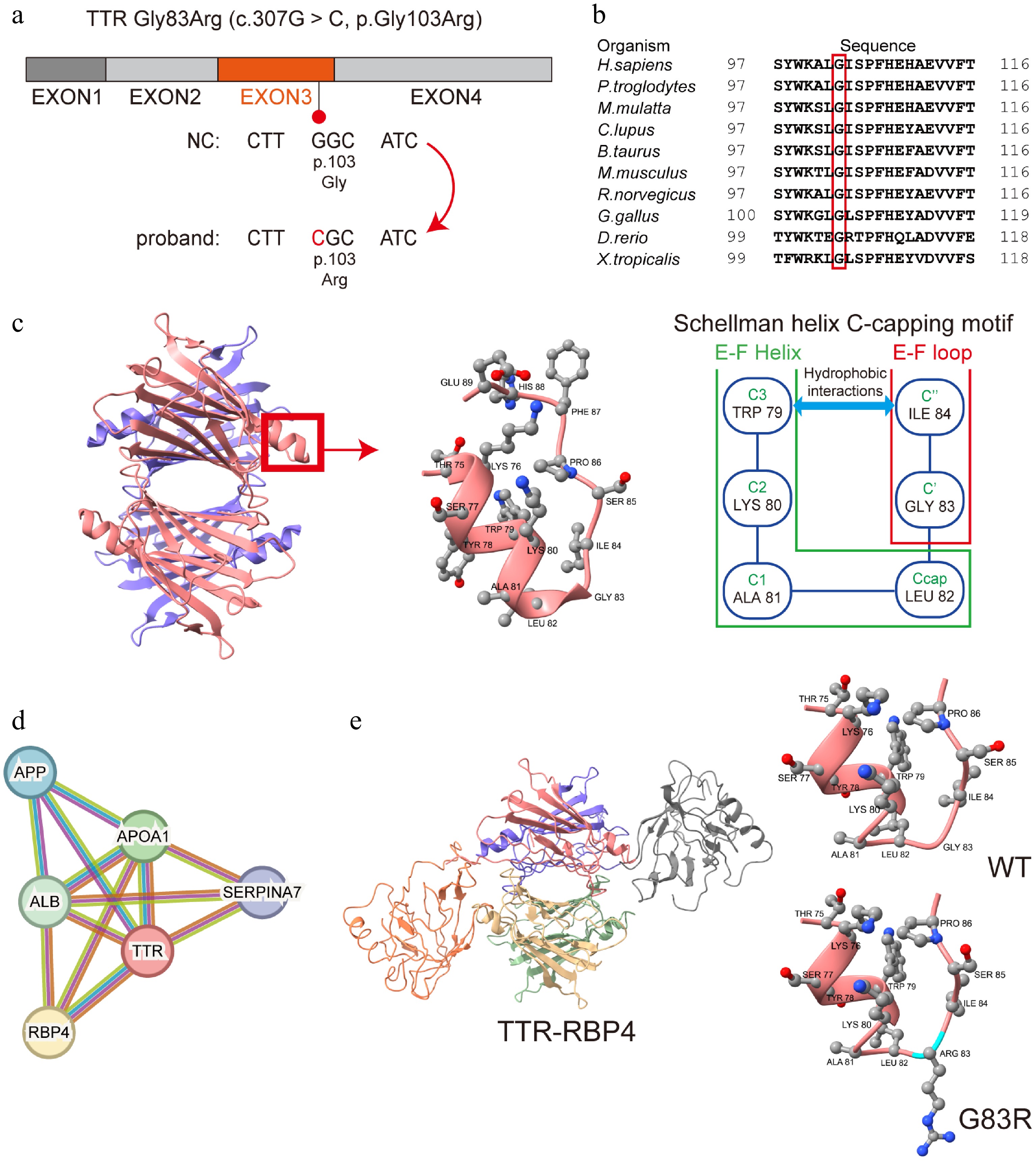
Figure 2.
Evolutionary conservation and structural prediction of the TTR Gly83Arg variant. (a) The schematic of the TTR gene and the c.307G > C mutation in Exon 3. (b) The glycine at Position 83 of TTR is highly conserved among humans and other organisms. (c) Structural graphic of the TTR protein's tetramer, EF helix (Thr75-Leu82), and EF loop (Gly83−Glu89) and a schematic of the Schellman helix C-capping motif described in a previous study[31]. (d) The PPI network shows a close relationship between the TTR protein and RBP4. (e) Graphic of TTR-RBP4 and the structural prediction of TTR Gly83Arg substitution. Disallowed phi/psi angles are detected. TTR, transthyretin; PPI, protein-protein interaction; RBP4, retinol-binding protein 4.
-
Transthyretin amyloidosis consists of wild-type transthyretin amyloidosis (ATTRwt) and hereditary transthyretin amyloidosis (ATTRv). ATTRwt manifests as heart involvement in males and could affect the lung, ligaments, and tenosynovium in senile individuals[29], whereas ATTRv manifests as involvement in various systems[7]. ATTRv is the most common type of autosomal-dominant inherited amyloidosis, with endemic distribution differences and disease penetrance[1,6]. Transthyretin cardiac amyloidosis with the variant TTR is associated with accumulated amyloid fibrils in the heart, resulting in ventricular wall thickening and severe diastolic dysfunction[33]. ATTRv also has a close relationship with neuro-ophthalmology, as some phenotypes could affect both the nervous system and eyes[34]. The peripheral nervous system's involvement includes carpal tunnel syndrome, upper limb numbness, dissociated sensory disturbance, or pain in the extremities[6]. Neurophysiological tests such as electromyograms are significant in detecting abnormal changes such as decreased neurosensory-motor conduction velocity or decreased action potential amplitude[18,35]. Patients with an affected autonomic nervous system could develop symptoms including urinary disturbance, erectile dysfunction, and orthostatic hypotension[6]. Central nervous system involvement, such as radiculopathy, subarachnoid and intraparenchymal hemorrhage, stroke-like episodes, and decreased consciousness, has been reported[34]. Renal impairment and gastrointestinal manifestations such as diarrhea, constipation, persistent nausea, vomiting, and inexplicable weight loss were also reported[36].
ATTRv with ophthalmologic manifestations was first reported in 1953[2]. Ocular involvement occurs in approximately 10% of ATTRv patients, and VO is the most common[20]. TTR variants mainly affecting the eyes include Arg34Gly (c.160A > G), Lys35Thr (c.164A > C), Trp41Leu (c.182G > T), Tyr69His (c.265T > C), Gly83Arg (c.307G > C), and Ile107Met (c.381T > G)[6,10]. There are complicated pathogeneses and genotype-phenotype correlations of ATTRv, as different missense point mutations in the same locus could result in different phenotypes[37,38].
Liver transplantation is useful for treating ATTRv-related peripheral neuropathy and autonomic neuropathy. However, it is not the first-line treatment, as about 10% of cases show liver transplantation-related morbidity and mortality[39]. Treatments for ATTRv or ATTRwt have advanced in recent years. The TTR-tetramer stabilizer Tafamidis showed clinical efficacy in reducing mortality related to hereditary transthyretin amyloid polyneuropathy (ATTRv-PN) and transthyretin amyloid cardiomyopathy (ATTR-CM) and improving quality of life[40,41]. Gene silencing has emerged as an ATTR therapy in recent years, including antisense oligonucleotide (ASO) and small interfering RNA (siRNA) silencing[39]. Inotersen is a kind of ASO that selectively binds to TTR mRNA and prevents subsequent TTR production in the liver, which was clinically effective for both ATTRv-PN and ATTR-CM[42,43]. Patisiran is a TTR-specific siRNA that targets the untranslated region of mRNA in primates, reducing TTR production[44]. However, ASO and siRNA do not efficiently cross the blood-brain barrier[39]. Although Tafamidis was detected in the vitreous bodies of patients using oral doses of Tafamidis during PPV, the concentration of Tafamidis in the vitreous body is about 0.6% of that in the plasma[45]. These newly developed drugs might have limited efficacy in ATTR-related VO.
ATTRG83R manifests as more than VO
-
To date, the TTR Gly83Arg variant has been discovered only in China. In the Chinese population, ATTRG83R is the most frequently reported[11] and most common genotype of ATTRv[12]. This variant might be a hot spot in ATTRv research in China. A positive family history was noted in all ATTRG83R patients. ATTRG83R amyloidosis mainly develops in patients in their 30s or 40s. Previous studies have pointed out that VO first appears in the area near the retinal vessels of the posterior pole[21]. VO is the only abnormality in some studies of ATTRG83R amyloidosis[17,23,25,27]. However, the reason why RAA or glaucoma was not found might be the relatively short follow-up period.
The multimodal imaging of VO includes the findings of slit lamp tests, fundus examinations, B-scan ultrasonography, and optical coherence tomography (OCT). A fundus examination may present various grades of VO, which appear in the posterior pole first. Anterior VO is called pseudopodia lentis, which has an attachment point on the posterior lens capsule. B-scan ultrasonography could reveal irregular strip-like and fleck-like hyper-echoic signals within the vitreous cavity. Recently, characteristic needle-shaped deposits on the retinal surface observed by OCT and curvilinear vitreous humor deposits strands captured by three-dimensional reconstruction were noted in ATTRv patients presenting with VO[46,47].
Complete removal of amyloidosis-related VO is essential during PPV. VO's recurrence might result from continuous intraocular TTR production or incomplete vitrectomy without posterior vitreous detachment (PVD)[23]. One study demonstrated that 25% (3/12) of ATTRG83R patients had not undergone complete PVD during PPV because of strong retinal-VO adherence and ultimately developed recurrent VO[24]. In a long-term follow-up study, 94.4% of ATTRG83R amyloidosis patients developed recurrent VO 8 years after PPV, indicating continuous intraocular TTR production[15]. Hence, regular ophthalmic follow-up every 3 months was recommended for asymptomatic ATTRv patients with VO[15]. For post-PPV patients, more frequent fundus examinations and intraocular pressure tests during follow-up were required to avoid further visual impairment[15]. Repeat PPV and phacoemulsification could improve visual acuity after recurrence, as most recurrent VO started from the peripheral retina and went to the retrolental vitreous area[15]. Panretinal laser photocoagulation (PRP) was reported to alleviate ocular ATTRv progression, as it damages the retinal pigment epithelium[48]. PPV combined with PRP might prevent the recurrence of amyloidosis[15].
RAA includes retinal microangiomas, retinal vessel sheaths, retinal ischemia, and vessel wall staining in fundus fluorescein angiography (FFA). As the disease progresses, severe complications such as neovascularization, retinal or vitreous hemorrhages, and even tractional retinal detachment might occur, leading to poor prognosis[13]. Long-term follow-up showed that all post-PPV patients who underwent FFA were found to develop RAA 3 years on average (range: 1−15 years) after disease onset[13]. Therefore, it was recommended to perform FFA every year for post-PPV ATTRv patients[15]. Wild-field optical coherence tomography angiography (OCTA) is useful and convenient in detecting microangiomas, retinal nonperfusion areas, and neovascularization in the mid-peripheral and peripheral retina, but it cannot fully replace FFA in visualizing vessel wall staining and dye leakage.
One major complication of ATTRG83R patients is secondary glaucoma. The incidence of this visual complication ranges from 17% to 24% in ATTRv patients[24]. A long-term study of ATTRG83R revealed that the prevalence rate of secondary glaucoma is 33%−36%[13]. Patients could develop secondary glaucoma 6−15 years after the onset of the symptoms of VO[24].
ATTRG83R patients could develop peripheral neuropathy or autonomic neuropathy years after disease onset, with an average of 7.2 years for sensorimotor polyneuropathy[18]. Only three ATTRG83R amyloidosis cases with cardiac involvement were reported[21,24]. It remains uncertain whether ATTRG83R amyloidosis has a low prevalence of cardiac involvement or takes a long time to lead to cardiomyopathy. The mechanism of amyloidosis deposits appearing in the vitreous body earlier than in other affected tissues remains to be elucidated. It was hypothesized that vitreous collagen fibers provide a scaffold for TTR, as TTR has a high affinity for both basement membranes and the vitreous matrix, both of which are predominantly composed of Type 2 collagen and share structural and biochemical similarities[7].
Fibrillogenic mechanisms of hereditary transthyretin amyloidosis
-
TTR, a tetramer of four identical protein subunits of 127 amino acids, is synthesized in the liver (approximately 90%), choroid plexuses, retinal pigment epithelial cells, and pancreas. It is widely distributed in the eye, and the majority of TTR within the eyes is locally produced by the retinal pigmentary epithelium cells[7,49]. Continuous progression of ocular manifestations in ATTRv patients after liver transplantation supports this view[7]. The physiological functions of TTR in the eye have not yet been completely understood. It may play a significant role in retinol metabolism in the interphotoreceptor matrix[49].
TTR fibrillogenesis follows a downhill polymerization mechanism, in which partial unfolding of the tetramer and misfolding of the monomers initiate the amyloid aggregation cascade[31]. Wild-type TTR is inherently amyloidogenic[29,50]. Cryo-electron microscopy revealed a side-by-side intertwining of additional protofilaments to form vitreous amyloid fibrils[9]. The pathogenesis of TTR Gly83Arg variant amyloidosis remains to be clarified. One study noted the Gly83 residue located at the EF loop, which functioned as a classic Schellman helix C-capping motif of TTR and helped stabilize the EF helix structure[31]. As one of the key sites of the Schellman motif, the Gly83 residue must be in the αL backbone conformation, with a positive φ angle[32]. Substitution of glycine by arginine in this highly conserved position would lead to an unfavorable αL conformation, which destabilizes TTR and results in a sixfold increase in the monomer aggregation rate[31]. Additionally, Gly83Arg mainly promotes amyloidosis by increasing the monomers' instability, as Gly83Arg and wild-type TTR had basically equivalent depolymerization rates[31].
Other potential mechanisms of ATTRv amyloidosis have been illustrated. Certain amino acid sites are critical for tetramers' stability as they have a specific electrostatic potential. A missense variant might cause a surface-level electrostatic alteration, resulting in enhanced electrostatic repulsion and a decrease in tetramer stability[51]. Glycine is a nonpolar neutral amino acid, whereas arginine carries a positive charge. Replacing glycine with arginine could also introduce surface electrostatic alterations. Whether this mechanism contributes to ATTRG83R amyloidosis requires further exploration. Moreover, a proteolytic cleavage pathway was found in ATTRv amyloidosis. TTR's Ser52Pro substitution increased the susceptibility of selective proteolytic TTR cleavage, and the polypeptide fragment would undergo a distinct pathway of TTR amyloidogenesis[33].
In this review, we found RBP4 to have a close relationship with TTR through protein-protein interactions. Structural damage of an impermissible phi/psi angle was detected in the Gly83Arg variant of TTR-RBP4. Retinol and RBP4 can stabilize the structure of the TTR tetramer through interactions[52]. The Gly83 residue of TTR is in contact with the Leu35 of RBP4, as well as with the hydroxyl group of retinol[53]. Replacing Gly83 with Arg83 might disrupt this interaction, resulting in an unstable compound structure and subsequent depolymerization. Further in vitro and in vivo experiments are required to confirm the significance of decreased TTR-RBP4 stability in ATTRG83R amyloidogenesis.
Given multimodal ophthalmic imaging and the theoretical knowledge presented above, we propose a hypothesis of amyloid VO formation in ATTRG83R. The variant TTR may be synthesized by retinal pigmentary epithelium cells and is secreted predominantly in an apical direction. With increasing monomer instability, amyloid TTR deposits could appear in the inner retinal layers. As the disease progresses, these insoluble amyloid fibrils reach the vitreous humor at some sites that are close to the retinal vessels in an unknown way. This could result in VO close to the retinal vessels, which might account for the strong adhesion between VO and the retina during vitrectomy. These amyloid deposits might also cause retinal angiopathy. If patients do not receive a proper intervention in time, more and more amyloid deposits would aggregate in the vitreous body and finally result in severe visual loss.
This work was supported by the Science and Technology Program of Guangzhou, China (Grant No. SL2022A03J00452); the Higher Education Quality and Teaching Reform Construction Project for Undergraduate Education in Guangdong Province, China (Grant No. [2024] 9); Sun Yat-sen University's 2023 University-level Undergraduate Teaching Quality Engineering Project (Grant No. 2023-394-39); the Bethune Lumitin Research Funding for Young and Middle-Aged Ophthalmologists (Grant No. BJ-LM2021014J); the Ophthalmology New Technology Incubation Fund (Y2025FH-YKYSJSJ07-08); the Projects of Research Center for Sharp Vision at The Hong Kong Polytechnic University (Grant No. P0057931); and the Health and Medical Research Fund – Research Fellowship Scheme, Health Bureau, Hong Kong (Grant No. 07210207).
-
Not applicable.
-
The authors confirm their contributions to the paper as follows: conceptualization: Liu Z, Ma J, Lin Y; data curation: Liu Z, Cui J; formal analysis: Liu Z, Cui J, Wu K; funding acquisition: Tan S, Lin Y; investigation: Liu Z, Cui J, Wu K, Liu X; methodology: Liu Z, Cui J, He J, Lian P, Leng F; project administration: Ma J, Lin Y; resources: Lin Y; supervision: Lian P, Leng F, Tan S, Ma J, Lin Y; validation: Cui J; visualization: Liu Z, Wu K, Liu X, He J, Song Y; writing – original draft: Liu Z; writing – review and editing: Tan S, Ma J, Lin Y. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article.
-
The authors have no conflict of interest.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Liu Z, Cui J, Wu K, Liu X, He J, et al. 2026. Hereditary transthyretin amyloidosis: a literature review of ATTRG83R. Visual Neuroscience 43: e019 doi: 10.48130/vns-0026-0018 

Hereditary transthyretin amyloidosis: a literature review of ATTRG83R
- Received: 16 November 2025
- Revised: 28 December 2025
- Accepted: 09 January 2026
- Published online: 28 April 2026
Abstract: Hereditary transthyretin amyloidosis (ATTRv) with Gly83Arg (G83R) variant is predominantly found in China and is characterized by prominent ocular involvement. This literature review, encompassing 80 reported cases, demonstrates that vitreous opacity (VO) is the most common initial manifestation (79/80, 98.8%), typically presenting in the 30–40 s range. Ocular involvement often extends beyond VO to include retinal amyloid angiopathy (RAA), secondary glaucoma (GLC), and tractional retinal detachment (RD). Systemic manifestations are not uncommon, with peripheral neuropathy (PN) being the most frequent extraocular feature, often developing years after the onset of ocular symptoms. In contrast, cardiac involvement is rare. Although pars plana vitrectomy improves vision, VO's recurrence is high. These findings establish ATTRG83R as an ocular-predominant ATTRv subtype, for which timely diagnosis and awareness of the potential for both progressive ocular complications and systemic neuropathy are crucial for management.