









-
Plant secondary metabolites represent an almost inexhaustible, diverse source of new drug candidates, which can serve as leads for the production of synthetic or semi-synthetic drugs. Natural products have been sources of inspiration for the design of new therapeutic agents[1], including highly successful plant-derived compounds with anti-cancer activity that have been approved for clinical use against various cancers such as paclitaxel, vincristine, irinotecan (inspired by camptothecin) and etoposide (semi-synthesised from podophyllotoxin)[2].
Plant natural products are a highly heterogeneous group of compounds often with diverse, molecular properties compared to synthetic compounds and drugs[3]. They may occupy parts of the chemical space largely unexplored by screening available libraries while at the same time adhering to Lipinski's rule-of-five or other ‘drug-likeness’ descriptors[3]. The pharmaceutical industry has focused much of its attention on easy-to-synthesise chemicals produced by combinatorial chemistry, selected to interact with a narrow range of disease targets, such as kinase inhibitors. These have proved highly valuable, although many of the lowest hanging fruit have now been picked[4]. While there remains interest in developing small molecule drugs for cancer[5], exploring large numbers and diverse types of drug-like compounds is a challenge. However, new chemical leads can still be identified from traditional medicines, where long-standing ethnobotanical applications support more detailed investigation. The meeting point between characterisation of natural plant products and subsequent refinements in production and therapeutic use could be an immensely productive area for exploration.
Colorectal cancer is the third most frequent malignant disease, globally (1.85 million new cases/year; 10.2% of total malignancies)[6]. Provided that current predictions are reliable, the global burden of colorectal cancer is expected to be 2.2 million new cases per year by 2030, a further 20% increase[7]. However, more than one‐half of all cases are attributable to modifiable risk factors and are consequently preventable[8]. Given the potentially preventable nature of colon and many other cancers, natural plant products may have an expanding role to play in treating human disease via chemoprevention. This represents a complementary approach in the battle against cancer[2], involving the ingestion of compounds to prevent or delay carcinogenic processes[2], by targeting emerging cancer cells[9]. Many chemopreventive secondary metabolites in plant extracts as well as purified molecules isolated from teas, herbs, spices, fruits and vegetables have been explored[2]. It is highly likely that many more remain to be discovered.
Scutellaria barbata D.Don is a key medicinal plant species in the Lamiaceae (the mint family) and the aerial parts have been used traditionally for treating cancer in many Asian countries, where it is known as Ban Zhi Lian (半枝莲). It produces copious amounts of specialized metabolites including diterpenoids (some containing nitrogen such that they are also classified as alkaloids), flavones, steroids, and polysaccharides[10]. Previous investigations have identified two main classes of bioactive constituents – clerodane diterpenoids and 4’-deoxyflavones[11].
Mechanistically, natural products should target tumorigenic processes to be beneficial as either therapeutic or preventative agents. Central to cancer development is the inactivation of apoptosis, considered one of its fundamental hallmarks[12]. Apoptosis is transduced by either intrinsic (mitochondrial) or extrinsic (death receptor) pathways[13]. Both pathways culminate in the activation of the effector caspase 3, a highly specific cysteine protease essential for the programmed destruction of the cell[14]. The apoptotic response is one that acts to cull cells that are proliferating aberrantly or that have suffered DNA damage, such as cells with checkpoint or repair defects[12]. Indeed, to become malignant, a cell must switch off the apoptotic pathway[12]. Once done, the susceptibility of cancer cells to apoptosis is severely compromised, and these cells are less receptive to chemotherapeutic agents[12]. Apoptosis is a tightly regulated process balancing both pro-apoptotic and pro-survival proteins[15]. Mechanisms are in place to regulate this, including inhibitors of apoptosis (IAPs)[15]. In cancer, pro-apoptotic factors are suppressed and pro-survival proteins, such as IAPs are upregulated, promoting uncontrolled cell division[16]. The IAP proteins represent a group of endogenous inhibitors of caspases and cell death which share BIR protein-protein interaction domains essential for their function[14,17]. Due to their prominent ability to control cell death and their elevated expression in a variety of cancer cell types, IAP proteins are prime targets for the development of new anti-cancer treatments[17,18]. Killing of tumour cells by cytotoxic approaches such as anticancer drugs, γ-irradiation, suicide genes, or immunotherapy are all mediated through induction of apoptosis in target cells[19]. Malignant cells can also acquire resistance to apoptosis[20]. Broadly, resistance mechanisms can be divided between disruptors of the balance between pro-apoptotic proteins (reduced in malignant cells) and pro-survival proteins (increased in malignant cells) and impaired death receptor signalling linked to reduced caspase expression[20].
Here we report characterisation of the major diterpenoid in S. barbata leaves, scutebarbatine A, and show that it is produced in specialized peltate trichomes, not previously described in species of Scutellaria. We show that scutebarbatine A induces apoptosis specifically in human colon cancer cells, and not in normal colonic epithelial cells, through the inhibition of IAPs. Methanolic extracts of S. barbata leaves are also cytotoxic to cancer cells but work by mechanisms that can not be accounted for by the activity of scutebarbatine A alone. This implies polyvalency, with scutebarbatine A promoting apoptosis together with other bioactives present in leaves of S. barbata. Comparisons of the bioactivity of methanolic extracts to traditional aqueous decoctions (Ban Zhi Lian) showed different compositional profiles of diterpenoids and flavonoids, although both contained scutebarbatine A. Our results establish leaves of S. barbata as an important source of anti-cancer metabolites which could be further developed as chemopreventive drug leads by breeding to increase diterpenoid production or by process refinement involving switching to alcohol based extraction to improve diterpenoid extraction. More broadly, our results illustrate how the understanding of plant specialized metabolism combined with traditional medicine can be harnessed to identify potential new chemo- or combination therapies for the treatment of cancer.
-
Traditional decoctions of Ban Zhi Lian are prepared from dried aerial tissues of S. barbata, from which two major classes of bioactives have been reported, clerodane diterpenoids and flavonoids, particularly 4’-deoxyflavones such as baicalein and wogonin. It is the latter, 4’-deoxyflavones, that confer the cytotoxic bioactivity of another medicinal species in the genus, S. baicalensis Georgi roots, known as Huang Qin[21,22]. We compared the 4’-deoxyflavone content of S. barbata in leaves and roots and found that like S. baicalensis, the specialised 4’-deoxyflavones were present almost exclusively in extracts from roots (Supplemental Fig. S1). Therefore, we focused on the clerodane diterpenoid bioactives which accumulate predominantly in aerial tissues of S. barbata.
Scutebarbatine A was identified in methanolic extracts of aerial tissues of S. barbata by comparison to a commercial reference standard, (Purifa, Chengdu, China) verified by NMR spectroscopy (Fig. 1a, Supplemental Fig. S2). Having identified scutebarbatine A in these extracts, the amounts in different tissues of S. barbata were determined, and it was found to be most abundant in leaves and flowers, with much lower concentrations occurring in roots and stem tissues (Table 1, Supplemental Fig. S3).
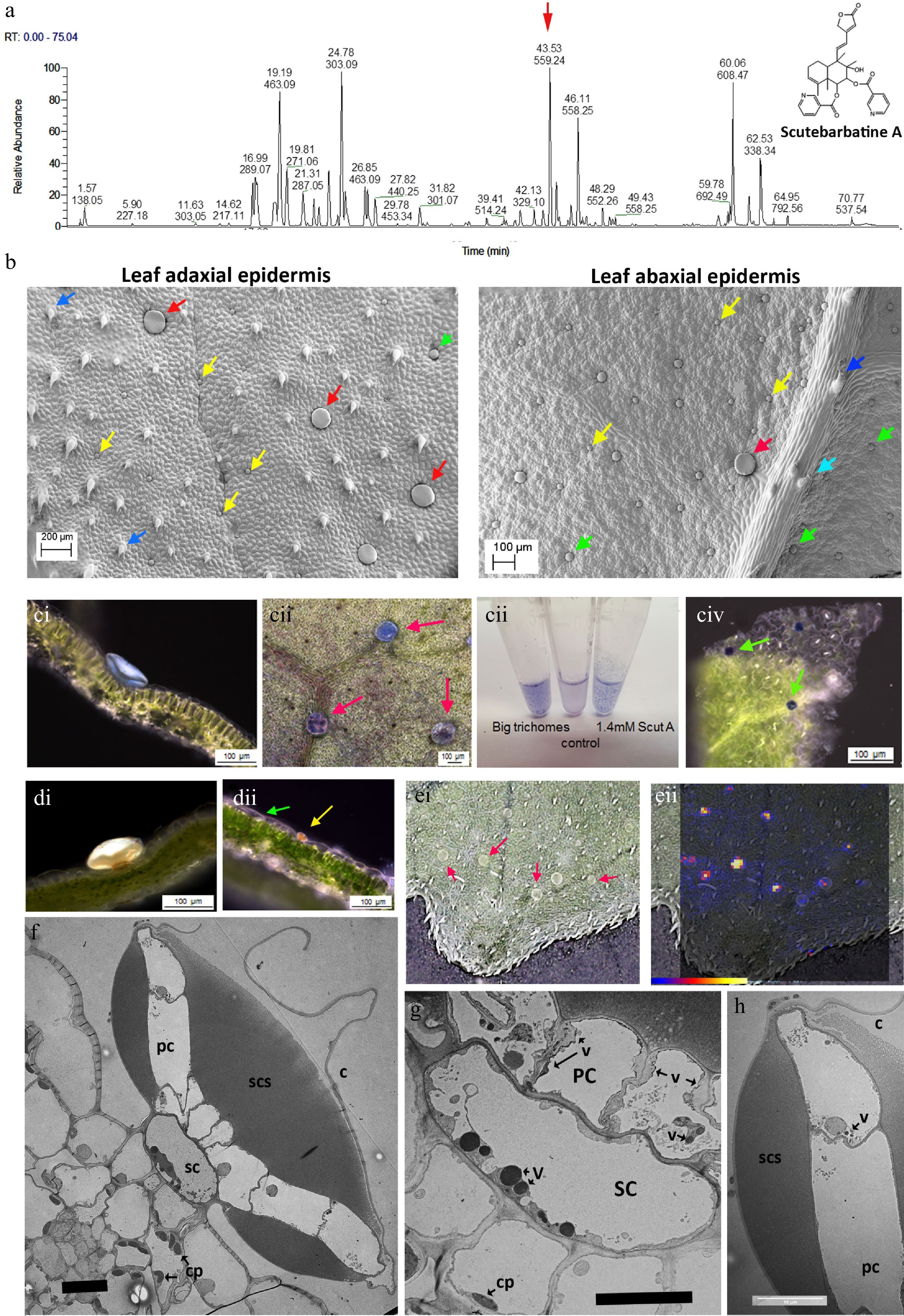
Figure 1.
Scutebarbatine A is the major clerodane diterpenoid and localises to a specialised type of peltate trichome on S. barbata leaves. (a) UV profile of methanolic extract of leaves from leaves of S. barbata. Each peak is labelled with its migration time and mass. The red arrow indicates the peak with mass 559.24, scutebarbatine A. Tentative identification of other peaks is provided in Supplemental Table S1, based on MS3 fragmentation profiles, accurate mass and in some cases NMR analysis. (b) Cryo-SEM micrographs showing the large (red arrows), medium (green arrows) and small (yellow arrows) peltate trichomes on the mature leaves of S. barbata. Images of the upper, adaxial leaf surface are shown on the left and on the lower abaxial leaf surface at 2x magnification on the right. Dark blue arrows indicate non-glandular trichomes and the pale blue arrow indicates a capitate glandular trichome. (c) NaDi staining of leaves of S. barbata for terpenoids, (c i) A large trichome on the adaxal leaf surface staining light blue. (c ii) Surface view of large trichomes stained with NaDi. The leaf has been decolorized in ethanol. (c iii) NaDi staining of large trichomes hand picked into methanol, methanol alone and purified scutebarbatine A. (c iv) Surface view of NaDi staining of medium trichomes. (d) Sudan IV staining of trichomes for lipids. (d i) A large trichome. (d ii) a small trichome. (e) MALDI-TOF images, showing location of mass 559. (e i) Shows a photograph of the leaf imaged with large trichomes indicated by red arrows. (e ii) shows scutebarbatine A (mass 559) localization indicated by red-yellow fluorescence on the leaf surface shown to be in the large trichome by overlaying the MALDI-image on the leaf surface photograph. Scale bar for signal intensity is shown on (e ii). (f−h) TEM micrographs showing a transverse sections of large peltate trichomes. (f) TEM of entire large trichome showing single stalk cell atop the adaxial leaf epidermis. The stalk cell (SC) supports a plate of cells (PC), one cell thick which exudes or secretes terpenoids into the sub-cuticular space (SCS) which lies on either side of the cell plate. This differs from the structure of the small and medium peltate trichomes which have sub-cuticular spaces above only the outer surface of the gland cells. C = cuticle of subcuticular space. (g) Detail of the stalk cell (SC) v = electron-dense vesicle, cp = chloroplast. (f) Displaying dense vacuolar material. (h) Detail of the outer cell of the plate of cells constituting the glandular head of a large peltate trichome. The contents of the subcuticular space adopt a honeycomb-like appearance in TEMs suggesting a solid or resinous structure rather than liquid contents, which does not diffuse after the cuticle has been ruptured. Scale bars for TEMs in (f), (g) and (h) represent 10 µm.
Table 1. Contents of scutebarbatine A in different organs of S. barbata.
Tissue Content (ng/g fresh weight) Root 0.38 Flower 105.26 Young stem 1.75 Old stem 1 Leaf (< 0.5 cm) 922.76 Leaf (0.5−1 cm) 953.84 Leaf (1−2 cm) 551.42 Leaf (> 2 cm) 408.90 Of all the specialized cell types in plants, trichomes are renowned for their ability to store and secrete specialized metabolites[23]. Among the different trichome types, glandular trichomes are akin to specialized metabolite factories[23]. These consist of differentiated basal, stalk and apical cells and can be found on approximately 30% of all vascular plants[24]. In S. barbata, leaves, stems and flowers are covered in multicellular trichomes, particularly glandular trichomes. Investigation of leaves and stems of S. barbata by cryo-scanning electron microscopy (SEM) identified one non-glandular type and four glandular multicellular trichome types on the leaves of S. barbata. Of the glandular trichome types one was capitate, with a relatively long multicellular stalk and a single-celled glandular head, and was most profuse around the leaf margins. The three peltate trichome types could be distinguished by their size; small (25−32 μm diameter), medium (45−55 μm diameter) and large (100−150 μm diameter) (Fig. 1b). Only small and medium-sized trichomes were observed on stems. The large peltate trichomes were localised predominantly on the adaxial surface of S. barbata leaves especially towards the leaf margins and there were fewer around the midrib. Although present on the abaxial leaf epidermis these large peltate trichomes are far less common on the under surface of the leaves.
To determine which trichomes, if any, synthesised clerodane diterpenoids, we stained leaf tissue with napthol + dimethylparaphenyldiamine (NaDi) stain (for terpenoids) and Sudan IV (for lipids, triglycerides and lipoproteins). NaDi stained the medium sized peltate trichomes the classic dark blue colour indicating the presence of terpenoids (Fig. 1c iv). NaDi also stained the large trichomes a distinct light blue colour, suggesting that they also contained terpenoids but different ones to the medium-sized trichomes (Fig. 1c i & 1c ii). We examined the staining of our purified standard, scutebarbatine A, and found that addition of stain produced the same light blue colour as the large trichomes (Fig. 1c iii). The large trichomes were big enough to allow them to be picked from leaves under a dissecting microscope and staining of these selected trichomes gave the same pale blue colour (Fig. 1c). Sudan IV stained the small peltate trichomes strongly and stained the large peltate trichomes weakly, confirming the metabolite differences between peltate trichomes of different sizes (Fig. 1d i & 1d ii). We also undertook matrix-assisted laser desorption/ionization (MALDI) imaging of leaves to identify the site of scutebarbatine A production and confirmed that an ion with the predicted m/z of 559.24 was present in the large peltate trichomes (Fig. 1e ii). When the large trichomes were picked from the leaves and extracted in either ethyl acetate or chloroform, the predominant signal was m/z 559.24 with fragments characteristic of the standard scutebarbatine A (Supplemental Fig. S4). MALDI-imaging indicated that a large number of compounds (predominantly clerodane diterpenoids) were also present in the large peltate trichomes. Some of these have been identified tentatively, based on accurate mass and MS2 fragmentation patterns in methanol extracts of these large trichomes (Supplemental Table S1).
Vibratome sections of leaves stained with NaDi showed that the pale blue material was localized in the subcuticular space of the large trichomes on either side of a central plate of cells, supported by a stalk cell (Fig. 1c i). Transmission electron micrographs (TEM) showed these trichomes to have a highly specialized structure with a single stalk cell atop epidermal pavement cells, supporting a plate of cells, one cell layer thick, which constituted the ‘glandular head’ of the trichome (Fig. 1f). The stalk cell contained large, electron-dense vesicles, suggestive of specialised metabolite synthesis and/or transport (Fig. 1g). The cells in the plate appeared to secrete material into a sub-cuticular space, both above and below the plate (Fig. 1f), which possibly occurred through the non-cuticularised cell walls, as evidenced by electron-dense vesicles in the plate cells (Fig. 1g & h). The bulk of the sub-cuticular material had a reticulate or semi-solid appearance in TEM (Fig. 1f & h), and NaDi staining suggested that it included scutebarbatine A (Fig. 1c i). These observations fitted well with the limited solubility of scutebarbatine A observed in aqueous solutions (Fig. 1c iii).
Scutebarbatine A induces apoptosis in a dose dependent manner
-
We investigated the effect of scutebarbatine A on human colon cancer Caco-2 cells. We incubated Caco-2 cells with scutebartine A or with a methanol-only control for 24 h. Scutebarbatine A at 60 µM caused a substantial induction of apoptosis, compared to the control (see Fig. 2b & f). The percentage of late apoptotic cells went from an average of 9.06% in the control to 31.57% in the 60 µM scutebarbatine A sample (see Fig. 2b). To quantify this effect further, we undertook a series of dose response experiments from 10-60 µM, and a clear dose-response was observed (Fig. 2).
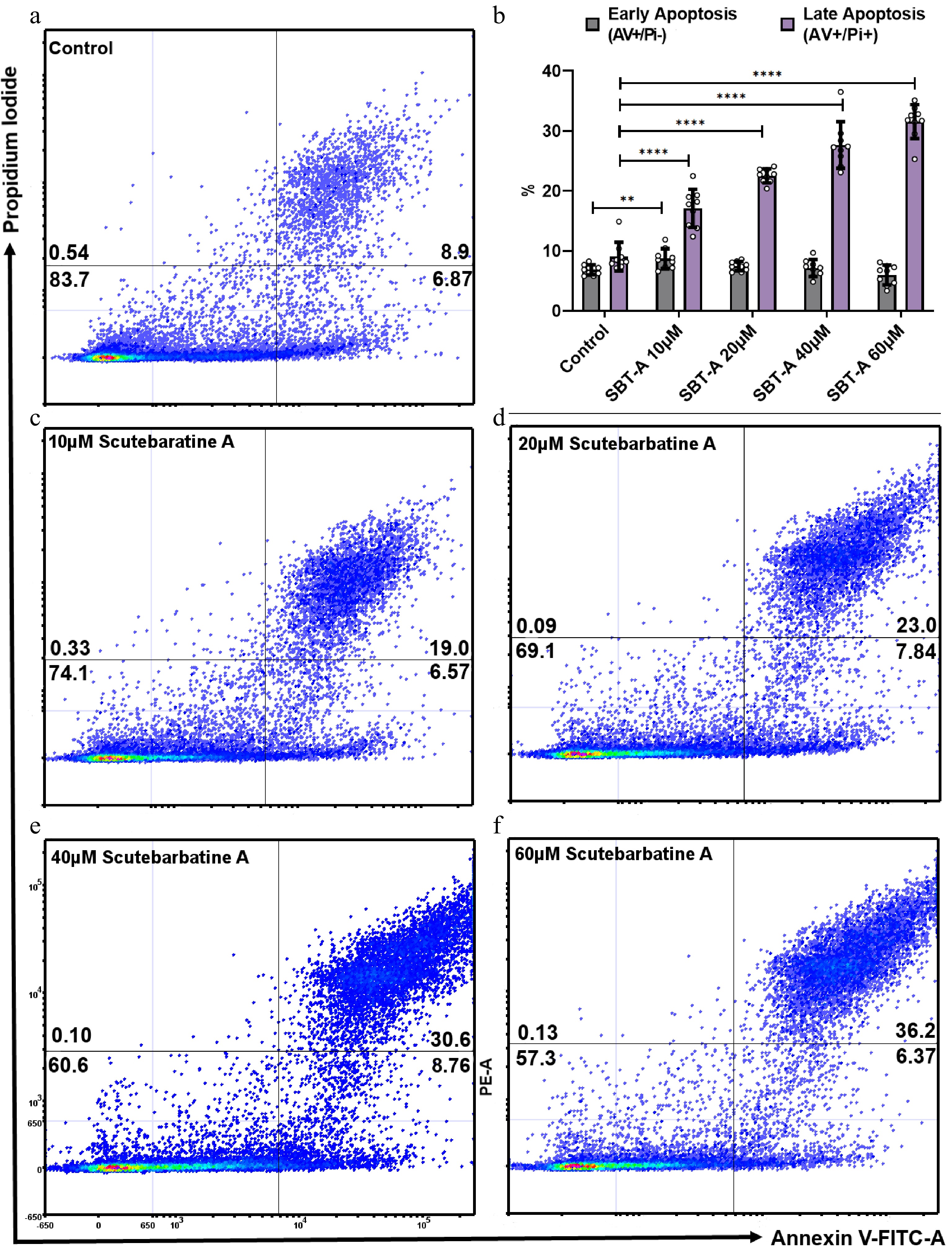
Figure 2.
Scutebarbatine A dose response effects on Caco-2 cancer cells. Cells were treated with low to high concentrations (10−60 µM). (a) & (c−e) show representative dot plots. (a) Control. (c−f) Increasing scutebarbatine A addition and a corresponding increase in late apoptosis. Annexin V positive cells are on the FITC-A (X axis) and Pi positive cells are on the PE-A (Y axis). Numbers in quadrats are %. Data are displayed as individual datapoints. (b) Shows the dose response as a bar chart. Horizontal bars represent the means and error bars SDev, (**** for P ≤ 0.0001, ** for P ≤ 0.01) n = 3 biologically independent experiments.
Scutebarbatine A has tumor selective cytotoxicity
-
To be considered a potential chemotherapeutic agent, any candidate should have greater selectivity for tumor cells than for normal cells, to minimise damage to healthy cells. Therefore, we investigated apoptosis in cancerous Caco-2 cells and in the directly comparable normal HCoEpiC colonic epithelial cell line. Flow cytometric analysis revealed normal levels of apoptosis in the HCoEpiC cell line with scutebarbatine A incubation (Fig. 3a ii). The effect of scutebarbatine A on Caco-2 cells was, however, dramatic ( Fig. 3b ii). Scutebarbatine A (60 µM) led to a three-fold increase in cells in apoptosis, compared to controls, after 24 h incubation ( Fig. 3d).
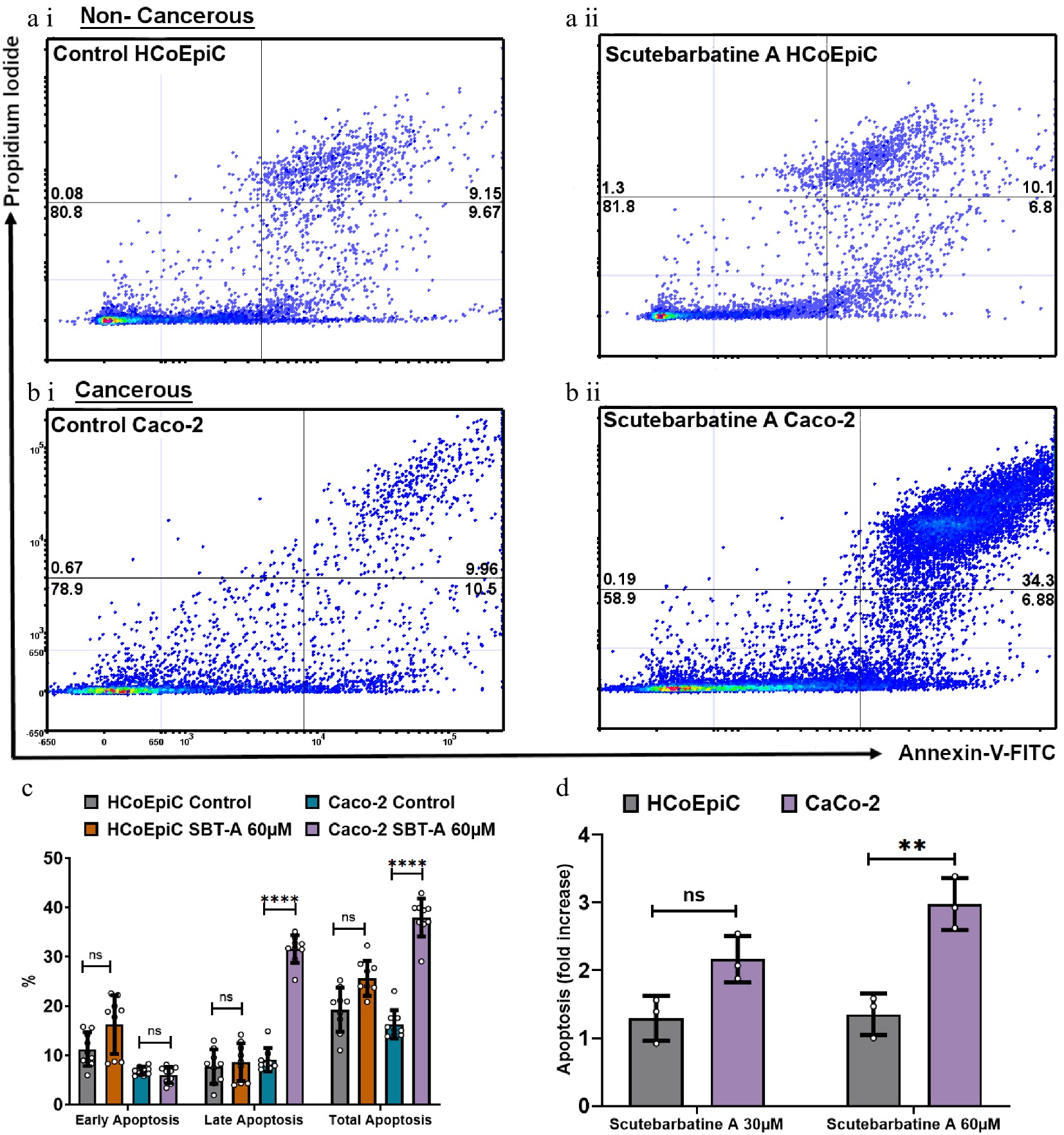
Figure 3.
Scutebarbatine A demonstrates selective cytotoxicity against cancer cells. (a1) Non-cancerous HCoEpiC cells with control. (a ii) HCoEpiC cells treated with Scutebarbatine A at 60 µM. (b i) Cancerous Caco-2 cells with control treatment. (b ii) Cancerous Caco-2 cells treated with Scutebarbatine A at 60 µM. Annexin V positive cells are on the FITC-A (X axis) and Pi positive cells are on the PE-A (Y axis). Numbers in quadrats are %. Data is displayed as individual dots. (c) Bar chart showing differences in the two different cell lines, statistical difference compared to respective controls (n = 3). (d) Fold difference in total apoptosis, annexin V+ cells and Annexin V/Pi+ cells. Data are displayed as individual datapoints. Horizontal bars represent the means and error bars SDev, (**** for P ≤ 0.0001, ** for P ≤ 0.01), n = 3 biologically independent experiments.
Protein profiling revealed scutebarbatine A primarily affects Inhibitors of Apoptosis and their direct interactors
-
To investigate the potential mechanism behind scutebarbatine A inducing cancer cell-specific apoptosis, we employed an antibody-based array to detect the relative expression/activation state of 43 human apoptotic protein markers. This gave a comprehensive picture of its effects on the key regulators of apoptosis. Half the proteins affected belonged to the inhibitors of apoptosis (IAP) class of proteins or were proteins known to interact directly with IAPs, such as IAP chaperones or antagonists (Fig. 4). Along with the induction of apoptosis, an induction of caspase-3 was seen with scutebarbatine A (Fig. 4a ii & b). Interestingly, up-regulation of only one other protein, the IGFB-6 tumour suppressor protein, was seen (Fig. 4a ii & b). The 12 proteins showing expression changes have been labelled on a representative array membrane and average optical density values from three independent replicate treatments are shown (Fig. 4). Although normal levels of apoptosis were seen in the HCoEpiC cell line, we investigated changes in markers of apoptosis to see if scutebarbatine A had any effects on apoptosis in healthy cells. Array results for the HCoEpiC cells showed no significant changes in expression of markers of apoptosis (Fig. 5) although, as expected, most markers assayed showed low levels of expression in healthy cells. The tumour suppressor, p21, stood out due to its high expression levels in HCoEpiC cells (Fig. 5), nearly twice those seen in the Caco-2 cell array (Fig. 4b). Interestingly, p21 decreased in the cancer cell line following scutebarbatine A treatment (Fig. 4b).
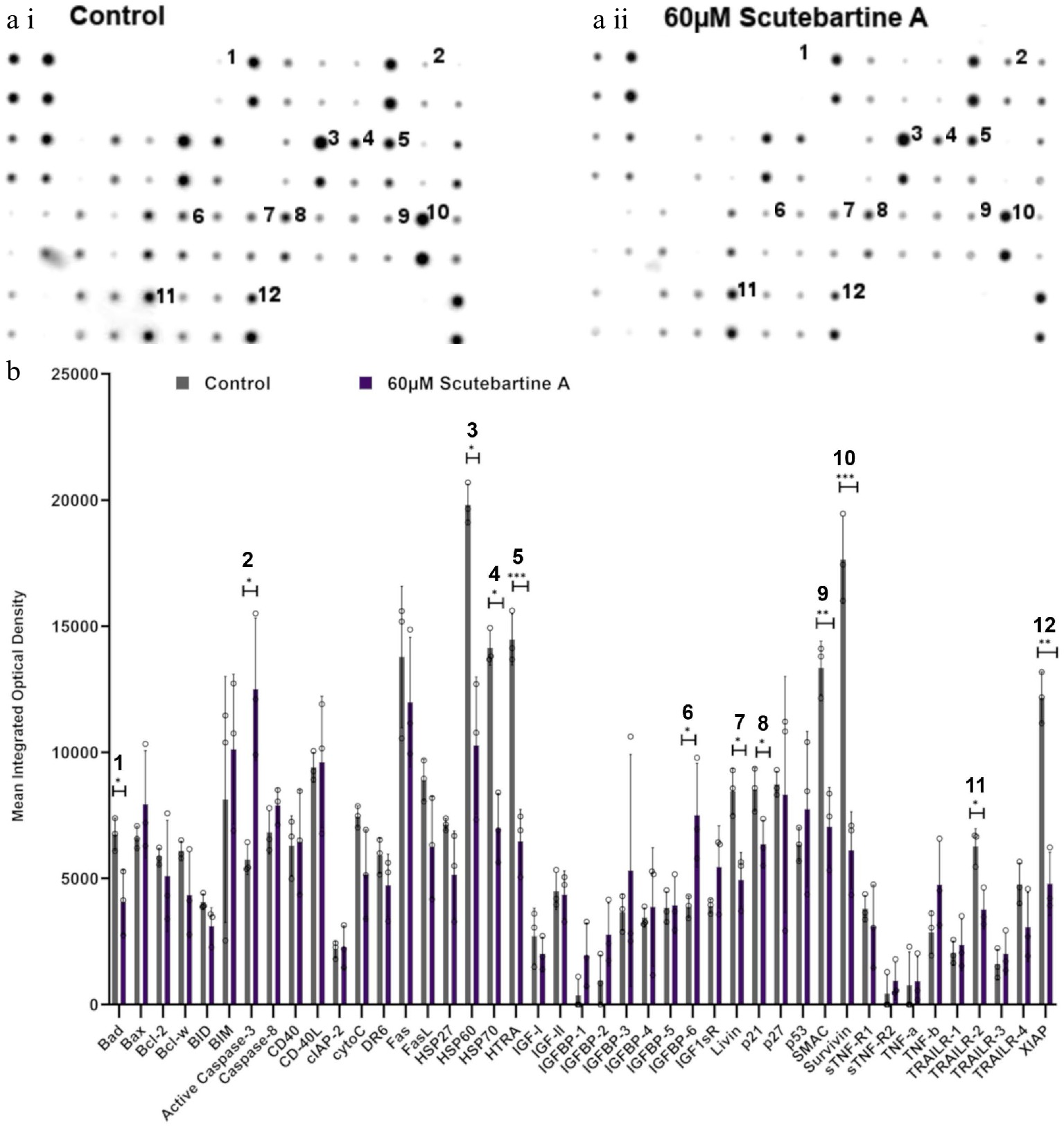
Figure 4.
Specific apoptotic protein changes in Caco-2 cancer cells, treated with 60 µM scutebartine A. (a i) and (a ii) show representative antibody array dot plots. (a i) shows the control treatment. (a ii) was treated scutebarbatine A (60 µM) array. Affected genes are labelled: 1-Bad, 2-Caspase 3, 3-HSP60, 4-HSP70, 5-HTRA2, 6-IGFBP-6, 7-Livin, 8-p21, 9-SMAC, 10-Survivin, 11-TRAILR-2, 12-XIAP. (b) Bar chart showing all 43 of the apoptotic genes assayed in the antibody array, * P ≤ 0.05, ** for P ≤ 0.01, *** P ≤ 0.001. Genes affected are: Bad, Caspase 3, HSP60, HSP70, HTRA2, IGFBP-6, Livin, p21, SMAC, Survivin, TRAILR-2, XIAP. Data are displayed as individual datapoints. Horizontal bars show significant changes and error bars show SDev, n = 3 biologically independent experiments.
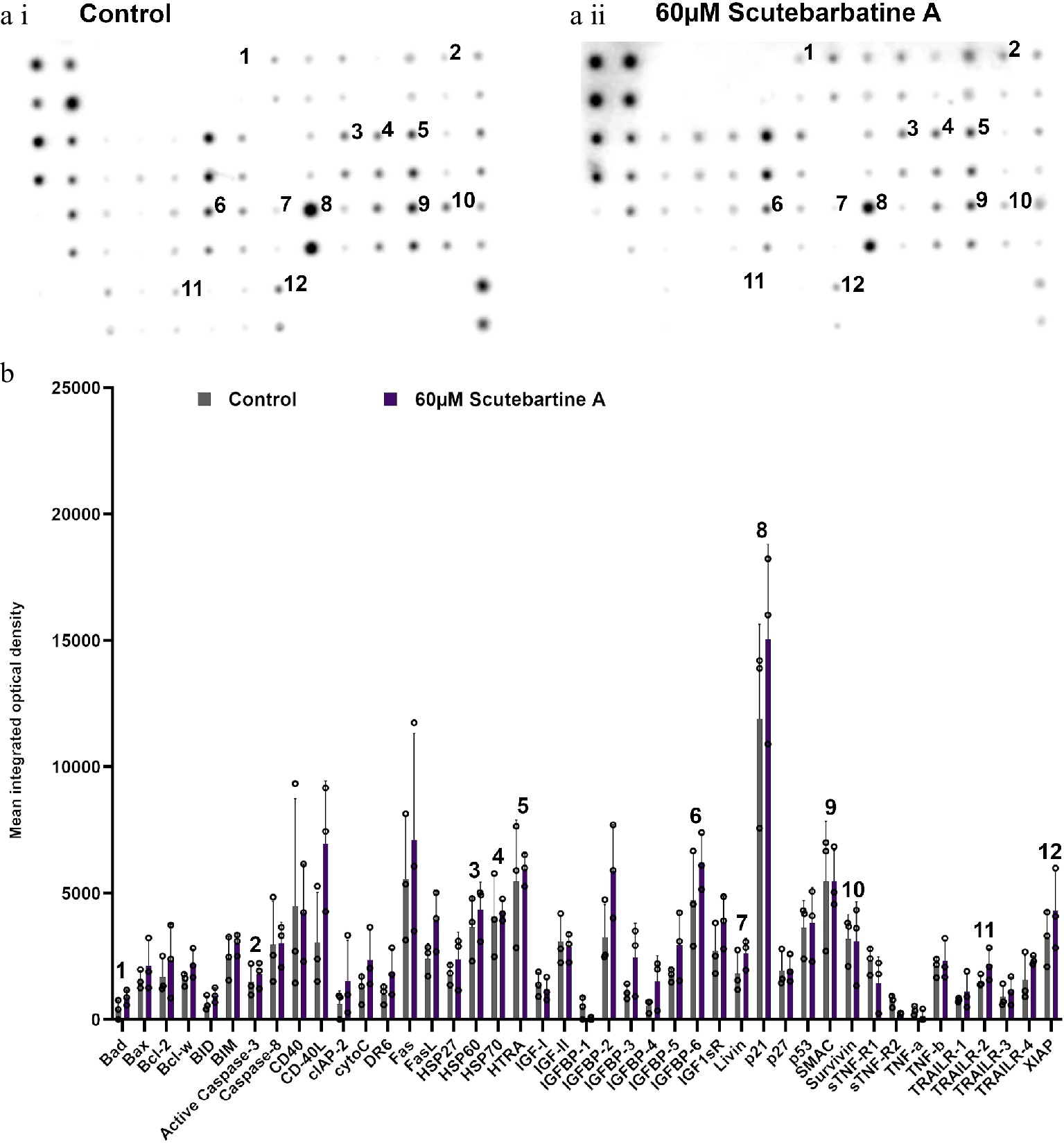
Figure 5.
Profile of changes in proteins associated with apoptosis in a non-cancerous cell line (HCoEpiC). (a i) & (a ii) representative antibody array dot plots. (a i) shows the control. (a ii) is the array from cells treated with scutebarbatine A (60 µM). Proteins affected in the Caco-2 cell lines are labelled for comparison: 1-Bad, 2-Caspase 3, 3-HSP60, 4-HSP70, 5-HTRA2, 6-IGFBP-6, 7-Livin, 8-p21, 9-SMAC, 10-Survivin, 11-TRAILR-2, 12-XIAP. (b) Bar chart showing all 43 of the apoptotic proteins assayed in the antibody array, mean ± S.D. 1-Bad, 2-Caspase 3, 3-HSP60, 4-HSP70, 5-HTRA2, 6-IGFBP-6, 7-Livin, 8-p21, 9-SMAC, 10-Survivin, 11-TRAILR-2, 12-XIAP. Protein levels remained unchanged with scutebarbatine A (60 µM) treatment. Mean ± S.D (n = 3). Data are displayed as individual datapoints. Horizontal bars show significant changes and error bars show SDev, n = 3 biologically independent experiments.
Comparison of the pro-apoptotic activity of a methanolic extract of S. barbata leaves to a traditional Ban-Zhi-Lian decoction.
-
To achieve maximum extraction of clerodane diterpenoids we used 70% methanol for extraction. However, in Traditional Chinese Medicine (TCM), typically aqueous decoctions are prepared from dried plant material. These different extraction methods provide more hydrophobic (methanolic extract) and hydrophilic (TCM decoction) extractions.
To ascertain the relative bioactivities of extracts of S. barbata prepared traditionally (hydrophilic) or using 70% methanol (hydrophobic), we purchased dried extract of Ban Zhi Lian, from a Chinese Pharmaceutical Supplier (Yang Yuen Tang Limited London, UK) and prepared a decoction (BZL) by infusing 0.16 g directly into 10 ml of 1% FCS DMEM at 4 °C overnight. This was used as a stock for addition to medium at concentrations between 1 and 3 mg/ml. We calculated that treatments with BZL of 3 mg/ml constituted 1 μM of scutebarbatine A. We compared this to the methanolic extract of S. barbata leaves, prepared as a stock solution from freeze-dried powder as described in the methods. We calculated that treatments of the methanolic extract of 3 mg/ml constituted 19 μM scutebarbatine A. Flavonoids were predominant in the hydrophilic extract and diterpenes in the hydrophobic, methanolic extract (Fig. 1a & 6a, Supplemental Fig. S5, Supplemental Table S2). We incubated Caco-2 cells and measured the numbers of apoptotic cells, after 24 h incubation, compared to a methanol-only control. The BZL extract induced a statistically significant increase in early and late apoptosis in a dose-dependent manner over the 24 h incubation period (Fig. 6c & d, Supplemental Fig. S6a & S6b), whereas the methanol extract induced a significant increase in late apoptosis in a dose dependent manner over the same incubation period (Fig. 6d, Supplemental Fig. S6a & S6b).
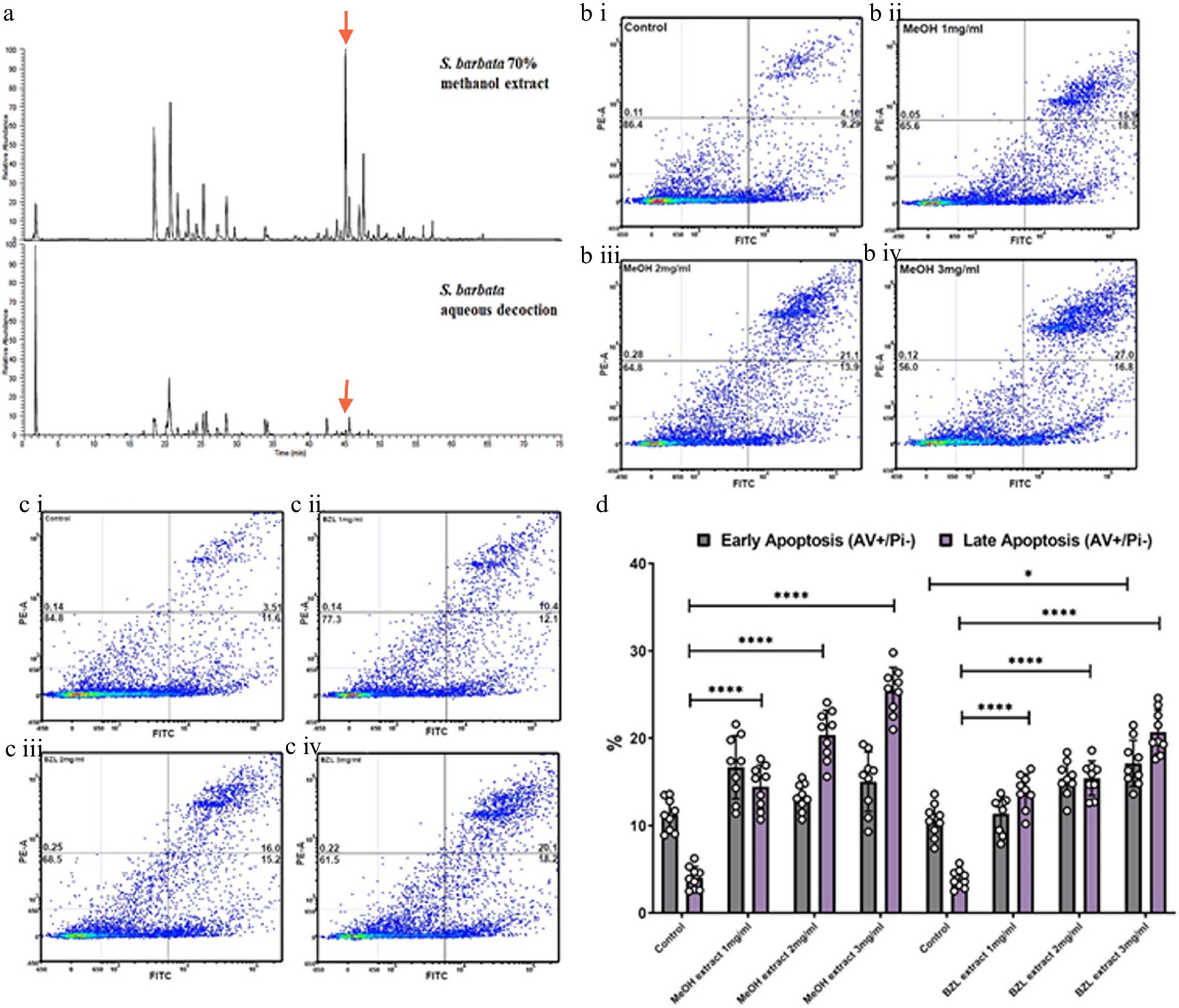
Figure 6.
Comparative analysis of hydrophilic and hydrophobic extractions of S. barbata on initiation of apoptosis in Caco-2 cancer cells. (a) Base ion chromatograms from LC-MSn (positive ESI) analysis of the S. barbata leaf 70% methanol extract and the BZL aqueous decoction. ↓ indicates m/z 559.24, scutebarbatine A. Compounds detected in both extracts have been assigned as detailed in Supplemental Table S2 and Supplemental Fig. S5. (b i)–(b iv) Caco 2 cells treated with MeOH S. barbata extract at 1, 2 and 3 mg/ml respectively. Annexin V positive cells are on the FITC-A (X axis) and Pi positive cells are on the PE-A (Y axis). Numbers in quadrats are %. (c i)–(c iv) .Caco 2 cells treated with BZL hydrophilic S. Barbata extract at 1, 2 and 3 mg/ml respectively. Annexin V positive cells are on the FITC-A (X axis) and Pi positive cells are on the PE-A (Y axis). Numbers in quadrats are %. (d) Bar chart showing differences in cells in early and late apoptosis following treatment with differing concentration of the hydrophobic methanolic extract and the hydrophilic BZL extract, (**** for P ≤ 0.0001, *** P ≤ 0.001, * for P ≤ 0.05), statistical difference compared to respective controls. Data are displayed as individual datapoints. Horizontal bars show significant changes and error bars show SDev, n = 3 biologically independent experiments.
The MeOH extract was more effective at inducing apoptosis than BZL in terms of the percentage of cells in late apoptosis after 24 h but it also enhanced significantly the proportion of cells in early apoptosis compared to pure scutebarbatine A (Supplemental Fig. S6c), suggesting that the presence of additional compounds (predominantly other clerodane diterpenoids) in the methanolic extract enhanced cytotoxicity. The BZL extract showed significant induction of both early and late apoptosis in Caco-2 cells over 24 h, in a dose dependent manner, although its lower content of scutebarbatine A (1 μM in 3 mg/ml BZL) suggested that the other components in BZL also contributed substantially to its cytotoxicity. We performed MTT cytotoxicity assays on Caco-2 and normal (MCF-10A) and cancerous breast cells (MCF-7) and compared the responses to those of Caco-2 cells. The results confirmed that the MeOH extract cytotoxicity is not cell line-specific and the MeOH extract has no effect on another normal cell line (Supplemental Fig. S7). In fact, the MCF-7 cell line was found to be four times more sensitive than the Caco-2 cell line (Supplemental Fig. S7).
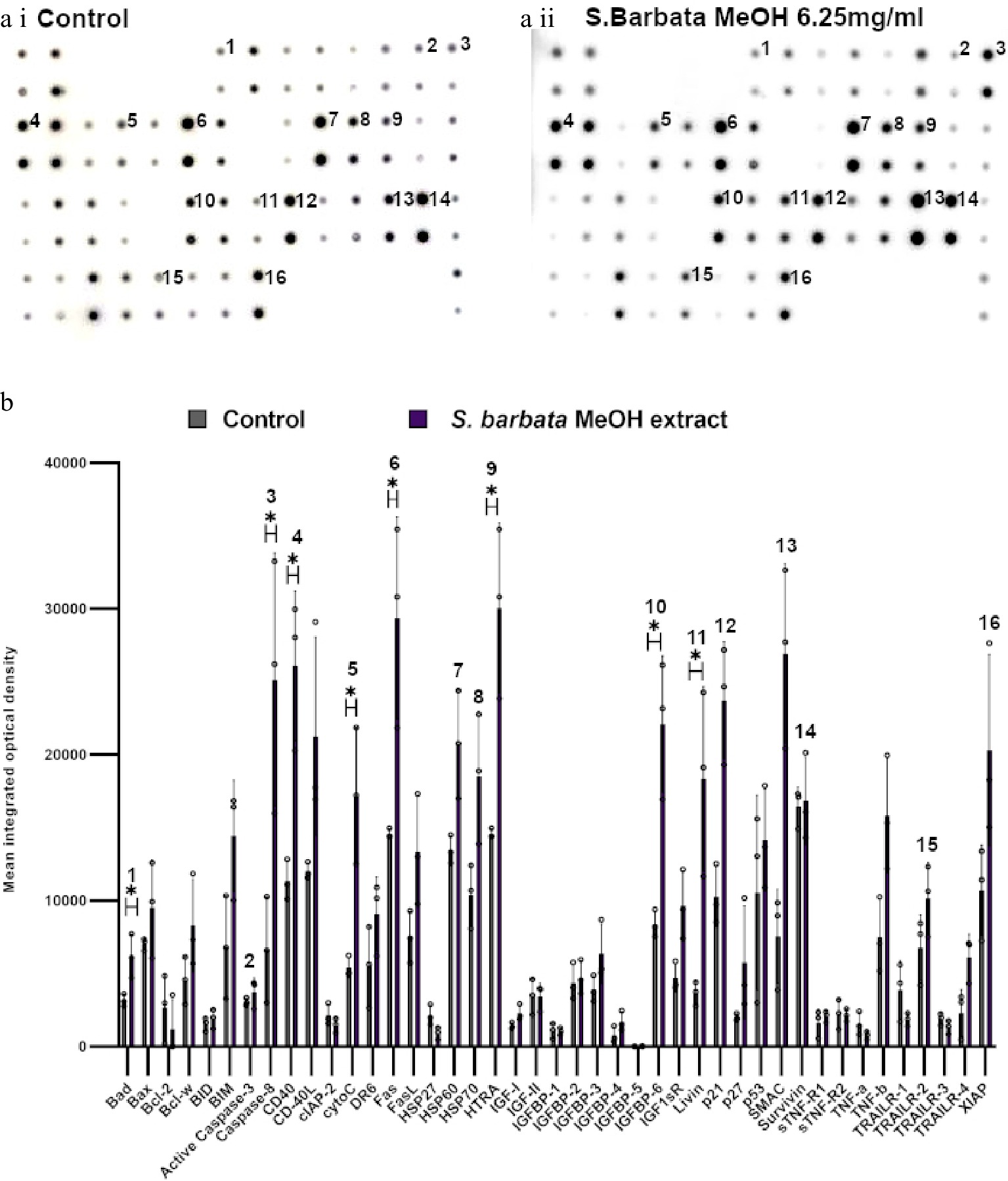
Figure 7.
Specific apoptotic proteins in Caco-2 cancer cells, treated with a methanolic extract from leaves of S. barbata. (a i) and (a ii) show representative antibody array dot plots. (a i) shows the control untreated cells. (a ii) shows the proteins of the apoptosis array treated the methanolic extract (3.0 mg/ml). (b) Bar chart showing all 43 of the apoptotic proteins assayed in the antibody array,, (* P < 0.05, n = 3). Genes affected are labelled: 1-Bad, 2-Caspase 3, 3-Caspase-8, 4-CD40, 5-CytoC, 6-Fas, 7-HSP60, 8-HSP70, 9-HTRA, 10-IGFB-6, 11-Livin, 12-p21, 13-SMAC, 14-Survivn, 15-TRAILR-2, 16-XIAP. Data are displayed as individual datapoints. Horizontal bars show significant changes and error bars show SDev, n = 3 biologically independent experiments * for P ≤ 0.05.
Mechanism of action of the methanolic extract of leaves
-
Examination of apoptosis-related proteins in Caco-2 cells treated with the methanolic extract of S. barbata indicated a distinct set of changes compared to treatment with scutebarbatine A alone (Fig. 7). Seven proteins on the apoptosis array changed significantly and all of them showed increased levels following 24 h treatment. These included the apoptosis inducer Bad, the initiator caspase, caspase 8 (although the apoptosis array could not distinguish between the pro-caspase and active forms of caspase 8), cytochrome C (the intrinsic pathway mediator), Fas (suggesting induction of apoptosis through the Death Inducing Signalling Complex (DISC) extrinsic pathway), HtrA2 and IGFB-6 (tumour suppressor protein). Levels of the mitochondrial protease, SMAC (an endogenous IAP inhibitor) and the tumour suppressor protein, p21 were also increased by treatment with the methanolic extract, although those increases were not statistically significant. Amongst the proteins induced, only IGFB-6 was in common with scutebarbatine A treatment, and higher levels of some of the inhibitors of apoptosis (Livin, Survivin and XIAP) were observed after treatment with the methanolic extract, although these were not statistically significantly higher than in untreated cells. These analyses suggested that while showing strong promotion of apoptosis in cancer cells, the methanolic extract induced apoptosis by a variety of mechanisms (polyvalency). The clear inhibition of inhibitors of apoptosis by scutebarbatine A may mitigate less-selective toxicity of other components of the methanolic extract, resulting in a potent extract that remains selective against cancer cells (Supplemental Fig. S6a & S6b).
-
From a complex and diverse mix of S. barbata diterpenoids extracted from leaves, we focused on scutebarbatine A, previously shown to be the most cytotoxic against four different tumour cell lines of 16 studied clerodane diterpenoids[25]. The most sought-after chemotherapeutic targets are those that are differentially active in cancer compared to healthy cells[26], and IAP antagonists represent promising candidates for restoring the apoptotic response to proapoptotic stimuli, particularly those introduced by standard cancer therapies[27]. To investigate selective cytotoxicity our study used both a human colonic epithelial adenocarcinoma (Caco-2) cell line and a non-cancerous colonic epithelial (HCoEpiC) cell line for direct comparison.
The Caco-2 cell line was chosen because the gastro intestinal tract is in direct topological contact with extracts shortly after oral intake[28], and epithelial cells are likely to receive high doses of bioactive natural products, prior to their metabolism and/or absorption. The Caco-2 cell line has proven drug permeability and models the intestinal epithelial barrier[28,29]. From our results, only the cancer cells showed a dramatic induction of apoptosis in response to scutebarbatine A. We have shown that purified scutebarbatine A affects the expression of some apoptotic proteins found only in cancer cells, offering a plausible explanation of its selective cytotoxicity. Interestingly, in non-cancerous HCoEpiC cells, the tumour suppressor protein, p21, remained highly expressed in treated cells. Although p21 was downregulated by scutebarbatine A in Caco-2 cancer cells, loss of p21 increases sensitivity of cells to apoptosis induced by chemotherapeutic agents[30]. Small molecules that eliminate p21 expression have been reported to improve the action of other anticancer drugs[30].
Of the 12 proteins affected by scutebarbatine A treatment, six act either directly as IAPs or interact with IAP proteins. Uninhibited, IAPs can suppress apoptosis and promote cell cycle progression. Therefore, it is unsurprising that cancer cells demonstrate significantly elevated expression of IAPs, giving rise to enhanced cell survival, tumour growth and subsequent metastasis[16]. Targeting IAPs has become an increasingly attractive strategy to re-sensitize cancer cells to chemotherapies[16]. In fact, direct manipulation of apoptotic pathways via IAP inhibition can offer safer, alternative chemotherapies because they have limited effects on apoptosis in non-cancer cells that do not highly express IAPs[16]. For example, recent attention has focused on mimics of second mitochondria-derived activator of caspase (SMAC) proteins because SMACs interact with and antagonise the activity of IAPs[27].
Three IAPs were affected in our study; XIAP, survivin and livin, all vital pro-survival proteins. Their normally high expression in Caco-2 cells prohibits apoptosis – they act as molecular brakes on the apoptosis machinery. With scutebarbatine A treatment, these effects were reduced dramatically, limiting cancer cells escaping cell death. All three individual IAPs have known oncogenic functions. For example, XIAP binds and inhibits caspase-3 by ubiquitinylation[17]. Therefore, lower levels will lead to less ubiquitinated caspase-3 and consequently increased apoptosis. Elevated levels of XIAP are correlated with disease progression, metastasis or poor survival in colon cancer patients[9]. XIAP has also been reported to be overexpressed in breast cancers, melanoma and clear-cell renal carcinoma[18]. Survivin, the smallest IAP, is also expressed at higher levels in tumour cells[18]. The most likely function of survivin is as a pro-survival protein, inhibiting caspase-9[31] and in colorectal cancer, overexpression of survivin is associated with poor patient prognosis[18]. Due to this, it has received significant attention as a potential oncotherapeutic target[26]. Survivin levels have been reported to increase in chemoresistant patients[32] suggesting that persistent chemotherapy may selectively kill chemosensitive cells and thereby enrich survivin-positive, chemoresistant cells. Therefore, extracts of S. barbata could offer a route to reverse multidrug resistance through combination with approved chemotherapies[16,33].
The pro-survival mechanism of livin is by direct inhibition of caspases-3, -7, and -9, and it is overexpressed in many cancer cell types including colon cancer[34]. Interestingly, livin expression is also associated with resistance to colon cancer chemotherapy[35], again emphasizing the potential of extracts of S. barbata in limiting multidrug resistance in cancer cells.
Our results provide additional evidence for the mechanistic effects of scutebarbatine A in mediating apoptosis, demonstrated for the first time in gastrointestinal tract cells. A previous study suggested that scutebarbatine A induces apoptosis via a caspase-dependent pathway in a human hepatocellular carcinoma cell line and concluded that scutebarbatine A inhibits cell proliferation and triggers apoptosis via activation of p38 mitogen-activated protein kinase (MAPK) and endoplasmic reticulum stress (ER) stress through the upregulation of protein kinase RNA-like ER kinase[36]. In a human lung carcinoma cell line (A549). Scutebarbatine A has been reported to up-regulate caspase-3 and -9 expression, and down-regulate Bcl-2, to trigger mitochondria-mediated apoptosis[37].
Scutebarbatine A treatment affected heat shock proteins (HSPs), which are strongly cytoprotective and can associate with multiple apoptotic partners and thereby block apoptosis at different levels[38]. HSP70 is one of the most powerful pro-survival proteins because it blocks almost all identified cell death pathways[38] and is used as a prognostic factor in malignant diseases[35,36,38]. Scutebarbatine A reduced levels of HSP60 which orchestrates a broad cell survival program centered on stabilisation of mitochondrial survivin[39]. Consequently, decreasing HSP60 adds to the effects of scutebarbatine A on inducing apoptosis, as not only is survivin expression greatly reduced, the survivin produced is more likely to be unstable and degraded, due to the lack of HSP60[39].
Scutebarbatine A treatment caused a significant increase in levels of just one protein, the apoptosis inducer - IGFBP-6 (insulin-like growth factor binding protein 6), illustrating the specific nature of the molecular changes caused by scutebarbatine A treatment. IGFBP-6 expression is normally lower in malignant cells, where it acts as an inhibitor of tumorigenic processes, by binding to and sequestering insulin-like growth factor II (IGF-II)[40]. IGF-II induces cell proliferation, differentiation and survival significantly[36]. The insulin/insulin-like growth factor (IGF) system is a major determinant in the pathogenesis and progression of cancer, including colorectal cancer[41]. Importantly, IGFBP-6 has been proposed as a potent inducer of apoptosis in cancer cells.
Along with decreased levels of pro-survival proteins, several pro-apoptosis proteins were affected by scutebarbatine A treatment. XIAP antagonists; HTRA2 and SMAC were down regulated. Bad also showed a significant decrease. These changes do not appear to have been enough to cause physiologically-relevant effects, and no increase in cytochrome C levels were observed. There were also reduced Trail-2 (DR5) levels in scutebarbatine A-treated Caco-2 cells[42].
Extracts of S. barbata have been widely used in experimental medicine where they have been shown to arrest tumour growth[43]. Studies with whole plant extracts have also been shown to promote apoptosis[44,45], while aqueous extracts of aerial parts of S. barbata have been tested in clinical trials for efficacy against cancer, and the aqueous preparation BZL101 showed promising efficacy in early phase trials for breast cancer[46−48]. In our study, we showed that both hydrophobic and their relative cytotoxicity, the hydrophobic extracts being more potent than traditional water-based extracts over a 24 h incubation period and this cytotoxicity was specific for cancer cells. Our results are in line with studies on other cancer cell lines where these extracts have induced apoptosis at similar concentrations. For instance, a hydrophobic fraction was used to treat the SW620 human colon cancer cell line, at 50 µg/ml[49]. Another study showed a similar range of bioactive concentrations, 20–80 µg/ml[40]. The BZL aqueous extract has been reported to exert apoptotic effects between 0.5 mg/ml[50] and 5 mg/ml[51] and has also been suggested to induce anti-proliferative gene expression responses in human breast and prostate cancer cells at similar concentrations[51].
Analysis using mass spectrometry revealed scutebarbatine A to be present in both hydrophobic and hydrophilic extracts (Fig. 6). However, it was clear (Fig. 6a) that the compositions of the hydrophilic and hydrophobic extracts were different. Furthermore, HPLC analysis showed that the methanolic extract contained 20-fold higher concentration of scutebarbatine A, than BZL (see Supplemental Table S2). The main bio-active components of BZL appear to be flavonoids[52] whereas in the hydrophobic extract the diterpenoids predominate (Supplemental Fig. S5, Supplemental Table S2). Compounds that likely contribute to the cytotoxicity of the methanolic extract are other clerodane diterpenoids. MALDI-imaging suggested that these accumulate in the large glandular trichomes of the leaves, probably produced by alternative decorations of the clerodane skeleton (Supplemental Table S1). Our results suggest that there is polyvalency (different constituents with different mechanistic effects relevant to anti-cancer bioactivities), within the methanolic extract which has greater capacity to induce early apoptosis than the main clerodane diterpenoid, scutebarbatine A, alone (Supplemental Fig. 6c). Indeed, compounds assigned as scutebarbatine G, 6,7-di-O-nicotinoylscutebarbatine G, scutehanine A, 6-O-(2-oxo-3-methylbutanoyl)scutehenanine A, 6-O-nicotinoylscutebarbatine G, barbatin C and scutebarbatine B, or isomers, which were detected in the methanolic extract (Supplemental Fig. S5, Supplemental Table S2), show cytotoxic activity in human cancer cell lines (HONE-1 nasopharyngeal, KB oral epidermoid carcinoma, HT29 colorectal carcinoma cells) with IC50 values in the range 2.1–8.5 µM[53−56]. Scutebata B and scutebata M, show moderate cytotoxic activity in different human cancer cell lines (SMMC-7721, A-549, MCF-7, SW480) with an IC50 range of 22.1– 31.4 µM[57,58].
Polyvalency is unlikely to be overcome by cancer cells developing resistance, and suggests that methanolic extracts could be particularly effective in combination therapies either with other IAP inhibitors (such as SMAC mimics) or with conventional chemotherapies, such as 5-fluorouracil[59]. Indeed, a combination of compounds with different modes of action for polyvalency is aligned with conventional cancer chemotherapy with cocktails of drugs that optimise efficacy and minimise resistance and adverse effects[60]. While our studies have focused on colon cancer cells, both hydrophilic and hydrophobic extracts of S. barbata have been shown to be selectively toxic to a range of cancer cell lines[43,61−64].
Positive indications have been reported using BZL extracts for Phase 1B clinical trials of BZL101 in combination with conventional chemotherapy on patients with stage IV breast cancer[46]. However, the main constituents identified in BZL101 were not clerodane diterpenoids, but flavonoids. In these trials, relatively large amounts (40 g/day) were administered, and high drop out rates were reported as a result of the unpleasant taste. Treatments based on smaller amounts from alcohol based extracts might be easier to administer as well as potentially more efficacious.
-
Our analyses provide new insights into the chemotherapeutic bioactivities of S. barbata extracts and demonstrate the untapped potential of clerodane diterpenoids as chemopreventive or chemotherapeutic agents. Complementing this, we identified the major site of diterpenoid synthesis in Scutellaria plants, which could accelerate future extraction protocols such as selective extraction of trichomes from fresh leaf tissue. Our studies could open new avenues for metabolic engineering through increasing bioactivity, by selecting for varieties with higher densities of large, peltate trichomes on their leaves, or by further modification of metabolic pathways for increased production of clerodane diterpenoids. This study presents the case for multidisciplinary investigation of traditional medicinal plants to realise the full potential of natural products for chemopreventive preparations for the treatment of cancer.
-
The dried extract (Ban Zhi Lian, Yang Yuen Tang Limited, London, UK) was prepared fresh each time as a 10X stock, by infusing 0.16 g directly into 10 ml of 1% FCS DMEM at 4 °C overnight (termed hydrophilic extract). Preparations were then centrifuged at 500 g for 5 min to clarify the infused media and sterilised by syringe filter (0.45 µm). This was then diluted in 1% FCS DMEM to the desired concentrations. The hydrophobic extract was prepared by freeze drying overnight 0.5 g of mature S. barbata leaves, then grinding by pestle and mortar in 10 ml of 70% methanol, ultrasonicating (Soniprep 150 plus, mseuk) at max amplitude for 10 min on ice. The preparation was then centrifuged at 12,000× g for 2 min and the supernatant aliquoted (25 mg/500 µl). Aliquots were dried down gradually in glass HPLC vials, via an 80 °C heat block and stored at −80 °C. When needed were resuspended in 70% methanol (1 mg/µl) for each assay to desired concentration. For scutebarbatine A, a 10 mg stock (purchased from Purifa, Chengdu, China) was resuspended to 60 mM in dichloromethane and then aliquoted to single use HPLC vials, vacuum dried (GeneVac EZ-2) and stored at −20 °C in single use aliquots. When needed they were resuspended in 70% methanol to 60 mM.
Cell lines
-
Caco-2 cells were obtained from Public Health England and maintained from passages 14−24, MCF-7 and MCF-10A were from ATCC. Caco-2 were cultured at 37 °C in a humidified atmosphere of 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum (FCS), 1% glutamate and 0.5% non-essential amino acids (Gibco). HCoEpiC primary cells were purchased from SciencCell Research Laboratories and maintained from passage 1-4 according to the supplier’s protocol. Briefly, poly-L-Lysine coated (to aid cell attachment) T-75 flasks, or 12-well plates were used, cells were cultured in complete CoEpiCM media (SciencCell Research Laboratories). A 0.05% trypsin/EDTA solution was used for gentle cell detachment. The company’s TNS and T/E neutralisation solution were used to aid cell recovery. The cells were cultured at 37 °C in a humidified atmosphere of 5% CO2. The human breast cancer cell line MCF-7 cells were cultured in DMEM containing 10% fetal bovine serum (FBS), 2 mM glutamine, penicillin (50 U/ml) and streptomycin (50µg/ml) (Gibco, ThermoFisher Scientific, UK) at 37 °C in a humidified atmosphere containing 5% CO2. Human breast cell line MCF-10A was cultured in DMEM/F12 (Gibco, ThermoFisher Scientific, UK) supplemented with 5% horse serum (Gibco, ThermoFisher Scientific, UK), EGF (20 ng/ml) (Peprotech), Hydrocortison (0.5 mg/ml) and insulin (10 μg/ml) (Sigma Aldrich, Gillingham, UK) at 37 °C, 5% CO2. Cells were routinely tested for mycoplasma contamination by nested PCR and their identity was verified by STR-profiling. HCoEpiC cells were originally characterised by SciencCell, by immunofluorescence with antibodies specific to CK18 and CK19.
-
MCF-7, MCF-10A and Caco-2 cells were seeded into 96-well plates (1.0 × 104 cells/well). When cells were at approximately ~80% confluence, different doses of MeOH S. barbata extracts (0.25-2 mg/ml MCF-7 and MCF-10A, 0.25-3 mg/ml Caco-2) were added with fresh medium; MeOH (2%) was used as controls (n = 6). After 24 h the medium was removed, 100 μl (5 mg/ml) MTT solution was added, and the mixture was incubated at 37 °C in a 5% CO2 incubator for 1 h to allow metabolism of MTT. The formazan formed was then re-suspended in 100 μl DMSO per well. The final absorbance was recorded using a microplate reader (BMG Labtech Limited, Aylesbury UK) at a wavelength of 570 nm and a reference wavelength of 670 nm.
Apoptosis analysis
-
For analysis of apoptosis, cells were cultured to ~70% confluence in 24 well plates. They were then washed twice with DPBS and cultured in a low serum media (1% FCS) with specified extracts at stated concentrations for 24 h, scutebarbatine A or methanol control (max 0.2% v/v). After incubation, the cells were detached with a gentle EDTA treatment (50 µM) until dissociated single cells were observed. Samples were then allowed to recover at 37 °C for 1 h. This was done to minimise any cell membrane damage during detachment. Combined Annexin V-FITC/PI staining (1:40 FITC Annexin V VAA-33 - eBioscience, propidium iodide at 1µg/ml - Merk) was performed according to the manufacturer's instructions (ThermoFisher). For all samples; 10,000 events were assayed in each sample event, unstained cells and single stain controls were run, to determine compensation values. Sample events were collected on BDflow cytometers (LSR Fortessa, BD Biosciences) and were analyzed using Weasel flow cytometry software (Frank Battye Flow Cytometry Consultancy). Unless otherwise stated 10,000 events were collected for each experiment.
Antibody array profiling
-
Caco-2 and HCoEpiC cells were grown to ~70% confluence in 12-well tissue culture plates, washed twice (DPBS), and for the Caco-2 cell line incubated with low serum DMEM media (1% FCS). To this the following extracts or compounds were added; 60 µM Scutebarbatine A, 3 mg/ml methanolic extract in methanol (25 μl) or methanol (0.2% v/v) for 24 h. Tissue culture plate wells were then washed twice with DPBS and lysed with the manufacturers lysis buffer (Abcam). Total protein concentrations were quantified using a BCA assay (ThermoFisher). Normalised cell lysates (400 µg/blot) were used for all antibody array incubations. The manufacturer’s protocol (Abcam) was followed for blot incubation, washes, development and CCD camera-based image capture (ImageQuant LAS500). Array blots were quantified densitometrically with the generated mean integrated density values used for analysis. Dot blot normalisation was performed using positive samples and background subtraction with blank and negative samples, using Image J software and following the manufacturer's instructions.
Statistical analysis
-
If not otherwise specified, statistical data analysis was performed using PRISM 8 (GraphPad Software Inc., CA, USA) on the raw data. If not otherwise specified in the figure legends, comparison of two groups in functional in vitro experiments was carried out using Student’s t-tests. These were parametric unpaired tests (the null hypothesis was rejected if the two-tailed p-value was < 0.05). Statistically significant results are indicated with an asterisk (**** for P ≤ 0.0001, *** P ≤ 0.001 ** for P ≤ 0.01, * for P ≤ 0.05) in the figures. If not otherwise specified in the figure legends, data are presented with the horizontal bar representing the mean and error bars representing the standard deviation around the mean. Individual data points are shown as symbols. The sample size for all experiments was chosen empirically. Unless otherwise specified, all cell culture experiments are three biologically independent experiments, each of which has three technical repeats.
Plant materials and growth conditions
-
Scutellaria barbata D. Don plants were grown in the greenhouses of the John Innes Centre with 26 ℃, light/dark 12/12 h cycle. Mature leaves were collected for SEM, TEM, MALDI-ToF analysis, NADI staining and methanol extraction.
Quantification of scutebarbatine A in different organs of S. barbata by LCMS
-
The content of scutebarbatine A in different tissues was determined using a Waters Xevo TQ-S (Massachusetts, USA) using Kinetex 2.6 μm EVO C18 (50 × 2.1 mm). Chromatographic separation was achieved using 0.1% formic acid (eluent A) and acetonitrile (eluent B) as mobile phase under the gradient program: starting with 2% B for 3.5 min; to 70% B at 3.6 min; to 2% B for at 5 min The flow rate was 0.6 ml/min. TargetLynx software (Waters) was used to conduct data acquisition and analyses and MRM was used to detect scutebarbatine A. Scutebarbatine A (standard) dissolved in methanol with a starting concentrations for the calibration curve to be set at 50 ng/ml and diluted twofold progressively according to their detection by the Waters Xevo TQ-S. For each concentration, three measurements were performed for the establishment of a calibration curve. Different tissues (roots, flowers, young stems, old stems, leaves (< 0.5 cm), leaves (> 0.5 cm & < 1 cm), leaves (> 1 cm & < 2 cm), leaves (> 2 cm)) were ground and then dissolved in methanol at a dilution ratio corresponding to 1 mg of plant material per 20 μl methanol. The samples were performed at three biological replicates.
Mass spectrometry analysis of S. barbata extracts
-
The S. barbata methanol leaf extract and BZL were analysed by LC-MS on a Synapt G2-Si mass spectrometer coupled to an Acquity UPLC system (Waters, Manchester, UK). Aliquots of 7 µl (aqueous extract, 0.16 g of BZL in DH20) or 3 µl (methanol) were injected onto an Acquity UPLC® BEH C18 column, 1.7 µm, 1 × 100 mm (Waters, Manchester, UK) and eluted with a linear gradient of acetonitrile (ACN) in 0.1% formic acid at a flow rate of 0.08 ml/min with a column temperature of 45 °C. The gradient started at 5% ACN, this was ramped up to 75% in 5 min, followed by a ramp to 99% ACN in 10 min. The mass spectrometer was controlled using Masslynx 4.1 software (Waters, Manchester, UK) and operated in positive MS-Tof and resolution mode with a capillary voltage of 3 kV and a cone voltage of 40 V in the m/z range of 100−1,200. Leu-enkephalin peptide (Waters, Manchester, UK) was infused at 10 µl/min as a lock mass and measured every 30 s.
Chromatograms and spectra were generated in MassLynx 4.1. Spectra were generated by combining several scans across the LC peak, and the peaks were centred using automatic peak detection with lock mass correction. Data were further analysed in Progenesis QI 1.1 (Nonlinear Dynamics, Newcastle upon Tyne, UK).
High performance liquid chromatography analysis of S. barbata extracts to estimate content of scutebarbatine A
-
Scutebarbatine A was quantified using a Vanquish UHPLC equipped with PDA and a QExactive Orbitrap MS (Thermo). Separation was on a 50 × 2.1 mm 2.6 μ Kinetex EVO column (Phenomenex) using 2-μL injections and the following gradient of acetonitrile (B) versus water (A), run at 40 °C and 600 μl/min: 0 min, 5% B; 6.4 min, 95% B; 7.4 min, 95% B; 7.5 min, 5 % B; 10 min, 5% B. The instrument collected UV/visible spectra from 200−600 nm at 20 Hz with a response time of 0.2 sec, from which chromatograms were extracted at 259−269 nm, and used only to confirm quantification by MS. Unfragmented (m/z 140−1,500; resolution 70,000) and 'all ions fragmentation' (AIF) spectra were collected using positive mode electrospray, and Scutebarbatine quantified relative to external standards in extracted mass chromatograms at m/z 559.2411−599.2467. Spray chamber conditions were 3,000 V spray voltage, 350 °C capillary temperature, 35 units sheath gas, 10 units aux gas. The instrument was calibrated according to the manufactufrer’s instructions.
More detailed analyses of the S. barbata methanolic leaf extract and BZL were performed on a Thermo Scientific system consisting of a 'Vanquish Flex' U-HPLC-PDA, and an 'Orbitrap Fusion' mass spectrometer fitted with an electrospray source (Thermo Scientific, Waltham, MA, USA). Chromatography was performed on 5 µl sample injections onto a 150 mm × 3 mm, 3 µm Luna C-18 column (Phenomenex, Torrance, CA, USA) using the following 400 µl/min mobile phase gradient of H2O/CH3OH/CH3CN +1% HCOOH: 90:0:10 (0 min), 0:90:10 (60 min), 0:90:10 (70 min), 90:0:10 (71 min), 90:0:10 (75 min). The ESI source was operated under standard conditions and the mass spectrometer was set to record high resolution (60 k resolution) MS1 spectra (m/z 125–2,000) in both positive and negative modes using the orbitrap; and data dependent MS2 and MS3 spectra in both modes using the linear ion trap. Detected compounds were assigned using the approach described by Schymanski et al.[65] and by comparison of accurate mass (ppm) and interpretation of available MSn and UV spectra, with reference to the Royal Botanical Garden at Kew’s in-house libraries of ion trap MS and UV spectra, as well as by comparison with available reference standards: wogonin, wogonoside (Sigma-Aldrich, Gillingham UK), scutebarbatine A (Purifa, Chengdu, China), scutebata F (Efebio, Shanghai, China)
Cryo-Scanning Electron Microscopy (SEM)
-
The fresh leaves were immediately cryopreserved by plunging into liquid nitrogen and transferred to the cryostage of an ALTO 2500 cryotransfer system (Gatan) attached to a cryo-system on an FEI Nova NanoSEM 450 (FEI UK Ltd, Cambridge, UK). Surface frost was sublimated at 95 °C for 3 min before sputter coating with platinum for 150 s at 10 mA whilst below 110 °C. Finally, the sample was moved onto the cryostage in the main chamber of the microscope, held at 125 °C, and viewed at 3.0 kV. Images were stored as TIFF files.
Transmission Electron Microscopy (TEM)
-
The leaves were cut into 1 mm2 pieces and immediately placed in a solution of 2.5% (v/v) glutaraldehyde in 0.05 M sodium cacodylate, pH 7.3 for fixation, and left overnight at room temperature. Samples were then placed in baskets and loaded into the Leica EM TP embedding machine (Leica, Milton Keynes, UK) using the following protocol. The fixative was washed out by three successive 15-min washes in 0.05 M sodium cacodylate and post-fixed in 1% (w/v) OsO4 in 0.05 M sodium cacodylate for 2 h at room temperature. The osmium fixation was followed by three, 15-min washes in distilled water before beginning the ethanol dehydration series (30%, 50%, 70%, 95% and two changes of 100% ethanol, each for 1 h). Once dehydrated, the samples were gradually infiltrated with LR White resin (London Resin Company, Reading, Berkshire, UK) by successive changes of resin:ethanol mixes at room temperature (1:1 for 1 h, 2:1 for 1 h, 3:1 for 1 h, 100% resin for 1 h then 100% resin for 16 h and a fresh change again for a further 8 h) then the samples were transferred into gelatin capsules full of fresh LR White and placed at 60oC for 16 h to polymerize.
The material was sectioned with a diamond knife using a Leica UC6 ultramicrotome (Leica, Milton Keynes, UK) and ultrathin sections of approximately 90 nm were picked up on 200 mesh copper grids which had been formvar and carbon coated. The sections were stained with 2% (w/v) uranyl acetate for 1 h and 1% (w/v) lead citrate for 1 min, washed in distilled water and air dried. The grids were viewed in a FEI Talos 200 °C transmission electron microscope (FEI UK Ltd, Cambridge, UK) at 200 kV and imaged using a Gatan OneView 4K x 4K digital camera (Gatan, Leicester, UK) to record DM4 files.
MALDI imaging
-
Leaves were fixed on glass slides using double-sided tape and dried under a moderate vacuum overnight. The samples were coated with DHB (2,5-dihydroxybenzoic acid) matrix (20 mg/ml in 80% methanol) using a SunCollect sprayer (SunChrome, Friedrichsdorf, Germany). Approximately 30 layers of the matrix were applied at a flow rate of 30 µl/min, at maximum sprayer distance aiming at a matrix density of ~ 0.5 µg/cm2.
MALDI imaging was performed on a Synapt G2-Si mass spectrometer (Waters) in sensitivity and positive ion mode using Masslynx 4.1 and HDI 1.4 software (Waters). The instrument was equipped with a MALDI source comprising of a 2.5 kHz neodymium-doped yttrium aluminum garnet (ND:YAG) solid state laser generating a laser beam with 30 µm diameter (full width half maximum (FWHM) resolution). External calibration was performed using red phosphorous cluster ions. The sample slides were scanned with a common flat-bed scanner to generate images for aligning the MALDI source. Selected sample areas were scanned at a spatial resolution of 15-45 μm (using oversampling) with a scan time of 0.5 s, a laser frequency of 1 kHz and a laser power of 200. The resulting raw files were processed in HDI 1.4 using a mass window (m/z) of 0.05 Da and an MS resolution of 10,000. For visualisation, intensities were normalized by TIC. Microscopic images of the selected areas of the original leaves were taken using a Canon 5D MIV camera with a Canon MP-E 65 mm f/2.8 1-5x Macro Photo lens. Those images were overlaid with the MALDI images to localise the origin of the MALDI signals.
NaDi staining
-
NADI (naphthol + dimethylparaphenylenediamine) reagent was made by mixing 0.5 ml of a 1% α-naphtol solution in 40% alcohol, 0.5 ml of 1% dimethyl-p-phenylenediamine chloride in water, and 49 ml of phosphate buffer 0.05 M (pH 7.2). The fresh leaves were put in NaDi reagent to stain overnight, and then the stained leaves were washed twice with distilled water. The washed leaves were fixed in 50 °C, 7% low-melt agar solution in the plastic molds (length × width: 2 cm × 1 cm). The fixed samples were cut into 50 μm slices using a vibrotome. The pictures of microsections were taken using a Leica DM6000 with bright field and 40× magnification.
-
This study involved the use of established human cell lines. The cell lines used in this research were obtained from Public Health England, UK and were used in accordance with institutional and national ethical standards. The cell lines have been previously published or validated, and no new human tissues were used in this study.
We acknowledge the Royal Society for a Newton Advanced Fellowship awarded to ECT (NAF\R2\192001) and CEPAMS Funding (Project CPM19) for support of a collaboration project 'Scutellaria Anticancer Metabolites' for E.C.T., C.M., M-J.R.H., J.F. and Q.Z. C.M., M.T., E.B., M.R., G.S., L.H.. and J.L. were also supported by the Institute Strategic Programme ‘Molecules from Nature’ (BB/ P012523/1) from the UK Biotechnology and Biological Sciences Research Council. MZ was supported by a CSC visiting scholarship and by the Natural Science Foundation of Zhejiang Province, China (LY21H280009). We thank CAS for the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB27020204) and for International Partnership Program of CAS (153D31KYSB20160074); we gratefully acknowledge the Ministry of Science and Technology for Foreign Expert Project 2019 (G20190113016), the Science and Technology Commission of Shanghai Municipality for Shanghai Talent Recruitment Program 2018 and funds from National Key Laboratory of Plant Molecular Genetics and Shanghai Institute of Plant Physiology and Ecology to ECT for support of this project.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Matthew L. Tomlinson, Man Zhao
- Supplemental Fig. S1 Content of flavones in leaves, roots and flowers of Scutellaria baicalensis and S. barbata. The scale on the left refers to levels (% dry weight) in leaves and roots and the scale on the right shows the levels (% dry weight) in flowers. The relative contents of the 4’deoxyflavones, wogonin and baicalein are shown in orange and their glycosides in blue in each tissue sample. The relative content of the major 4’ hydoxyflavone, scutellarein is shown in orange and its glycoside (scutellarin) is shown in blue. Both S. baicalensis and S. barbata produce very low levels of the bioactive 4’deoxy flavones and their glycosides in leaves but accumulate significant amounts of these specialized flavones in their roots and flowers. Scutellarein and scutellarin accumulate predominantly in aerial tissues of both species.
- Supplemental Fig. S2 Confirmation of identity of m/z 559.24 as scutebarbatine A by MS2.
- Supplemental Fig. S3 Standard curve for calculating content of scutebarbatine A by LCMS.
- Supplemental Fig. S4 Analysis of scutebarbatine A in large peltate trichomes, selected and extracted in methanol and detected by MALDI Autoflex.
- Supplemental Fig. S5 nalysis of methanol (A) and aqueous BZL (B) S. barbata extracts. Base ion chromatograms from LC-MSn (positive ESI) analysis of the Scutellaria barbata leaf extracts.. Assigned compounds for the main detected peaks: 1 6-Hydroxyluteolin 7-hexuronide; 2 Tetrahydroxyflavanone; 3 Tetrahydroxyflavanone; 4 Scutellarein 7-hexuronide; 5 Trihydroxyflavone; 6 Hydroxyapigenin; 7 Isoscutellarein 7-hexuronide; 8 Scutellarein 6-hexuronide; 9 Apigenin hexuronide; 10 Tetrahydroxyflavone methyl ether hexuronide; 11 1-Octen-3-ol pentosyl dihexoside; 12 Scutellarein; 13 Isoscutellarein 8-hexuronide; 14 Tetrahydroxyflavone; 15 Scutellarein Me ether; 16 2ʹ,5,7-Trihydroxyflavone; 17 Barbatin C or isomer; 18 Scutolide I or isomer; 19 Scutebata F; 20 Salvigenin; 21 Scutebarbatine A; 22 Scutebata E or R or Scutolide G; 23 Scuetbata O; 24 Scutebarbatine B or Y or Scutebartine F; 25 Scutebarbatolide C or isomer; 26 6-O-(2-oxo-3-methylbutanoyl)scutehenanine A or isomer. For additional compound assignments see Supplementary Table S2.
- Supplemental Fig. S6 Methanolic extract of S barbata leaves showed no cyto-toxicity on a normal cell line. Ai) -Non-cancerous HCoEpiC cells with control treatment, Aii - AiV) - HCoEpiC cells treated with MeOH S. barbata extract at 1 mg/ml, 2 mg/ml and 3 mg/ml respectively. Annexin V positive cells are on the FITC-A (X axis) and Pi positive cells are on the PE-A (Y axis). Numbers in quadrats are %,. n=3s biologically independent experiments. B) Bar chart showing the effects of the hydrophobic MeOH extract on HCoEpiC cells,, **** for P ≤ 0.0001, *** P ≤ 0.001, statistical difference compared to control (n=3). Data is displayed as individual dots. Horizontal bars show significant changes and error bars show SDev, n=3 biologically independent experiments. C) Apoptotic induction by purified scutebarbatine A (20 µM) and methanol extract of S. barbata(calculated at 19 µM scutebarbatine A) on CaCo 2 cells. Bar chart shows differences, **** for P ≤ 0.0001, statistical difference compared to respective controls (n=3). Data are displayed as individual datapoints. Horizontal bars show significant differences and error bars show SDev, n=3 biologically independent experiments.
- Supplemental Fig. S7 Cytotoxicity (MTT assay) of the hydrophobic S. barbata extract on human breast cancer MCF-7 cells, normal breast MCF-10A cells and colon cancer Caco-2 cells. A) Shows the dose response for both cancerous MCF-7 and normal MCF-10A breast cells, as a bar chart. B) Shows the dose response for Caco-2 cells. All cells were treated with the MeOH extract from S. barbata at 0.25 mg/ml, 0.5 mg/ml, 1 mg/ml, 2 mg/ml (3 mg/ml only for Caco-2). Statistical differences determined by Student’s t tests compared to respective controls. Horizontal bars represent the means and error bars SDev (**** for P ≤ 0.0001, *** P ≤ 0.001), n=6 biologically independent experiments.
- Supplemental Table S1 m/z values of compounds located in large peltate trichomes of leaves of S. barbata, together with tentative identification of some based on accurate mass and MS2 data from methanolic extracts of trichomes. m/z values of compounds identified in medium and small leaf trichomes and in stem trichomes.
- Supplemental Table S2 Assigned compounds detected in the 70% methanol extract of Scutellaria barbata leaves and the aqueous BZL decoction by LC-PDA-MSn analysis.
- Copyright: © 2022 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Tomlinson ML, Zhao M, Barclay EJ, Li J, Li H, et al. 2022. Diterpenoids from Scutellaria barbata induce tumour-selective cytotoxicity by taking the brakes off apoptosis. Medicinal Plant Biology 1:3 doi: 10.48130/MPB-2022-0003 

Diterpenoids from Scutellaria barbata induce tumour-selective cytotoxicity by taking the brakes off apoptosis
- Received: 25 February 2022
- Accepted: 23 May 2022
- Published online: 30 June 2022
Abstract: Medicinal plants are an excellent source of structurally diverse, bio-active compounds with potential in the fight against cancer. One of the most promising is Scutellaria barbata, prescribed traditionally for the treatment of cancers. Scutebarbatine A is the major diterpenoid, produced in specialized large, peltate trichomes on leaves of S. barbata. It induces dose-dependent apoptosis, specifically in cancer cells. The major class of proteins down-regulated are pro-survival proteins, the Inhibitors of Apoptosis (IAPs), and IAP regulating proteins. We propose that scutebarbatine A works by releasing the molecular brakes (the IAPs) on apoptosis in cell death-evading cancer cells. Comparison between the cytotoxicity of methanolic extracts of S. barbata leaves and decoctions (Ban Zhi Lian) prepared traditionally, showed substantially different chemical compositions and differential induction of apoptosis. Analyses suggest polyvalency between the constituents in both extracts, and ways to produce enhanced chemopreventive preparations for the treatment of cancer.
-
Key words:
- Scutellaria barbata /
- Clerodane diterpenoids /
- Traditional Chinese medicine /
- Apoptosis /
- Cancer /
- Inhibitors of apoptosis /
- IAPs