










-
Long non-coding RNAs (lncRNAs) are RNA molecules ≥ 200 nucleotides (nt) long that lack the potential to encode proteins[1]. LncRNAs regulate the expression of neighboring genes via cis-regulation or perform distal regulatory functions as scaffolds via trans-regulation by facilitating the assembly and targeting of chromatin-modifying complexes to specific genomic loci to regulate gene expression[2−5]. LncRNAs are classified into four groups based on their location relative to the nearest protein-coding genes (PCGs): long intergenic non-coding RNAs (lincRNAs) located and transcribed from intergenic regions, intronic non-coding RNAs (incRNAs) transcribed from the introns of PCGs, long sense non-coding RNAs (lsncRNAs) overlapping with PCGs in sense orientation, and long antisense non-coding RNAs (lancRNAs) transcribed from the antisense strands of PCGs. Emerging evidence reveals roles of lncRNAs in regulating key processes in plants, such as flowering time control, reproduction, seedling photomorphogenesis, and environmental adaptation[6−10].
LncRNAs have been identified in numerous plants, such as Arabidopsis (Arabidopsis thaliana), maize (Zea mays), carrot (Daucus carota), soybean (Glycine max), and rice (Oryza sativa)[11−15] . With the continuous discovery of lncRNAs and accumulating knowledge of their functions in plants, many databases have been constructed in the past few years to provide platforms for exploring plant lncRNAs. NONCODEV6 is mainly used to annotate lncRNAs in animals and plants[16]; PLncDB V2.0, AlnC, and GreeNC 2.0 provide lncRNA information for multiple species[17−19]; LncPheDB is a systematic resource of genome-wide lncRNA-phenotype associations for variants of nine species[20]; RiceLncPedia systematically characterizes rice lncRNAs, with expression profiles and multi-omics data, to promote research on rice lncRNAs[21]. However, there lacks a well-curated database of lncRNAs in the staple crop wheat (Triticum aestivum).
Seed development is an important biological process in plants and a major focus of crop breeders since it is directly associated with crop yields and quality. LncRNAs play vital roles in seed development[22]. For example, the lncRNA MSTRG.86004 from rapeseed (Brassica napus) delays seed development and positively regulates fatty acid biosynthesis genes to promote oil accumulation[23]. The rice lncRNA MISSEN (MIS-SHAPEN ENDOSPERM) negatively regulates seed development, as its overexpression leads to increased numbers of shriveled seeds[24]. The lncRNA wheat seed germination-associated RNA (WSGAR) is targeted by miR9678, which induces the generation of phased small interfering RNAs (siRNAs) to inhibit seed germination[25]. The wheat lncRNA TCONS-00130663 participates in resistant starch biosynthesis which was considered to be beneficial to health by negatively regulating starch branching enzyme IIb (TaSBEIIb)[26].
Wheat is one of the most widely grown crop plants worldwide, providing the majority of calories in the human diet. LncRNAs participate in wheat growth and development, such as root and spike development, as well as flowering and stress responses[27−30]. In this study, we performed strand-specific RNA sequencing (ssRNA-seq) of wheat endosperm at 10 and 15 d after pollination (DAP). Based on the ssRNA-seq data, together with 545 published RNA sequencing (RNA-seq) data sets, we identified wheat lncRNAs at the genome-wide level and developed wLNCdb, a comprehensive database for systematically characterizing wheat lncRNAs. wLNCdb integrated the expression profiles of wheat lncRNAs, their epigenetic modification patterns, their target genes in cis and trans, sequence variations, and lncRNA-microRNA (miRNA) associations to facilitate research on wheat lncRNAs. As proof of concept, we focused on lncRNAs that regulate target genes encoding enzymes involved in starch biosynthesis and seed storage proteins (SSPs). We determined that TraesLNC1D26001.1 negatively regulates seed germination, as its overexpression in wheat delayed seed germination. Our findings provide insight for further understanding the regulatory mechanisms of wheat lncRNAs during seed development. In addition, wLNCdb provides valuable information for discovering important lncRNAs for wheat improvement.
-
All wheat (Triticum aestivum) plants were cultivated in the experimental field of China Agricultural University in Beijing, China (40°08′15″ N, 116°11′24″ E) during the normal growing season and in a greenhouse at a relative humidity of 40% and 26 °C/20 °C day/night temperatures under a 16h-light/8h-dark cycle, with a light intensity of 6,000 lux (Master GreenPower CG T 400W E40; Philips). Wild-type (WT), OE1, OE2, and OE3 seeds were planted in a block containing 1.5-m-long rows at a spacing of 20 cm in the field. Three identical blocks were planted. For gene expression analysis, WT, OE1, OE2, and OE3 endosperm at 10, 15, 20, and 25 d after pollination (DAP) was harvested from plants grown in the field. The leaves, stems, roots, young spikes (2 cm), and developing seeds (at 2, 10, and 15 DAP) were also collected from WT plants grown in the field. For each sample, endosperm was collected from three different plants and mixed to represent one biological replicate. At least three technical replicates were performed. The samples were immediately frozen in liquid nitrogen and stored at −80 °C until use.
Strand-specific RNA sequencing
-
Total RNA was isolated with TRIzol reagent (Invitrogen) and treated with DNaseI (Invitrogen) to eliminate contaminating DNA. Ribosomal RNA was removed using a Ribo-Zero™ rRNA Removal Kit (Illumina). Subsequently, random primers from TruSeq® Stranded Kit (Illumina) were used to synthesize cDNA with the templates of fragmented RNAs. The cDNA library was obtained by PCR amplification and sequenced on a HiSeq 6000 instrument (Illumina) that generated paired end reads of 150 bp. Sequence files were deposited at the National Centre for Biotechnology Information (NCBI) Sequence Read Archive under BioProject ID PRJNA974952.
Collection of public transcriptomic datasets
-
A total of 549 available RNA-seq datasets were downloaded from the SRA repository of the National Center for Biotechnology Information (NCBI). These transcriptome profiles cover diverse studies, developmental stages, tissues, and growth conditions (Supplemental Table S1). The fastq-dump tool was used to convert the files from SRA to FASTQ format. Publicly available isoform sequencing (Iso-Seq) datasets from three studies were also collected and used to predict lncRNA models[31−33].
LncRNA identification
-
The quality of the RNA-seq datasets was checked using FastQCv0.11.5 with default parameters. All reads were processed and filtered using fastp[34] 0.19.4 with default parameters. The resulting reads were mapped to the wheat reference genome (IWGSC RefSeq v.1.1) using STAR[35] 2.5.3a with default parameters. SAMtools[36] 1.3.1 were used to extract reads with unique hits.
All reads from Iso-seq were mapped to wheat reference genome (IWGSC RefSeq v.1.1) using GMAP[37] with default parameters. All transcripts from Iso-seq and RNA-seq data were combined using StringTie 1.3.3b with parameter 'merge' into a pool of candidate transcripts.
A strict pipeline was used to identify lncRNAs in wheat, as previously described[38,39]. StringTie[40] 1.3.3b with default parameters was used to assemble and combine all transcripts from Iso-Seq and RNA-seq data into a pool of candidate transcripts. The transcripts with Transcripts Per Million (TPM) > 0.5 and length > 200 bp were retained. The transcripts that overlapped with protein-coding genes and transposable elements were excluded from further analysis. The wheat protein databases CPC2 v1.0.1[41] and CNCI[42] with default parameters were used to remove the transcripts with protein-coding potential. Transcripts that passed the above filtering steps were annotated as lncRNAs. The lncRNA transcripts were classified according to the reference annotation file using GffCompare[43] v0.11.2 with default parameters. The lncRNAs were categorized into four subclasses based on their class codes: sense lncRNAs (class codes 'j' and 'o'), antisense lncRNAs (class code 'x'), intron lncRNAs (class code 'i'), and intergenic lncRNAs (class code 'u').
Gene expression analysis
-
All RNA-seq datasets were mapped to the protein-coding genes (PCGs) and lncRNA transcripts using kallisto[44]. The tximport tool was used to summarize expression values from the transcript level to the gene level. Only lncRNAs with TPMs > 1 in at least two samples were considered to be expressed, resulting in 20,893 expressed lncRNAs, accounting for 98.0% of annotated lncRNAs. Principal component analysis (PCA) was conducted to assess the representativeness of these RNA-seq datasets (Supplemental Fig. S1).
LncRNA target prediction
-
Since lncRNAs can affect gene expression in cis and trans, lncRNAs with evidence of both effects were identified. For cis-target genes, the neighboring genes of each lncRNA were examined. The genes were identified as cis-regulated target genes when (i) the mRNA loci were within 50 kb upstream or downstream of the given lncRNA and (ii) the Pearson correlation coefficient (PCC) of lncRNA-mRNA expression values were > 0.9. Trans-target genes were identified using custom scripts. If a gene was significantly co-expressed with a given lncRNA (PCC > 0.99) and its locus was not within 50 kb upstream or downstream of the given lncRNA, it was annotated as a trans-target gene.
Implementation of wLNCdb platform
-
The wLNCdb platform contains 23000 lncRNAs and implements seven versatile analysis and visualization tools for the community to explore functional lncRNAs in wheat. The detail is shown in the supplemental information.
RT-qPCR
-
Total RNA was extracted from leaves, stems, roots, young spikes (2 cm), and developing seeds at 2, 10, and 15 DAP and germinating seeds at 1 d after imbibition (DAI) using TransZol Plant reagent (TransGen Biotech, ET121-01), and first-strand cDNAs were synthesized using HiScript II Q RT SuperMix (Vazyme Biotech, R223-01) according to the manufacturer's instructions. For RT-qPCR, Taq Pro Universal SYBR qPCR Master Mix (Vazyme Biotech, Q712-02/03) was used. The reaction mixture was composed of 5 µL SYBR Master Mix, 1 µL cDNA template, and 0.2 mM primers in a final volume of 10 µL. Amplification was performed using a CFX96 real-time system (BioRad), and TaACTIN (TraesCS5B02G124100) was used as the internal control gene. Differences in relative transcript levels were calculated using the CT (2−ΔCT) method[45]. Each experiment was performed with three biological replicates and three technical replicates. Statistical significance was determined by performing a Student's t-test at p < 0.05. Primers are listed in Supplemental Table S2.
Plasmid construction and plant transformation
-
To generate the overexpression constructs, the TraesLNC1D26001.1 transcript was amplified and inserted into the BamHI sites of the pMWB110 vector. The recombinant plasmid was transferred into Agrobacterium tumefaciens strain EHA105 and transformed into wheat cultivar Fielder by Agrobacterium-mediated transformation.
Reverse-phase high-performance liquid chromatography (RP-HPLC)
-
The high-molecular-weight glutenins (HMW-GSs), low-molecular-weight glutenins (LMW-GSs), and gliadins content of the samples were examined by RP-HPLC as previously described with some modifications[46]. The Agilent Technologies 1260 Infinity II RP-HPLC system and an Agilent ZORBAX 300SB-C18 column (150 mm × 4.6 mm, 5 μm) were used. The elution solvents were water containing 0.6 mL·L−1 trifluoroacetic acid and acetonitrile containing 0.6 mL·L−1 trifluoroacetic acid. The total amounts of HMW-GSs, LMW-GSs, and gliadins were estimated by integrating the relevant RP-HPLC peaks in the chromatograms. Three replications were performed per sample.
Germination test
-
To evaluate the germination rate, the seeds were washed with 1% sodium hypochlorite for 20 min and rinsed three times with sterile water. The seeds were placed in a 50-mL centrifuge tube and imbibed in 30 mL sterile water for 12 h. For each replicate, 50 seeds were sown in a 10-cm Petri dish containing 8 mL distilled water. All germination tests were performed in three independent replicates. The Petri dishes were incubated in the dark for 3 d and transferred to a growth chamber under a 16h-light/8h-dark cycle at a constant temperature of 22 °C. Seeds were considered to be germinated when the germ reached half the length of the seed; germination percentages were calculated at daily intervals. For ABA sensitivity assay of WT and TraesLNC1D26001.1 overexpression lines (OE1, OE2, and OE3) seeds, 0, 0.6, 1 and 2 mg·L−1 ABA were used in germination test. For each replicate, 30 seeds were sown in a 6-cm Petri dish.
-
To construct a comprehensive atlas of lncRNAs in bread wheat at the genome-wide scale, we performed ssRNA-seq using endosperm from grains at 10 and 15 d after pollination (DAP) and collected 545 publicly available RNA-seq datasets covering diverse developmental stages and tissues (Supplemental Table S1). Principal component analysis (PCA) demonstrated that these RNA-seq datasets are sufficiently representative and that redundant datasets have been removed (Supplemental Fig. S1a & S1b). The pipeline used to identify lncRNAs is based on a previous study[38] with three key steps and some additional modifications, including mapping, assembly, and filtering. Step 1: We mapped all clean reads from the RNA-seq datasets to the reference genome (IWGSC RefSeq v.1.0). Step 2: These sequences were used to perform de novo transcript assembly with StringTie[40], which resulted in 83,623 expressed loci with 98,444 transcript isoforms. Step 3: We excluded transcripts that overlapped with PCGs. We used Pfam and the wheat protein databases CPC2[41] and CNCI[42] to remove the transcripts with protein-coding potential. Finally, we identified a total of 20,893 lncRNAs in wheat (Supplemental Table S3).
To investigate the characteristics of lncRNAs in wheat, we compared the transcript features of lncRNAs and PCGs. The average transcript length of lncRNAs was 900 bp, which was much shorter than that of PCGs (2,329 bp) (Fig. 1a). The lncRNAs contained fewer exons than PCGs, and the majority (48%) of the lncRNAs consisted of a single exon (Fig. 1b). Transposable elements were enriched in the genomic regions of lncRNAs, which is consistent with previous findings[47]. Among lncRNAs, lincRNAs showed the highest transposable element density (Fig. 1c). The types of transposable elements that overlapped with lncRNAs differed from those that overlapped with PCGs. More long terminal repeat retrotransposons (LTRs, 41.40%) overlapped with lncRNAs, whereas more DNA transposons (62.86%) overlapped with PCGs (Fig. 1d). In addition, lncRNAs showed higher tissue specificity than PCGs (Fig. 1e). In general, wheat lncRNAs exhibit shorter transcript lengths, fewer exons, and higher tissue-specific expression than PCGs, which are consistent with the typical features of lncRNAs identified in soybean, carrot, and codling moth (Cydia pomonella)[13,14,47].
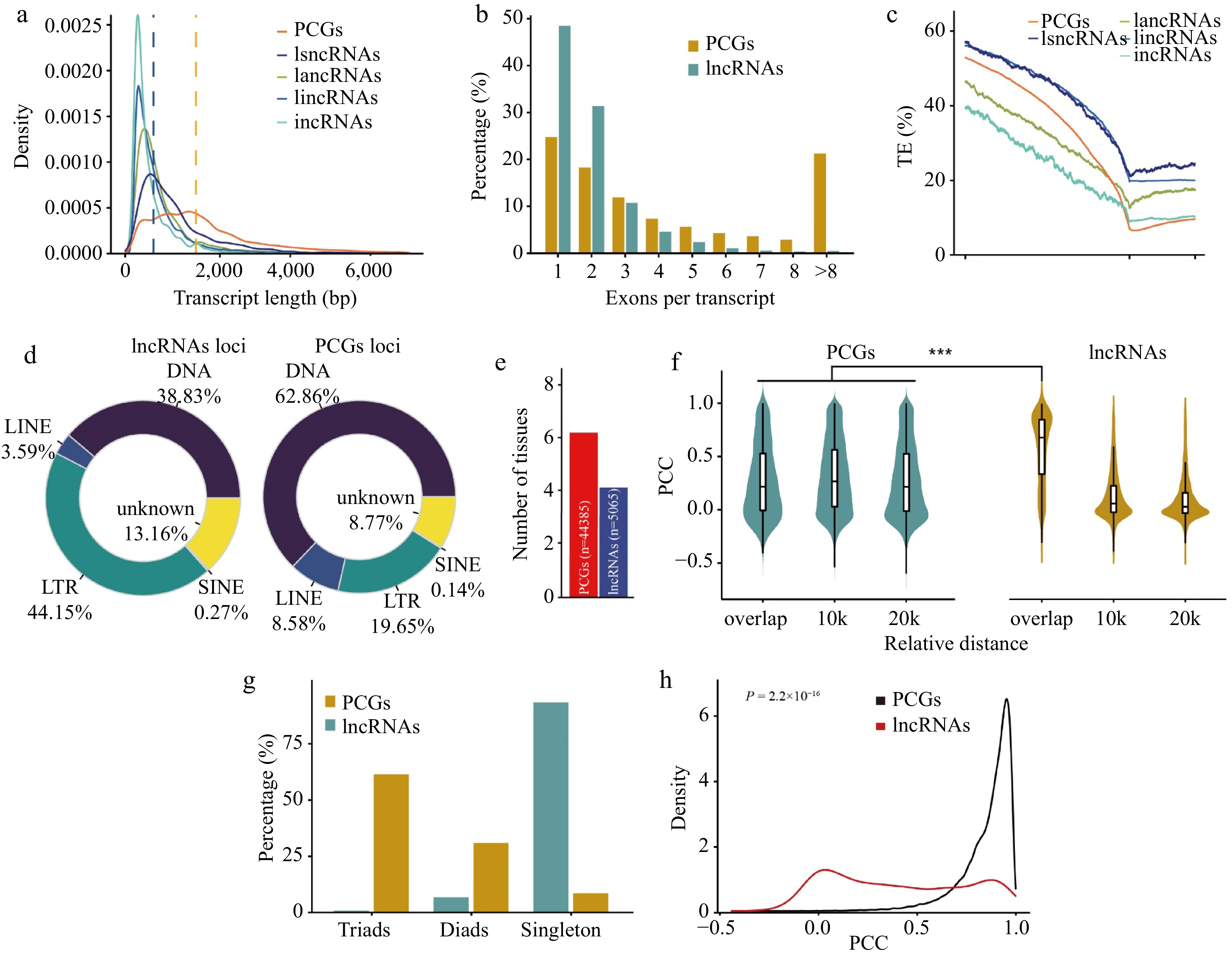
Figure 1.
Comparison of protein-coding genes and lncRNAs in wheat. (a) Distribution of transcript length of PCGs and lncRNAs. Each line represents a type of transcript, including transcripts from protein-coding genes (PCGs), long sense non-coding RNAs (lsncRNAs), long antisense non-coding RNAs (lancRNAs), long intergenic non-coding RNAs (lincRNAs), and intronic non-coding RNAs (incRNAs). The y-axis shows kernel density distribution values. (b) Number of exons in transcripts of PCGs and lncRNAs. (c) Transposable element (TE) density in PCGs, lsncRNAs, lancRNAs, lincRNAs, and incRNAs calculated using a sliding window approach over 100-bp intervals across the 5-kb promoter region and gene body. TSS: transcription start site; TTS: transcription termination site. The density of transposable elements from 5-kb upstream of the transcription termination site of each lncRNA and PCG was calculated in sliding windows of 100 bp. (d) Pie charts showing the distribution patterns of different classes of transposable elements overlapping with PCGs and lncRNAs. The classes of transposable elements include long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), long terminal repeats (LTRs), and DNA transposons (DNA). (e) Number of tissues in which 50% of lncRNAs or PCGs are highly expressed. For lncRNAs and PCGs, the median expression level for each tissue was calculated and genes were binned as having high (above the median) or low expression levels in each tissue. Genes were ranked based on the number of tissues in which they were highly expressed, and the median was plotted for PCGs (blue) and lncRNA genes (red). (f) Violin chart showing the correlation of expression patterns between neighboring (overlap, 10 kb, 20 kb) gene pairs and lncRNA-PCGs gene pair. (g) Number of homoeologous alleles including singletons, dyads, and triads in PCGs and lncRNAs. The homoeologous lncRNA pairs between the A, B, and D subgenomes were identified using MCscan with default parameters. (h) Distribution of Pearson correlation coefficients (PCC) calculated for homoeologous lncRNAs (red) and PCGs (black) using gene expression values.
To explore the function of lncRNAs in gene regulation, we examined whether lncRNAs and their adjacent genes are concordantly or discordantly expressed. We calculated the Pearson correlation coefficient (PCC) between the different types of lncRNAs and their adjacent protein-coding genes (PCGs). The PCC values between adjacent PCG pairs were calculated in parallel for comparison. We found that the PCC values of overlapping lncRNA/PCGs pairs were significantly higher than the values of overlapping protein-coding pairs (Fig. 1f), suggesting that lncRNAs have a stronger tendency to have positively correlated expression patterns with their adjacent PCGs.
Wheat is an allohexaploid (BBAADD) and composed of three subgenomes A, B, and D. To investigate the relationships of lncRNAs between different subgenomes, we identified the homoeologs of lncRNAs across the A, B, and D subgenomes. Whereas 90.7% of the lncRNAs were present in a single subgenome, only 1.0% were shared by the three subgenomes (Fig. 1g), which is much lower than the triplet ratio of homoeologous PCGs (55.1%) previously identified in wheat[48]. In addition, the correlation between the expression levels of homoeologous PCGs is higher than that of homoeologous lncRNAs (p = 2.2 × 10−16) (Fig. 1h). These results indicate that compared with PCGs, lncRNAs showed different evolutionary fates. Half of PCGs were in triads from three subgenome and likely correspond to highly conserved and evolutionary constrained genes[49], and lncRNAs preferred to present in single copy originated from one subgenome, which belonged to dispensable genes involved in functions such as biotic stress response and development.
wLNCdb: a comprehensive database of lncRNAs in wheat
-
To efficiently utilize the extensive information about the annotated wheat lncRNAs, we developed wLNCdb (
http://wheat.cau.edu.cn/wLNCdb/ ), a wheat lncRNA database integrated comprehensive information and versatile analysis tools for decoding functional lncRNAs (Fig. 2a). wLNCdb provides versatile tools for browsing and searching multiple features of a given lncRNA. These comprehensive features include expression patterns, co-expression networks, cis- (relative position of lncRNA with PCGs < 50 kb and Pearson correlation coefficient with PCGs > 0.95) and trans-regulation (relative position with PCGs > 50 kb and Pearson correlation coefficient with PCGs > 0.99), epigenomic states, miRNA-lncRNA interactions, genomic variations, and functional annotations. In wLNCdb, we assigned each lncRNA to a unique identifier, which can be used to query in the Search page interactively in a pangenomic manner, and provided an application programming interface (API) for convenient integration with other modules or databases.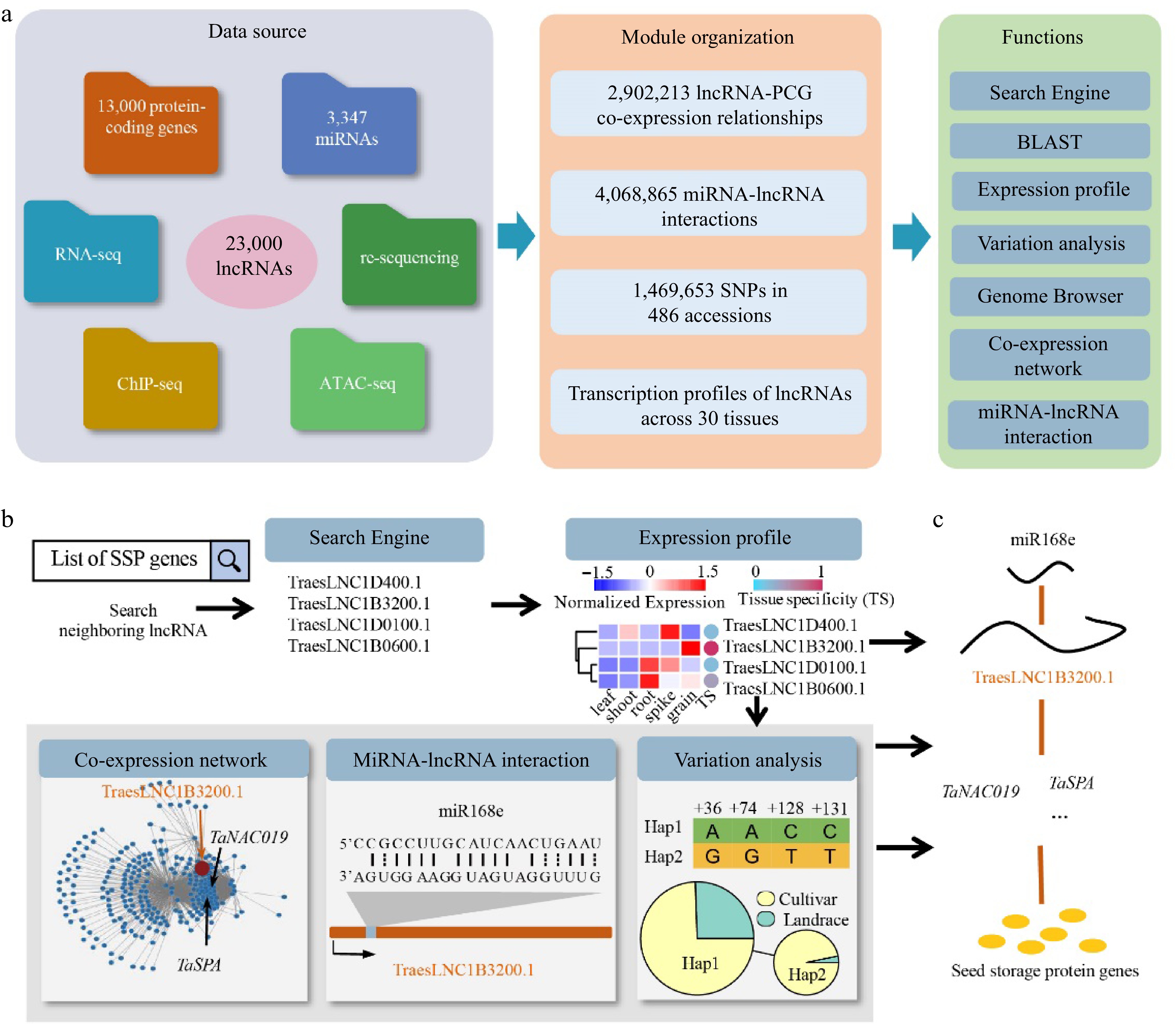
Figure 2.
Overview of wLNCdb and its application in exploring lncRNAs. (a) The framework of wLNCdb, including multi-omics datasets as a resource database for construction (left panel), module organization (middle panel), and interactive analysis and visualization tools (right panel). (b) A case study showing how wLNCdb can be used to explore functional lncRNAs. The 'Search Engine' module used a list of seed storage protein (SSP) genes as the input and identified SSP-associated lncRNAs. The 'Expression profile' module shows the normalized expression levels (Z-score) and tissue-specificity scores of the identified lncRNAs. For each gene or lncRNA, the tissue-specificity score was calculated by dividing the average TPM in a tissue by the sum of the average TPM values of all tissues. The 'Co-expression network' module shows protein-coding genes (blue nodes) co-expressed with TraesLNC1B3200 (orange node), together with their regulatory relationships (edges) acquired from wGRN[38]. The 'miRNA-lncRNA interaction' module shows the predicted interactions between miR168e and TraesLNC1B3200. The 'Variation analysis' module shows the variation profiles and haplotype distribution of TraesLNC1B3200 across 125 Chinese wheat accessions. (c) Proposed model of the role of TraesLNC1B3200 in regulating grain development and end-use quality, supported by wLNCdb.
wLNCdb contains seven interactive modules for exploring wheat lncRNA repertoires (Fig. 2a). On the home page, the 'Expression profile' module allows users to interactively visualize gene expression profiles across diverse tissues and identify tissue-specific genes with the assistance of specially designed scores, as described previously[50]. The 'Genome Browser' module allows users to interactively visualize the transcripts of lncRNAs and interrogate epigenomic signals around the lncRNA locus, such as histone modifications and open chromatin profiles[38]. The 'Blast' module based on SequenceServer[51] is also provided for searching lncRNA sequences against genome assemblies.
We developed the 'Co-expression network' module to identify the potential targets of lncRNAs and the pathways involving these lncRNAs. Users can input a lncRNA of interest to query lncRNA-PCG co-expression networks and filter the resulting co-expression relationships according to a variable parameter, the Pearson correlation coefficient. To expand our understanding of post-transcriptional regulation, we also designed the 'miRNA-lncRNA interaction' module to identify miRNA precursors and miRNA-lncRNA interactions. Two user-friendly functions are provided, allowing users to predict targets of a specific miRNA by selected lncRNA transcripts and to predict the targets of small RNAs against user-provided transcripts.
The 'Genome variation' module allows users to explore the genetic variations of lncRNAs across wheat populations based on whole-genome sequencing datasets for 681 wheat accessions[52] (
http://wheat.cau.edu.cn/WheatUnion/ ) and 1,469,653 single nucleotide polymorphisms (SNPs) across 21,793 lncRNA transcripts, with an average of 10 SNPs per lncRNA transcript. Users can download a table of SNPs and insertions/deletions (InDels) around lncRNA loci using the VarTable function and visualize the variations in multiples ways, such as constructing haplotype networks using HapNet, exploring SNP-based population structure using PhyloTree, and visualizing the geographical distribution of haplotypes on a world map using HapMap. The SeqMaker function can be used to create a consensus sequence by substituting identified SNPs and InDels against the reference genome.Exploring wLNCdb: a case study
-
Here, we present a case study for exploring lncRNAs using wLNCdb. Taking the gene encoding ω-gliadin (associated with the end-use quality of wheat) as an example (Fig. 2b), we used a list of SSP genes as input in the 'Search' module to search for neighboring lncRNAs. This search identified the candidate lncRNA TraesLNC1B3200, which is located adjacent to the ω-gliadin gene (TraesCS1B02G041928). Using the 'Expression' module, we determined that TraesLNC1B3200 is specifically expressed in grain, pointing to its potential role in controlling wheat end-use quality.
To investigate the regulatory network involving TraesLNC1B3200, we used the 'Co-expression network' module and determined that it is co-expressed with two previously characterized genes, NAC transcription factor (no apical meristem/Arabidopsis transcription activation factor/cup-shaped cotyledon) (TaNAC019) and wheat storage protein activator (TaSPA), encoding proteins that regulate starch and protein biosynthesis in wheat[53,54]. Using the 'miRNA interaction' module, we speculated that this lncRNA might be downregulated by miR168e, a miRNA involved in grain development[55]. To investigate the haplotype distribution of TraesLNC1B3200, we used the 'Variation' module and identified its two major haplotypes. These haplotypes showed significantly different frequencies between wheat cultivars and landraces (p = 8.0 × 10−3, Fisher's exact test), pointing to possible selection pressure during modern wheat breeding. These results reveal a complex network involving TraesLNC1B3200 that might contribute to grain development and the regulation of end-use quality (Fig. 2c). These findings highlight the power of using wLNCdb to discover regulatory mechanisms associated with complex phenotypic traits. In summary, this case study demonstrated that wLNCdb can provide valuable information to identify important lncRNAs for wheat improvement.
Characterizing lncRNAs associated with grain development using a network-based approach
-
To further explore the functions of the lncRNAs, we calculated the correlation of expression levels between genes and lncRNAs among 37 wheat samples covering diverse developmental stages and tissues to identify lncRNA-PCG pairs with similar expression patterns. Since correlated genes are predicted to play similar roles in development, pathway enrichment analysis of correlated clusters/modules can shed light on the potential functions of lncRNAs in correlated modules. The 'Co-expression' module in wLNCdb provides a list of co-expressed genes for each lncRNA. Here, we focused on lncRNAs that are highly co-expressed with PCGs related to wheat grain development.
We collected a list of 267 PCGs related to wheat grain size and weight as well as those involved in starch metabolism (Supplemental Table S4). Eleven PCGs were predicted to be highly co-expressed with lncRNAs, including Brittle 1 (TaBT1-6D), two cell wall invertase genes (TaCwi-5D and TaCwi-4A), a sucrose transporter gene (TaSUT-1A), Sucrose non-fermenting 1-related protein kinase 2 (TaSnRK2.10-4A), three Staurosporine and temperature sensitive 3 homoeologs (TaSTT3-1A, TaSTT3-1B, and TaSTT3-1D), two homoeologous beta-amylase genes (TraesCS2A02G215100 and TraesCS2B02G240100), and Granule-bound starch synthase II (TaGBSSII) (Table 1, Supplemental Table S5). These lncRNAs are candidates for functional analysis of the regulation of wheat grain size and weight.
Table 1. Genes co-expressed with lncRNAs involved in wheat grain size and weight and starch metabolism.
Gene ID Gene name Function Number of co-expressed lncRNAs TraesCS6D02G168200 TaBT1-6D Adenine nucleotide transporter BT1, chloroplastic/amyloplastic/mitochondrial 8 TraesCS5D02G552900 TaCwi-5D Beta-fructofuranosidase, insoluble isoenzyme 3 213 TraesCS4A02G321700 TaCwi-4A Beta-fructofuranosidase, insoluble isoenzyme 3 210 TraesCS4A02G235600 TaSnRK2.10-4A Serine/threonine-protein kinase SAPK10 21 TraesCS2B02G390700 TaGBSSII Granule-bound starch synthase 1b, chloroplastic/amyloplastic (Fragment) 42 TraesCS2B02G240100 Beta-amylase Beta-amylase Tri a 17 26 TraesCS2A02G215100 Beta-amylase Beta-amylase Tri a 17 5 TraesCS1D02G342400 TaSTT3a-1D Dolichyl-diphosphooligosaccharide-protein glycosyltransferase subunit STT3A 2 TraesCS1B02G352700 TaSTT3a-1B Dolichyl-diphosphooligosaccharide-protein glycosyltransferase subunit STT3A 2 TraesCS1A02G340400 TaSTT3a-1A Dolichyl-diphosphooligosaccharide-protein glycosyltransferase subunit STT3A 1 TraesCS1A02G134100 TaSUT-1A Sucrose transport protein SUT 5 To identify lncRNAs that regulate SSP accumulation, we searched for lncRNAs that are co-expressed with SSP genes. We analyzed 102 SSP genes encoding high-molecular-weight glutenins (HMW-GSs), low-molecular-weight glutenins (LMW-GSs), or gliadins in wheat[53]. Thirty-three SSP genes were highly co-expressed with lncRNAs (Table 2, Supplemental Table S6). Notably, six of these lncRNAs are transcribed from the adjacent SSP genes (Fig. 3, upper panel of each figure part). Similar to SSP genes, these lncRNAs are also exclusively expressed in grains at 2, 10, and 15 DAP (Fig. 3, middle panels), and their expression is significantly correlated with the expression of adjacent SSP genes (Fig. 3, lower panels). For example, the antisense lncRNA TraesLNC1D26001.1 (which is transcribed from the promoter of the HMW-GS gene TaGLU-1Dy) and TraesLNC1D26001.1 showed high co-expression with TaGLU-1Dy (Fig. 3a). These results suggest that these lncRNAs potentially regulate the expression of these SSP genes. Due to the important contribution of TaGLU-1Dy to wheat flour processing quality, we performed functional analysis of TraesLNC1D26001.1 to explore the regulatory relationship between TraesLNC1D26001.1 and TaGLU-1Dy.
Table 2. Seed storage protein genes that are co-expressed with lncRNAs.
Gene ID Annotation Co-expressed lncRNAs TraesCS1D02G317301 HMW-GSs
(TaGLU-1Dy)TraesLNC1D26001.1 TraesCS1D02G008600 LMW-GSs TraesLNC1D600.1 TraesCS1D02G007626 LMW-GSs TraesLNC1D500.1 TraesCS1D02G007400 LMW-GSs TraesLNC1D500.1 TraesCS1A02G007934 LMW-GSs TraesLNC7D23000.1 TraesCS1D02G000582 ω-gliadin TraesLNC1D200.1 TraesCS1D02G000531 ω-gliadin TraesLNC1D200.1,
TraesLNCUn18600.1TraesCS1B02G041928 ω-gliadin TraesLNC1B3200.1 TraesCS1B02G011484 ω-gliadin TraesLNC1B700.2 TraesCS1B02G011467 ω-gliadin TraesLNC1B700.2 TraesCS1B02G011463 ω-gliadin TraesLNC1B700.2 TraesCS1B02G011460 ω-gliadin TraesLNC1B700.1 TraesCSU02G182638 ω-gliadin TraesLNC1D32300.1,
TraesLNC1A31800.1,
TraesLNC3B43500.1TraesCS1D02G001300 δ-gliadin TraesLNC7A20900.1,
TraesLNC7D23000.1,
TraesLNCUn39000.1TraesCS1D02G001400 γ-gliadin TraesLNC1B700.1 TraesCS1B02G011000 γ-gliadin TraesLNC1B700.1 TraesCS1B02G010900 γ-gliadin TraesLNC7D23000.1 TraesCS6B02G066100 α-gliadin TraesLNC6B6900.1,
TraesLNCUn39000.1TraesCS6B02G066000 α-gliadin TraesLNC6B6800.1,
TraesLNC6B6900.1TraesCS6A02G049400 α-gliadin TraesLNC7A20900.1 TraesCS6A02G049200 α-gliadin TraesLNC6A4700.1,
TraesLNC1B700.1TraesCS6A02G049100 α-gliadin TraesLNC6A4700.1 TraesCSU02G108100 α-gliadin TraesLNC1B700.1 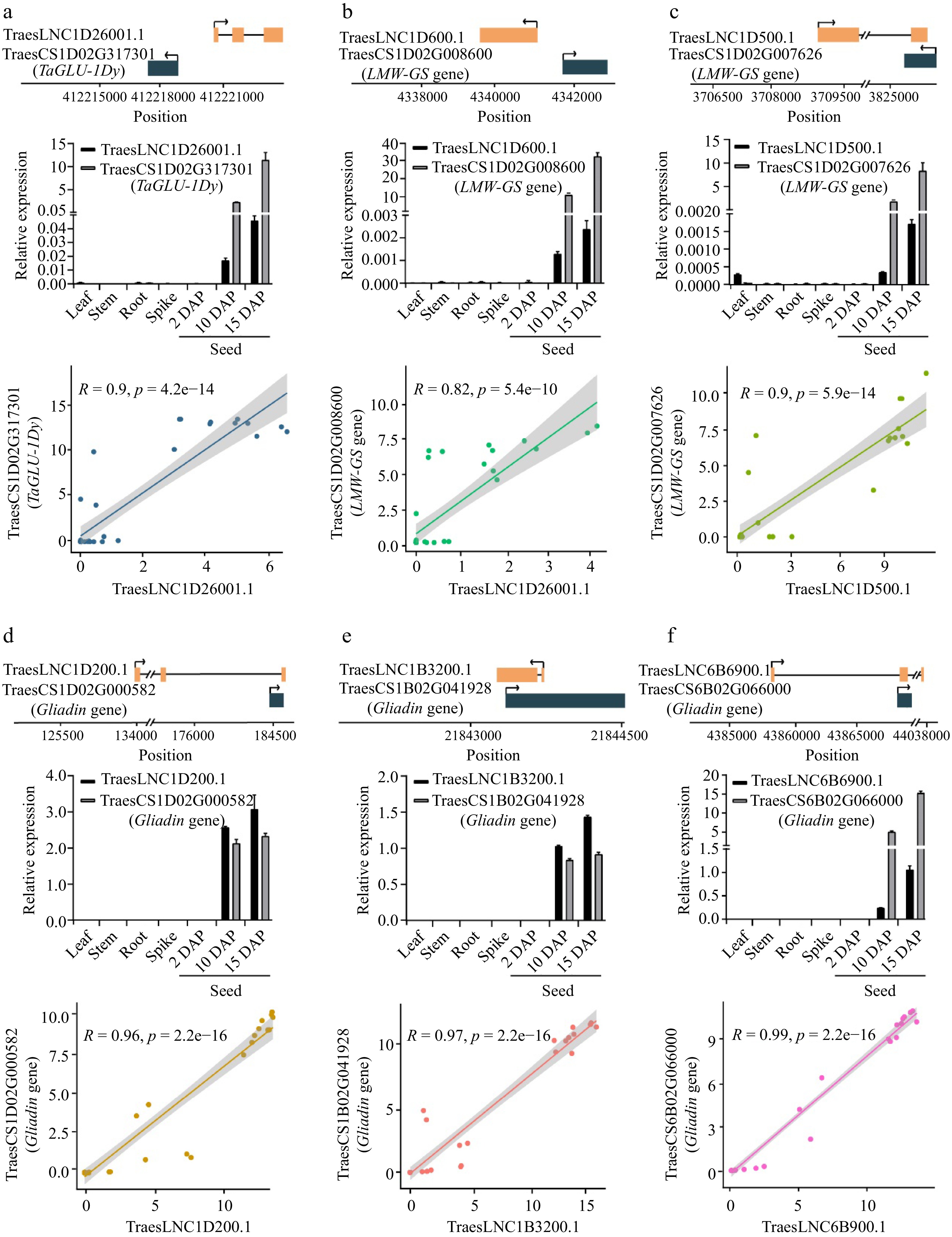
Figure 3.
The relative position, expression, and Pearson correlation coefficient between the expression levels of lncRNAs and cis-regulated SSP genes. (a) TraesLNC1D26001.1 and TraesCS1D02G317301, (b) TraesLNC1D0600.1 and TraesCS1D02G008600, (c) TraesLNC1D0500.1 and TraesCS1D02G007626, (d) TraesLNC1D0200.1 and TraesCS1D02G000582, (e) TraesLNC1B3200.1 and TraesCS1B02G041928, (f) TraesLNC6B6900.1 and TraesCS6B02G066000. For example, (a) shows the relative position, expression, and Pearson correlation coefficient between the expression level of TraesLNC1D26001.1 and TraesCS1D02G317301. The upper panel shows the gene structure and physical locations of TraesLNC1D26001.1 and TraesCS1D02G317301. The middle panel shows TraesLNC1D26001.1 and TraesCS1D02G317301 expression in leaves, stems, roots, young spikes (2 cm), and seeds at 2, 10, and 15 d after pollination (DAP), as revealed by RT-qPCR. Data were normalized to wheat TaACTIN and are shown as mean ± SD from three biological replicates. The bottom panel shows the Pearson correlation coefficient between the expression levels of TraesLNC1D26001.1 and TraesCS1D02G317301. R, Pearson’s correlation coefficient of transcription leves of TraesLNC1D26001.1 and TraesCS1D02G317301 across 37 samples. Each point represents a tissue, with the x-axis and y-axis representing the log2(TPM + 1) of TraesLNC1D26001.1 and TraesCS1D02G317301 in that tissue, respectively.
TraesLNC1D26001.1 is involved in wheat seed germination
-
Since TraesLNC1D26001.1 is located near to TaGLU-1Dy, we would determine their regulatory relationship. We overexpressed this lncRNA in wheat cultivar Fielder driven by the maize UBIQUITIN promoter. Three independent positive transgenic lines OE1, OE2, and OE3 showed higher TraesLNC1D26001.1 expression levels compared to the wild type (WT) in the T3 generation seeds (Fig. 4a). We investigated the TaGLU-1Dy expression level in the OE transgenic lines and WT using endosperm at the watery ripe stage (10 DAP), milky stage (15 DAP) and soft dough stage (20 and 25 DAP). TaGLU-1Dy expression did not significantly differ between the WT and OE lines (Fig. 4b). We then measured the accumulation of HMW-GSs in mature seeds via reverse-phase high-performance liquid chromatography (RP-HPLC), finding no significant change in the level of any subunit, including 1Bx, 1By, 1Dx, and 1Dy (Fig. 4c). In addition, there were no significant differences in LMW-GS or gliadin levels between the OE lines and the WT (Fig. 4d & e). These results suggest that TraesLNC1D26001.1 does not regulate its nearby gene in cis at the transcriptional or translational level and might be able to sequester proteins from their targets of action.
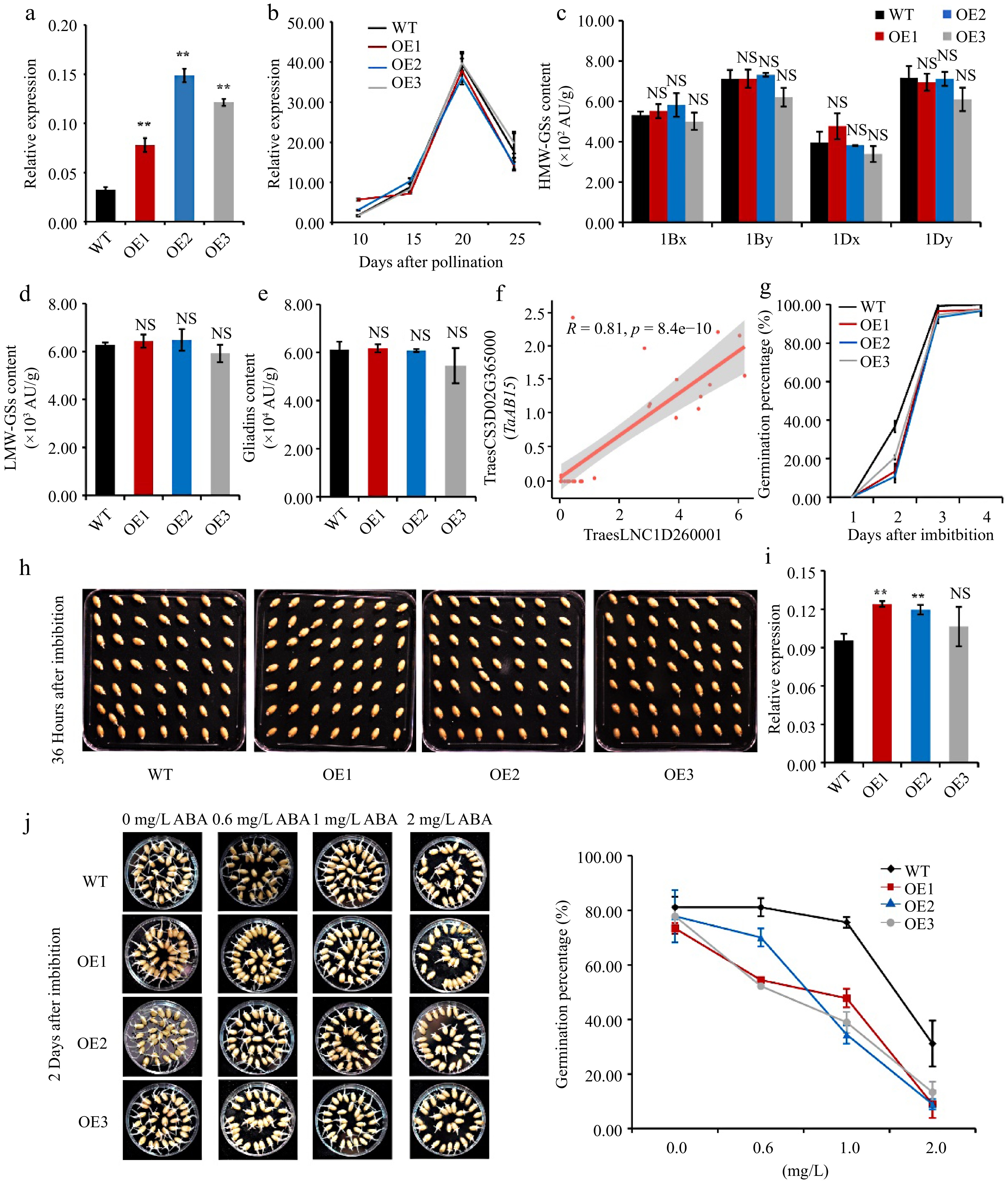
Figure 4.
Overexpressing TraesLNC1D26001.1 delays germination in wheat. (a) TraesLNC1D26001.1 expression levels determined by RT-qPCR in three independent TraesLNC1D26001.1-overexpression lines (OE1, OE2, and OE3) and the wild type (WT). Data were normalized to TaACTIN and are shown as mean ± SD from three biological replicates. Significant differences compared to the WT are indicated by **, p < 0.01, as determined by Student's t-test. NS, no significant difference. The statistical analyses described here applied to all other statistical analyses in this figure. (b) TaGLU-1Dy expression determined by RT-qPCR in OE1, OE2, OE3, and the WT using RNA extracted from whole seeds at 10, 15, 20, and 25 DAP. Data were normalized to TaACTIN and are shown as mean ± SD from three biological replicates. (c) High-molecular-weight glutenins (HMW-GSs), (d) low-molecular-weight glutenins (LMW-GSs), and (e) gliadin content in OE1, OE2, OE3, and the WT, as determined by reverse-phase high-performance liquid chromatography (RP-HPLC) analysis. Three biological replicates were carried out for each sample. (f) Pearson correlation coefficient between the expression levels of TraesLNC1D26001.1 and TraesCS3D02G365000 (TaABI5). R and p represent the Pearson correlation coefficient and p-value of TraesLNC1D26001.1 and TaABI5 expression in 37 samples, respectively. Each point represents a tissue, with the x-axis and y-axis representing the log2 (TPM+1) of TraesLNC1D26001.1 and TaABI5 in that tissue, respectively. (g−h) Germination percentages of OE1, OE2, OE3, and WT seeds. (g) Data are represented as a line connecting the average of three data points. (h) Representative photographs of OE1, OE2, OE3, and WT seeds at 36 h after imbibition (HAI) are also shown. (i) TaABI5 expression determined by RT-qPCR analysis in OE1, OE2, OE3, and the WT using RNA extracted from whole seeds at 1 d after imbibition (DAI). Data were normalized to TaACTIN and are shown as mean ± SD from three biological replicates. (j) Representative photographs and germination percentages of OE1, OE2, OE3, and WT seeds at 2 d after imbibition with 0, 0.6, 1 and 2 mg·L−1 ABA treatment, respectively. Data are represented as a line connecting the average of three data points.
Then we analyzed the putatively trans-regulated genes that were co-expressed with TraesLNC1D26001.1 using wLNCdb, among which we identified ABSCISIC ACID-INSENSITIVE 5 (TaABI5) (Fig. 4f) which involved in seed maturation, germination and ABA response[56]. Compared to the WT, TaABI5 expression was significantly higher in OE1 and OE2 at 1 DAI by RT-qPCR (Fig. 4i). We compared the germination ability of OE and WT seeds and the results indicated that OE1, OE2, and OE3 seeds exhibited substantially reduced germination percentages at 2 and 3 days after imbibition (DAI) compared to the WT, indicating that TraesLNC1D26001.1 inhibits seed germination (Fig. 4g & h). We further compared the germination rate of WT and OE lines in response to ABA. Under normal condition, seeds of OE lines showed slower germination rates compared to WT, while the germination rate of OE seeds strikingly decreased with the increased ABA concentrations, suggesting that overexpression of TraesLNC1D26001.1 enhanced ABA sensitivity during seed germination (Fig. 4j). In Arabidopsis, abi5 mutation causes insensitivity to ABA-dependent seed germination and overexpression of ABI5 results in enhanced sensitivity to ABA[57]. In addition, ectopic overexpression of wheat TaABI5 in Arabidopsis was also hypersensitive to ABA in seed germination[56]. These results suggest that TraesLNC1D26001.1 plays an important role in seed germination and ABA response by regulating TaABI5 expression.
-
In this study, we constructed the wheat lncRNA database wLNCdb, which provides the following information: (i) integrated data about lncRNAs at the whole-genome scale and their expression patterns in various samples, including different tissues and developmental stages; (ii) epigenetic histone modifications on lncRNA-encoding loci and their flanking genomic regions; (iii) SNPs in lncRNAs among 486 wheat accessions; (iv) PCGs that are co-expressed with lncRNAs. Although many plant lncRNA databases have been developed[18,20,21], the current plant lncRNA databases have some limitations, including small sample sizes and the lack of comprehensive information. wLNCdb is the first comprehensive database specifically developed for wheat lncRNAs. Compared to other plant lncRNA databases such as PLncDB V2.0 and AlnC, which mainly provide information about lncRNAs[18,19], we designed wLNCdb to investigate transcriptional regulation by wheat lncRNAs; wLNCdb contains three useful tools: the 'Genome variation', 'Co-expression', and 'miRNA' modules. Users can query a certain lncRNA by its transcript-ID or sequence using the BLAST function. Its co-expressed putative target genes, miRNA precursors, or interactions between miRNAs and lncRNAs can be predicted, and the sequence variations of lncRNAs can be queried on the 'Genome variation' page. Thus, wLNCdb is a powerful platform that provides greater possibilities for exploring the roles of lncRNAs in every aspect of biology. In the future, we will expand the lncRNA expression data to include more biotic and abiotic stress conditions and develop new features such as phenotype-associated lncRNAs and variants and expression quantitative trait loci (eQTLs) in lncRNAs to provide comprehensive insights for lncRNA-related research.
Using wLNCdb, we systematically analyzed and identified wheat lncRNAs associated with grain development and predicted lncRNAs related to genes encoding SSPs and genes involved in regulating grain size and weight, as well as starch biosynthesis, including cis- or trans-regulation. Among these, the lncRNA TraesLNC1D26001.1 was specifically expressed during seed development. The expression levels of its nearby gene TaGLU-1Dy and accumulation of HMW-GSs showed no change in its overexpression lines OE1, OE2 and OE3, suggesting that TraesLNC1D26001.1 does not regulate TaGLU-1Dy in close genomic proximity at the transcriptional or translational level. In plants, several lncRNAs have similar functional mechanisms to that of TraesLNC1D26001.1. For example, MISSEN (MIS-SHAPEN ENDOSPERM) in rice negatively affects endosperm development without influencing its nearby genes within region of 10 kb upstream and downstream of the MISSEN locus, and hijacks the helicase protein HeFP in trans-manner to impair cytoskeletal polymerization during endosperm development[24]. Similarly, the lncRNA ALTERNATIVE SPLICING COMPETITIVE FACTOR (ASCO) binds nuclear speckle RNA-binding protein (NSR) splicing factors and competes with the alternative splicing targets of NSRs46[58]. Thus, TraesLNC1D26001.1 might be able to sequester proteins from their targets of action. TaABI5 expression was higher in TraesLNC1D26001.1-OE lines than in the WT (Fig. 4i), which could explain the reduced seed germination of the OE lines. ABI5 is a basic leucine zipper (bZIP) transcription factor that responds to abscisic acid (ABA) signals, and Arabidopsis abi5 mutants show weak seed dormancy[57]. While TaABI5 was predicted to be the target gene trans-regulated by TraesLNC1D26001.1, we suggest that TraesLNC1D26001.1 delays seed germination by positively regulating TaABI5 expression during ABA signaling. Enhancer-associated lncRNAs are transcribed from genomic regions highly enriched with monomethylation of histone H3 lysine 4 (H3K4me1) and are recruited to the transcriptional machinery to remodel the three-dimensional chromatin loops to promote target gene expression[59,60]. We suggest that TraesLNC1D26001.1 positively regulates TaABI5 expression by acting as an enhancer-associated lncRNA and interacting with transcription factors to facilitate the initiation of RNA polymerase II-mediated transcription and elongation at the TaABI5 transcriptional start site. It will be important to analyze the regulatory mechanism between TraesLNC1D26001.1 and TaABI5, as well as the ABA contents in TraesLNC1D26001.1 OE lines and the WT during seed germination. Such studies would provide more direct evidence for the mechanism linking TraesLNC1D26001.1 to germination.
-
In summary, we provide a comprehensive atlas of wheat lncRNAs. We developed wLNCdb, a database with comprehensive information and visualization tools for exploring lncRNAs in wheat, including sequences, expression patterns, co-expression networks, cis- and trans-regulation, epigenomic states, miRNA-lncRNA interactions, genomic variation, and functional annotations, providing valuable information for future discoveries and functional characterization of lncRNAs. As proof of concept, we focused on lncRNAs related to wheat grain size and weight and SSPs and identified TraesLNC1D26001.1 as a regulator of seed germination.
This work was supported by the National Key Research and Development Program of China (2022YFD1200203), National Natural Science Foundation of China (Grant no. 32125030), Hainan Yazhou Bay Seed Lab (no. B21HJ0502), Frontiers Science Center for Molecular Design Breeding (no. 2022TC149).
-
The authors declare that they have no conflict of interest.
-
# These authors contributed equally: Zhaoheng Zhang, Ruijie Zhang
- Supplemental Fig. S1 The RNA-seq datasets used represent diverse tissues. PCA of the expression of lncRNAs and PCGs was performed.
- Supplemental Table S1 Accession numbers of wheat RNA-seq datasets.
- Supplemental Table S2 The list of LncRNAs identified in this study.
- Supplemental Table S3 Genes involved in wheat grain size and weight and sharch metablish proces.
- Supplemental Table S4 Genes involved in wheat grain size and weight and sharch metablish process co-expressed with lncRNAs.
- Supplemental Table S5 SSPs genes co-expressed with lncRNAs.
- Supplemental Table S6 Primers used for genes expression and vectors construction.
- Supplemental Table S7 Accession numbers of open chromatin and ChIP-seq datasets.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press on behalf of Hainan Yazhou Bay Seed Laboratory. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang Z, Zhang R, Meng F, Chen Y, Wang W, et al. 2023. A comprehensive atlas of long non-coding RNAs provides insight into grain development in wheat. Seed Biology 2:12 doi: 10.48130/SeedBio-2023-0012 

A comprehensive atlas of long non-coding RNAs provides insight into grain development in wheat
- Received: 19 May 2023
- Accepted: 17 July 2023
- Published online: 04 September 2023
Abstract: Long non-coding RNAs (lncRNAs) are key regulators of many biological processes, but our knowledge of lncRNAs associated with wheat (Triticum aestivum) grain development is limited. Here, to generate a comprehensive atlas of lncRNAs in wheat, we performed strand-specific RNA sequencing (ssRNA-seq) using wheat endosperm at 10 and 15 d after pollination and collected 545 publicly available transcriptome datasets. We identified 20,893 wheat lncRNAs and developed the comprehensive database wLNCdb (http://wheat.cau.edu.cn/wLNCdb) to provide their sequences, expression patterns, co-expressed genes, and single nucleotide polymorphisms among wheat accessions. To investigate the functions of lncRNAs, we focused on lncRNAs predicted to regulate genes related to grain size and weight and seed storage proteins via cis- or trans-regulation. We identified a lncRNA TraesLNC1D26001.1 negatively regulates seed germination, as its overexpression delayed wheat seed germination by upregulating Abscisic acid-insensitive 5 (TaABI5). These results suggest wLNCdb as a useful resource for discovering the functions of lncRNAs in wheat during seed development and other processes.
-
Key words:
- Triticum aestivum /
- lncRNA /
- Database