






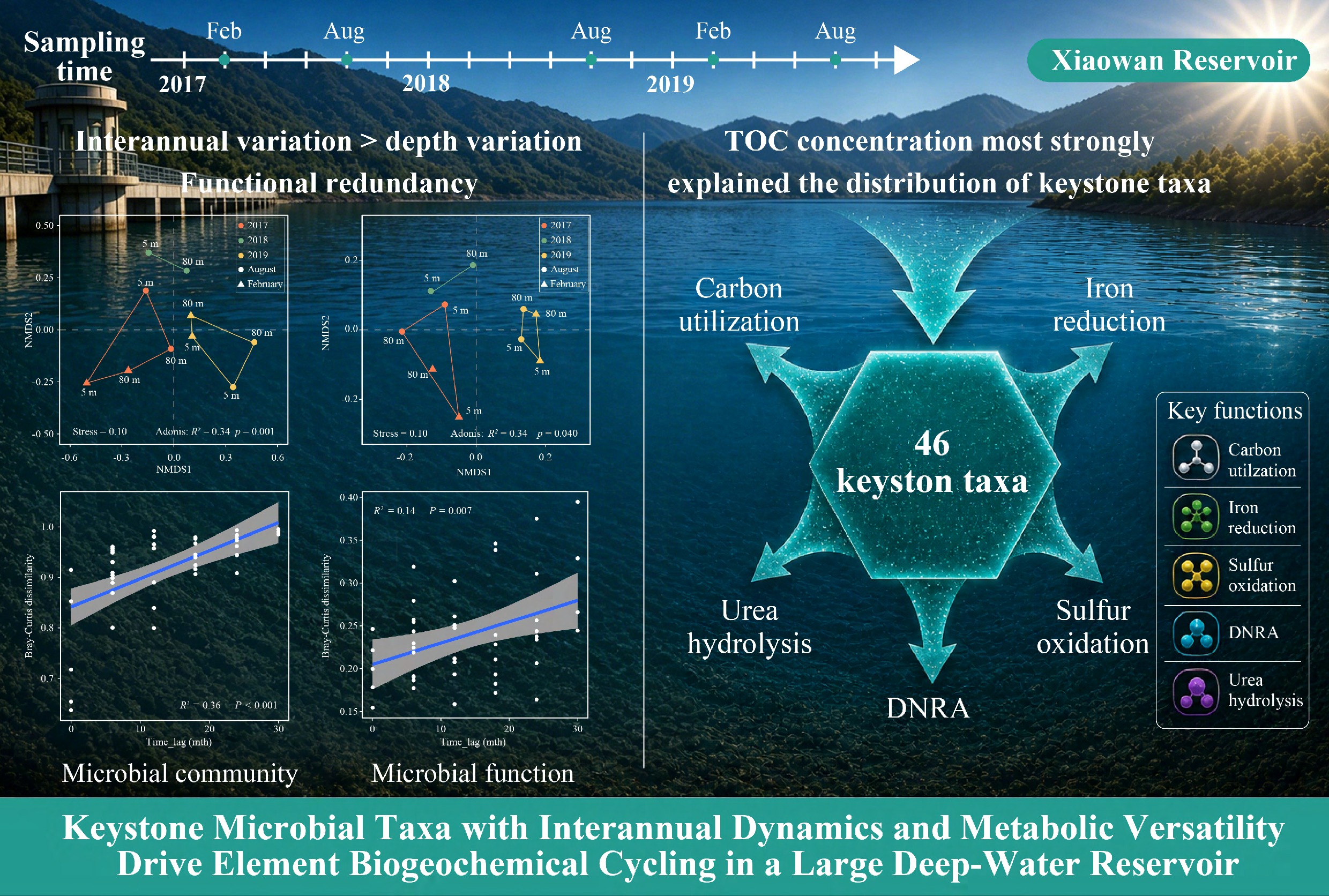



-
Freshwater reservoirs represent critical components of the global water infrastructure, serving multiple functions including water supply, hydroelectric power generation, flood control, and fisheries production[1]. As artificial lentic systems, large deep-water reservoirs exhibit unique hydrodynamic and biogeochemical characteristics that distinguish them from other freshwater ecosystems. These engineered aquatic ecosystems are characterized by pronounced seasonal stratification, creating distinct physicochemical gradients that profoundly influence microbial community structure, function, and biogeochemical cycling[2]. Microbial communities drive fundamental processes governing carbon, nitrogen, sulfur, and iron transformations, thereby regulating water quality and ecosystem functioning[3−5]. Understanding the temporal dynamics of microbial community structure and metabolic functions in these systems is essential for predicting ecosystem responses to environmental perturbations and optimizing reservoir management strategies[6]. However, the temporal dynamics of microbial communities and their metabolic capabilities in large deep-water reservoirs remain poorly understood, particularly at interannual scales.
Microbial communities in aquatic ecosystems exhibit temporal variations across multiple scales, ranging from diel cycles to interannual shifts. Previous studies have predominantly focused on seasonal variations in microbial communities within reservoir ecosystems, revealing that nutrients, thermal stratification, and associated changes in oxygen availability are primary drivers of microbial succession[7−9]. However, interannual variability, shaped by long-term environmental changes, hydrological management, and external nutrient inputs, has received far less attention. In particular, in large deep-water reservoirs, where hydrological residence times are extended and environmental conditions undergo progressive changes following impoundment. Previous studies have indicated that interannual differences in microorganisms were stronger than seasonal repeat patterns, suggesting that unpredictable interannual variation in human activities and climate change may impact microbial communities more than their resilience does[10−12]. Whether similar patterns occur in large deep-water reservoir ecosystems and how keystone microbial taxa contribute to the biogeochemical cycle across years remains largely unexplored.
Biogeochemical cycling in large deep-water reservoirs is largely driven by depth-dependent gradients in oxygen availability, light penetration, temperature, and nutrient concentrations[13]. These vertical gradients create distinct ecological niches that support specialized microbial assemblages adapted to specific redox conditions and resource availabilities[14]. The epilimnion typically sustains aerobic heterotrophic and phototrophic communities, while the hypolimnion may harbor microorganisms capable of anaerobic respiration, fermentation, and chemolithoautotrophy[15]. Nitrogen cycling in reservoir ecosystems is of particular concern due to its implications for eutrophication and water quality deterioration. Dissimilatory nitrate reduction, including denitrification and DNRA, represents a critical branch in the nitrogen cycle. While denitrification facilitates nitrogen removal, DNRA promotes nitrogen retention and potentially exacerbates eutrophication[16]. Similarly, sulfur cycling plays a key role in modulating redox conditions and trace metal bioavailability in aquatic ecosystems[17]. The relative contributions of different metabolic guilds to nutrient cycling likely vary temporally as environmental conditions evolve. However, the mechanisms linking environmental drivers to microbial metabolic strategies over interannual timescales remain incompletely understood.
Large deep-water reservoirs in tropical and subtropical regions face particular challenges related to thermal stratification, eutrophication risk, and anthropogenic nutrient inputs from intensive agriculture and aquaculture activities[18]. The Xiaowan Reservoir, located on the Lancang River (upper Mekong River) in Southwest China, represents one of the world's highest dams with a maximum depth exceeding 200 m[19]. This large deep-water reservoir experiences pronounced seasonal stratification and receives allochthonous organic carbon from its forested catchment. The agricultural and aquacultural activities of the reservoir in the surrounding areas further introduce anthropogenic nutrients. The combination of considerable depth, extended hydraulic retention time, and significant allochthonous inputs creates a unique biogeochemical environment where microbial communities mediate the transformation and fate of multiple elements. Despite the ecological and socioeconomic importance of this reservoir system, comprehensive investigations of its microbial community dynamics and metabolic capabilities, particularly over interannual timescales, have been lacking.
To address these knowledge gaps, our study investigated the interannual dynamics of microbial community structure, functional potential, and biogeochemical cycling in the Xiaowan Reservoir through integrated 16S rRNA gene sequencing and metagenomic analyses conducted over a three-year period (2017–2019). Compared with previous reservoir studies that mainly emphasized seasonal stratification effects, this study focused on interannual microbial succession and the metabolic potential of keystone taxa over a three-year period by integrating metagenome-assembled genomes (MAGs) with co-occurrence network analysis. The objectives were: (1) to characterize interannual variations in microbial community composition and functional profiles; (2) to reconstruct the metabolic capabilities of keystone microbial taxa; and (3) to elucidate the effects of the environmental traits on community turnover and keystone microbial taxa. We hypothesized that microbial community structure and function in this large deep-water reservoir exhibit stronger interannual than depth-related variation, and that keystone taxa harbor metabolic versatility enabling them to respond to changing environmental conditions. Our study aimed to provide novel insights into the microbial dynamics of large deep-water reservoirs on interannual timescales and to advance our understanding of how keystone microbial lineages mediate biogeochemical cycling in similar ecosystems.
-
Field sampling was conducted at the Xiaowan Reservoir (Supplementary Fig. S1). Briefly, water samples were retrieved using a Niskin bottle (Kanghua Inc., China) from depths of 5 and 80 m during February and August between 2017 and 2019, except for February 2018. A total of 10 L of composite water was obtained from each depth. Physicochemical parameters, including pH, temperature, dissolved oxygen (DO), oxidation-reduction potential (ORP), nitrate (NO3−), nitrite (NO2−), ammonia (NH4+), sulfate (SO42−), sulfide (S2−), total organic carbon (TOC), total phosphorus (TP), and Chlorophyll a (Chla) were determined following the protocols described in our previous study[20]. Subsequently, 1 L subsamples were filtered through 0.22 μm pore-size polycarbonate membranes (Millipore Inc., USA), and the filters were stored at −80 °C until DNA extraction.
DNA extraction and amplicon sequencing
-
Genomic DNA was extracted from the filters using the FastDNA SPIN Kit for Soil (MP Biomedicals, USA) following the manufacturer's protocol. The V4 region of the prokaryotic 16S rRNA gene was amplified using the primers 515F (GTGCCAGCMGCCGCGGTAA) and 806R (GGACTACHVGGGTWTCTAAT). PCR amplicons were purified, quantified, and pooled following standard protocols. Sequencing was performed on the Illumina MiSeq platform, and bioinformatics analysis was conducted as described in a previous study[21].
Metagenome sequencing, assembly, and functional annotation
-
The extracted genomic DNA was used for shotgun metagenome sequencing via the Illumina HiSeq 4000 platform (paired-end 150-bp mode) at Majorbio Bio-Pharm Technology Co., Ltd (Shanghai, China). Raw reads were dereplicated and quality-filtered using Sickle (v1.33) with parameters "-q 30 -l 50"[22]. High-quality reads were co-assembled into scaffolds using SPAdes (v3.15.2) with parameters "-k 33, 55, 77, 99 -meta". Prodigal (v2.6.3)[23] was employed to predict open reading frames (ORFs) and translated protein sequences with the "-p meta" option for scaffolds > 500 bp. ORFs from all samples were pooled and dereplicated at 95% nucleotide identity using CD-HIT-EST (v4.8.1)[24]. The relative abundance of ORFs was calculated as reads per kilobase per million mapped reads (RPKM) by mapping reads via BBMap (v0.12.7) with parameters "minid=0.97, local=t"[25]. Translated protein sequences of the dereplicated ORFs were functionally annotated using eggnog-mapper (v2.1.3) against the eggNOG database (v5.0) and METABOLIC (v4.0) with default parameters. METABOLIC identifies metabolic and biogeochemical potential by integrating hidden Markov model (HMM) databases, including KOfam (containing KEGG HMMs), TIGRfam, Pfam, and custom HMM databases[26].
Genome-resolved metagenomic analysis
-
Scaffolds > 1,500 bp were processed using the MetaWRAP pipeline (v1.3.2) for genome binning with default parameters. Genome binning was performed using MetaBAT2 (v2.12.1) and MaxBin2 (v2.2.5). The resulting bins were refined using the BIN_REFINEMENT module of Binning_refiner (v1.2). The completeness and contamination of MAGs were evaluated using CheckM (v1.0.12). Only medium- and high-quality MAGs (> 50% completeness and < 10% contamination) were retained for downstream analysis[27]. Taxonomic classification of MAGs was assigned using GTDB-Tk[28]. A maximum-likelihood phylogenetic tree was constructed, using IQ-TREE (v2.2.3) based on the concatenation of marker genes identified by GTDB-Tk and visualized using iTOL (v6.3).
Statistical analysis
-
All data were analyzed using Origin 2022, SPSS 27, and R software. Non-metric multidimensional scaling (NMDS) based on Bray–Curtis dissimilarity was used to visualize the beta diversity of microbial community composition and functional profiles. Permutational multivariate analysis of variance (PERMANOVA, implemented as the adonis function in the R 'vegan' package) was used to assess significant differences in microbial communities or functions among groups. Co-occurrence networks were constructed based on MAG abundance. Pairwise Spearman's rank correlations were calculated. Only robust (Spearman's r > 0.6) and significant (Benjamini–Hochberg corrected p < 0.05) correlations were retained[29], and exported for network visualization in Gephi[30]. To identify keystone MAGs and evaluate their topological roles within the network, the within-module connectivity (Zi) and among-module connectivity (Pi) were calculated for each node. Based on standard topological thresholds, nodes were classified into four categories: peripherals (Zi ≤ 2.5 and Pi ≤ 0.62), connectors (Zi ≤ 2.5 and Pi > 0.62), module hubs (Zi > 2.5 and Pi ≤ 0.62), and network hubs (Zi > 2.5 and Pi > 0.62)[20]. Pearson's correlations between the abundances of keystone MAGs and environmental parameters were calculated. The variance inflation factor (VIF) was calculated using the R 'car' package, and environmental variables with VIF values less than 10 were retained for further analysis. The explicit VIF values for the retained environmental parameters are provided in Supplementary Table S1. Redundancy analysis (RDA) was used to explore relationships between the keystone MAGs and environmental parameters.
-
The geochemical profiles of the Xiaowan Reservoir exhibited temporal fluctuations and vertical heterogeneity across the sampling period (2017–2019) (Fig. 1). Thermal stratification was observed in August, with surface temperatures (5 m) ranging from 23.8 ± 1.6 to 26.3 ± 1.3 °C, markedly higher than those at the bottom (80 m) (18.9 ± 0.8–20.7 ± 1.1 °C), while the water column remained relatively well mixed in February. Consistent with the temperature distribution, DO decreased with depth, reaching 4.39 ± 0.83–5.58 ± 0.91 mg/L at 80 m in August, contrasting with the oxygen-rich surface waters (6.01 ± 0.54–9.34 ± 0.77 mg L−1). The observed fluctuations in ORP (147.6 ± 4.1–269.3 ± 8.5 mV) likely reflect a transitional redox state, which was related to site-specific hydrodynamic and geochemical conditions. The pH was weakly alkaline (8.03 ± 0.7–9.01 ± 0.8) throughout the water column. Chl a concentration peaked in the surface layer during August (5.04 ± 0.56–8.34 ± 1.28 μg L−1), reflecting enhanced phytoplankton productivity, which likely contributed to the elevated TOC levels observed during the same periods. Additionally, TOC concentrations at the surface and bottom of Xiaowan Reservoir were higher in 2017 (3.46 ± 0.98–4.80 ± 0.80 mg L−1) than in 2019 (1.17 ± 0.59–3.45 ± 0.97 mg L−1). This trend may reflect increased mineralization as microbial communities adapted to the reservoir environment. All nutrient dynamics did not show significant differences with depth, but showed complex temporal dynamics. Polymodal peaks of NO3− were observed throughout the water column (0.18 ± 0.11–0.76 ± 0.19 mg L−1). NO2− remained consistently low except for a notable increase in February 2019 (0.07 ± 0.02–0.12 ± 0.05 mg L−1), while NH4+ tended to accumulate in the deeper layer (80 m) during August (0.12 ± 0.02–0.95 ± 0.05 mg L−1), potentially due to dissimilatory nitrate reduction or mineralization of organic matter under low-oxygen conditions. SO42− concentrations ranged from 33.5 ± 1.9 to 98.1 ± 4.7 mg L−1 with increasing trends toward 2019, while S2− concentrations remained low (< 5 µg L−1) throughout most periods. TP concentrations varied from 0.18 ± 0.11 to 0.76 ± 0.19 µg L−1, with the highest values recorded at 80 m in August 2018 (0.09 ± 0.03 µg L−1). These geochemical patterns suggested that the large deep-water reservoir experienced seasonal thermal stratification, with associated changes in redox conditions and nutrient dynamics that likely influenced microbially mediated elemental biogeochemical cycling.
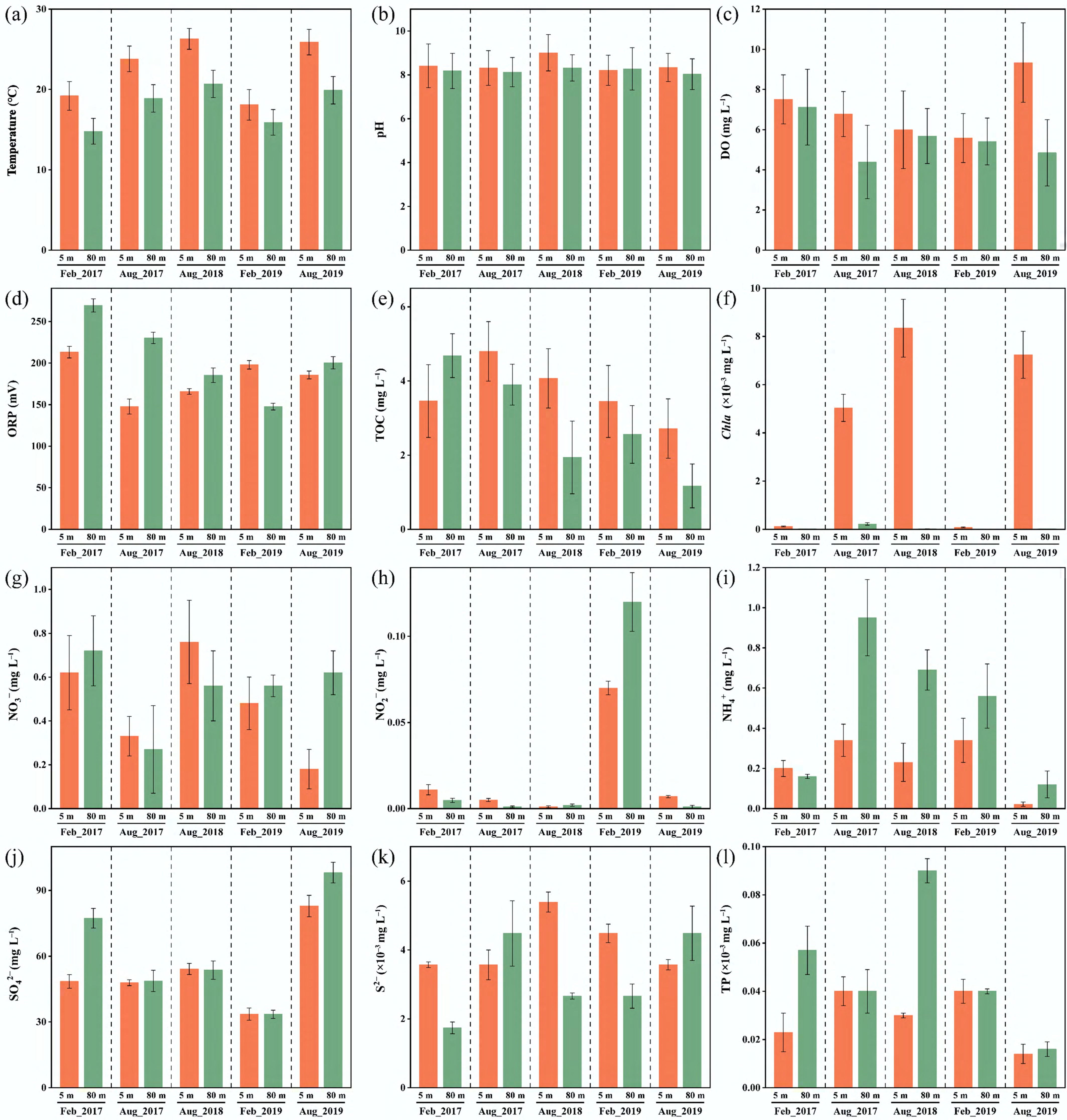
Figure 1.
Environmental variables of water samples in the Xiaowan Reservoir. (a) Temperature (T), (b) pH, (c) dissolved oxygen (DO), (d) oxidation-reduction potential (ORP), (e) total organic carbon (TOC), (f) Chlorophyll a (Chla), (g) nitrate (NO3−), (h) nitrite (NO2−), (i) ammonium (NH4+), (j) sulfate (SO42−), (k) sulfide (S2−), and (l) total phosphorus (TP).
Microbial community diversity and composition in Xiaowan Reservoir
-
A total of 376,500 high-quality 16S rRNA gene sequences were obtained from the 10 water samples. The rarefaction curves for most samples tended toward saturation (Supplementary Fig. S2), indicating that the current sequencing depth adequately captures the majority of microbial taxa. Microbial alpha diversity analysis revealed that microbial richness and diversity at different depths did not differ significantly, while the ACE and Shannon indices exhibited temporal fluctuations (Fig. 2a). Partitioning of beta diversity further indicated that interannual variation, rather than depth variation, dominated differences in microbial communities (Fig. 2b). Communities sampled in 2017 exhibited a distinct compositional structure compared to those from 2018 and 2019 (Adonis, R2 = 0.34, p = 0.001). Time-lag regression revealed that community dissimilarity increased continuously, indicating directional succession over the sampling period (R2 = 0.36k, p < 0.001) (Fig. 2c). Consistent directional shifts in microbial communities have also been observed in other large reservoirs and lakes[31,32]. In addition, another study had shown that microbial communities exhibit resilience over longer time intervals (> 5 years)[12].
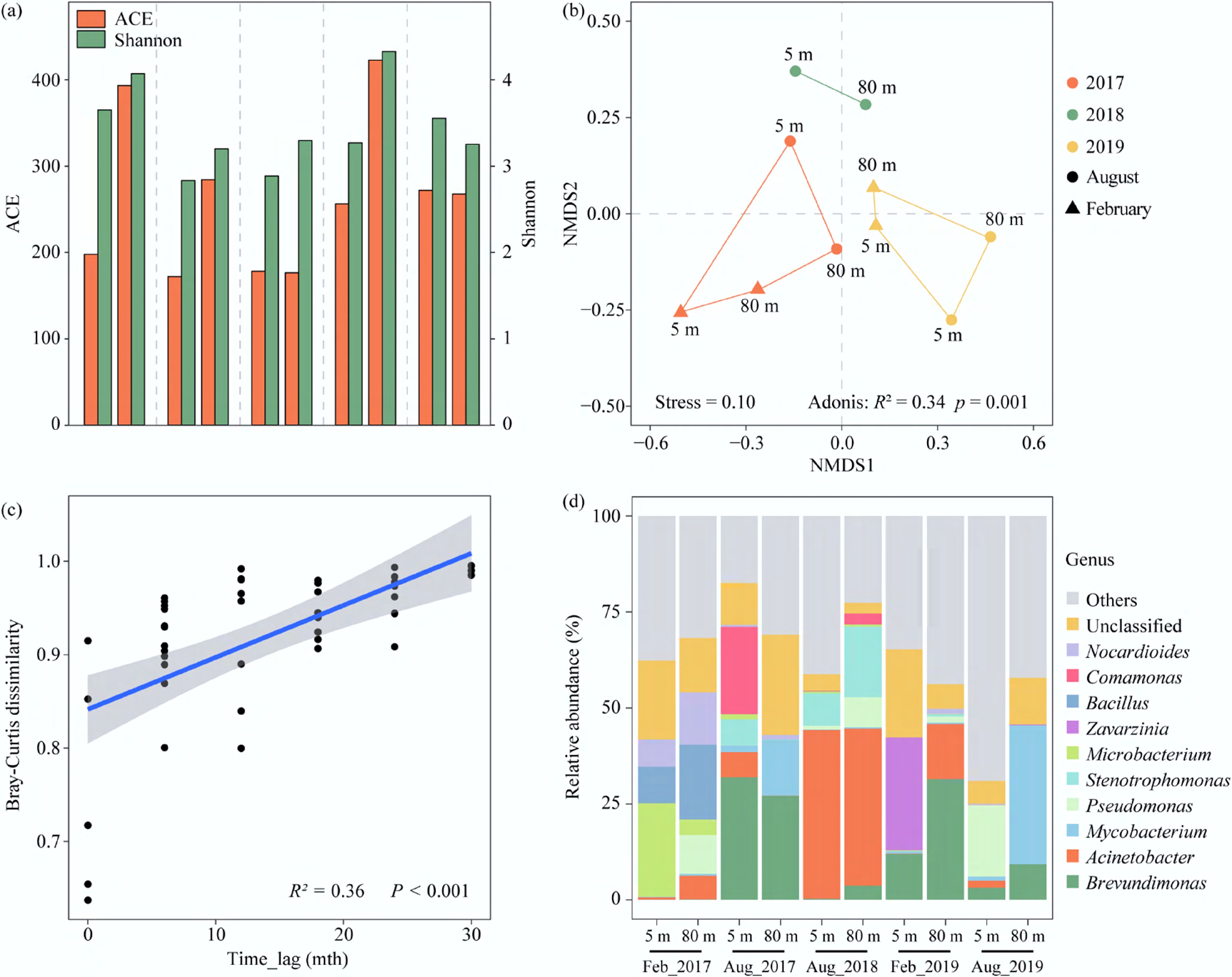
Figure 2.
Microbial community structure in the Xiaowan Reservoir. (a) Alpha diversity of microbial community; (b) non-metric multidimensional scaling (NMDS) based on Bray-Curtis distances of the relative abundances of microbial community; (c) Bray-Curtis similarity of microbial community against time lag; (d) microbial composition at the genus level.
For microbial community composition, both Proteobacteria (44.6%–73.4%) and Actinobacteriota (6.9%–35.6%) showed significant preferences at the phylum level in all samples (Supplementary Fig. S3). Additionally, Firmicutes was the third most abundant phylum in 2017 and 2018; Bacteroidota occupied this rank in 2019. At the genus level, microbial community composition varied more over time (Fig. 2d). Specifically, Brevundimonas dominated the microbial community in 2017 (14.8%) and 2019 (14.0%), while the relative abundance of Acinetobacter maximized in 2018 (42.4%). Both Brevundimonas and Acinetobacter are Gram-negative bacteria widely distributed in freshwater environments[33]. Furthermore, these genera were involved in the transformation of nitrogen and phosphorus compounds and played an important role in lowering the risk of algal blooms[34,35]. The dominance of these bacterial genera indicated that a healthy microbial community could respond to environmental changes, and their metabolic activities contributed to the removal of excess nutrients, thereby playing a significant role in preventing eutrophication. Considering the well-developed agricultural and fishing activities around the reservoir area, potential public health concerns warranted further attention.
Microbial functional profile in Xiaowan Reservoir
-
NMDS analysis of metabolic profiles from all functional genes revealed interannual clustering similar to that of community composition (Adonis, R2 = 0.34, p = 0.04) (Fig. 3a). Furthermore, time-lag patterns of functional dissimilarity were evaluated. As shown in Fig. 3b, microbial functional dissimilarity exhibited a marginally significant increasing trend over time (R2 = 0.14, p = 0.007). Notably, taxonomic divergence was substantially greater than functional divergence during the sampling period. This may be attributed to functional redundancy, where identical metabolic functions are encoded by taxonomically distinct microorganisms[36]. Overall, both taxonomic and functional compositions in the Xiaowan Reservoir clustered primarily by interannual variability. These findings provide statistical support for the hypothesis that microbial community structure and function exhibit stronger interannual variation than depth-related variation in this large deep-water reservoir.
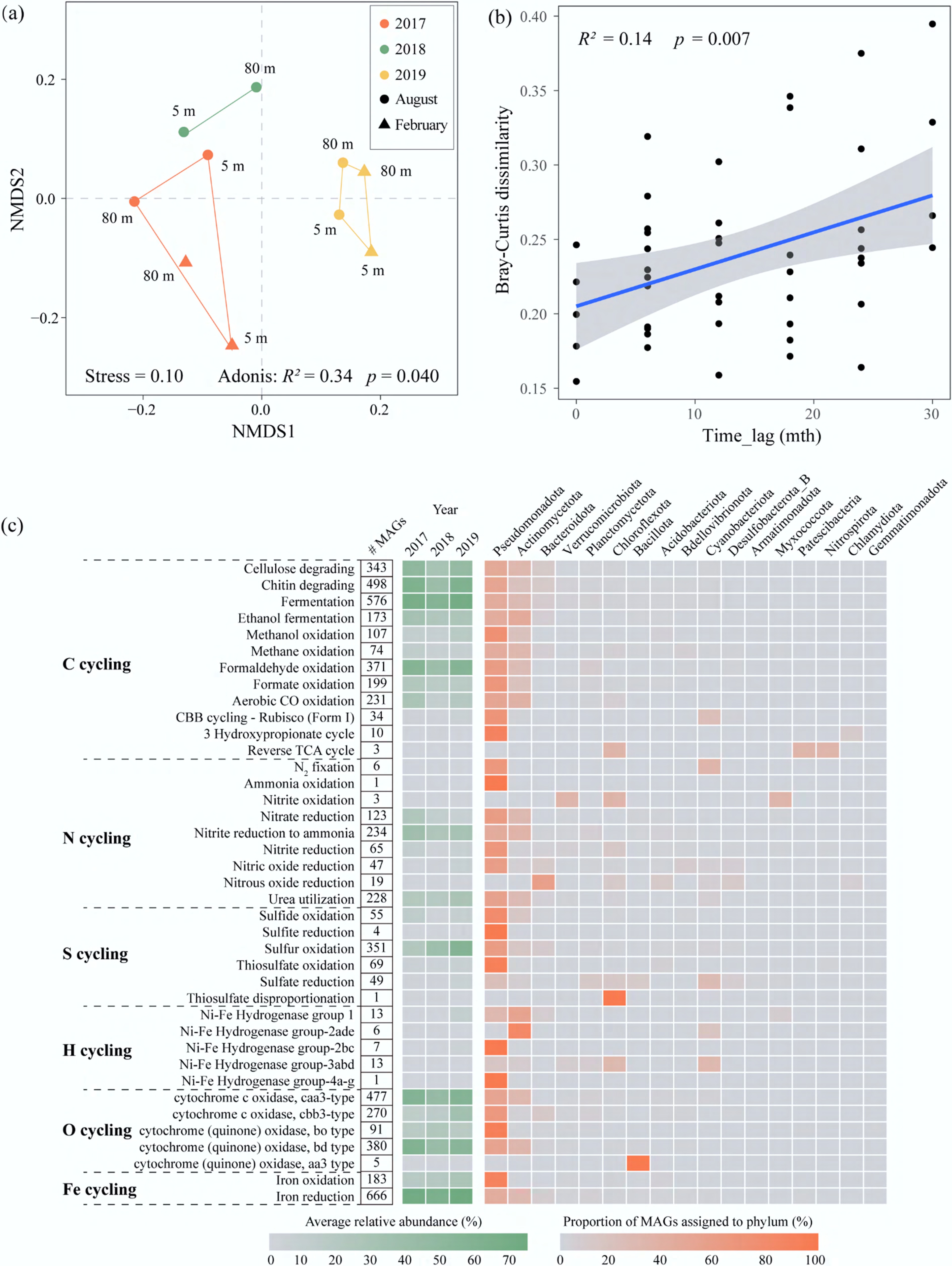
Figure 3.
Microbial function in the Xiaowan Reservoir. (a) Non-metric multidimensional scaling (NMDS) based on Bray-Curtis distances of the relative abundances of microbial functional genes; (b) Bray-Curtis similarity of microbial function against time lag; (c) key metabolic and biogeochemical cycling traits of MAGs in the Xiaowan Reservoir. From left to right: the numbers of MAGs that carry genetic markers for each functional trait are shown as numerals; the average relative abundance for corresponding MAGs across each year are displayed in the green heatmap; and the proportion of MAGs assigned to each phylum is shown in the orange heatmap.
The genome-resolved metagenomic analysis was performed to gain better insight into the metabolic potential of the key and abundant microbial species in different years. A total of 671 medium- to high-quality MAGs with at least 50% completeness and less than 10% contamination were retrieved (Supplementary Fig. S4 and Supplementary Table S2). These MAGs were phylogenetically classified into 17 phyla, among which Pseudomonadota (36.6%), Actinomycetota (26.2%), Bacteroidota (2.7%), Patescibacteria (1.2%), and Planctomycetota (0.9%) had predominant abundances across all samples.
Gene-based functional analysis indicated that the number of MAGs with genetic potential for each key metabolic reaction varied considerably (Fig. 3c). For carbon metabolism, fermentation, chitin degradation, and formaldehyde oxidation were the most common carbon metabolic pathways in the Xiaowan Reservoir, identified in 576, 498, and 371 MAGs, respectively. These main carbon-related functions were largely encoded by MAGs affiliated with Pseudomonadota and Actinomycetota. This pattern suggested that reservoir carbon cycling was primarily supported by the transformation of exogenous and indigenous organic matter. In particular, the widespread occurrence of fermentation genes indicated that fermentation may represent a key microbial route linking organic matter decomposition to overall organic carbon mineralization. Through fermentation, complex organic substrates could be converted into smaller intermediates, such as acetate, H2, and CO2, which may subsequently fuel other heterotrophic and respiratory metabolisms. Moreover, these results were also consistent with the lower TOC concentrations observed in 2019 relative to 2017, which may reflect progressive microbial processing of organic carbon. In comparison, some MAGs participated in CO oxidation and autotrophic carbon fixation, which may enhance metabolic flexibility under fluctuating environmental conditions. These results suggested that the Xiaowan Reservoir microbiome employed both heterotrophic and autotrophic metabolic strategies, with heterotrophs likely serving as the predominant community despite substantial genetic potential for CO oxidation and autotrophic carbon fixation, predominantly driven by allochthonous organic carbon inputs.
For the most part, nitrogen and sulfur cycling pathways were carried out by Pseudomonadota (Fig. 3c). Genes associated with DNRA and urea utilization were detected in 234 and 228 MAGs, respectively, suggesting that both processes appear to be important pathways driving nitrogen transformation in the Xiaowan Reservoir (Fig. 3c). The relative abundance of MAGs involved in urea utilization processes gradually increased over the three years (from 18.90% to 28.69%), indicating that these clades might be able to use urea instead of ammonia as the sole energy source, as has been shown in other freshwater ecosystems[37]. Urea typically originated from agricultural fertilizers, phytoplankton decomposition, and microbial metabolism of nitrogenous substrates[38,39]. Our results indicated that urea provided the most important components of bioavailable nitrogen for the Xiaowan Reservoir, and its contribution increased annually. Similarly, sulfur oxidation (sdo) processes dominated the sulfur cycle and were also increasing annually (increasing from 22.01% in 2017 to 36.49% in 2018, and 57.19% in 2019). Sdo encoded a sulfur deoxygenase that oxidized S-sulfanylglutathione under aerobic conditions[17]. Sulfite was the initial product of sdo-mediated S-sulfanylglutathione oxidation, which subsequently led to the non-enzymatic formation of sulfate[40]. Therefore, this sulfur oxidation potential may contribute to sulfate production in the Xiaowan Reservoir. The annual increase in urea utilization and sulfur oxidation pathways represented the functional adaptation to shifting environmental drivers. Given the extensive agricultural and aquacultural activities in the catchment of the reservoir, surface runoff introduced substantial quantities of urea-based fertilizers and complex organic sulfur compounds. Taxa possessing the urea utilization ability gained a selective advantage by exploiting anthropogenic urea as an alternative nitrogen source. Furthermore, the enrichment of the sulfur oxidation process was likely driven by the increased availability of organic sulfur intermediates derived from anthropogenic runoff. Together, these functional shifts illustrated how anthropogenic nutrient loading exerted selective pressure on the microbial community and shaped the overall biogeochemical cycling in the reservoir.
Additionally, multiple respiratory pathways were identified, mainly including the Caa3-type cytochrome c oxidase functioning under high oxygen tension, the Cbb3-type cytochrome c oxidase functioning under microaerobic conditions, and the bd-type cytochrome oxidase operating in microaerobic/low-oxygen environments[41]. This indicated that the microbes in the Xiaowan Reservoir had diverse respiratory capabilities, which may be related to the oxygen gradient in the vertical water column. Iron reduction could be coupled to the oxidation of H2, CH4, NH4+, and organic carbon to support respiratory metabolism for energy production[42]. Therefore, iron reduction was an important part of the biogeochemical cycle in the Xiaowan Reservoir over the three years.
Metabolic potentials of the keystone MAGs
-
Co-occurrence network analysis was performed to explore associations among MAGs and to identify putative keystone MAGs based on their topological roles in the network (Fig. 4a). This network captured associations between 522 MAGs (nodes) connected via 5,805 edges, with 20.3% representing negative associations and 79.7% representing positive associations. Furthermore, the network showed a strong modular structure (modularity = 0.66), with six modules identified. A total of 46 MAGs highly linked to different modules were identified as connectors (Fig. 4b), potentially acting as bridges within cross-functional network modules[43]. These keystone MAGs exhibited high completeness (> 70%) and low contamination (< 2%) (Supplementary Table S2), suggesting limited mis-binning bias and potential contamination. These MAGs were phylogenetically assigned to seven phyla, among which Pseudomonadota, Actinomycetota, and Bacteroidota had predominant abundances in all samples, respectively accounting for 45.7%, 30.4%, and 8.7% of the total MAGs (Fig. 5a). Among Pseudomonadota, five MAGs (bin.555, bin.559, bin.341, bin.788, and bin.584) belonged to genus Brevundimonas and one MAG (bin.246) belonged to genus Acinetobacter (Supplementary Table S2). Brevundimonas and Acinetobacter were the two most abundant taxa across all samples, further indicating their potentially important roles in the community.
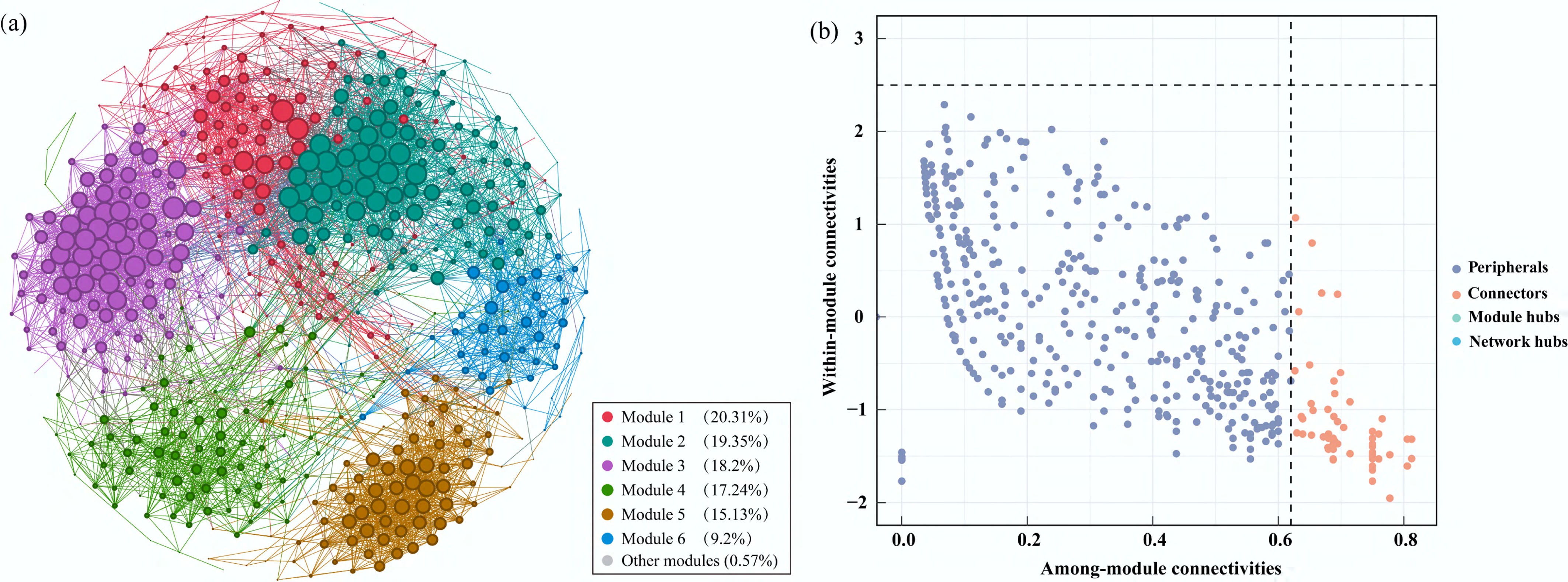
Figure 4.
Co-occurrence network analysis. (a) Biological interactions of MAGs in the Xiaowan Reservoir; (b) the scatter plot showed the criteria for selecting the keystone MAGs. Nodes represent MAGs and are colored by module. Node size corresponds to the number of interactions.
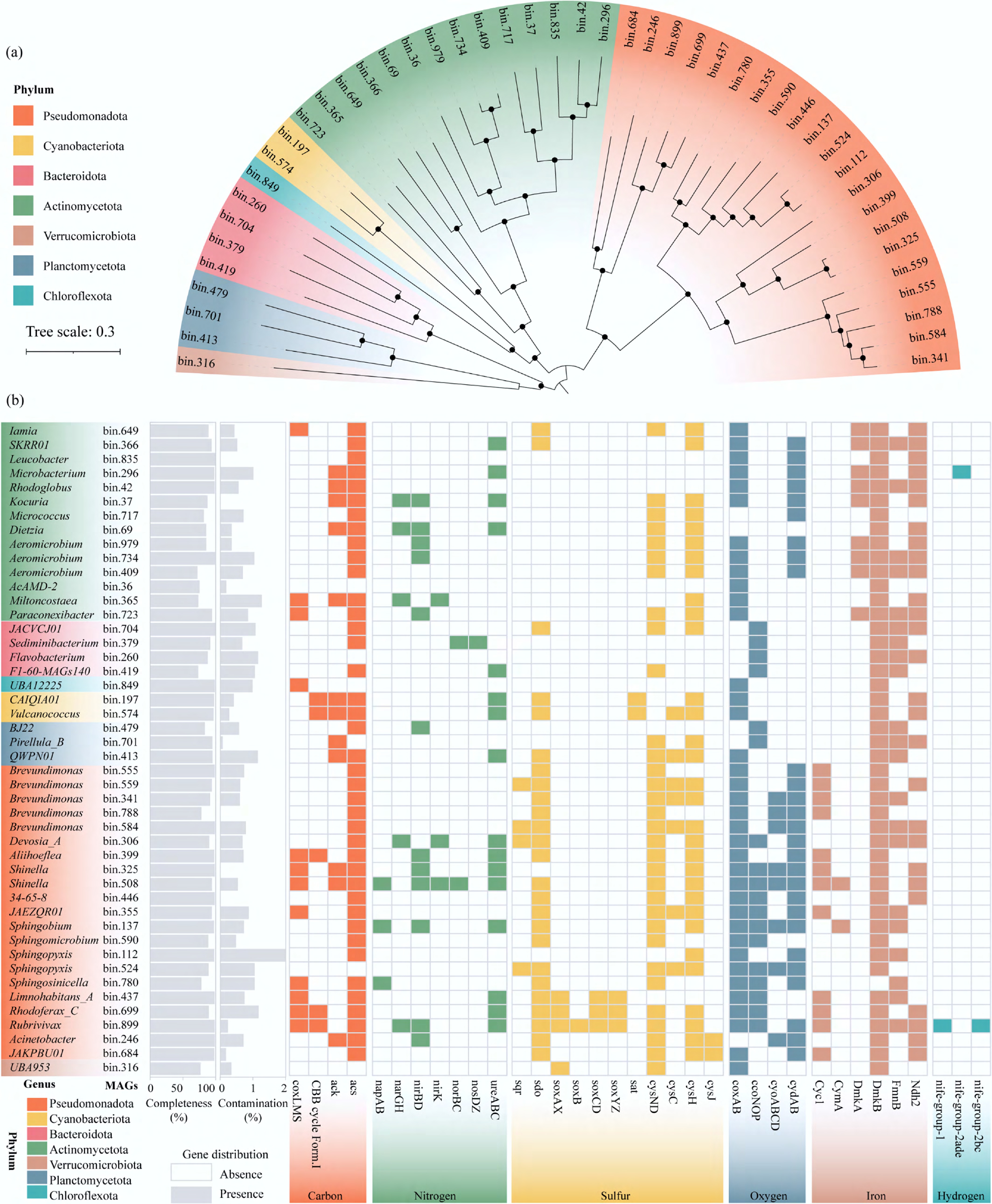
Figure 5.
Genomic characterization of keystone metagenome-assembled genomes (MAGs). (a) Phylogenetic tree, and (b) metabolic traits of the 46 metagenome assembled genomes (MAGs) (completeness > 70%, and contamination < 2%).
We furthermore evaluated the metabolic potentials of these keystone MAGs. Metabolic annotation revealed that the coding gene acs, responsible for synthesizing acetyl coenzyme A (acetyl-CoA), was present in almost keystone MAGs (Fig. 5b). Acetyl-CoA was reported to be either converted to acetate or used anabolically[44]. Therefore, our results suggested that keystone MAGs in the Xiaowan Reservoir had the capacity for endogenous carbon utilization and storage. Twelve MAGs were identified as possessing coxLMS genes encoding aerobic CO dehydrogenase, indicating that they harbor the genetic capacity for aerobic CO oxidation, potentially supporting CO2 fixation and energy generation. A similar carbon metabolism pathway had been reported in lacustrine environments[45]. Furthermore, several MAGs also contained carbon fixation pathways of the Calvin-Benson-Bassham (CBB) cycle. Studies have shown that the CBB cycle was commonly found in mixotrophic microorganisms, suggesting that these MAGs may also possess mixotrophic metabolic potential[46]. Importantly, most MAGs harbored at least one gene encoding cytochrome oxidase (coxAB, ccoNOP, cyoABCD, and cydAB), indicating their ability to reduce oxygen for respiration.
For nitrogen metabolism, genes involved in dissimilatory nitrate reduction were widely distributed across all keystone MAGs. However, no single keystone MAG contained the complete gene set for nitrate reduction from NO3− to N2. This pattern may indicate potential interspecies cooperation among community members, but it may also partly reflect limitations of MAG reconstruction, such as incomplete genome recovery or missing gene cassettes during assembly and binning. Additionally, urease genes (ureABC) were identified in 16 MAGs, indicating their ability to utilize urea as a carbon and a nitrogen source through the hydrolysis of urea into NH4+ and CO2. Totally, 25 of the 46 keystone MAGs harbored the sdo gene, while only a few MAGs were annotated to sqr, sat, and SOXsystem genes. These results highlighted the important role of sulfur oxidation processes in the Xiaowan Reservoir. In addition, a MAG affiliated with Rubrivivax (bin.899) was found to harbor complete SOX systems (soxAX, soxB, soxCD, and soxYZ), indicating the potential for thiosulfate oxidation within the same organisms. Interestingly, two hydrogenase genes were identified in the bin.899, suggesting the potential for H2 metabolism. Additionally, genes related to assimilatory sulfate reduction were found in these genomes, consistent with a previous study showing that the potential for assimilatory sulfate reduction was relatively more abundant in oligotrophic-mesotrophic freshwater ecosystems[47]. Iron oxidation genes (Cyc1) were present in 12 MAGs within the Pseudomonadota, indicating their potential for chemoautotrophic metabolism[48]. Meanwhile, diverse iron reduction genes (DmkA, DmkB, FmnB, and Ndh2) were detected in the majority of keystone MAGs, indicating widespread iron-reducing capacity among microbes in the Xiaowan Reservoir.
The relationships between geochemical properties and keystone MAGs
-
RDA was performed to explore the relationships between geochemical properties and keystone MAGs. As shown in Fig. 6a, RDA1 and RDA2 explained 31.8% and 21.9% of the variance of the keystone MAGs, respectively. Specifically, most keystone MAGs were simultaneously influenced by TOC, ORP, NO3−, pH, NO2−, and SO42−. Importantly, TOC concentration explained the largest proportion of variation in keystone MAGs distribution (14.3%, p < 0.01) (Fig. 6b). A similar study has shown that organic carbon levels play an important role in shaping the microbial communities in low-nutrient freshwater ecosystems[49]. Organic carbon from terrestrial sources may be regarded as a significant carbon source for microbial communities in such ecosystems. Our results suggested that TOC acted as a central ecological regulator of microbial metabolism in the Xiaowan Reservoir. Elevated organic carbon could stimulate heterotrophic respiration and organic matter mineralization, and promote the formation of microaerobic niches, especially in deeper waters during stratified periods. TOC in the Xiaowan Reservoir likely originates from both terrestrial allochthonous inputs and in-reservoir primary production. In addition, agricultural and aquacultural activities in the surrounding watershed may indirectly influence TOC distribution and nutrient stoichiometry by increasing external nutrient loading, stimulating phytoplankton production, and subsequent organic matter turnover.
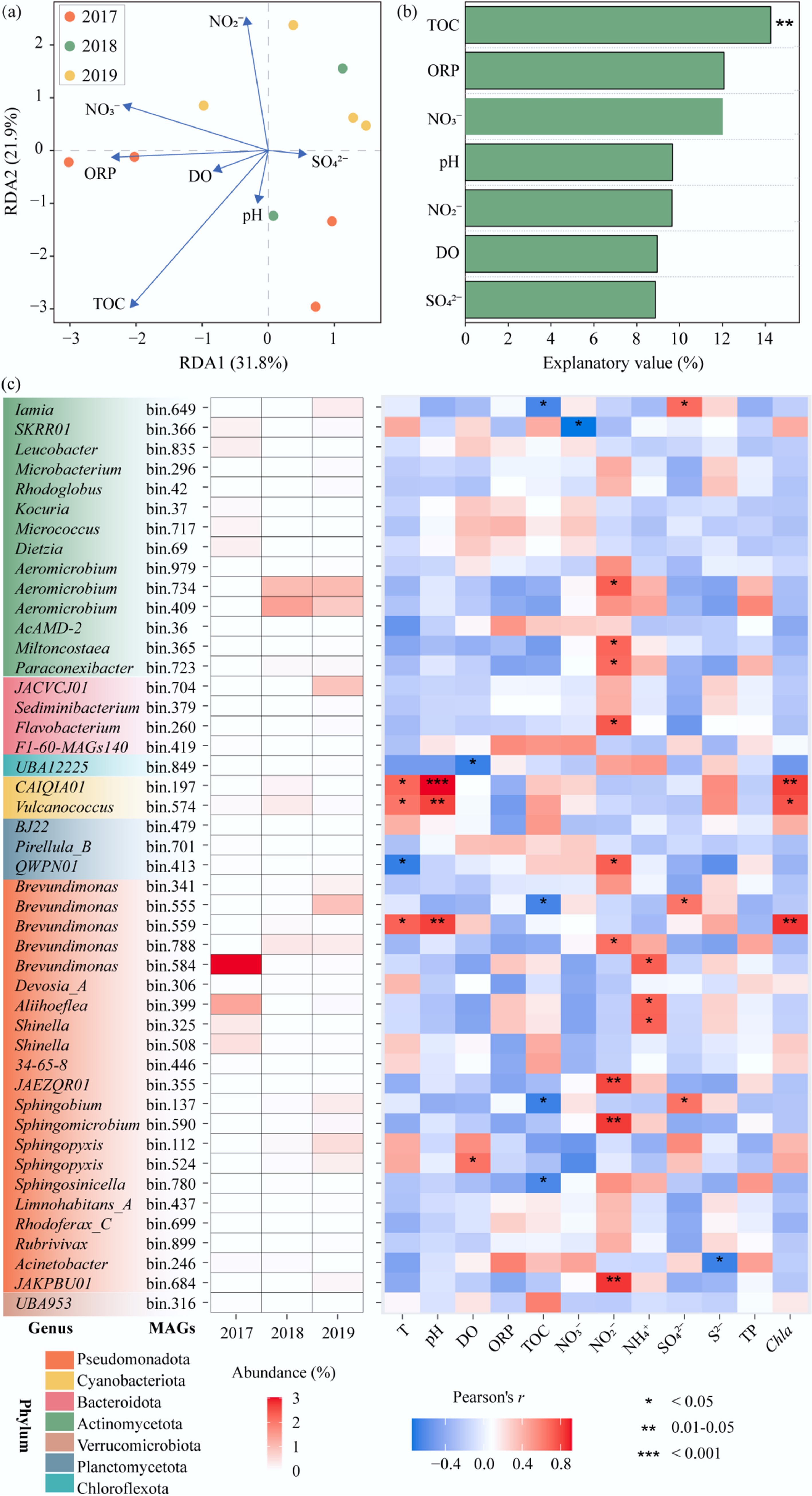
Figure 6.
The effects of geochemical properties on keystone MAGs. (a) Redundancy analysis (RDA) of the relationship between geochemical properties and keystones MAGs; (b) the proportion of variation in keystone MAGs distribution explained by each geochemical property; (c) heatmap showing the relative abundance of keystone metagenome-assembled genomes (MAGs) among different years and their pearson correlations with geochemical properties.
Since the distribution of keystone MAGs differed across years, environmental traits may influence keystone MAGs in different or complex ways in the Xiaowan Reservoir. The interactions between keystone MAGs and geochemical properties were further explored (Fig. 6c). Brevundimonas sp. bin.584 (3.03%), Aliihoeflea sp. bin.399 (1.37%), and Shinella sp. bin.325 (0.36%), affiliated with Pseudomonadota, were enriched in samples from 2017. Their abundances were all positively correlated with NH4+, revealing the intimate relationship with NH4+ production. The capacity for dissimilatory nitrate reduction (nirBD) was inferred in Aliihoeflea sp. bin.399, and Shinella sp. bin.325. Despite lacking the nirBD gene, Brevundimonas sp. bin.584 had a relatively high abundance (3.03%) as a connector in the network, potentially coordinating nitrogen metabolism across different network modules. Additionally, bin.734 and bin.409 belonging to the genus Aeromicrobium had higher relative abundances in 2018 (1.02% and 1.43%) and 2019 (1.05% and 0.81%) than in 2017 (0.02% and 0.01%). Among them, Aeromicrobium bin.734 was positively correlated with NO2−. Aeromicrobium has been reported to be widely involved in nitrogen cycling in lake environments[50]. Negative correlations were also observed between TOC and Brevundimonas sp. bin.555 and Sphingobium sp. bin.137, which were enriched in 2019 (0.95% and 0.31%, respectively) and harbored genes involved in synthesizing acetyl-CoA, indicating that these keystone MAGs may preferentially utilize simple organic compounds such as acetate. While our metagenomic analysis revealed the metabolic potential of keystone taxa, actual gene expression and substrate utilization may vary due to environmental constraints, and would require complementary approaches like metatranscriptomics or stable isotope probing for validation.
-
This study investigated interannual shifts in microbial community composition and function in the Xiaowan Reservoir over three consecutive years. Microbial communities exhibited stronger interannual than depth-related variation, whereas functional profiles remained comparatively stable, indicating that functional redundancy buffers ecosystem-level processes against taxonomic turnover. By reconstructing 671 metagenome-assembled genomes spanning 17 phyla, we revealed a predominantly heterotrophic but metabolically versatile framework, in which fermentation, degradation of complex organic matter, and formaldehyde oxidation co-occurred with substantial genetic potential for chemolithoautotrophic carbon monoxide oxidation and autotrophic carbon fixation. Nitrogen cycling was characterized by the persistent importance of dissimilatory nitrate reduction to ammonium and an increasing potential for urea utilization, suggesting a growing role of organic nitrogen as a bioavailable source. Sulfur oxidation pathways increased annually and likely contributed to sulfate production in surface waters, while iron cycling functions were broadly distributed among key taxa. Network analysis identified 46 putative keystone genomes that acted as connectors and encoded versatile metabolic potential for carbon utilization, nitrogen transformations, sulfur oxidation, and iron cycling; total organic carbon emerged as the primary environmental driver of keystone taxa distribution. Overall, our findings demonstrate that the metabolic versatility of keystone taxa may contribute to functional stability despite substantial taxonomic turnover. These results advance mechanistic understanding of microbial-mediated biogeochemical cycling in thermally stratified large deep-water reservoirs. These insights provide a foundation for predicting ecosystem responses to long-term environmental changes and optimizing management strategies for similar engineered aquatic systems globally.
-
It accompanies this paper at: https://doi.org/10.48130/ebp-0026-0006.
-
The authors confirm contributions to the paper as follows: Jiaxin Shi: writing – original draft, funding acquisition; Wenzhe Hu: writing – original draft; Shu Huang: validation; Jun Liu: writing – review & editing, supervision; Baogang Zhang: writing – review & editing, funding acquisition, conceptualization. All authors reviewed the results and approved the final version of the manuscript.
-
All data had been submitted to the National Genomics Data Center (NGDC) with the accession number of PRJCA058399.
-
The study was supported by the National Natural Science Foundation of China (NSFC) (No. 42407332, and 42525704), the Natural Science Foundation of Shandong Province (ZR2023QD049) and the start funding support from Qingdao University (DC2300000756).
-
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
-
# Authors contributed equally: Jiaxin Shi, Wenzhe Hu
Full list of author information is available at the end of the article. - The supplementary files can be downloaded from here.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Shi J, Hu W, Huang S, Liu J, Zhang B. 2026. Keystone microbial taxa with interannual dynamics and metabolic versatility drive element biogeochemical cycling in a large deep-water reservoir. Environmental and Biogeochemical Processes 2: e011 doi: 10.48130/ebp-0026-0006 

Keystone microbial taxa with interannual dynamics and metabolic versatility drive element biogeochemical cycling in a large deep-water reservoir
- Received: 27 February 2026
- Revised: 10 April 2026
- Accepted: 09 May 2026
- Published online: 27 May 2026
Abstract: Thermally stratified, large, deep-water reservoirs are biogeochemical hotspots; however, their interannual microbial dynamics and the roles of keystone taxa remain poorly resolved. Here, we combined 16S rRNA amplicon sequencing with genome-resolved metagenomics to investigate microbial community turnover and metabolic potential in the Xiaowan Reservoir (upper Mekong River, China) from 2017 to 2019. Microbial community composition exhibited stronger interannual than depth-related variation (Adonis, R2 = 0.34, p = 0.001), with taxonomic dissimilarity increasing continuously over time (Adonis, R2 = 0.36, p < 0.001). In contrast, functional dissimilarity showed weaker temporal divergence (R2 = 0.14, p = 0.007), indicating that functional redundancy buffered ecosystem-level metabolic capabilities. We reconstructed 671 metagenome-assembled genomes (MAGs) and identified 46 keystone taxa through network analysis. These keystone taxa harbored versatile metabolic capacities for carbon utilization, dissimilatory nitrate reduction to ammonium (DNRA), urea hydrolysis, sulfur oxidation, and iron reduction. Total organic carbon concentration most strongly explained the distribution of keystone taxa (14.3%, p < 0.01). Notably, urea utilization and sulfur oxidation pathways increased annually, reflecting enhanced metabolic versatility. Collectively, our findings demonstrate that the metabolic versatility of keystone taxa may help maintain functional stability despite environmental fluctuations, providing mechanistic insights into microbial-mediated biogeochemical cycling in deep, stratified freshwater ecosystems under interannual environmental variability.