









-
In eukaryotes, DNA is packaged into chromatin, a dynamic structure that governs genetic accessibility. The basic repeating unit of chromatin is the nucleosome, consisting of DNA wrapped around histone octamers (H2A, H2B, H3, and H4)[1]. By organizing DNA and regulating its accessibility, nucleosomes modulate DNA-templated processes, including transcription, replication, and repair[1].
Chromatin states are modulated by a complex array of covalent modifications. These epigenetic marks primarily include DNA methylation at the C5 position of cytosine[2] and a variety of post-translational modifications (PTMs) on histone proteins[3]. Histone PTMs represent a sophisticated layer of control, primarily concentrated on the disordered N-terminal tails extending from the nucleosome core and include the acetylation, methylation, ubiquitination, and SUMOylation of lysine; the methylation and citrullination of arginine; and the phosphorylation of serine and threonine[4]. Mass spectrometry has also revealed additional histone acylations, including crotonylation, butyrylation, and propionylation, further expanding the regulatory capacity of the histone-based signaling system[5].
The functional consequences of these modifications are often explained by the "histone code" hypothesis, which proposes that specific marks, either alone or in combination, recruit effector proteins to drive distinct biological outcomes[6]. This system is maintained by three functional groups of proteins: The "writer" that catalyzes the addition of modifications, the "reader" that recognizes these marks, and the "eraser" that ensures the reversibility of the process[7]. Together, these factors dynamically shape the chromatin states, thereby influencing fundamental processes ranging from transcription to DNA repair and cell cycle progression[8,9]. Among these PTMs, histone methylation is distinguished by its unique biochemical impact and combinatorial complexity[9,10]. Unlike lysine acetylation, which neutralizes the positive charge and can directly alter chromatin's compaction, methylation preserves the formal charge of the lysine or arginine side chains. Instead, it alters the hydrophobicity and steric profile of the residue, creating specific surfaces for the docking of reader domains[11]. Furthermore, lysine residues can be mono-, di-, or trimethylated (me1/me2/me3), whereas arginine residues can be monomethylated or symmetrically/asymmetrically dimethylated, providing a nuanced mechanism for fine-tuning gene expression.
In fungi, particularly plant pathogens, histone methylation has been identified as a key regulator of developmental transitions, environmental sensing, and infection cycles. Here, we synthesize current knowledge of the histone methylation status in fungi, with a specific focus on the major phytopathogens. We begin by categorizing the biochemical properties of histone and nonhistone methylation, highlighting their context-specific roles in gene regulation. We next discuss the conserved and divergent features of the "writer–reader–eraser" machinery, including fungus-specific mechanisms, such as the noncanonical reading of H3K27me3. Subsequently, we examine how specific methylation marks (H3K4, H3K9, H3K27, H3K36, H3K79, H4K20, and arginine methylation) influence pathogenic development, virulence, and the transcriptional control of secondary metabolite biosynthetic gene clusters (BGCs). Finally, we evaluate the feasibility of targeting histone methylation pathways to enhance fungicides' efficacy and attenuate fungal virulence, and propose future directions for integrating multi-omics and genetic approaches to model the dynamic epigenetic regulation of fungal BGCs and pathogenicity.
-
Protein methylation involves the covalent transfer of a methyl group (-CH3) to nitrogen atoms, targeting two main substrate categories: Histone proteins, where lysine (Lys, K) and arginine (Arg, R) residues are the principal acceptor sites[12], and various nonhistone proteins.
Histone lysine methylation
-
In fungi, histone lysine methylation predominantly occurs on specific residues of the histones H3, H4, and H2B. The modifications on H3K4, H3K9, H3K27, H3K36, and H4K20 have been extensively characterized; however, recent studies have identified additional methylation sites. These include the mono-methylation of H2BK122 and H2BK130, as well as the di-methylation of H2BK122 in Penicillium oxalicum[13]. These marks exhibit distinct genomic distributions linked to specific transcriptional states and are generally classified according to their regulatory roles, as described below.
Activating marks (Fig. 1a). H3K4 methylation is a canonical indicator of transcriptional activation and shows a spatially partitioned pattern: H3K4me3 is sharply enriched at the promoters of active genes, whereas H3K4me2 extends into the 5' coding region[14,15]. H3K36 methylation, particularly H3K36me3, is deposited cotranscriptionally across gene bodies, where it facilitates transcriptional elongation and suppresses cryptic initiation to maintain transcriptional fidelity[16]. Furthermore, H3K79 methylation, enriched in euchromatin and open reading frames (ORFs), helps sustain an accessible chromatin state in Saccharomyces cerevisiae, specifically by preventing the expansion of silencing at subtelomeric regions[17,18].
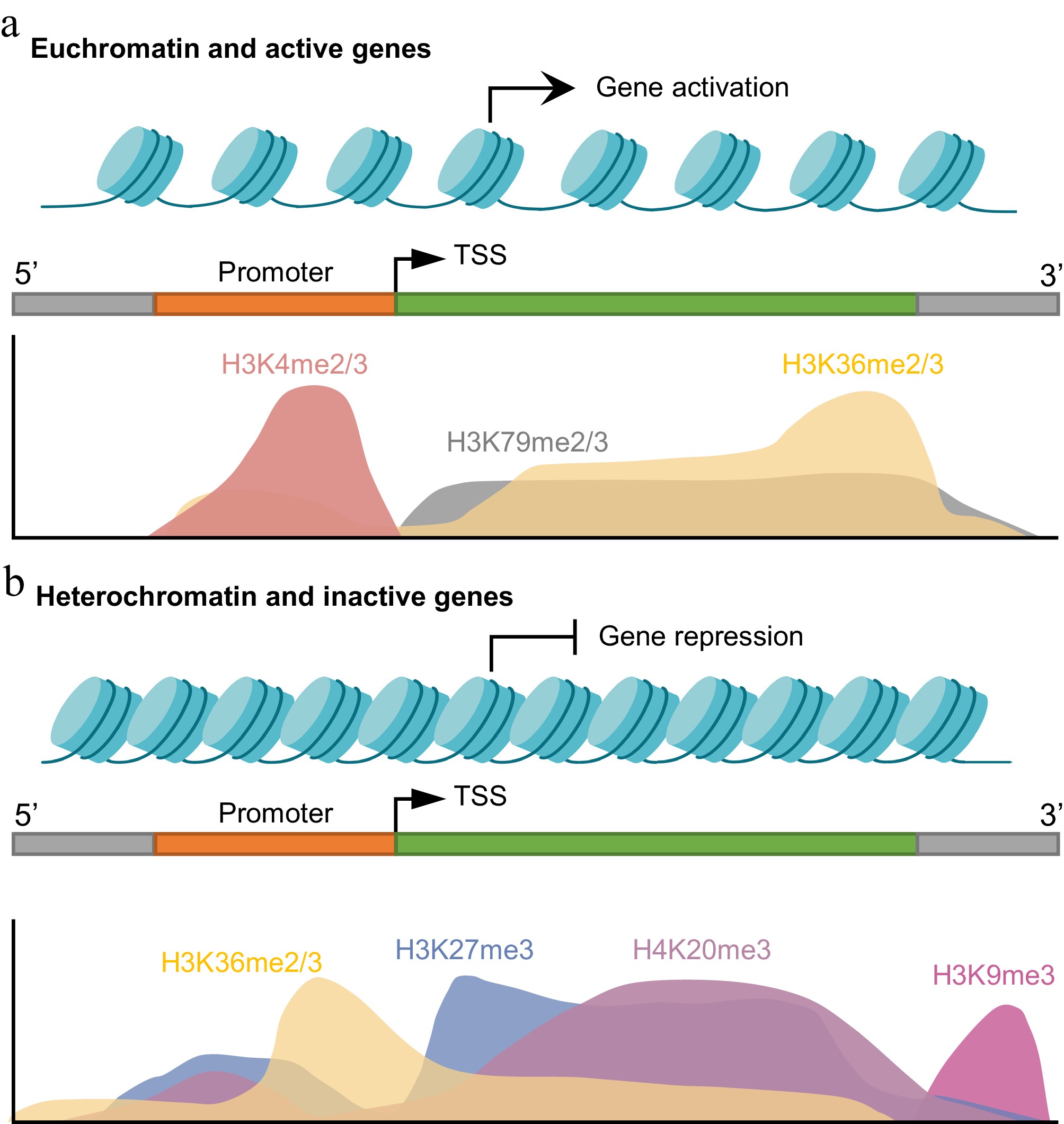
Figure 1.
Spatial distribution of histone lysine methylation marks in active and repressed genes. (a) In euchromatic regions linked to active transcription, H3K4me2/3 is enriched at the promoters and transcription start sites (TSSs), whereas H3K79me2/3 and H3K36me2/3 are found across gene bodies and facilitate transcriptional elongation. (b) Transcriptionally repressed regions in heterochromatin are marked by broad domains of H3K27me3 and H4K20me3, with H3K9me3 being primarily concentrated in constitutive heterochromatin, including telomeric and repeat-rich regions. In these repressive regions, H3K36me2/3 levels are reduced across gene bodies but may be locally enriched near the TSS. Created in BioRender. Xingmin, H. (2026)
https://BioRender.com/807zrla .Repressive marks (Fig. 1b). H3K9 methylation is a hallmark of constitutive heterochromatin and cooperates with DNA methylation to maintain the genome's stability[19]. In contrast, H3K27me3 marks facultative heterochromatin and mediates dynamic, reversible gene silencing. In phytopathogenic fungi, this modification is critical for the temporal regulation of developmental genes and secondary metabolite BGCs, enabling rapid adaptation to environmental stimuli[20,21]. H4K20 methylation is typically linked to transcriptional repression and contributes to the compaction of heterochromatin[22,23]. Notably, the functions of certain marks are context-dependent. For instance, although H3K36me2 is generally an activating mark, it has been associated with transcriptional repression in the rice blast fungus Magnaporthe oryzae[24,25].
In summary, the diversity of methylation sites, methylation states (mono-, di-, or tri-methylation), and their combinatorial arrangements create a complex epigenetic framework. This framework offers regulatory flexibility for fungal growth, development, environmental adaptation, and pathogenesis.
Histone arginine methylation
-
Arginine methylation is an evolutionarily conserved modification that occurs in three primary forms: Mono-methylarginine (MMA), symmetric di-methylarginine (sDMA), and asymmetric di-methylarginine (aDMA)[26,27]. In mammals, aDMA at residues such as H3R2 and H3R17 is typically associated with transcriptional activation[26], whereas sDMA at H3R8 and H4R3 more commonly correlates with transcriptional repression[28]. Although less well-characterized in fungi, emerging evidence suggests that specific marks, including H3R2 and H4R3 methylation, play a role in regulating chromatin's architecture and gene expression[27,29,30]. These modifications are associated with essential biological functions, including virulence, RNA splicing, and metabolic adaptation[29−33]. Despite advances in the study of model eukaryotes, the systematic identification of arginine methylation sites and their associated regulatory networks across the fungal kingdom remains incomplete.
Proteomic studies further reveal that the protein methylome includes a broad range of nonhistone substrates. Fungal methyltransferases can modify a variety of nonhistone substrates, including transcription factors, RNA-binding proteins, and signaling components[26]. This methylation can influence proteins' stability, subcellular localization, and interaction networks, thus affecting key cellular processes such as gene expression and DNA repair[12,34]. Recent studies have begun to elucidate the physiological relevance of these nonhistone modifications. For instance, in P. oxalicum, the arginine methyltransferases Prmt2 and Prmt3 cooperatively regulate the expression of cellulase by modulating the methylation status and nuclear localization of the transcription factor CxrA[31]. Similarly, in Aspergillus spp., enzymes such as RmtA regulate toxins' synthesis and development via nonhistone targets[32,33]. These studies highlight nonhistone methylation as an important regulatory layer in fungal pathogenesis and metabolism. Relative to lysine methylation, arginine methylation in fungi, especially in phytopathogens, remains much less comprehensively mapped at both the site and genome-wide levels, and many functional inferences still rely on a limited set of protein arginine methyltransferase (PRMT) mutants.
-
The deposition, interpretation, and removal of histone methylation involve the coordinated activities of writers, readers, and erasers[10]. In fungi, these factors primarily target the N-terminal histone tails extending from the nucleosome core, where they modulate chromatin's accessibility and gene expression through both conserved and species-specific mechanisms (Fig. 2, Supplementary Table S1).
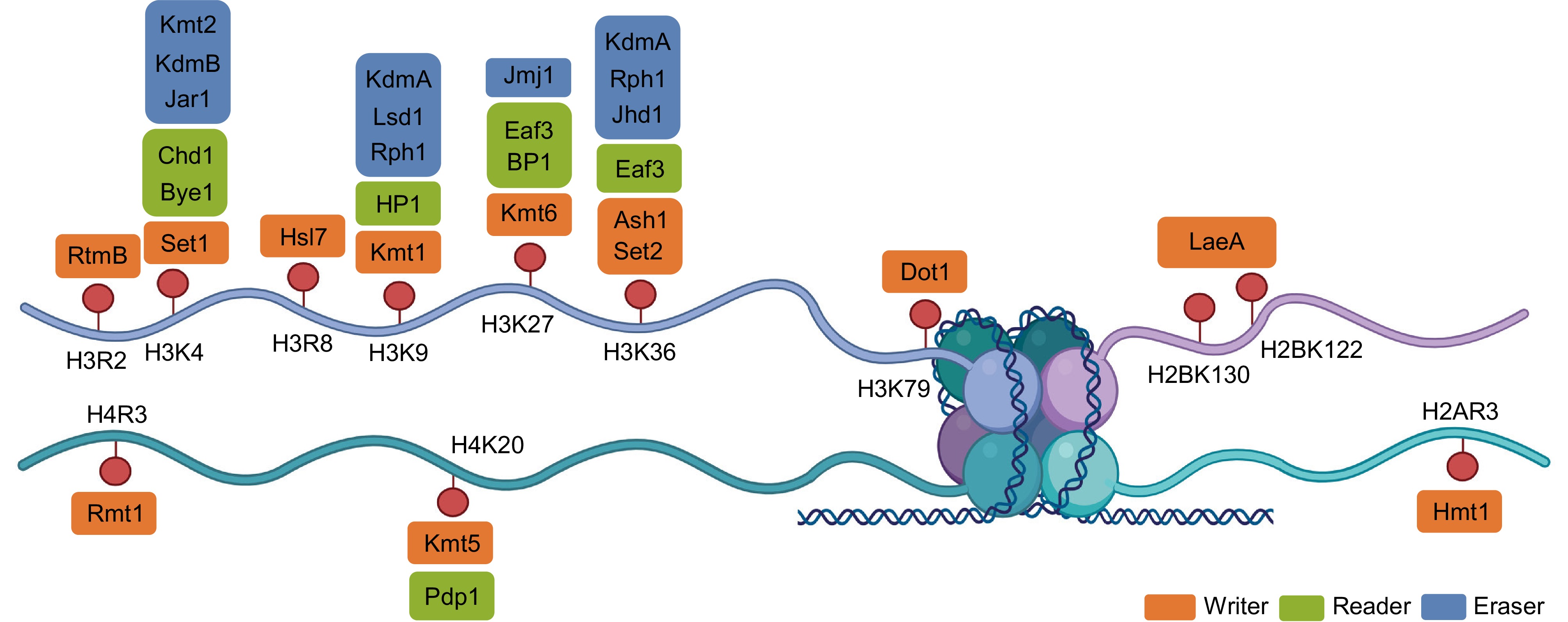
Figure 2.
Structural organization of histone methylation sites and the associated writer–reader–eraser proteins in fungi. Schematic representation of the core histone tails (H3, H4, H2A, and H2B) showing major lysine and arginine methylation sites, including H3R2, H3K4, H3K9, H3K27, H3K36, H3K79, H4K20, H2BK122/K130, and H2AR3. Each modification is presented alongside representative fungal methyltransferases (writers), demethylases (erasers), and reader proteins. Writer proteins (orange) include Set1 (H3K4), Kmt1 (H3K9), Kmt6 (H3K27), Set2 and Ash1 (H3K36), Dot1 (H3K79), Kmt5 (H4K20), LaeA (H2B), and Rmt1/Hmt1 (arginine). Reader proteins (green), such as HP1, Chd1, Bye1, Eaf3, and BP1, recognize methylated histones to mediate downstream chromatin remodeling and transcriptional changes. Erasers (blue) comprise Lsd1 and JmjC-domain-containing demethylases acting on distinct lysine residues. This diagram summarizes the coordinated protein machinery that maintains the state of the fungal epigenome. Created in BioRender. Xingmin, H. (2026)
https://BioRender.com/3r8vxo7 .Writer: histone methyltransferases
-
Fungal histone methyltransferases (HMTs) catalyze the transfer of a methyl group from S-adenosylmethionine (SAM) to specific lysine or arginine residues. According to their catalytic domains and substrate specificity, they are categorized into three main families: The SET-domain family, the Dot1/Dot1L-like family, and the PRMT family.
SET-domain proteins represent the major class of lysine methyltransferases (KMTs) in fungi. The SET domain, named after the Drosophila factors Su(var)3-9, Enhancer of zeste, and Trithorax, is an evolutionarily conserved catalytic motif[35]. Key fungal KMTs include Kmt2/Set1, the catalytic subunit of the COMPASS (complex of proteins associated with Set1) complex that deposits H3K4 methylation, a canonical mark of transcriptional activation[36]. Kmt1/Clr4/Dim-5 catalyzes H3K9 methylation and is essential for constitutive heterochromatin formation. Notable examples include Clr4 in Schizosaccharomyces pombe and Dim-5 in N. crassa. Kmt6/Ezh2, the catalytic subunit of the polycomb repressive complex 2 (PRC2), mediates reversible gene silencing via H3K27 methylation. Fungal PRC2 typically includes the Suz12 and Eed subunits. Though the accessory subunit Npf is dispensable in certain contexts, it is required for telomeric H3K27me3 deposition in N. crassa[20]. Kmt6 homologs are conserved across diverse fungal lineages[21,37,38]. Kmt3/Set2 and Ash1 catalyze H3K36 methylation. In Saccharomyces cerevisiae, Set2 is the primary H3K36 methyltransferase[39], whereas in many filamentous fungi, Set2 and Ash1 often function coordinately to methylate H3K36 across distinct chromatin regions[25,40−42]. Kmt5/Set9 catalyzes the di-, and tri-methylation of H4K20[23,43,44].
Unlike SET-domain enzymes, Dot1/Dot1L-like methyltransferases specifically methylate H3K79. In yeast, H3K79 methylation prevents the nonspecific binding of Sir proteins, thus maintaining silencing boundaries at subtelomeric regions[17,18]. In Aspergillus flavus, Dot1 regulates development, aflatoxin biosynthesis, and virulence[45]. Notably, the HMTs' repertoire varies significantly across species. For example, S. cerevisiae lacks the machinery for H3K9 and H3K27 methylation, whereas the core PRC2 components are absent in Aspergillus species[46].
Arginine methylation is catalyzed by PRMTs, which modify histones (e.g., H3R2 and H4R3) as well as numerous nonhistone substrates[26,34]. According to their products, PRMTs are classified into three types: Type I and Type II enzymes generate MMA as an intermediate and then produce aDMA or sDMA, respectively, whereas Type III enzymes generate only MMA[47,48]. Characterized fungal PRMTs include Hmt1 (Type I), Hsl7 (Type II), and Sfm1 (Type III) in S. cerevisiae, as well as Rmt2, which selectively methylates the δ-nitrogen of arginine[49]. In Aspergillus species, RmtA/B/C play central roles in development and secondary metabolism[33], and in P. oxalicum, Prmt2 and Prmt3 regulate the transcription factor CxrA[31].
Reader: interpreters of the histone code
-
Histone PTMs influence chromatin's dynamics through two main mechanisms: By directly altering the physical interactions between histones and DNA, or by recruiting reader proteins that recognize specific marks to assemble regulatory complexes[11]. Fungal readers recognize methylated lysine via evolutionarily conserved modules, including the bromo-adjacent homology (BAH) domain, chromodomain (CD), the plant homeodomain (PHD) finger, the PWWP (Pro-Trp-Trp-Pro) domain, and the Tudor domain, which display specificity for particular sites and methylation states (Fig. 2)[50].
H3K4 methylation is commonly recognized via PHD fingers or chromodomains. In S. cerevisiae, the PHD finger of Bye1 binds H3K4me3 at the promoters during the initiation of transcription and associates with RNA polymerase II during elongation[51]. In N. crassa, the Chd1 homolog NcChd1 utilizes tandem chromodomains to recognize H3K4me3, linking this mark to chromatin remodeling and transcriptional regulation[52]. A canonical reader of H3K9 methylation is HP1, which binds H3K9me2/3 via its chromodomain. In N. crassa, HP1 directs the formation of heterochromatin by recruiting the DNA methyltransferase Dim-2 and histone deacetylases to reinforce stable gene silencing[19,53,54]. H4K20 methylation is often recognized by PWWP domains. For instance, Pdp1 in S. pombe reads H4K20 methylation to regulate DNA repair and cell cycle checkpoints[55,56].
A distinctive feature of fungal epigenetic regulation is its divergence from the canonical Polycomb pathway in mammals. In mammals, gene silencing is mediated by a hierarchical PRC2–PRC1 cascade, in which PRC1 recognizes H3K27me3 through chromobox (CBX) chromodomain proteins and catalyzes the deposition of H2AK119ub1[57]. In contrast, most fungi lack a canonical PRC1 complex. Recent studies have identified alternative mechanisms for H3K27me3 recognition: (1) BP1, a BAH–PHD protein, binds H3K27me3 and interacts with PRC2. Notably, an intrinsically disordered region (IDR) in BP1 mediates liquid–liquid phase separation, thereby anchoring the silencing machinery to chromatin[58,59]. (2) Eaf3, a dual-function reader characterized in M. oryzae, contains a chromodomain for H3K27me2/3 and an MORF4-related gene (MRG) domain for H3K36me2. This integration of these opposing marks is essential for the maintenance of facultative heterochromatin[24].
Eraser: histone demethylases
-
The discovery of lysine-specific demethylase established the dynamic nature of histone methylation[60,61]. These erasers give plasticity to chromatin states through two distinct mechanisms: (1) Amine oxidation, exemplified by the LSD1 family of flavin adenine dinucleotide (FAD)-dependent oxidases, which demethylate mono- and dimethyl-lysine (me1/me2) but cannot process trimethylated lysine residues[62,63]; and (2) hydroxylation, catalyzed by Jumonji C (JmjC) domain-containing enzymes. These Fe(II)- and α-ketoglutarate (α-KG)-dependent dioxygenases can demethylate mono-, di-, and trimethyl-lysine[10].
LSD1 orthologs show functional diversity in fungi. In S. pombe, Swirm1 and -2 exist in stable complexes but show no detectable demethylase activity[64]. Conversely, in N. crassa, Lsd1 associates with Phf1 and Bdp-1 to restrict heterochromatin spreading by demethylating H3K9[65].
The JmjC family is the largest group of fungal histone demethylases (HDMs), targeting H3K4 (e.g., Jhd1/2, Rph1), H3K9/H3K36 (e.g., KdmA), and H3K27 (e.g., Jmj1). In S. cerevisiae, Jhd2 antagonizes Set1-mediated H3K4me3 to regulate subtelomeric silencing[66,67]. In A. fumigatus and A. nidulans, KdmA and KdmB target methylation of H3K36 and H3K4, respectively[68,69]. In M. oryzae, the H3K27me3 demethylase Jmj1 interacts with the chromatin remodeler Rsc1 to establish precise heterochromatin boundaries[70]. To date, all confirmed fungal HDMs target lysine methylation; an arginine demethylase has not yet been biochemically validated in fungi.
-
Histone methylation is a key regulatory layer that coordinates major transitions throughout the fungal infection cycle. By modulating chromatin's accessibility, these marks govern processes ranging from the switch between vegetative growth and the formation of specialized infection structures to the precise temporal execution of effector programs. In general, activating marks are associated with gene induction, morphogenesis, and metabolic reprogramming. In contrast, repressive marks establish heterochromatin to impose stage-specific silencing of dispensable or temporally restricted genes, supporting the genome's stability and transcriptional fidelity. Collectively, these modifications function as an integrated system that couples chromatin's organization to developmental stage, metabolic status, and host–pathogen interactions (Fig. 3).
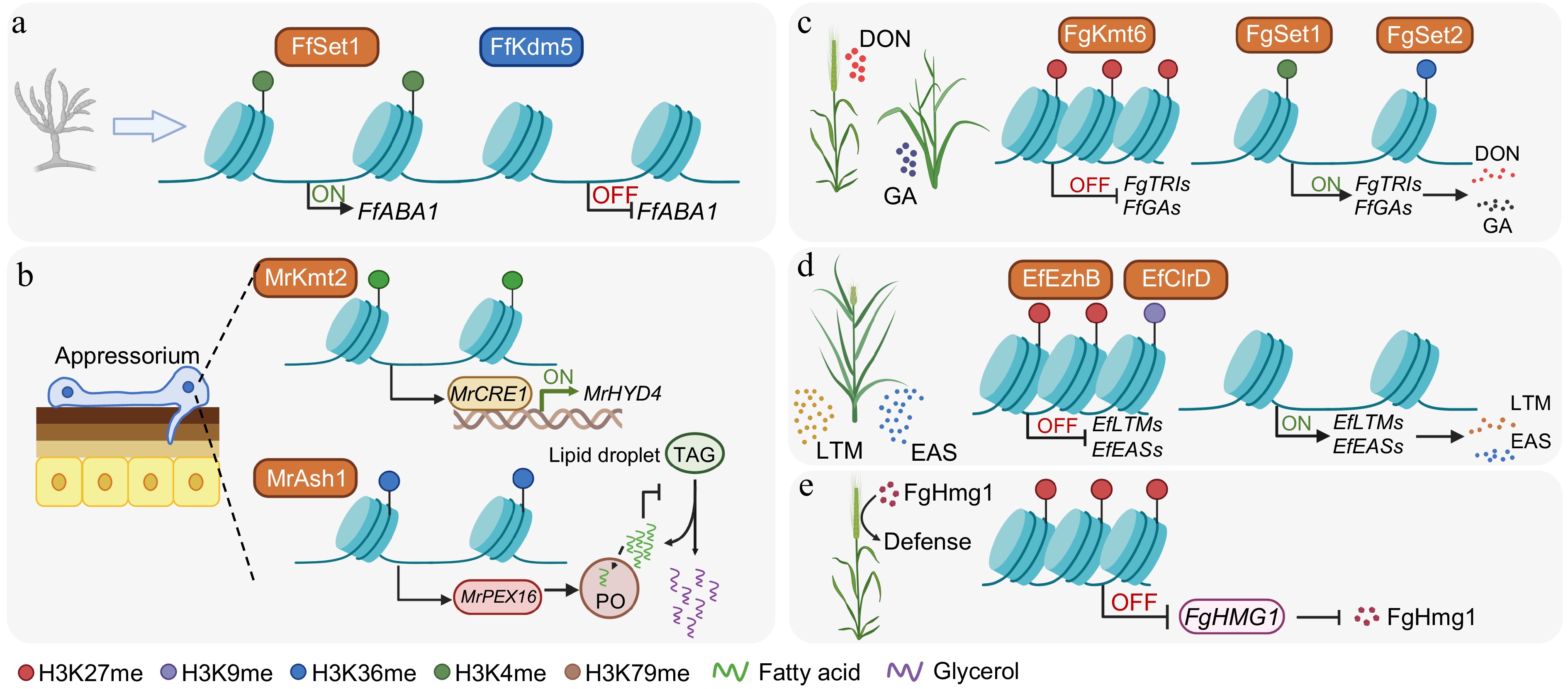
Figure 3.
Histone methylation-dependent mechanisms regulating fungal development, metabolism, and pathogenicity. (a) Regulation of asexual development by activating and repressive marks. In F. fujikuroi, Set1 deposits H3K4 methylation at the promoter of FfABA1, a central regulator of conidiation, to promote its transcription. Conversely, the removal of H3K4 methylation by the demethylase Kdm5 reduces FfABA1 expression. (b) Control of the formation of the infection structure and lipid metabolism in M. robertsii. During host infection, Kmt2-catalyzed H3K4me3 activates the transcription factor MrCRE1, which, in turn, induces the hydrophobin gene MrHYD4 to facilitate appressorium formation and surface adhesion. Simultaneously, Ash1-mediated H3K36me2 activates MrPEX16, supporting peroxisome (PO) biogenesis and fatty acid β-oxidation. This process prevents excessive lipid accumulation, promotes triacylglycerol (TAG) hydrolysis within lipid droplets, and supports glycerol production necessary for appressorial turgor generation and penetration. (c) In F. graminearum and F. fujikuroi, Kmt6-dependent H3K27me3 establishes repressive chromatin over the deoxynivalenol (DON) and gibberellic acid (GA) biosynthetic clusters, respectively. This repression is counteracted by Set1-mediated H3K4 methylation and Set2-mediated H3K36 methylation, which sustain the transcriptional activation of these clusters during pathogenesis. (d) Heterochromatin-mediated silencing and host-induced derepression of alkaloid gene clusters in E. festucae. The methyltransferases EzhB and ClrD catalyze H3K27me3 and H3K9me3, respectively, to cooperatively silence the lolitrem (LTM) and ergot alkaloid (EAS) clusters during axenic growth. During symbiosis, this dual heterochromatic repression is relieved, leading to the activation of LTM and EAS clusters and the production of protective alkaloids. (e) In F. graminearum, PRC2/FgKMT6-dependent H3K27me3 establishes a facultative repressive chromatin state at the pathogen-associated molecular pattern gene FgHMG1, which encodes a secreted glycoside hydrolase. This H3K27me3-mediated repression reduces host recognition and immune activation, thereby attenuating its immune elicitation and facilitating successful infection. Created in BioRender. Xingmin, H. (2026)
https://BioRender.com/lmnbvna .H3K4 methylation
-
As a well-established activating mark, H3K4 methylation is closely linked to gene induction, development, and virulence in diverse pathogens. In Fusarium fujikuroi, the methyltransferase Set1 and the demethylase Kdm5 antagonistically regulate H3K4me3 at the promoter of ABA1, encoding a master transcription factor, to control conidiation[71] (Fig. 3a). In Metarhizium robertsii, a Kmt2–Cre1–Hyd4 regulatory axis has been described: Kmt2-dependent H3K4me3 upregulates the transcription factor Cre1, which, in turn, activates the hydrophobin gene HYD4 to promote appressorium formation and host adhesion[72] (Fig. 3b). A similar pathway operates in the entomopathogen Beauveria bassiana[73]. H3K4 methylation also plays an important role in secondary metabolism. In Fusarium graminearum, Set1-mediated H3K4 methylation is required for the activation of the deoxynivalenol (DON) biosynthetic cluster[74] (Fig. 3c). Similarly, in F. fujikuroi, Set1 promotes H3K4me2/3 deposition at the gibberellic acid (GA) biosynthetic gene cluster, driving GA production[71] (Fig. 3c). In Colletotrichum higginsianum, the COMPASS subunit CclA is required for genome-wide H3K4me3 deposition, supporting spore germination, hyphal growth, asexual reproduction, and host penetration, and also constraining expression of specific terpenoid clusters[75]. Conversely, in Botrytis cinerea, deletion of the demethylase JAR1 causes the aberrant accumulation of H3K4me3, which paradoxically suppresses conidiation and appressorium formation, thereby impairing virulence[76]. These findings emphasize that homeostasis of H3K4me3, rather than static accumulation, is indispensable for normal fungal development and pathogenicity.
H3K36 methylation
-
H3K36 methylation coordinates transcriptional elongation and helps maintain chromatin's homeostasis, frequently exhibiting distinct spatial partitioning across the genome. Many filamentous fungi possess two nonredundant H3K36 methyltransferases, Set2 and Ash1. In F. fujikuroi, B. bassiana, and A. flavus, these enzymes establish divergent H3K36me3 distribution patterns with specific impacts on morphogenesis and mycotoxin biosynthesis[40−42]. During infection by M. robertsii, Ash1-catalyzed H3K36me2 activates PEX16, which promotes peroxisome biogenesis and lipid hydrolysis. The subsequent accumulation of glycerol generates the turgor pressure required for appressorial penetration[77] (Fig. 3b). In E. festucae, SetB is required for host colonization by regulating CRBA, a carbohydrate-binding protein gene[78]. Moreover, in N. crassa, Set2 maintains moderate acetylation at the circadian gene FRQ to ensure a chromatin state that is permissive for rhythmic transcription and development[79]. Regarding erasers, a Kdm4 homolog in B. cinerea demethylates H3K36me3 to modulate light-responsive genes, thereby influencing virulence and stress responses, highlighting HDMs as key integrators of environmental cues and infection fitness[41,80,81].
H3K9 methylation
-
H3K9 methylation is a hallmark of constitutive heterochromatin, in which transposable elements and accessory genes are silenced to maintain the genome's stability and pathogenicity. In B. bassiana and B. cinerea, loss of the H3K9 methyltransferase Dim5 leads to downregulation of the genes involved in host invasion, cell wall architecture, and mycotoxin biosynthesis, resulting in significantly reduced virulence[82]. In F. verticillioides, deletion of DIM5 impairs pathogenicity while inducing melanin biosynthesis, potentially as a compensatory response to osmotic stress[83]. In Leptosphaeria maculans, the absence of DIM5 triggers an RNAi response and induces chromatin decompaction; the loss of either HP1 or DIM5 derepresses numerous genes, including those encoding small secreted proteins[84]. In the endophyte Epichloë festucae, H3K9me3 (catalyzed by Clr4) and H3K27me3 (catalyzed by EzhB) collaboratively silence the lolitrem (LTM) and ergot alkaloid (EAS) clusters (Fig. 3d). This dual repression is specifically relieved during host symbiosis, allowing for precise, environment-dependent metabolic control[85].
H3K27 methylation
-
H3K27 methylation marks facultative heterochromatin and provides a reversible mechanism for silencing genes associated with development and virulence[86]. Disruption of PRC2 in pathogens such as F. graminearum, M. oryzae, and Ustilaginoidea virens abolishes H3K27me3 and typically causes aberrant growth, reduced conidiation, and the widespread misexpression of secondary metabolites and effector genes, ultimately compromising virulence[37,58,87]. During the early infection stage of Dothistroma septosporum, H3K27me3 decreases specifically at the dothistromin toxin cluster. This decrease is accompanied by coordinated chromatin transitions at the pathway regulator AflR, including reduced H3K9me3 and increased H3K9 acetylation, which facilitate transcription and ensure timely mycotoxin production[88]. In F. graminearum, H3K27me3-mediated facultative silencing can also contribute to immune evasion (Fig. 3e). A recent study reported that PRC2-associated H3K27me3 represses the immunogenic gene FgHMG1, which encodes a secreted glycoside hydrolase, reducing host recognition and promoting infection[89]. This example illustrates that Polycomb-dependent chromatin states can regulate not only secondary metabolite clusters but also specific host interaction loci.
Other histone and arginine methylation marks
-
Although less extensively studied, Dot1-mediated H3K79 methylation contributes to development and secondary metabolism in P. oxalicum and A. flavus[45,90]. H4K20 methylation, catalyzed by Kmt5, is primarily associated with genome maintenance and responses to DNA damage. In Zymoseptoria tritici, deletion of KMT5 increases sensitivity to genotoxic stress[23]. Notably, in F. graminearum, and F. fujikuroi, deletion of KMT5 has minimal effects on vegetative growth or virulence but significantly alters the secondary metabolite profile[43]. This suggests that H4K20 methylation may support long-term environmental adaptation rather than being a direct requirement for pathogenic fitness.
Methylation of arginine by PRMTs regulates pathogenic traits through the modification of both histone and nonhistone substrates. In A. flavus, deletion of RMTA disrupts the BrlA/AbaA/WetA regulatory axis, eliminating sclerotia formation and reducing aflatoxin production[32,33]. In F. graminearum, Prmt1/Amt1 controls hyphal growth and infection; mutants are hypersensitive to oxidative and membrane stress and produce less DON[30]. In P. oxalicum, Prmt2 and Prmt3 modulate the methylation status and nuclear localization of the transcription factor CxrA, thereby controlling the expression of plant polysaccharide-degrading enzymes[31]. Although mechanistic evidence in phytopathogens remains limited, these findings support a model in which PRMT-dependent methylation of key regulators links environmental sensing to host-associated gene expression programs.
Collectively, histone methylation serves as a central regulatory hub in pathogenic fungi. The dynamic antagonism and cooperation between activating and repressive marks specify virulence-related gene expression. Decoding the regulatory logic of the writer–reader–eraser machinery clarifies the molecular basis of fungal pathogenicity and may reveal potential vulnerabilities for developing epigenetics-based antifungal strategies.
-
Fungal secondary metabolites are critical for environmental adaptation, niche competition, and host–pathogen interactions. Although the production of secondary metabolites is not essential for viability, it is metabolically costly; accordingly, the BGCs encoding these pathways are subjected to stringent regulation. Histone methylation provides a major epigenetic layer that dictates BGCs' transcriptional status. This regulation relies on a dynamic balance between activating marks (e.g., methylation of H3K4 and H3K36) and repressive marks (e.g., methylation of H3K27 and H3K9), which together govern the transitions of BGCs from chromatin-mediated silencing to active transcription[91,92].
In fungi with a functional PRC2, including Fusarium spp., Z. tritici, and M. oryzae, H3K27 methylation is a principal repressive mechanism acting on BGCs. H3K27me3 establishes facultative heterochromatin that keeps BGCs in a "poised" yet silent state under noninducing conditions (Fig. 4a). This repression is reversible: Upon exposure to specific developmental cues or environmental stimuli, the removal of H3K27me3 triggers rapid transcriptional activation. Genetic disruption of the PRC2 catalytic subunit Kmt6 in F. graminearum and F. proliferatum results in genome-wide loss of H3K27me3 and derepresses numerous otherwise silent BGCs[21]. Similarly, in Z. tritici, H3K27me3 is enriched on accessory chromosomes harboring effectors and BGCs; its loss compromises growth and virulence, highlighting its role in defining transcriptionally repressed genomic compartments[93]. In M. oryzae, reduced H3K27me3 results in broad defects in development and infection, and indirectly remodels the secondary metabolome. Thus, H3K27me3 can precisely tune the timing and expression of BGCs by maintaining clusters in a reversibly silent state.
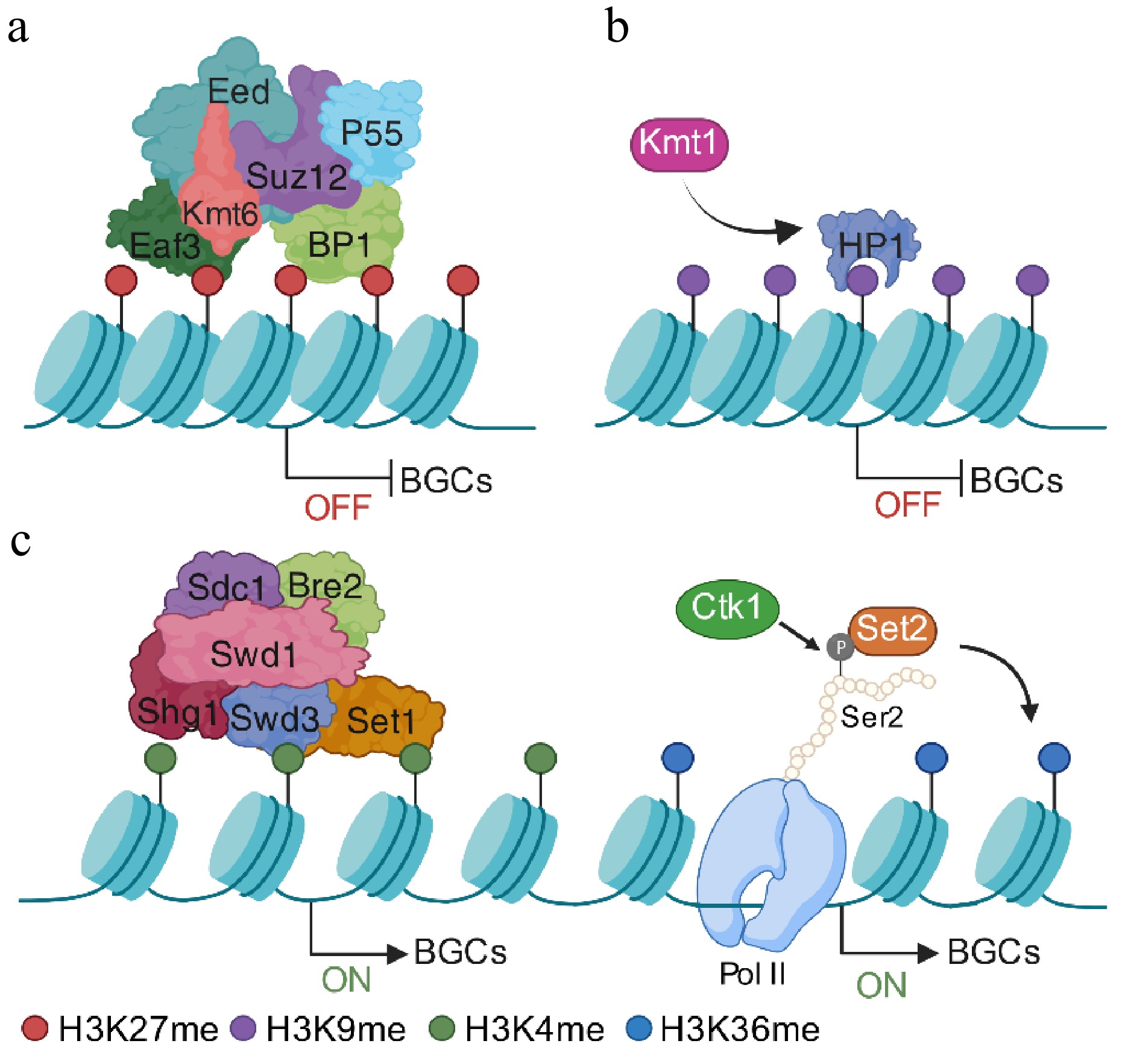
Figure 4.
Epigenetic regulation of fungal secondary metabolites' BGCs via histone methylation. (a) Under noninducing conditions, BGCs are maintained in a transcriptionally silent yet reversible state. In fungi possessing a functional PRC2, the catalytic subunit Kmt6 deposits H3K27me3 to establish facultative heterochromatin over BGC regions. (b) Alternatively, in fungi lacking PRC2, Kmt1-mediated H3K9 methylation cooperates with the reader protein HP1 to stabilize compact chromatin and restrict transcriptional accessibility. (c) Upon pathway induction, the removal or redistribution of repressive markers enables chromatin relaxation. The COMPASS complex, including Set1, deposits H3K4 methylation at BGC promoters to facilitate transcriptional initiation. Following derepression, active BGC expression is sustained by Set2-mediated H3K36 methylation across gene bodies. This mark is coupled to RNA Polymerase II elongation via Ser2 phosphorylation of the C-terminal domain, ensuring efficient transcriptional processivity and robust secondary metabolite production. Created in BioRender. Xingmin, H. (2026)
https://BioRender.com/55u0m5b In fungi lacking a functional PRC2 complex, such as Aspergillus species, H3K9 methylation functions as the dominant repressive mark. H3K9me3 typically marks constitutive heterochromatin and cooperates with HP1 to silence BGCs (Fig. 4b). The activation of these clusters often requires global regulators, such as the loss of aflR expression A (LaeA)/Velvet complex, to counteract this repressive chromatin. For instance, in A. fumigatus, LaeA partly relieves repression by reducing the local H3K9me3, thereby transitioning the chromatin toward a permissive state[91,94]. Notably, H3K9 methylation can be context-dependent. In the mango pathogen F. mangiferae, deletion of the H3K9 methyltransferase Kmt1 abolishes the production of fusapyrone, indicating that H3K9 methylation can, in some settings, function as a positive regulator[95].
H3K4 methylation, deposited by the COMPASS complex, generally functions as an initiating signal for the activation of BGCs (Fig. 4c). In Fusarium species, Set1-mediated H3K4 methylation is required to produce mycotoxins and pigments, including DON and aurofusarin[71,74]. Similarly, in Penicillium expansum, Set1 activates the patulin gene cluster both directly and indirectly via the regulation of LaeA[96]. H3K4 methylation also contributes to metabolic homeostasis by preventing inappropriate activation. In A. fumigatus, reduced H3K4 methylation derepresses normally silent BGCs and leads to excessive gliotoxin accumulation[75]. Together, these observations support the dual role for H3K4 methylation in activating productive pathways while suppressing spurious transcription.
H3K36 methylation is tightly coupled to transcriptional elongation. Set2-mediated H3K36me3 marks the coding regions of active BGC genes and facilitates RNAPII's processivity (Fig. 4c). This mechanism promotes the synthesis of fumonisin B1 in F. verticillioides[81] and sustains the expression of symbiotic BGCs in E. festucae[78]. In Aspergillus, where H3K27 methylation is absent, H3K36 methylation cooperates with H3K9me3 and histone acetylation to define BGCs' chromatin states[68]. Recent findings in F. graminearum further suggest a division of labor between two H3K36 methyltransferases[97]. Ash1 deposits H3K36me3 at secondary metabolite gene promoters, where it colocalizes with H3K27me3 to maintain repression; deletion of ASH1 reduces both marks, leading to the derepression of BGCs. In contrast, Set2 is recruited to gene bodies by the Ctk1-phosphorylated C-terminal domain of RNAPII to catalyze H3K36me3 and ensure efficient elongation (Fig. 4c).
Beyond these core modifications, H4K20 methylation appears to influence secondary metabolism, although mechanisms remain less clear. In F. graminearum, deletion of the H4K20 methyltransferase Kmt5 markedly alters the secondary metabolome without strongly affecting vegetative growth[43]. The direct contributions of H3K79 methylation and arginine methylation to the regulation of BGCs also remain incompletely defined.
Across phytopathogenic fungi, several principles appear to be broadly conserved. Methylation of H3K4 and H3K36 is frequently associated with active transcriptional programs that support development, infection, and metabolite biosynthesis, whereas H3K9me3 and/or H3K27me3 constrain lineage-specific effector genes and BGCs. However, the dominant repressive strategy is lineage-dependent. PRC2/H3K27me3 is prevalent in many pathogens, whereas PRC2 is absent in Aspergillus, where H3K9me3/HP1 and global regulators (e.g., LaeA/Velvet) play central roles in BGCs' repression and activation. Moreover, reader innovations (e.g., BP1, Eaf3) and the context-dependent behaviors of specific marks (e.g., H3K36me2-associated repression in M. oryzae) exemplify lineage-specific adaptations.
-
Phytopathogenic fungi impose substantial global crop losses and economic instability. Continued reliance on conventional fungicides has accelerated the evolution of resistance in major pathogens, such as Fusarium and Colletotrichum spp., necessitating the identification of novel fungicide targets. As a core regulator of stress adaptation and pathogenicity, histone methylation offers an intervention point distinct from conventional targets. Targeting methylation pathways could function through two strategies: Enhancing the efficacy of existing fungicides through sensitization, and directly attenuating virulence by suppressing effector and mycotoxin programs. Findings from human fungal pathogens provide a useful framework for translating epigenetic targeting concepts into plant protection.
Evidence from yeast models suggests that reducing H3K4 methylation can increase susceptibility to azole. In Candida glabrata, Set1-mediated H3K4 methylation is required for inducible transcription of the ergosterol biosynthesis pathway (including ERG11 and ERG3) under antifungal stress. Under azole treatment, H3K4me3 increases at the ERG loci to help maintain ergosterol's homeostasis; consequently, SET1 deletion abolishes this adaptive transcriptional response[98]. Although deletion of SET1 in S. cerevisiae also increases sensitivity to azole[98], the downstream transcriptional effects are species-specific, involving reduced expression of the efflux pump PDR5 rather than ERG11 induction[98]. Despite mechanistic variations, Set1 activity consistently supports drug tolerance, suggesting that inhibitors of H3K4 methyltransferases could significantly increase pathogens' sensitivity to azoles, potentially allowing for reduced chemical dosages and improved field efficacy.
In contrast to H3K4 methylation, H3K36 methylation can show an opposing relationship with azole resistance, dictating a different intervention strategy. In C. glabrata, deletion of SET2 (reducing H3K36 methylation) increases azole resistance, whereas deletion of the H3K36 demethylase RPH1 enhances sensitivity to the drug[99]. These effects are accompanied by the altered expression of PDR1-associated drug resistance genes, indicating a role for histone methylation in the response to azole[99]. These findings argue that to enhance sensitivity to fungicides, the objective should be to maintain high H3K36 methylation levels. Consequently, H3K36 demethylases (e.g., Rph1 homologs) may represent more suitable inhibitory targets, as inhibiting Set2 methyltransferases might inadvertently promote resistance.
Currently, several small-molecule inhibitors of histone-modifying enzymes have demonstrated antifungal potential. JIB-04, an inhibitor of Jumonji family demethylases, displays activity against Cryptococcus neoformans[100]. In addition, recent investigations into the biotransformation of capsaicin have identified lead compounds that inhibit the Lsd1 demethylase, offering potential for both anticancer and antifungal applications[101]. Overall, selectively targeting specific writers or erasers could disrupt fungal stress responses and virulence-associated transcriptional programs. In particular, inhibiting H3K4 methyltransferases or H3K36 demethylases represents a rational strategy to counter resistance to azole, motivating efforts to identify fungus-selective inhibitors and to evaluate their synergy with existing agricultural fungicides.
Translating histone methylation pathways into safe and durable fungicide targets faces several challenges. Because many HMTs and HDMs are conserved across eukaryotes, achieving pathogen selectivity while minimizing off-target effects on crops, beneficial fungi, and other nontarget organisms will be critical. Resistance could also arise through mutations at the target site, pathway rewiring, or enhanced efflux. These considerations support prioritizing fungus-selective chemotypes, targeting fungal-specific protein–protein interfaces, and deploying epigenetic inhibitors in rational combinations to reduce the risk of resistance.
-
The study of fungal histone methylation has progressed from static catalogs of individual marks to a dynamic view of the "writer–reader–eraser" system as a foundational regulatory layer in fungal biology. Recent work has revealed key lineage-specific features, including noncanonical H3K27me3 readers, such as BP1 and Eaf3 that maintain facultative heterochromatin in the absence of a mammalian-like PRC1 complex. In addition, studies linking the methylation of H3K4 and H3K36 to antifungal responses have provided a mechanistic connection between epigenetic states and drug tolerance. Finally, the expansion of the methylome beyond lysine residues, encompassing arginine methylation and widespread methylation of nonhistone proteins, points to a broad regulatory network coordinating fungal development, virulence, and metabolic adaptation.
Despite these advances, key gaps remain in understanding how methyl markers are integrated into the broader context of cellular and infection stage. A priority is the systematic identification of the machinery governing arginine methylation, particularly the search for candidate arginine demethylases and dedicated reader domains. In phytopathogenic fungi, many conclusions still rely on the deletion of global methyltransferases and on ChIP-based profiling, both of which have important limitations. Deletion of writers or erasers can cause broad developmental defects and indirect transcriptional changes that confound locus-level causality. ChIP-seq's outcomes also depend strongly on antibody specificity, the normalization strategy, and culture conditions, challenges that are amplified in stage-specific infection samples. Emerging approaches, including cleavage under targets and tagmentation (CUT&Tag), cleavage under targets and release using nuclease (CUT&RUN), site-specific histone point mutagenesis, and integrating high-throughput chromosome conformation capture (Hi-C) with real-time imaging, should help resolve locus-specific chromatin effects and place regulatory events within the three-dimensional nuclear organization.
Future studies should also move beyond single-mark analyses to define the spatiotemporal dynamics of combinatorial chromatin states. This will require the integration of high-resolution epigenomic profiling with transcriptomics, chromatin accessibility assays such as assay for transposase-accessible chromatin using sequencing (ATAC-seq), and metabolomics. These multi-omic strategies will be essential to reconstruct epigenetic trajectories at BGCs and virulence loci as they transition from stable silencing to "poised" intermediates, and to full activation. The next frontier in fungal epigenetics will be to model heterochromatin's dynamics with sufficient precision to inform sustainable disease control strategies. Rather than treating chromatin as a simplistic "on/off" switch, future work should elucidate how repressive domains are nucleated, spread, and constrained by the coordinated actions of readers, erasers, and chromatin remodelers. The contrasting roles of histone methylation pathways in drug resistance highlight the need for specificity. Inhibiting H3K4 writers may sensitize fungi to existing fungicides, whereas inhibiting H3K36 writers could inadvertently promote resistance. Accordingly, antifungal development should focus on inhibitors that exploit the structural divergence of fungal enzymes or disrupt fungal-specific protein–protein interfaces. By integrating structural biology with functional genomics, these epigenetic insights could be translated into practical strategies for crop protection and resistance management.
This work was supported by the National Natural Science Foundation (32172356), the China National Key Research and Development Program (2022YFD1400100), China Agriculture Research System (CARS-3-1-15), and the Fundamental Research Funds for the Central Universities (226-2024-00213, 226-2024-00070).
-
The authors confirm their contributions to the paper as follows: study conception and design, data interpretation: Chen Y, Han X; data collection: Han X, Xu C, Ren Y, Guo M, Yan J, Wang X; manuscript drafting: Han X; manuscript revision: Han X, Chen Y, Ma Z. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing is not applicable to this article, as no new datasets were generated or analyzed in this study.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 Methylation writers, readers, and erasers mentioned in this manuscript.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Han X, Xu C, Ren Y, Guo M, Yan J, et al. 2026. Histone methylation in the pathogenesis and secondary metabolism of phytopathogenic fungi. Epigenetics Insights 19: e006 doi: 10.48130/epi-0026-0003 

Histone methylation in the pathogenesis and secondary metabolism of phytopathogenic fungi
- Received: 31 December 2025
- Revised: 14 February 2026
- Accepted: 27 March 2026
- Published online: 28 April 2026
Abstract: Histone methylation is a crucial epigenetic modification of chromatin that regulates gene expression and chromatin architecture in eukaryotes. This review consolidates current knowledge on the histone methylation status of fungi, with a specific focus on major plant pathogens. We first describe the biochemical diversity of lysine and arginine methylation on histone and nonhistone substrates, along with the enzymatic framework of the writer–reader–eraser machinery. We also evaluate the roles of specific methylation marks in regulating fungal morphogenesis, genomic integrity, host infection, and the transcriptional control of secondary metabolite biosynthetic gene clusters. Finally, we explore the feasibility of targeting histone methylation pathways to reduce fungal pathogenicity and mitigate fungicide resistance. Collectively, this work provides a systematic analysis of the epigenetic mechanisms driving fungal virulence and offers insights into utilizing these pathways for sustainable control of crop diseases.
-
Key words:
- Histone methylation /
- Phytopathogenic fungi /
- Virulence /
- Secondary metabolism /
- Fungal disease control