-
The hydrazine-based fuel/nitrogen tetroxide (N2O4) bipropellant combinations are widely used in liquid rocket propellant systems in rocket engines worldwide. Among commonly used hydrazine-based fuels are hydrazine (N2H4), and its methyl derivatives, notably unsym-dimethylhydrazine ([CH3]2NNH2,UDMH), and methylhydrazine (CH3NHNH2). Characterizing the combustion chemistry of the hydrazine-based fuel/N2O4 systems, especially the comprehensive elucidation of elementary reactions, such as fuel unimolecular decomposition reactions, H-abstraction reactions, and ensuing β-scission and isomerization reactions, is crucial for optimizing liquid rocket engine performance and improving combustion efficiency. However, the hydrazine-based fuel/N2O4 systems involve particularly complex reaction processes with numerous reaction pathways[1], making it essential to obtain accurate combustion reaction kinetic parameters for a deeper understanding of their combustion mechanisms, and for constructing precise kinetic models.
Previous investigations are mainly dedicated to N2H4 and CH3NHNH2, including the experimental measurements[2−9] and theoretical calculations[10−16]. Catoire et al.[5,8] used a shock tube to determine ignition characteristics of CH3NHNH2/O2/Ar, and thermal decomposition of CH3NHNH2 across various temperatures, pressures, and equivalence ratios, and further examined the effects of N2 and H2 doping on the CH3NHNH2/O2/Ar system[6]. Liu et al.[7] introduced small amounts of CH3NHNH2 and NO2 along with large amounts of N2 diluent into a gold-plated reaction chamber, and found that CH3NHNH2 and NO2 still reacted upon contact, with uncertainties in the mixing process significantly affecting experimental results. As for theoretical calculations, Sun et al.[10] utilized quantum Rice-Ramsperger-Kassel (RRK) theory, master equation analysis, and high-level ab initio methods to examine CH3NHNH2 thermal decomposition kinetics, identifying N-N and C-N bond dissociation reactions as the dominant channels for CH3NHNH2 decomposition under ambient conditions. Sun et al.[17] performed theoretical calculations on the H-abstractions of CH3NHNH2 with H atoms, and identified the abstraction at the terminal amine site producing the trans-CH3NHNH as the most significant pathway within the various abstraction processes. Moreover, Ren et al.[18] systematically investigated the H-abstractions of CH3NNH2 involving several species, including NO2, OH, H, and so on. The results suggest that the effects of molecular structural distinction between trans-CH3NHNH2 and cis-CH3NHNH2 on H-abstraction reactions cannot be ignored.
Comparatively, the investigations into the combustion reaction kinetics of UDMH are relatively rare due to its highly toxic nature and extreme reactivity[19]. For fundamental combustion experimental investigations, Gray et al.[20] measured flame speeds and flammability limits of UDMH under different oxidizer atmospheres (O2 and N2O), using a constant volume combustion vessel, with the highest flame speed reported at 3,320 cm/s. Kumar and Bhaskaran[21] obtained ignition delay times (IDTs) of UDMH/CH4/O2/Ar using a shock tube. These experiments covered temperature ranges from 1,600−2,100 K, a pressure of 3 atm, equivalence ratios of 0.5−2.0, and a UDMH concentration of ~4% relative to methane. They found that IDTs decreased with increasing concentrations of UDMH, which may be caused by thermal effects or formation of CH3 and H. Additionally, Bhaskaran et al.[22] measured IDTs of UDMH/H2 under air conditions and found that small additions (< 1%) of UDMH can shorten the IDTs of hydrogen-air mixtures, while higher concentrations (> 3%), prolong them. For the rate constant measurement, Vaghijiani[3] employed laser photolysis and UV absorption spectroscopy to determine the rate constants for H-abstractions of UDMH with H attacking, when the temperature was 295 K. The measurements in this work yielded total second-order H atom rate coefficients as (13.48 ± 2.02) × 10−13 cm3/molecule/s respectively for (CH3)2NNH2 at ambient temperature and 26.0 Torr He. Subsequently, Vaghijiani[4] obtained the rate constants for UDMH + OH reactions across 254−633 K by using a flow reactor and photolysis apparatus. As indicated in the above review, the available experimental studies were mostly performed either in the presence of other fuels or under standard conditions, which complicates the investigations of pure UDMH combustion chemistry.
Against this background, theoretical calculations of the reaction rate constants facilitate further model development, and advance an insight into combustion chemistry of UDMH. Researchers have made theoretical efforts to compute the H-abstraction reactions of UDMH. Tang et al.[23] derived the rate constants for the UDMH + OH reactions at the BMC-CCSD//B3LYP/6-311G(d,p) level. They found that the HNN(CH3)2 + H2O product pair forms more favorably than the CH3N(CH2)NH2 + H2O pair, in the competition among H-abstraction reactions involving OH attack. Wang et al.[13] employed the canonical variational transition state theory with the small-curvature tunneling correction (CVT/SCT), to calculate the rate constants of the UDMH + H/D reactions on a potential energy surface (PES) computed at the MCG3-MPWPW91//MPW1K/6-311G(d,p) level. Using transition state theory (TST) combined with several tunneling correction methods, Kanno et al.[24] examined H-abstraction reactions of UDMH, by H attacking at the CBS-QB3//DSD-BLYP-D3(BJ)/def2-TZVP level. They found that the NHN(CH3)2 + H2 formation path was dominant in the UDMH + H reactions at low temperatures. Bai et al.[25] investigated the H-abstractions of UDMH by H, OH, NO2, HO2, CH3, CH3O, and CH3O2 radicals at the M06-2X/6-311++G(d,p), and CCSD/cc-pVXZ (where X = T, Q) levels. These findings reveal that the reactions with H, OH, and CH3O, dominate at 300–700 K, while CH3O2, HO2, and NO2 play minor roles. In summary, current research on the rate constants for UDMH reactions is quite insufficient. Specifically, existing studies on UDMH have primarily focused on H-abstraction reactions. At the same time, studies on H-abstraction reactions involving species such as CH3, NO2 remain relatively scarce. However, these reactions are of great importance, as CH3 and NO2 are major decomposition products of UDMH and N2O4, respectively. It is worth mentioning that previous studies[26−28] have also explored reactions between other fuels and NO2, demonstrating the unique characteristics of NO2 as an attacking species in H-abstraction reactions. Furthermore, investigation into the unimolecular decomposition reactions of UDMH is relatively scarce. Detailed studies on various key reactions within UDMH/N2O4 systems are urgently needed.
In this work, the unimolecular decomposition and H-abstraction reactions of UDMH were theoretically and comprehensively calculated. The corresponding PES, and rate constants were obtained. Meanwhile, the competitive characteristics among different reaction pathways were compared and analyzed. In addition, a detailed comparative analysis of the H-abstraction reaction characteristics was performed between UDMH calculated in this work and CH3NHNH2 obtained from previous studies.
-
In this work, the structural optimization and frequency calculations for UDMH and its corresponding products of unimolecular decomposition and H-abstraction reactions were performed using the M06-2X method[29], in conjunction with the def2-TZVP basis set[30]. This method was evaluated by Xing et al.[31], and shown to have a lower mean unsigned error, accounting for all forward energy barriers and reaction energies compared with other density functional theory (DFT) methods, such as M06-2X/6-311+G(2df,2p), M06-2X/jun-cc-pVTZ, and so on. In addition, the M06-2X method has been widely employed in calculations involving nitrogen-containing systems[32,33]. The zero-point energy correction factor was 0.971[34], and the frequency correction factor was 0.984[34]. The intrinsic reaction coordinate (IRC) approach was utilized to verify the accuracy of the transition state structures, and obtain the corresponding reactants and product geometries. The corresponding results are depicted in Supplementary Figs S1−S4. Hindrance potentials are obtained by performing dihedral angle scans of the specified rotational group in 10° increments, using the same approach employed for structural optimization. The relative energy for the rotation of the amine group exhibits two stable molecular structures (A and B), for the UDMH molecule (Fig. 1). According to calculated Boltzmann distributions depicted in Supplementary Fig. S5, it can be found that conformer A is dominant at low temperatures, while its relative population decreases with increasing temperature, and becomes comparable to that of conformer B at 2,500 K. In this work, the structure with the lowest energy only served as the reference geometry for optimizing geometry and analyzing frequency, while the contribution of NH2 internal rotation was explicitly accounted for through a hindered rotor treatment in the partition function.
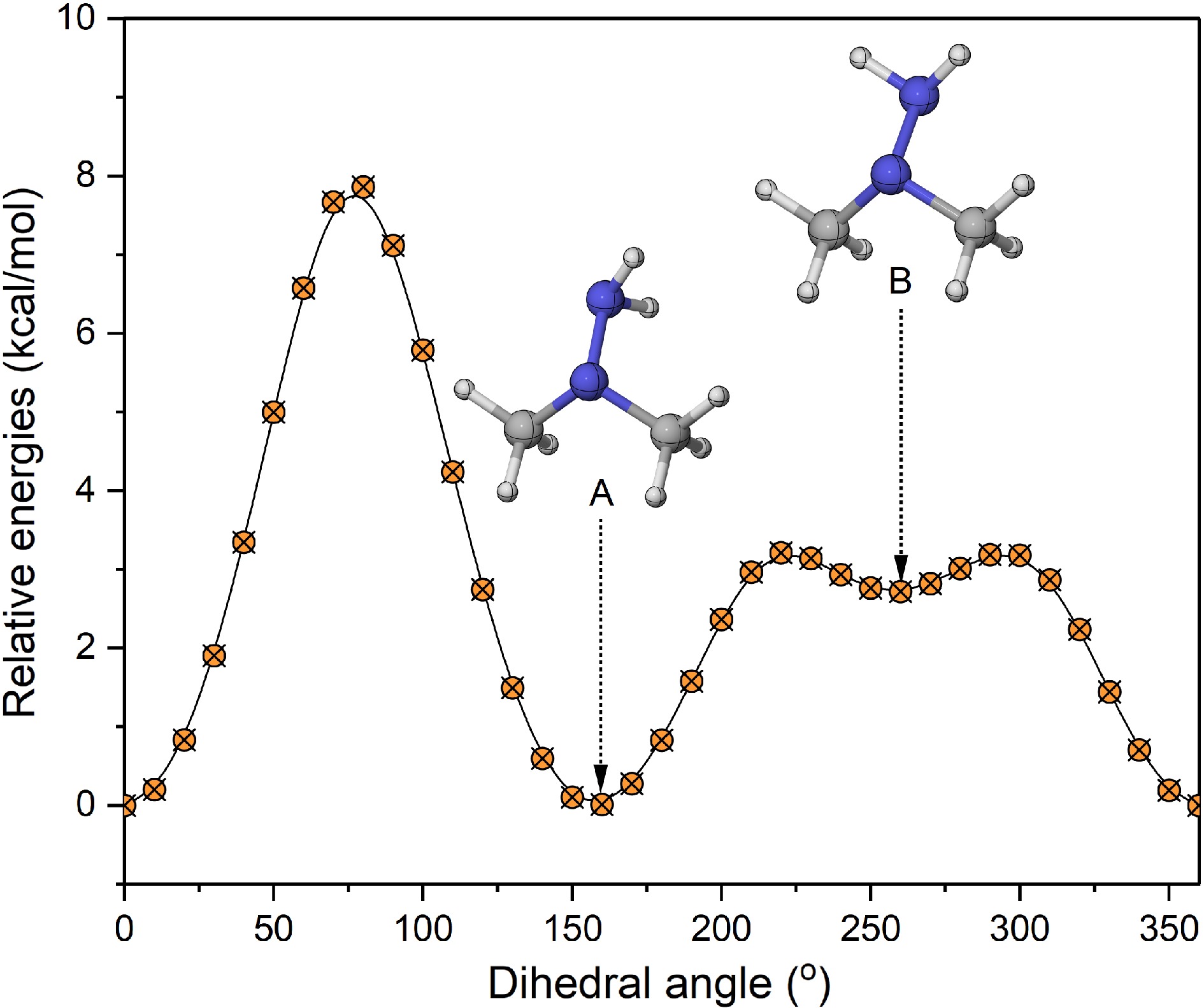
Figure 1.
Relative energy of the amine group rotation in the UDMH molecule (unit: kcal/mol).
For high-precision single-point energy (SPE) calculations of the stable molecules and transition states, coupled cluster methods with the cc-pVDZ and cc-pVTZ basis sets were employed for the energies calculation, and then the complete basis set energies were extrapolated using Eq. (1)[35,36]. To assess convergence, the larger basis sets (cc-pVQZ and cc-pVTZ, as shown in Eq. [2]) were employed[37]. As illustrated in Fig. 2, the calculated SPE's for the UDMH decomposition reactions at the CCSD(T)/CBS(T + Q) level are within 1 kcal/mol of those obtained at the CCSD(T)/CBS(D + T) level, indicating that the chosen theoretical approach is feasible. Lee & Taylor[38] suggested that T1 values of all reactant species, product radicals, and transition states should not exceed 0.025, 0.035, and 0.044, respectively. T1 diagnostics studied herein all met these requirements (Supplementary Table S1), which confirms that the single reference method can accurately evaluate the wave function. All quantum chemical calculations for geometry optimization, frequency analysis, rotational potential scans, and thermodynamic data were conducted with the Gaussian 09 program[39].
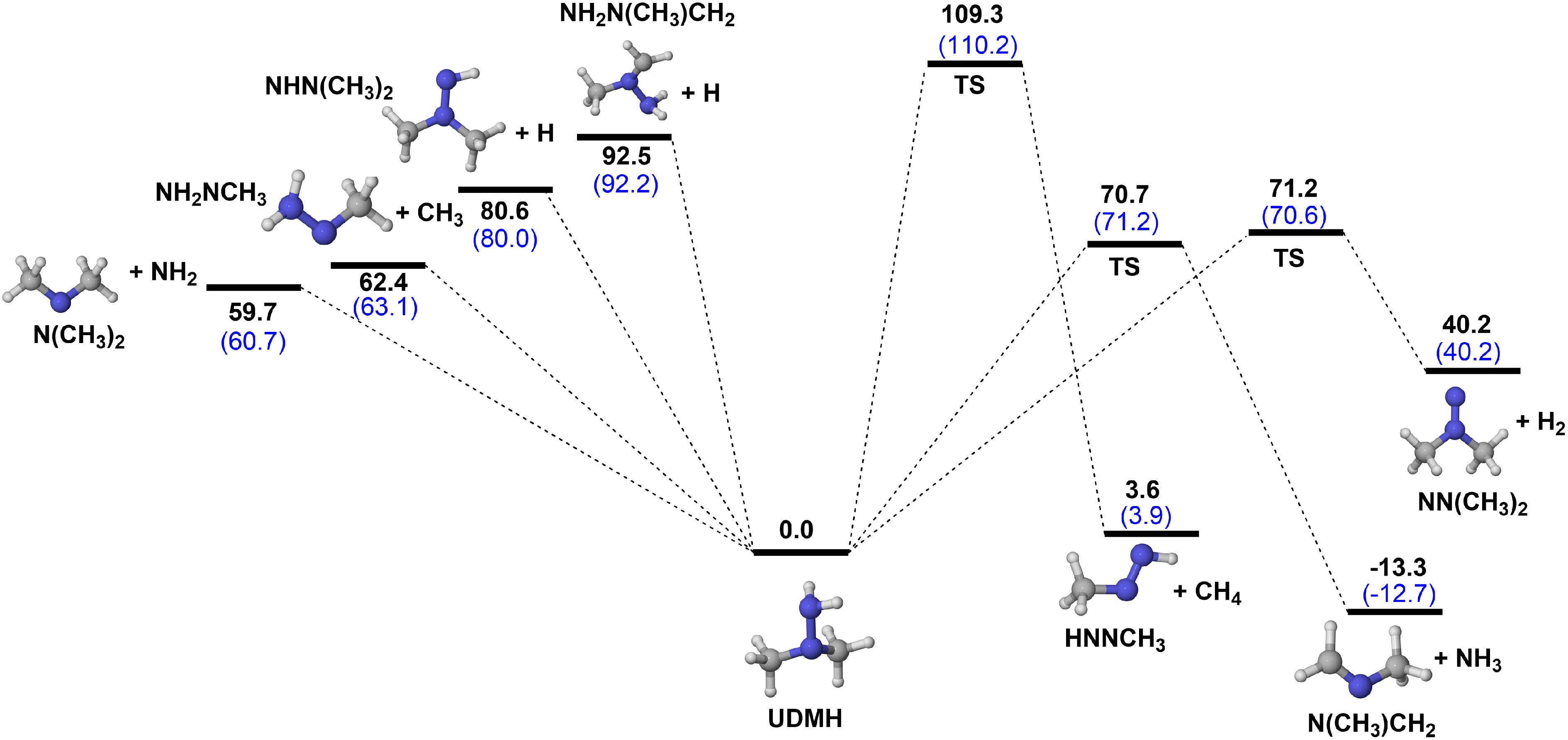
Figure 2.
PESs for the unimolecular decomposition reactions of UDMH at CCSD(T)/CBS(D+T)//M06-2X/def2-TZVP (black), and CCSD(T)/CBS(T+Q)//M06-2X/def2-TZVP (blue) level (unit: kcal/mol).
$ \begin{split}E_{\mathrm{CCSD}(\mathrm{T})/\mathrm{CBS}(\mathrm{D}+\mathrm{T})}=\; & E_{\mathrm{CCSD}(\mathrm{T})/\mathrm{cc}\text{-}\mathrm{pVTZ}}+(E_{\mathrm{CCSD}(\mathrm{T})/\mathrm{cc}\text{-}\mathrm{pVTZ}}- \\ \; & E_{\mathrm{CCSD}(\mathrm{T})/\mathrm{cc}\text{-}\mathrm{pVDZ}})\times 3^4/\left(4^4-3^4\right)\end{split} $ (1) $ \begin{split}E_{{\mathrm{CCSD}}({\mathrm{T}})/{\mathrm{CBS}}({\mathrm{T}}+{\mathrm{Q}})} =\;& E_{{\mathrm{CCSD}}({\mathrm{T}})/{\mathrm{cc}}\text{-}{\mathrm{pVQZ}}} + (E_{{\mathrm{CCSD}}({\mathrm{T}})/{\mathrm{cc}}\text{-}{\mathrm{pVQZ}}} -\\ \;& E_{{\mathrm{CCSD}}({\mathrm{T}})/{\mathrm{cc}}\text{-}{\mathrm{pVTZ}}}) \times {4^4}/{(5^4 - 4^4)}\end{split} $ (2) Due to the pronounced multi-reference characteristics, typically exhibited by barrierless bond dissociation reactions in the transition state region, configurations along the minimum energy path (MEP) for the bond breaking process were optimized using the complete active space second-order perturbation theory (CASPT2) method, combined with the cc-pVDZ basis set. For CH3 group dissociation reactions, an active space (6e, 6o) was employed. The six orbitals were the σ and σ* orbitals of the N-N, and the two N-C bonds. For NH2 dissociation reactions, a larger active space (10e, 8o) was used to consider the influence derived from the lone pair electrons of nitrogen. For N-H and C-H bond dissociation reactions, the selected active space was (2e, 2o), where the two orbitals were the σ and σ* orbital of the elongated N-H or C-H bonds. Utilizing the optimized geometries at the CASPT2 level, single point energies were calculated at a higher level using the multi-reference configuration interaction method (MRCISD), in conjunction with the cc-pVDZ and cc-pVTZ basis sets. These energy values were subsequently extrapolated to the CBS level by Eq. (3)[40].
$ \begin{split} E_{\text{MRCISD/CBS}} = \;& E_{\text{MRCISD/cc-pVTZ}} + (E_{\text{MRCISD/cc-pVTZ}} -\\ \;&E_{\text{MRCISD/cc-pVDZ}}) \times {2^3}/{(3^3 - 2^3)}\end{split} $ (3) The calculated MEP results were shown in Supplementary Fig. S6. Here, multi-reference calculations were conducted employing the Molpro 2015 program[41].
Leveraging the obtained PES results, the rate constants across 300−2,500 K, and 0.01−100 atm were determined. For the UDMH dissociation reactions with barrierless characteristics, the rate constants were determined based on the variational transition state theory (VTST) approach, based on the obtained MEP. Additionally, the formation of pre-reaction van der Waals complexes (RC), and the decomposition of product complex (PC) in H-abstraction reactions by NO2, also exhibit the barrierless characteristics[18] under long-range interaction. Phase space theory[42] was utilized to estimate the microcanonical rate constants, with the potentials along the reaction coordinate given by V = 10/R6, where 10 has units of Bohr6[43]. To calculate the rate constant for the unimolecular elimination and H-abstraction reactions of UDMH with a tight energy barrier, conventional transition state theory (cTST) was employed. For the H-abstraction reactions by H atoms and CH3 radicals, which do not involve the evident formation of RC and PC, their high-pressure-limit (HPL) rate constants were determined. The partition functions of reactants and transition states were approximated using the rigid rotor harmonic oscillator (RRHO) model, with the low-frequency torsional mode treated as a one-dimensional (1D) hindered rotor[44]. The details, including hindered rotor potentials, can be found in the MESS input files in the Supplementary Materials. Tunneling effects were accounted for using the 1D asymmetric Eckart model[45] when calculating the rate constant. The effects of hindered rotor and tunneling treatments on the calculated rate constants were comprehensively analyzed (Supplementary Figs S7−S10), with a detailed discussion provided in the following section. In addition, the anharmonicity contributions to rate coefficients of the main reactions are found to be negligible, based on an evaluation of the effect of considering the harmonic frequency scale factor (Supplementary Figs S11−S13). Because the unimolecular elimination and H-abstraction reactions investigated in this work possess tight energy barriers, recrossing effects on their rate constants are usually subtle.
The RRKM theory with the ME method[46] was utilized to obtain the pressure-dependent rate constants. The collisional energy transfer model was treated using the empirical formula: <∆E>down = 200(T/300)0.85[47]. The Lennard-Jones parameters σ = 4.4, Å and ε = 440 cm−1, were employed for all intermediates[47]. The bath gas in this work was Argon (Ar), with σ = 3.47 Å, and ε = 78.88 cm−1[48]. The rate constants were computed employing the MESS kinetic code[49]. The pressure-dependent and HPL rate constants are sub sequently described through a modified Arrhenius Eq. (4),
$ \mathit{k} \mathrm{(} \mathit{T} \mathrm{)} \mathit{=AT} ^{ \mathit{n} } \mathrm{exp(-} \mathit{E} _{ \mathit{a} } \mathit{/RT} \mathrm{)} $ (4) where, A denotes the pre-exponential factor, n the temperature exponent, and Ea the reaction activation energy. Furthermore, two-parameter fittings are used for the reactions with non-Arrhenius characteristics. The rate constants for the unimolecular decomposition and H-abstraction reactions of UDMH obtained in this work are presented in Supplementary Tables S2, S3.
Uncertainty quantification for the rate constants was also conducted. The commonly and widely expected uncertainty for CCSD(T)/CBS calculation is less than 1 kcal/mol[32,50,51]. Using reaction R10 as an example, varying the barrier height by ±1 kcal/mol introduced errors of about a factor of two at 700 K, and a factor of five at 300 K (Supplementary Fig. S14), demonstrating greater sensitivity at lower temperatures. Regarding the collisional parameters, the influence of σ was negligible for R2 (Supplementary Fig. S15). Additionally, the uncertainty in the rate constants from harmonic frequency scaling was small (within approximately 5%), and considered negligible (Supplementary Figs S11−S13). Consequently, harmonic frequency scaling contributes substantially less uncertainty to the calculated rate constants than single-point energy variations.
-
According to the calculated PESs presented in Fig. 2, the dissociation energy of the C-H bond of UDMH is 92.5 kcal/mol, higher than the 80.6 kcal/mol observed for the N-H bond. Additionally, the dissociation energy for the CH3 groups in UDMH is 62.4 kcal/mol, while that for the amine group is 59.7 kcal/mol. The required energy of CH3 and NH2 groups dissociation reactions (R1 and R2) are significantly lower than the required energies of H atom dissociation reactions. Compared to CH3NHNH2, the BDEs of C-H and N-H bonds in UDMH are similar to those in CH3NHNH2 calculated by Ren et al.[18]. The BDE of the C-H bond in CH3NHNH2 is 91.45 kcal/mol, exceeding that of the N-H bond on the amine group (79.91 kcal/mol). Furthermore, the BDE for the C-N bond is 63.72 kcal/mol, marginally higher than that of the N-N bond, which is 62.28 kcal/mol. Based on the calculated BDEs of UDMH, it can be inferred that CH3 and NH2 group dissociation reactions (R1 and R2) dominantly contribute to the UDMH unimolecular decomposition. It should be noted that the BDEs of C-H and N-H bonds of UDMH are related to the efficiency of H-abstraction reactions, which will be discussed in more detail later.
$ \mathrm{UDMH=NH}_{ \mathrm{2}} \mathrm{NCH}_{ \mathrm{3}} \mathrm{+CH}_{ \mathrm{3}} $ (R1) $ \mathrm{UDMH=N(CH}_{ \mathrm{3}} \mathrm{)}_{ \mathrm{2}} \mathrm{+NH}_{ \mathrm{2}} $ (R2) $ \mathrm{UDMH=NHN(CH}_{ \mathrm{3}} \mathrm{)}_{ \mathrm{2}} \mathrm{+H} $ (R3) $ \mathrm{UDMH=NH}_{ \mathrm{2}} \mathrm{N(CH}_{ \mathrm{3}} \mathrm{)CH}_{ \mathrm{2}} \mathrm{+H} $ (R4) $ \mathrm{UDMH=N(CH}_{ \mathrm{3}} \mathrm{)CH}_{ \mathrm{2}} \mathrm{+NH}_{ \mathrm{3}} $ (R5) $ \mathrm{UDMH=NN(CH}_{ \mathrm{3}} \mathrm{)}_{ \mathrm{2}} \mathrm{+H}_{ \mathrm{2}} $ (R6) $ \mathrm{UDMH=HNNCH}_{ \mathrm{3}} \mathrm{+CH}_{ \mathrm{4}} $ (R7) In addition to the four bond dissociation reactions, there are three types of elimination reactions caused by intramolecular H-migration to form N(CH3)CH2 + NH3 (R5), NN(CH3)2 + H2 (R6), and HNNCH3 + CH4 (R7), with energy barriers of 70.7, 71.2, and 109.3 kcal/mol (Fig. 2). However, for the production of NN(CH3)2 + H2, and HNNCH3 + CH4, energy absorption of 40.2 and 3.6 kcal/mol are needed, while the reaction for the production of N(CH3)CH2 + NH3, releases 13.3 kcal/mol of energy. Among these seven unimolecular decomposition reactions, CH3 and NH2 group dissociation reactions have relatively low energy barriers.
Figure 3 presents the rate constants and branching ratios of seven unimolecular decomposition reactions of UDMH at HPL. It is obvious that the CH3 and NH2 group dissociation reactions (R1 and R2), have relatively higher rate constants than the other reactions. Consequently, UDMH decomposition is dominated by these two reactions, which is in agreement with the PES results. Furthermore, Fig. 3b depicts that the NH2 group dissociation reaction (R2) is dominant, while the contribution of the CH3 group dissociation reaction (R1) increases with temperature and reaches ~40% at 2,500 K. Furthermore, a comparison with the experimental data from Eberstein & Glassman[2], as shown in Fig. 3a, demonstrates excellent convergence with the calculated results. In addition, the rate constants for unimolecular decomposition reactions of UDMH at different pressures were calculated and presented in Supplementary Fig. S16. The rate constants of these seven reactions all exhibit a slight pressure dependence, especially R3, R4, R5, R6, and R7. As depicted in Supplementary Fig. S7, the hindered rotor treatment has less effect on the rate constants of the dominant unimolecular decomposition reactions.
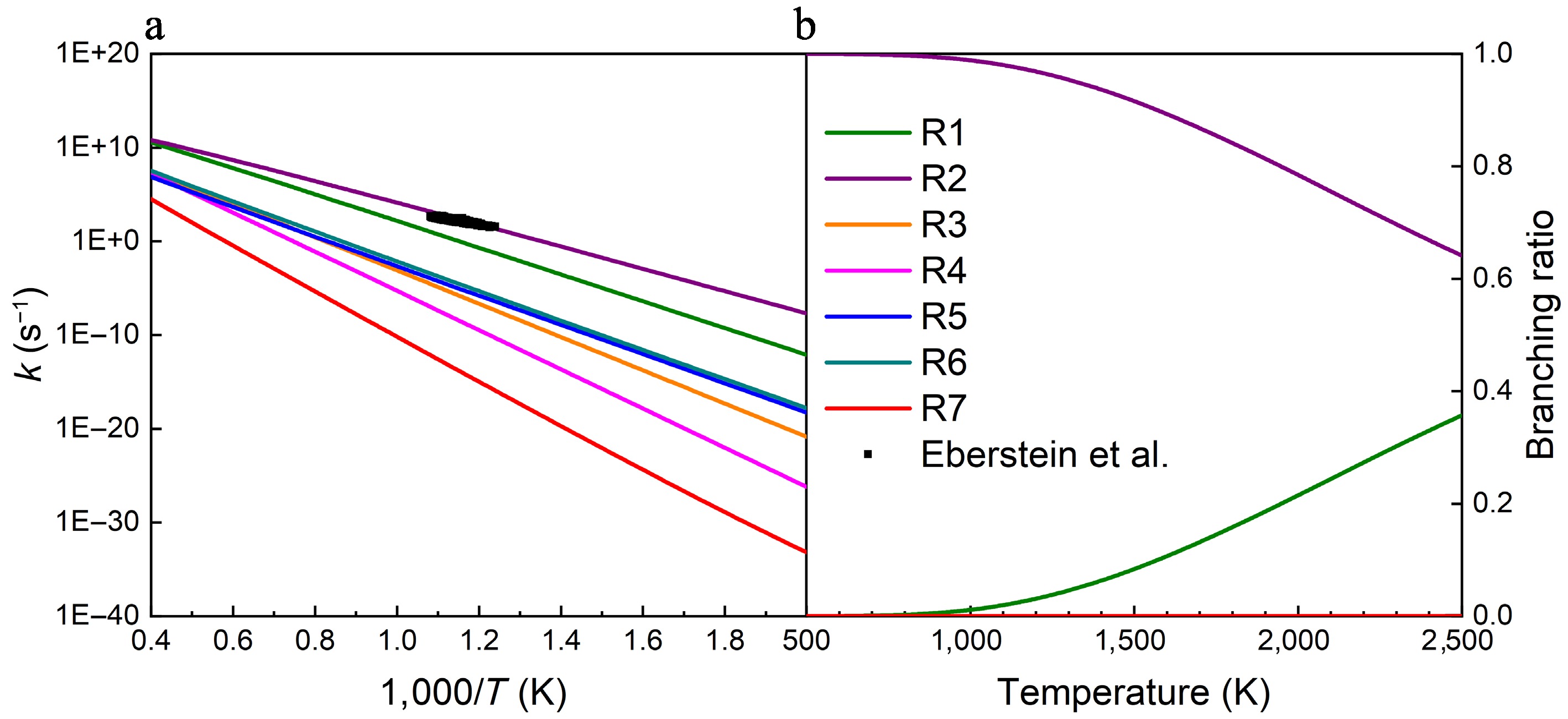
Figure 3.
(a) Rate constants, and (b) branching ratios for the unimolecular decomposition reactions of UDMH at HPL. The black squares represent the total rate constants for the UDMH decomposition reaction measured by Eberstein and Glassman at atmospheric pressure[2].
H-abstraction reactions of UDMH
NO2 attacking
-
The PESs for the H-abstraction reactions of UDMH by NO2 were calculated and the corresponding results are shown in Fig. 4. There are three kinds of H-abstraction sites, HNH2, Hcis-CH3, and Htrans-CH3 (Supplementary Fig. S17). It should be noted that differences in the CH3 group (cis-CH3 and trans-CH3), may influence the H-abstraction behavior, which will be discussed next. When NO2 attacks the amine group (HNH2) (Fig. 4a), there are three different reaction pathways. When the N atom of NO2 serves as the attacking atom, a product complex (PC) is produced through a transition state (TS), with a 9.7 kcal/mol energy barrier. Subsequently, an energy absorption of 12.9 kcal/mol is required in PC decomposition to produce NHN(CH3)2 and HNO2 radicals (R8). Notably, the products exhibit the energy of 1.1 kcal/mol, which is higher than that of the transition state. When the O atom of NO2 attacks HNH2 sites, there are two different reaction channels, NHN(CH3)2 + trans-HONO formation (R9), and NHN(CH3)2 + cis-HONO formation (R10). For these two reactions, the energy barriers stand at 9.9 and 6.0 kcal/mol, respectively. Then the PCs with relatively low energy of −9.6 and −8.2 kcal/mol, are formed. Finally, these PCs undergo decomposition reactions to form corresponding products.
$ \mathrm{UDMH+NO}_{ \mathrm{2}} \mathrm{=NHN(CH}_{ \mathrm{3}} \mathrm{)}_{ \mathrm{2}} \mathrm{+HNO}_{ \mathrm{2}} $ (R8) $ \mathrm{UDMH+NO}_{ \mathrm{2}} \mathrm{=NHN(CH}_{ \mathrm{3}} \mathrm{)}_{ \mathrm{2}} +{trans\text{-HONO}} $ (R9) $ \mathrm{UDMH+NO}_{ \mathrm{2}} \mathrm{=NHN(CH}_{ \mathrm{3}} \mathrm{)}_{ \mathrm{2}} +{cis\text{-HONO}} $ (R10) $ \mathrm{UDMH+NO}_{ \mathrm{2}} ={cis\text{-NH}}_{ \mathrm{2}} \mathrm{N(CH}_{ \mathrm{3}} \mathrm{)CH}_{ \mathrm{2}} \mathrm{+HNO}_{ \mathrm{2}} $ (R11) $ \mathrm{UDMH+NO}_{ \mathrm{2}}= {cis\text{-NH}}_{ \mathrm{2}} \mathrm{N(CH}_{ \mathrm{3}} \mathrm{)CH}_{ \mathrm{2}} +{trans\text{-HONO}} $ (R12) $ \mathrm{UDMH+NO}_{ \mathrm{2}}={cis \text{-NH}}_{ \mathrm{2}} \mathrm{N(CH}_{ \mathrm{3}} \mathrm{)CH}_{ \mathrm{2}} +{cis\text{-HONO}} $ (R13) $ \mathrm{UDMH+NO}_{ \mathrm{2}}={ trans\text{-NH}}_{ \mathrm{2}} \mathrm{N(CH}_{ \mathrm{3}} \mathrm{)CH}_{ \mathrm{2}} \mathrm{+HNO}_{ \mathrm{2}} $ (R14) $ \mathrm{UDMH+NO}_{ \mathrm{2}} ={trans\text{-NH}}_{ \mathrm{2}} \mathrm{N(CH}_{ \mathrm{3}} \mathrm{)CH}_{ \mathrm{2}} +{trans\text{-HONO}} $ (R15) $ \mathrm{UDMH+NO}_{ \mathrm{2}} ={trans\text{-NH}}_{ \mathrm{2}} \mathrm{N(CH}_{ \mathrm{3}} \mathrm{)CH}_{ \mathrm{2}}+ {cis\text{-HONO}} $ (R16) Furthermore, when NO2 attacks the H atom on the primary carbon sites (Hcis-CH3 and Htrans-CH3), the corresponding products after H-abstraction reactions are cis-H2NN(CH3)CH2 (Fig. 4b), and trans-H2NN(CH3)CH2 (Fig. 4c). Similar to the H-abstraction reaction on the HNH2 sites, when the N atom functions as the attacking atom, the products are cis-/trans-H2NN(CH3)CH2, and HNO2 (R11 and R14). The energies of products of these two reactions (22.8 and 23.3 kcal/mol), are both higher than the corresponding energy barriers (17.3 and 17.0 kcal/mol). When the O atom attacks primary carbon sites of UDMH, reactions R12 and R15 involving trans-HONO formation require transition states with energies of 19.8, and 27.7 kcal/mol. Notably, the products formed in R8, R11, and R14 possess higher energies than those of their transition states. A similar situation was also observed for the H-abstraction reaction at the primary carbon sites of MMH[18], suggesting that this is a specific multi-well characteristic of both UDMH, and MMH. This behavior might be derived from the strong vdW interaction in their product complexes. To better characterize the interactions, an iso-surface map analysis, based on the Interaction Region Indicator (IRI) function was performed (Supplementary Fig. S18). It can be found that a covalent bond-like interaction between the H and the N atom introduces strong stabilizing interactions within these vdW complexes, except for the hydrogen bonding effect, which leads to a lower overall energy.
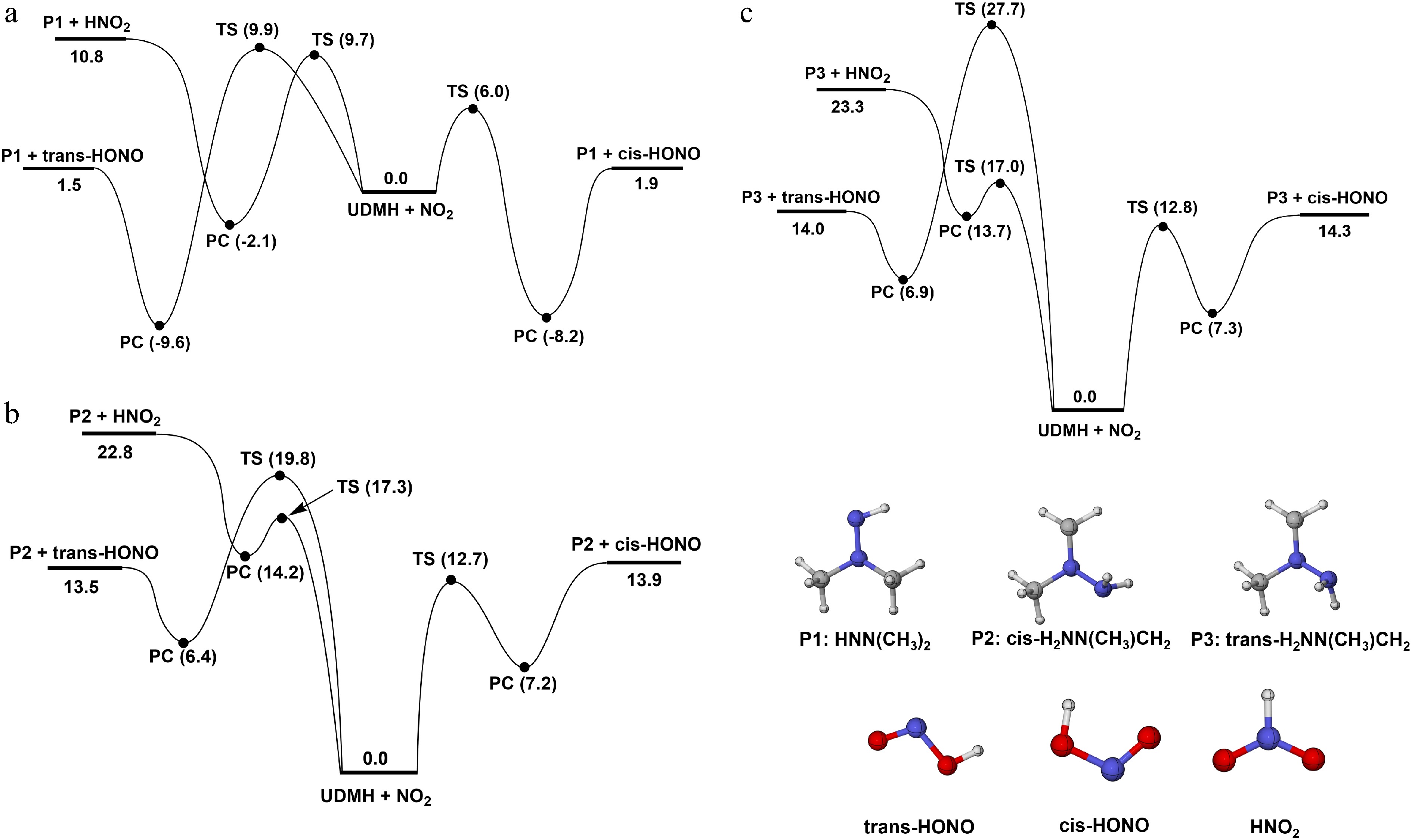
Figure 4.
PESs for the H-abstraction of UDMH on (a) HNH2, (b) Hcis-CH3, and (c) Htrans-CH3 sites, with NO2 attacking at the CCSD(T)/CBS//M062X/def2-TZVP level (unit: kcal/mol).
In this work, these reactions were treated as multi-well reactions in kinetic calculations. With energy barriers of 12.7 and 12.8 kcal/mol, reactions R13 and R16 lead to cis-HONO formation exhibit significantly lower values than R12 and R15, indicating that attacking direction plays a considerable role. From an overall perspective, the H-abstraction reactions at the primary carbon site have higher energy barriers than those at the amine group. This trend agrees with the BDE trend of C-H and N-H bonds.
Figures 5−7 depict the calculated rate constants for the H-abstraction reactions of UDMH with NO2 attacking at 100 atm based on the PES results. When NO2 attacks the HNH2 sites of the amine group, the rate constant of the reaction R10 is higher than those of reactions R8 and R9, owing to its energy barrier advantage. As presented in Fig. 5b, the contributions of R8 and R9 generally rise with temperature, while R10 is the major contributor across the entire temperature range investigated. Specifically, the contribution of R8 reaches 30%~40%, and that of R10 decreases to 60%~70% at temperatures above 1,000 K. When NO2 attacks the primary carbon sites (Hcis-CH3, and Htrans-CH3), the results of rate constants and branching ratios show similar trends to those when NO2 attacks the HNH2 sites (Figs 6, 7). That is, the reactions R13 and R16 have larger rate constants and branching ratios, playing dominant roles when H-abstraction reactions occur on the primary carbon sites, which are caused by the lower energy barriers. Additionally, no pressure dependence was observed for reactions R9−R16, whereas R8 exhibited only a weak dependence at low temperatures (Supplementary Fig. S19).
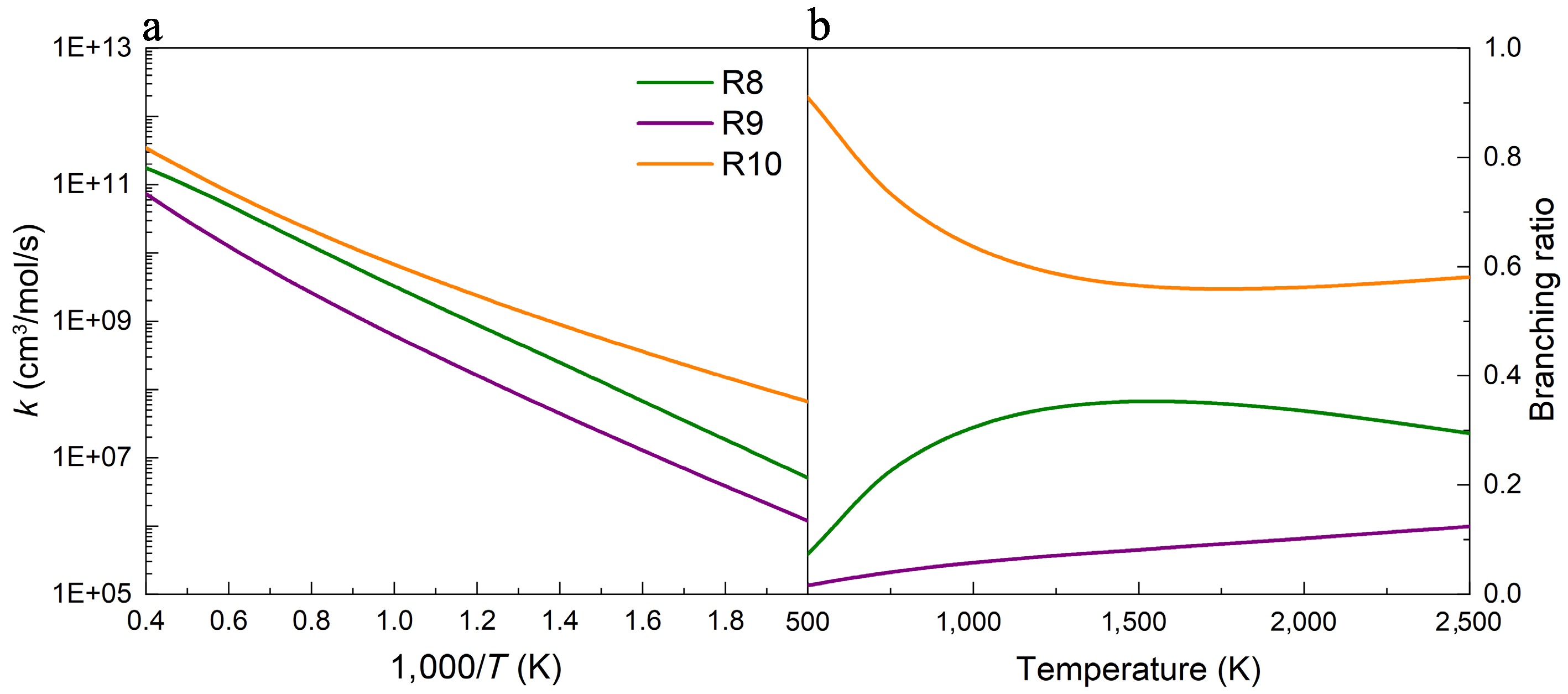
Figure 5.
(a) Rate constants, and (b) branching ratios for the H-abstraction reactions of UDMH on the HNH2 sites, with NO2 attacking at 100 atm.
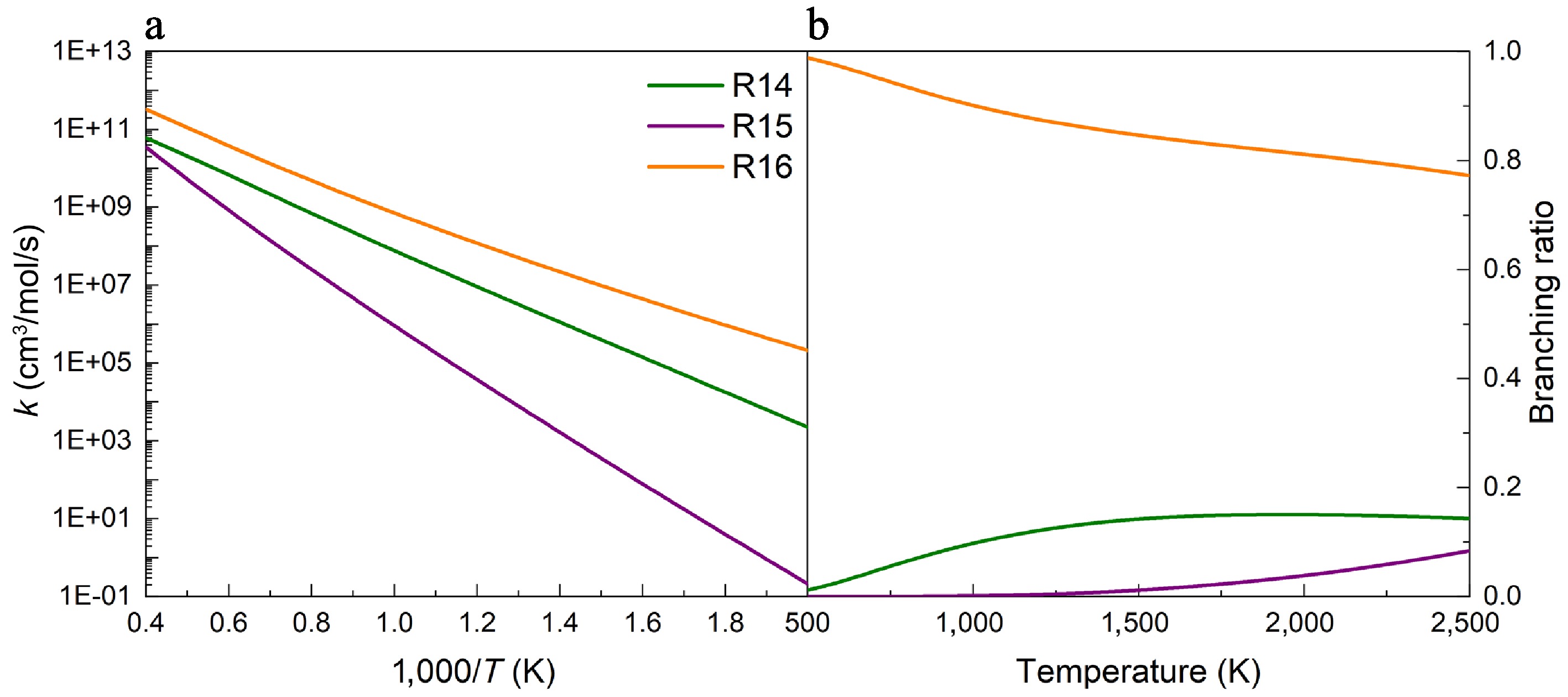
Figure 7.
(a) Rate constants, and (b) branching ratios for the H-abstraction reactions of UDMH on the Htrans-CH3 sites, with NO2 attacking at 100 atm.
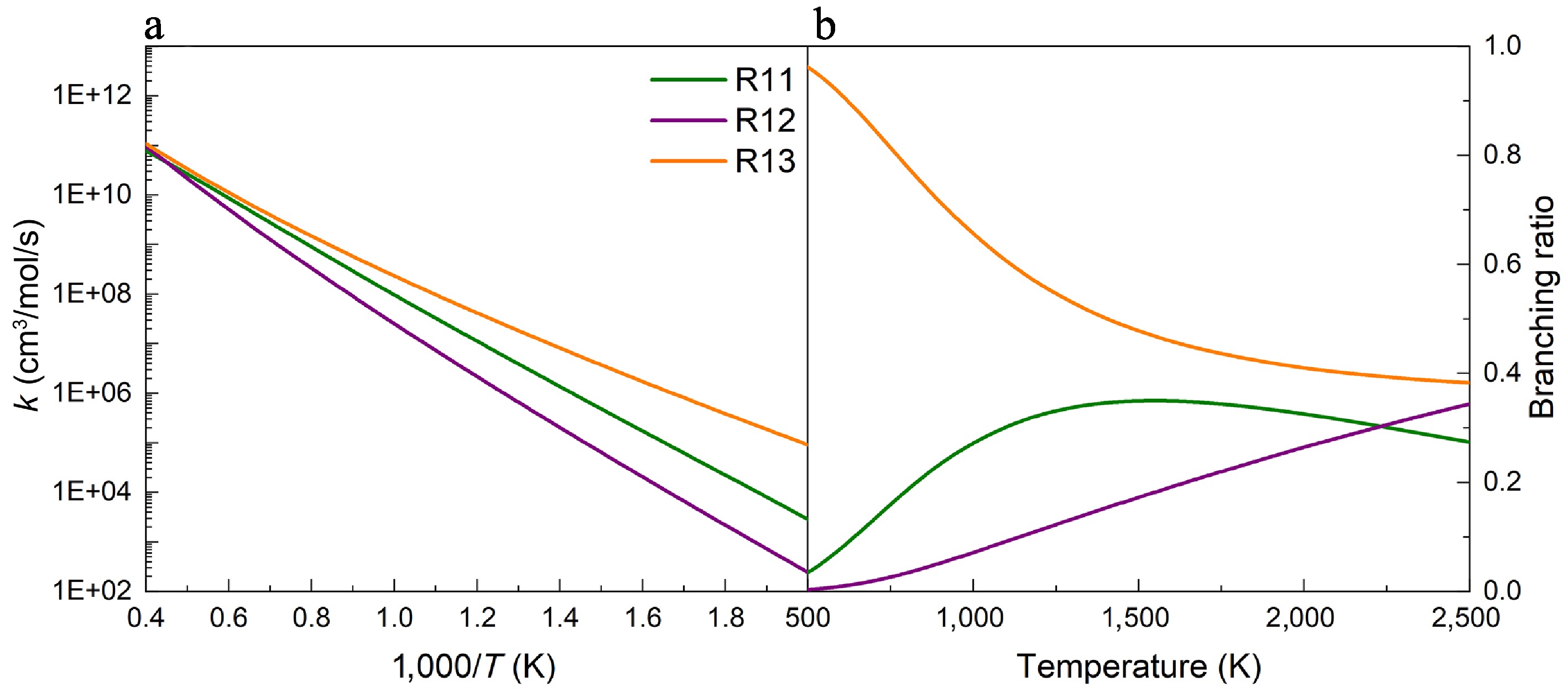
Figure 6.
(a) Rate constants, and (b) branching ratios for the H-abstraction reactions of UDMH on the Hcis-CH3 sites, with NO2 attacking at 100 atm.
Meanwhile, the rate constants for the H-abstraction reactions of UDMH on different H atom sites (HNH2, Hcis-CH3, and Htrans-CH3), with NO2 attacking at 100 atm were compared (Supplementary Fig. S20). It is obvious that, regardless of whether the attacking atom is the O or N atom in NO2, when the attack occurs at the HNH2 site, the corresponding H-abstraction reactions have much higher rate constants than those at the Hcis-CH3 and Htrans-CH3 sites. As shown in Supplementary Fig. S8, the hindered rotor treatment has less impact on the rate constants for H-abstraction reactions at primary carbon sites with NO2 and H attacking, whereas its effect on H-abstraction reactions at the amine group is relatively evident. For the tunneling treatment, its influence on the H-abstraction reactions with NO2 attacking can be neglected.
H attacking
-
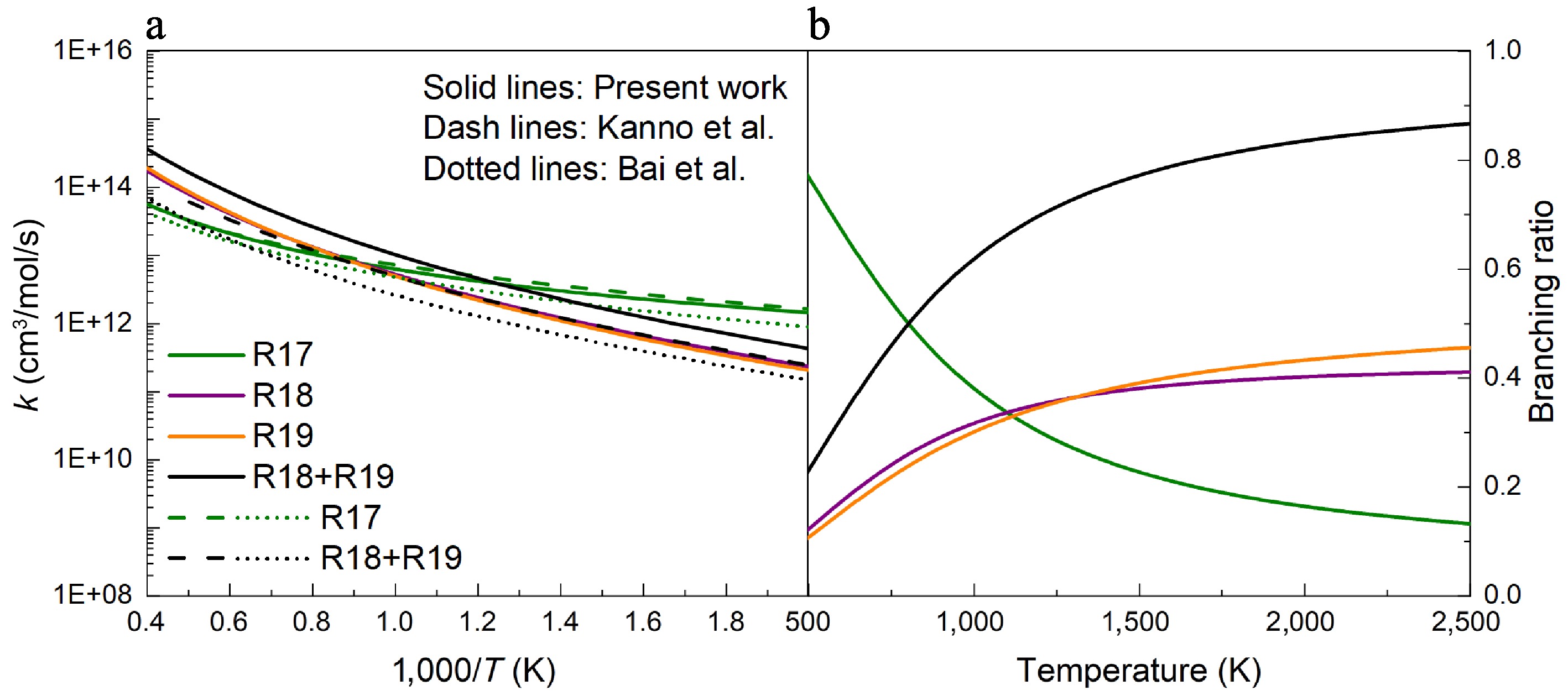
Apart from the H-abstraction reactions with NO2, the H-abstraction reactions of UDMH involving common species such as H and CH3, were calculated. As shown in Supplementary Fig. S17, there are three types of H atoms: Hinside, Houtside, and Hplane. Hydrogen atoms in different chemical environments are chemically distinct, even within the same group. The calculations reveal that the energy barriers for H-abstraction at the Hinside atom are significantly lower than those at the Houtside, and Hplane atoms (Supplementary Table S4). Consequently, the rate constants for abstractions involving the Hinside atom are higher than those involving the Houtside, and Hplane atoms (Supplementary Fig. S21). In this work, the total rate constants for each site were calculated as k(NH2) = k(Hinside) + k(Houtside), and k(CH3) = k(Hinside) + k(Houtside) + k(Hplane). A comparison of the energy barriers for H-abstraction reactions at Hinside atoms of different groups was performed. Figure 8 demonstrates that the energy barrier of H-abstraction reactions at the amine group is 1.9 kcal/mol, and releases 23.9 kcal/mol of heat to produce HNN(CH3)2, and an H2 product pair. When an H atom attacks the primary carbon sites (Hcis-CH3 and Htrans-CH3), these two H-abstraction reactions exhibit close energy barriers, i.e., 5.2 and 5.4 kcal/mol, which are higher than those of the amine group. The slight PES difference between different primary carbon sites suggests that the molecular conformer effects on H-abstraction reactions with the H atom attacking are negligible. Based on the calculated PES, it should be noted that the energy barrier trend for the H-abstraction reaction by H atoms is consistent with the BDE trends. As illustrated in Fig. 9a, the rate constant of R17 intersects with those of R18 and R19 at about 1,100 K. At low temperatures, the rate constant of R17 exceeds those of R18 and R19, whereas at high temperatures, it falls below them. Meanwhile, the calculations in this work were compared with previous results presented by Kanno et al.[24] and Bai et al.[25]. Both the magnitude and trend of the rate constants for the H-abstraction reactions at the NH2 sites are generally consistent with previous studies. In contrast, our calculated results for the reactions involving the CH3 sites are higher than the previous results. Correspondingly, Fig. 9b depicts that R17 exhibits a higher branching ratio at low temperatures, whereas R18 and R19 dominate at higher temperatures. UDMH prefers H-abstraction reactions to occur at primary carbon sites.
$ \mathrm{UDMH+H=NHN(CH}_{ \mathrm{3}} \mathrm{)}_{ \mathrm{2}} \mathrm{+H}_{ \mathrm{2}} $ (R17) $ {\mathrm{UDMH}}+{\mathrm{H}}={cis\text{-NH}}_{ \mathrm{2}} \mathrm{N(CH}_{ \mathrm{3}} \mathrm{)CH}_{ \mathrm{2}} +{\mathrm{H}}_{ \mathrm{2}} $ (R18) $ {\mathrm{UDMH}}+{\mathrm{H}}={trans\text{-NH}}_{ \mathrm{2}} \mathrm{N(CH}_{ \mathrm{3}} \mathrm{)CH}_{ \mathrm{2}} \mathrm{+H}_{ \mathrm{2}} $ (R19) 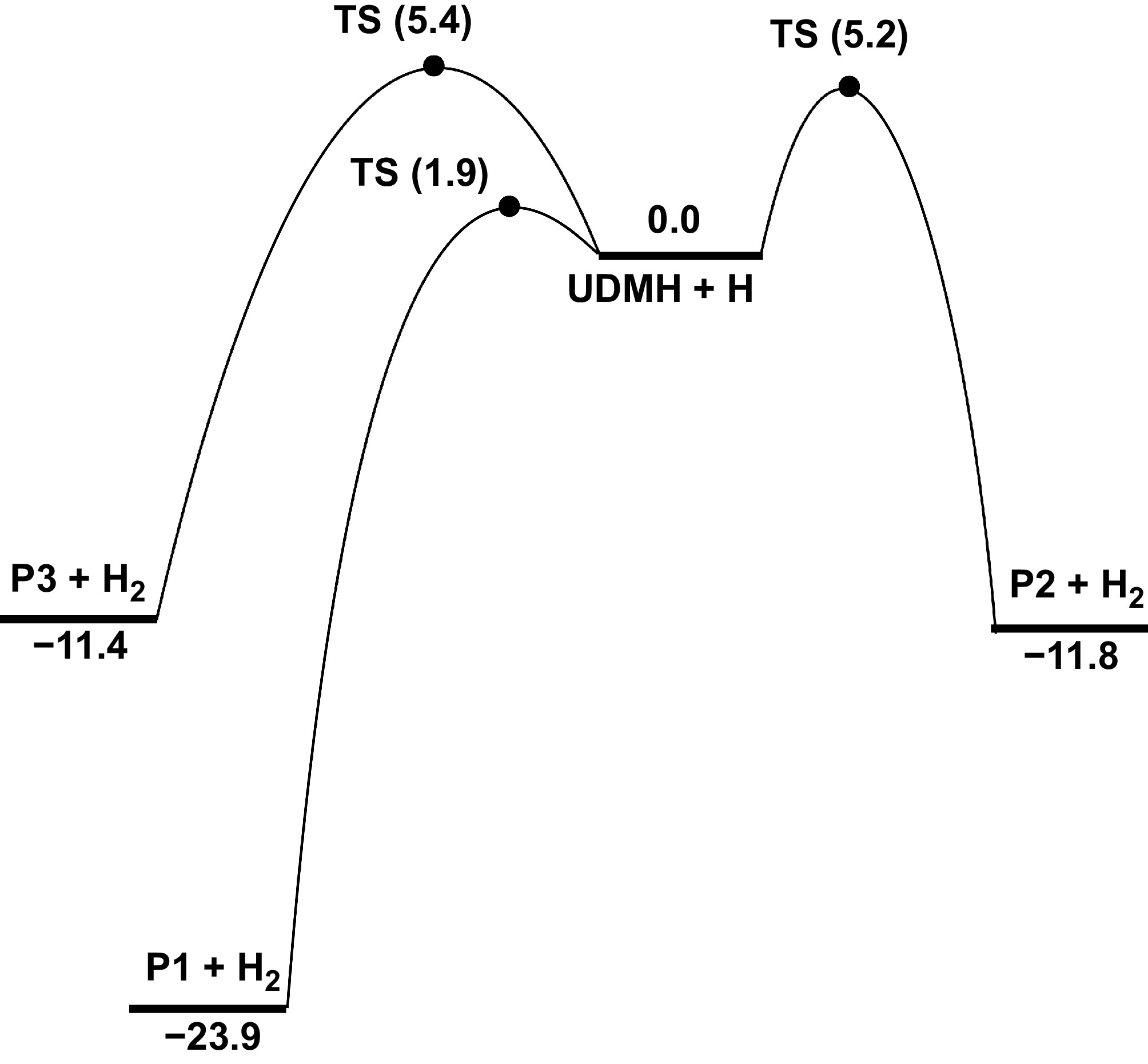
Figure 8.
PESs for the H-abstraction at Hinside atoms of UDMH with H attacking at CCSD(T)/CBS//M062X/def2-TZVP level (unit: kcal/mol).
CH3 attacking
-
From the above analysis of UDMH unimolecular decomposition, it is prone to producing CH3 through N-C bond dissociation. Figure 10 shows the PESs for the H-abstraction reactions at Hinside atoms of UDMH by CH3. When the CH3 radical attacks the HNH2 sites, the energy barrier is 6.4 kcal/mol, and releases 23.0 kcal/mol of heat to produce the HNN(CH3)2, and CH4 pair. For the primary carbon sites (Hcis-CH3 and Htrans-CH3), the energy barriers of H-abstraction reactions by CH3 are also close (11.1 and 11.2 kcal/mol), and higher than that of the H-abstraction reaction at the HNH2 sites, which is similar to the situation of H-abstraction reaction by the H atom. Meanwhile, R20 exhibits a higher rate constant and a larger branching ratio at low temperatures, whereas the rate constants of R21 and R22 surpass that of R20 at high temperatures, and become dominant (Fig. 11). A similar trend is identified for the H-abstraction reactions initiated by H atoms. Compared with the previous results from Bai et al.[25], the results involving primary carbon sites are slightly lower, and our results for R20 are approximately four times lower than those in previous work. In the lower temperature range, Supplementary Figs S9, S10 show that the tunneling treatment exerts a pronounced influence on the rate constants of H-abstraction reactions with H and CH3 attacking, in contrast to the H-abstraction reactions by NO2. For the hindered rotor treatment, the effects on the H-abstraction reactions targeting the NH2 group are relatively pronounced compared to CH3 groups.
$ \mathrm{UDMH+CH}_{ \mathrm{3}} \mathrm{=NHN(CH}_{ \mathrm{3}} \mathrm{)}_{ \mathrm{2}} \mathrm{+CH}_{ \mathrm{4}} $ (R20) $ \mathrm{UDMH+CH}_{ \mathrm{3}}={cis \text{-NH}}_{ \mathrm{2}} \mathrm{N(CH}_{ \mathrm{3}} \mathrm{)CH}_{ \mathrm{2}} \mathrm{+CH}_{ \mathrm{4}} $ (R21) $ \mathrm{UDMH+CH}_{ \mathrm{3}}= {trans\text{-NH}}_{ \mathrm{2}} \mathrm{N(CH}_{ \mathrm{3}} \mathrm{)CH}_{ \mathrm{2}} \mathrm{+CH}_{ \mathrm{4}} $ (R22) 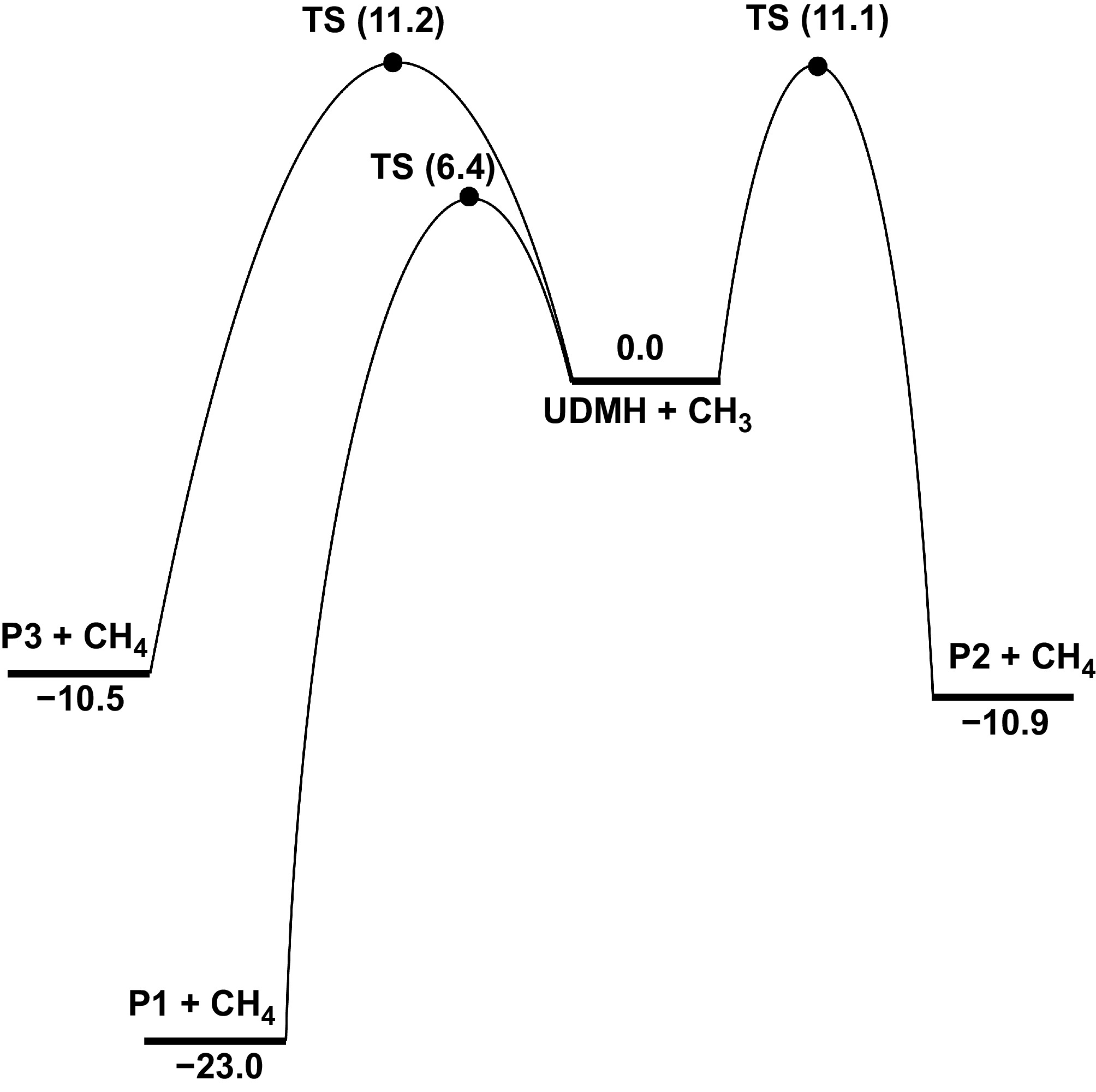
Figure 10.
PES for the H-abstraction at Hinside atoms of UDMH, with CH3 attacking at CCSD(T)/CBS//M062X/def2-TZVP level (unit: kcal/mol).
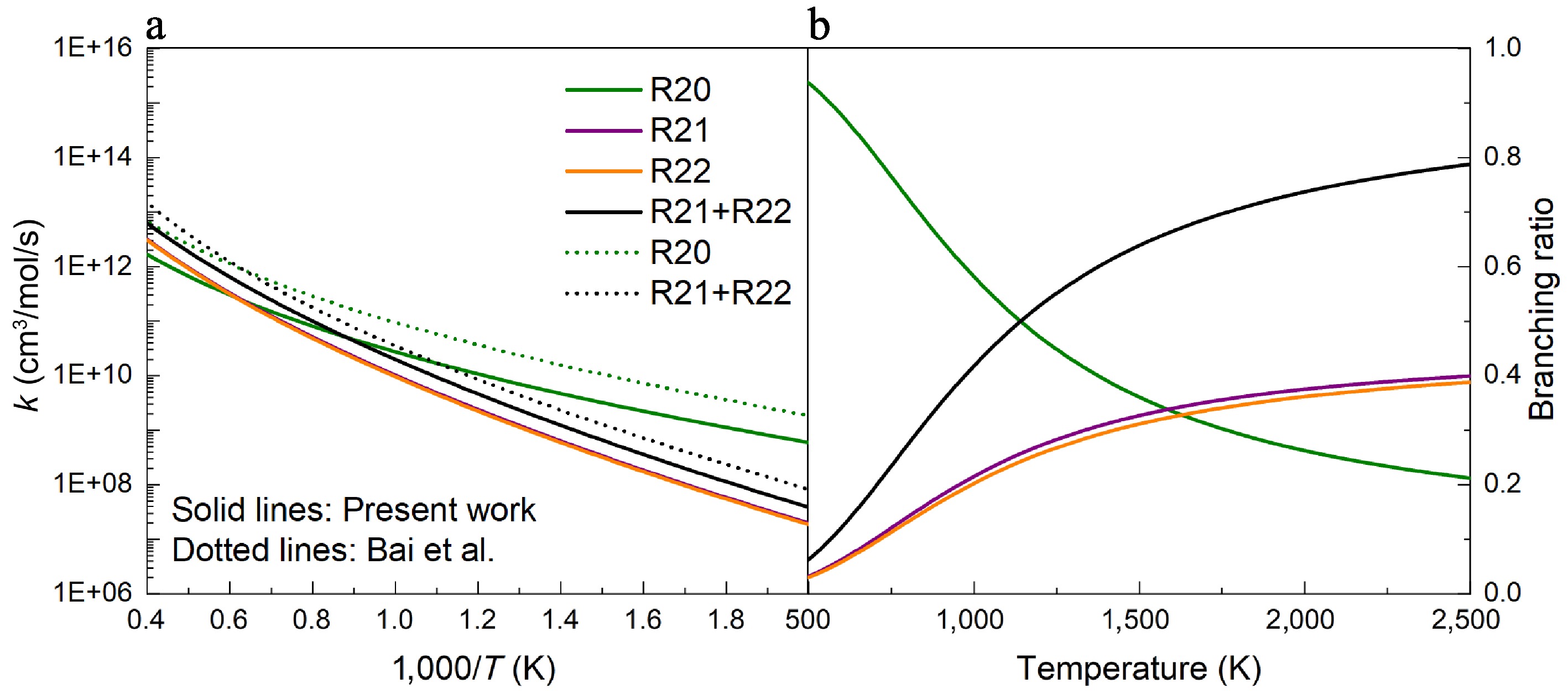
Figure 11.
(a) Rate constants, and (b) branching ratios for the H-abstraction reactions of UDMH with CH3 attacking at HPL calculated in this work and by Bai et al.[25].
Comparison between the H-abstraction reactions of UDMH and CH3NHNH2
-
CH3NHNH2 and UDMH are both hydrazine-based fuels containing N-N bonds and CH3 groups, and their structural similarity suggests the possibility of analogous H-abstraction reactions. Accordingly, a comparative analysis of rate constants considering molecular configuration and degeneracy is conducted in this work. In addition, since only theoretical calculations on the H-abstraction reactions of CH3NHNH2 with NO2 and H have been reported by the existing literature[18], this further comparison focuses on H-abstraction reactions involving these two species, excluding those induced by CH3 radical.
NO2 attacking
-
A comparative analysis was conducted between the rate constants for H-abstraction reactions of UDMH with NO2 attacking calculated in this work, and those of CH3NHNH2 previously reported[18], as illustrated in Figs 12 and 13. When NO2 attacks the amine group with the N atom serving as the attacking atom, the rate constants of H-abstraction reactions of CH3NHNH2 are nearly one order larger than those of UDMH (Fig. 12a), especially at 300 K and 2,500 K. However, if the O atom serves as the attacking species, the rate constants of H-abstraction reactions of CH3NHNH2 are on a comparable level with those of UDMH, when the product includes trans-HONO, and two rate constant curves intersect at around 500 K (Fig. 12b). When the product includes cis-HONO, the H-abstraction reaction of CH3NHNH2 shows a lower rate constant, and the difference between the two curves becomes larger at lower temperatures (Fig. 12c).
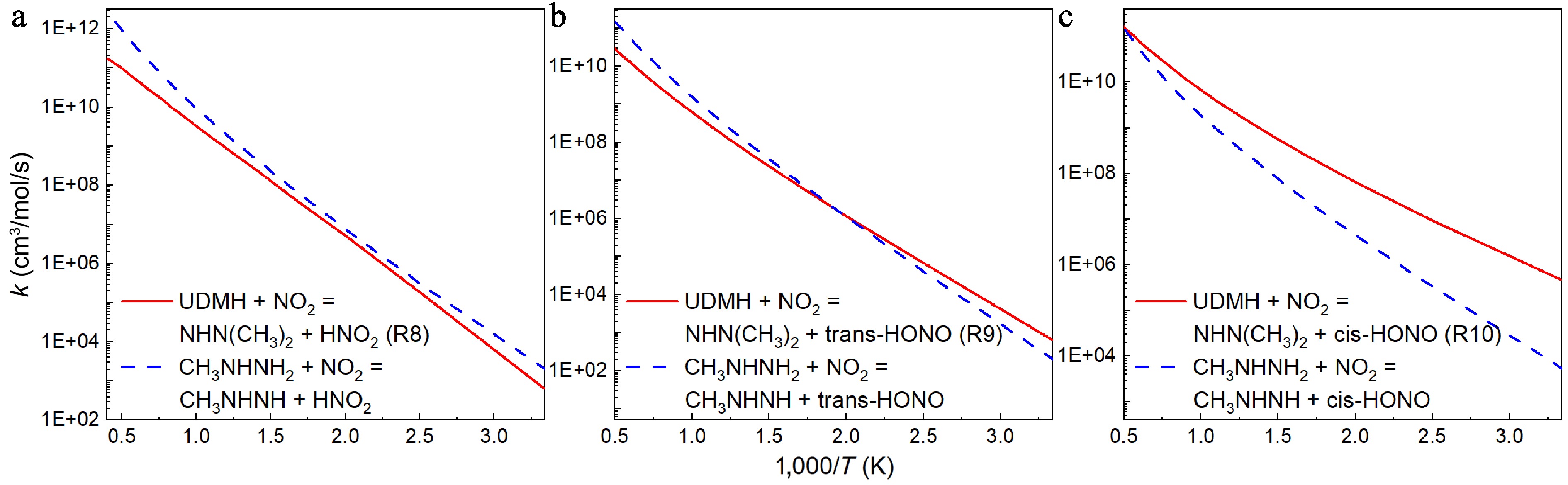
Figure 12.
Comparison between the rate constants for the H abstraction reactions at the NH2 group of UDMH ([a] R8, [b] R9, and [c] R10) and CH3NHNH2[18] with NO2 attacking.
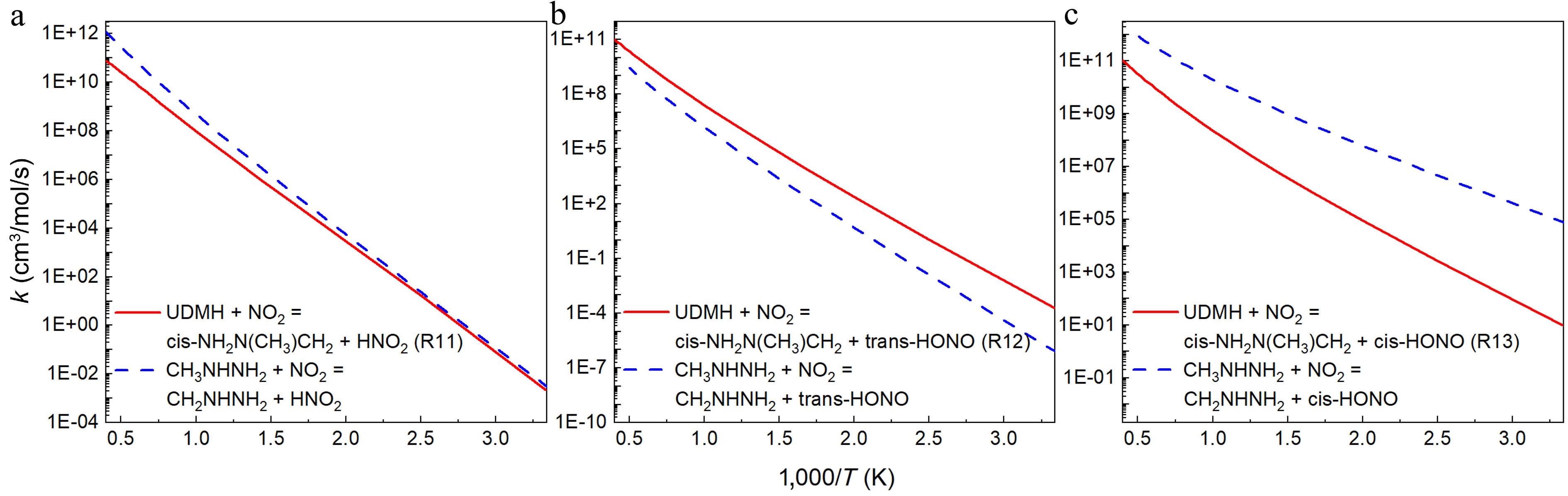
Figure 13.
Comparison between the rate constants for the H abstraction reactions at per CH3 group of UDMH ([a] R11, [b] R12, and [c] R13) and CH3NHNH2[18] with NO2 attacking.
When NO2 attacks the primary carbon sites with the N atom as the attacking atom (Fig. 13a), the rate constants for H-abstraction reactions of UDMH are a little lower than those of CH3NHNH2, and these two reactions present similar PES characteristics, as illustrated in Fig. 4b, and in the previous research, calculated by Ren et al.[18]. When the O atom plays the role of the attacking agent, the reactions produced trans-HONO (Fig. 13b), and the reactions that produced cis-HONO (Fig. 13c) exhibited an inverse numerical relationship between UDMH and CH3NHNH2. When the products include trans-HONO, the H-abstraction reaction of UDMH has a higher rate constant than that of CH3NHNH2. This is mainly caused by a higher energy barrier of H-abstraction reaction of CH3NHNH2 (CH3NHNH2: 22.26 kcal/mol[18], UDMH: 19.8 kcal/mol). In contrast, when the products include cis-HONO, the H-abstraction reaction of UDMH has a lower rate constant than that of CH3NHNH2, and the rate constant difference can differ by as much as five orders of magnitude at 300 K. This is the result of the dominant multi-well reaction pathway of UDMH, although the reaction energy barrier of CH3NHNH2 is slightly higher (CH3NHNH2: 15.33 kcal/mol[18], UDMH: 14.2 kcal/mol). Furthermore, the rate constants of three reactions involving CH3NHNH2 in Fig. 13 are compared in Supplementary Fig. S22. Three reactions exhibit the following rate constant order: CH3NHNH2 + NO2 = CH2NHNH2 + cis-HONO > CH3NHNH2 + NO2 = CH2NHNH2 + HNO2 > CH3NHNH2 + NO2 = CH2NHNH2 + trans-HONO, which is consistent with the order of R13 > R11 > R12 for UDMH.
H attacking
-
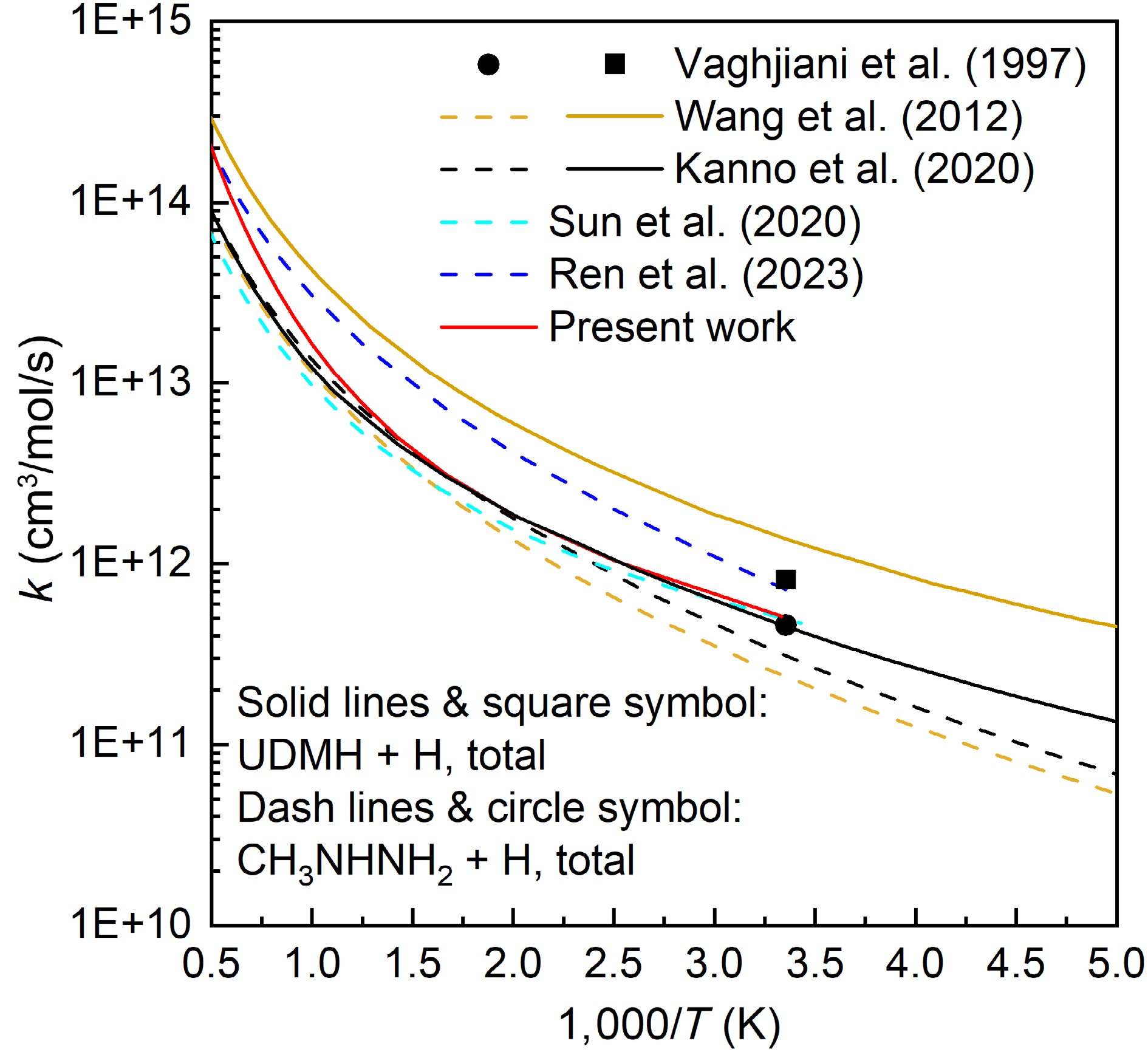
A comparative analysis was also carried out between the rate constants for the H-abstraction reactions of UDMH with H attacking calculated in this work, and those of CH3NHNH2 with H attacking, in previous reports, as illustrated in Fig. 14. Previous results from Ren et al.[18], Sun et al.[17], and Kanno et al.[24] demonstrated that the rate constants for CH3NHNH2 + H = CH3NHNH + H2, and CH3NHNH2 + H = CH2NHNH2 + H2, exhibited a consistent decreasing trend with temperature. For H abstraction from the amine group (HNH2), the rate constants are lower than those of CH3NHNH2 across the entire temperature range, and exhibit a shallower slope. This may be attributed to the structural differences between UDMH and CH3NHNH2. When H atoms attack the primary carbon sites, the rate constants for the H-abstraction reactions of UDMH are similar in numerical value to that for CH3NHNH2, calculated by Sun et al.[17]. However, notable discrepancies were observed between the calculated results of these three studies. That is, the calculations of Ren et al.[18], and Sun et al.[17] for CH3NHNH2 are lower than those of UDMH, while the results of Kanno et al.[24] are higher.
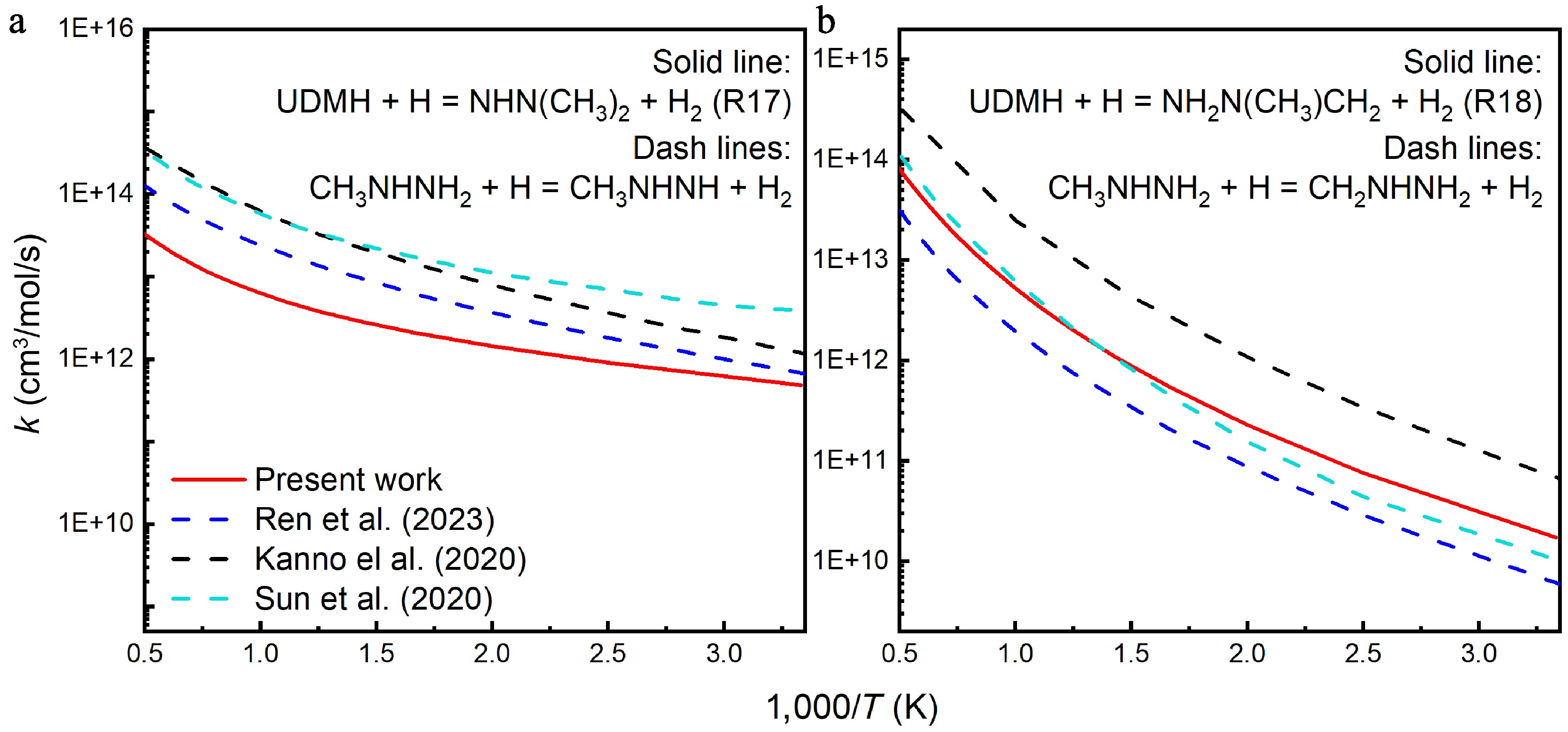
This work also compares the total rate constants of the UDMH + H reaction with those of the CH3NHNH2 + H reaction (Fig. 15). The rate constants of the UDMH + H reaction calculated herein, approach the results of Kanno et al.[24], and are slightly lower than the experimental data at 298 K measured by Vaghjiani et al[3]. For the CH3NHNH2 + H reaction, the rate constant calculations of Wang et al.[13], Kanno et al.[24], and Sun et al.[17] are close to each other, with the results of Sun et al.[17] at 298 K being the most consistent with the experimental result obtained by Vaghjiani et al[3]. In contrast, the results of Ren et al.[18] are higher than those of all the aforementioned studies. Overall, the calculated rate constants for the UDMH + H reaction tend to be higher than those for the CH3NHNH2 + H reaction.
-
In this work, the unimolecular decomposition and H-abstraction reactions of UDMH were systematically and theoretically investigated. The PESs of these reactions were characterized, and the corresponding rate constants covering various pressures, including HPL over 300~2,500 K were determined. Principal conclusions are summarized as follows:
(1) For UDMH, the unimolecular decomposition is dominated by the dissociation reactions on the NH2 and NH3 groups, owing to their relatively low energy barriers among the bond dissociation channels. The elimination reactions make less contribution to UDMH because of their high energy barriers. Furthermore, between these two reactions, the NH2 group dissociation reaction plays the most important role at low- and mid- temperature ranges, while the CH3 group dissociation reaction becomes more significant at higher temperatures.
(2) For H-abstraction reactions of UDMH, when NO2 serves as the attacking subject, the H-abstraction reactions at the primary carbon sites exhibit higher energy barriers than those at the amine group, and the reactions involving cis-HONO formation are easier to occur. When H or CH3 functions as the attacking subject, the H-abstraction reactions at the amine group are more significant at low temperatures, while those at the primary carbon sites are dominant at high temperatures. Totally, the trends in energy barriers for H-abstraction by NO2, H, and CH3 all correlate with the trends in BDE's of C-H and N-H bonds of UDMH.
(3) A comparison is made between the H-abstraction reactions of UDMH and CH3NHNH2, initiated by NO2 and H radicals. The results show that, due to differences in reaction energy barriers and molecular structures, the rate constants of various types of H-abstraction reactions differ between UDMH and CH3NHNH2.
-
The authors confirm their contributions to the paper as follows: formal analysis, investigation, writing − original draft preparation: Zhang Y, Fang Q; investigation: Han S, Fang J; conceptualization, revised manuscript: Li W. All authors reviewed the results and approved the final version of the manuscript.
-
All theoretically computed and analytically derived data supporting this study is contained within this article.
-
Not applicable.
-
The authors declare that they have no conflict of interest.
-
Authors contributed equally: Yi Zhang, Qilong Fang
- Supplementary Materials MESS
- Supplementary Table S1 Diagnostic values of key species at the CCSD(T)/cc-pVTZ level and zero-point energy without correction.
- Supplementary Table S2 Pressure-dependent rate constants for the unimolecular decomposition reactions of UDMH.
- Supplementary Table S3 High-pressure-limit rate constants for the H-abstraction reactions of UDMH.
- Supplementary Table S4 Relative energy for the H-abstraction reactions at different H atoms of HNH2, Hcis-CH3 and Htrans-CH3 sites of UDMH by H and CH3 at CCSD(T)/CBS//M062X/def2-TZVP level (unit: kcal/mol).
- Supplementary Fig. S1 IRC results for the elimination reactions of UDMH.
- Supplementary Fig. S2 IRC results for the H-abstraction reactions of UDMH with NO2 attacking.
- Supplementary Fig. S3 IRC results for the H-abstraction reactions of UDMH with H attacking.
- Supplementary Fig. S4 IRC results for the H-abstraction reactions of UDMH with CH3 attacking.
- Supplementary Fig. S5 Calculated Boltzmann distributions of A and B conformers over 300-2500 K.
- Supplementary Fig. S6 The calculated MEP results for the unimolecular bond dissociation reactions of UDMH.
- Supplementary Fig. S7 Effects of hindered rotor treatment on the rate constants for R1 and R2.
- Supplementary Fig. S8 Effects of tunnelling treatment and hindered rotor treatment on the rate constants for R8-R16.
- Supplementary Fig. S9 Effects of tunnelling treatment and hindered rotor treatment on the rate constants for the H-abstraction reactions involving Hinside atoms by H.
- Supplementary Fig. S10 Effects of tunnelling treatment and hindered rotor treatment on the rate constants for the H-abstraction reactions involving Hinside atoms by CH3.
- Supplementary Fig. S11 The influence of the harmonic frequency scale factor on the calculated rate constants of R10, R13, R16.
- Supplementary Fig. S12 The influence of the harmonic frequency scale factor on the calculated rate constants for the H-abstraction reactions involving Hinside atoms by H.
- Supplementary Fig. S13 The influence of the harmonic frequency scale factor on the calculated rate constants for the H-abstraction reactions involving Hinside atoms by CH3.
- Supplementary Fig. S14 The influence of barrier heights at 300-2500 K and 100 atm. The barrier heights were changed by 1 kcal/mol for the uncertainty analysis.
- Supplementary Fig. S15 The influence of collisional parameters at 300-2500 K and 0.1-10 atm. σ was changed by 10% for the uncertainty analysis.
- Supplementary Fig. S16 Pressure-dependent rate constants of the unimolecular decomposition reactions of UDMH.
- Supplementary Fig. S17 H-atom abstraction sites in UDMH.
- Supplementary Fig. S18 Iso-surface map of the product complex (PC) structure.
- Supplementary Fig. S19 Pressure-dependent rate constants of the H-abstraction reactions of UDMH with NO2 attacking.
- Supplementary Fig. S20 High-pressure-limit rate constants for the H-abstraction reactions of UDMH with NO2 attacking.
- Supplementary Fig. S21 Rate constants for the H-abstraction reactions at Hinside, Houtside, and Hplane atoms of UDMH by H and CH3.
- Supplementary Fig. S22 High-pressure-limit rate constants for the H-abstraction reactions of CH3NHNH2 on the CH3 group with NO2 attacking.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang Y, Fang Q, Han S, Fang J, Li W. 2026. Ab initio kinetic study on the unimolecular decomposition and H-abstraction reactions of unsym-dimethylhydrazine. Progress in Reaction Kinetics and Mechanism 51: e007 doi: 10.48130/prkm-0026-0001 

Ab initio kinetic study on the unimolecular decomposition and H-abstraction reactions of unsym-dimethylhydrazine
- Received: 01 July 2025
- Revised: 13 December 2025
- Accepted: 16 January 2026
- Published online: 30 March 2026
Abstract: The unsym-dimethylhydrazine ((CH3)2NNH2,UDMH)/nitrogen tetroxide (N2O4) combination is a widely used high-energy propellant in liquid propulsion systems, and understanding its combustion kinetics is essential to optimize ignition and combustion properties. In this work, the unimolecular decomposition and H-abstraction reactions of UDMH were comprehensively and theoretically investigated. Their potential energy surfaces (PESs) were calculated at the CCSD(T)/CBS//M06-2X/def2-TZVP level of theory. The minimum energy path (MEP) of barrierless bond dissociation reactions was obtained using the multi-reference method of MRCISD/CBS//CASPT2/cc-pVDZ. Pressure-dependent rate constants at 300−2,500 K were obtained by RRKM/Master Equation calculations. For the unimolecular decomposition of UDMH, the dissociation energies of the NH2 and CH3 groups are relatively low among the bond dissociation channels, resulting in NH2 and CH3 group dissociation reactions dominating the unimolecular decomposition of UDMH, while the elimination reactions have less contribution caused by their high energy barriers. For the H-abstractions by NO2, reactions involving cis-HONO formation occur more easily at all three sites of UDMH. Additionally, the H-abstraction reactions at the NH2 group possess the greatest rate constants because of lower energy barriers. Both the H-abstraction reactions initiated by H atoms and CH3 radicals at the amine group, play important roles under low temperature conditions, and those at the primary carbon sites are dominant at high temperatures. Additionally, the H-abstraction reactions of UDMH and monomethylhydrazine initiated by NO2 and H, are compared. The detailed kinetic parameters of unimolecular decomposition, and H-abstraction reactions of UDMH obtained in this work aid model development, and enhance the understanding of its combustion chemistry.