








-
In the grand landscape of modern drug research and development, natural products (NPs) have always been a core source of innovative drug molecules. According to statistics, nearly half of the drugs approved in the past 40 years directly or indirectly originate from NPs[1]. As a distinguished repository of such natural resources, Traditional Chinese Medicine (TCM) provides rich chemical diversity and a long history of clinical application. However, the modernization and internationalization of TCM has long faced the dual challenges of an ambiguous pharmacological basis and obscure mechanisms of action. TCM formulas or isolated active components often exhibit pharmacological characteristics of 'multiple components, multiple targets, and low affinity', making target deconvolution a key bridge connecting traditional medicine with modern molecular medicine and a necessary path for drugs to move from empirical application to precision therapy[2].
In the early stages of drug research and development, identifying the target proteins of active molecules not only helps in understanding efficacy and off-target effects but also provides a prerequisite for guiding the structure-activity relationship (SAR) optimization of lead compounds. However, NPs have complex structures, diverse targets, and weak or moderate binding affinity, creating substantial challenges for traditional target identification techniques[3,4]. Specifically, for active molecules that function through weak interactions or transient binding, conventional methods often fail to capture them, leaving the mechanism of action for many TCM active ingredients as a 'black box'.
Methodological limitations of traditional target identification techniques
-
Before the emergence of targeted protein degradation (TPD)-based chemoproteomic strategies, target identification of bioactive NPs relied primarily on classical chemical proteomics and label-free biophysical approaches, including affinity chromatography, activity-based protein profiling (ABPP), and stability-based methods such as cellular thermal shift assay (CETSA), thermal proteome profiling (TPP), and drug affinity responsive target stability (DARTS). These methods have collectively established the conceptual and technical foundation of modern target deconvolution and have enabled numerous important discoveries in NPs research[5−11]. However, NPs are often structurally complex, modification-intolerant, multi-targeted, and frequently engage their targets with only moderate or even relatively weak affinity, which substantially magnifies the intrinsic limitations of conventional methods. In practice, classical strategies often fail for one of two reasons: either the compound cannot be derivatized without compromising activity, or ligand binding does not induce a sufficiently strong biochemical or biophysical signal for reliable detection.
Affinity chromatography
-
Affinity chromatography, also known as matrix-based affinity capture, is the earliest and most straightforward 'target fishing' strategy. In this approach, the bioactive molecule would be immobilized on a solid phase material and used to enrich interacting proteins from cell or tissue lysates, followed by washing, elution, and proteomic identification[5−7]. This approach has the advantages of rapid and relatively high-throughput enrichment of candidate target proteins. An early landmark study employed an affinity matrix containing the immunosuppressive drug FK506 to isolate its binding protein, FK506-binding protein 12 (FKBP12), thereby providing proof-of-principle for compound-centric target identification[12]. The representative studies on NPs further demonstrated the universality of this strategy. For example, immobilization-based target isolation revealed prohibitin 1 (PHB1) as the key binding partner of aurilide, thereby uncovering a mitochondria-dependent apoptotic mechanism[13]. Similarly, artesunate-coupled solid support enabled the identification of gephyrin as a specific interacting protein of artemisinins[14], while ECH-conjugated epoxy-activated Sepharose beads were used to capture CK2α' as the direct cellular target of echinacoside[15]. These studies collectively demonstrate that affinity chromatography technology holds significant historical importance in the identification of NPs targets, and still holds importance to this day.
Despite its conceptual simplicity, affinity chromatography is severely limited by the amenability of the parent compound to probe derivatization. Many NPs lack a suitable derivatization site or tolerate only minimal structural modification; the introduction of a linker or affinity group may alter the native pharmacophore, reduce binding affinity, and even abolish biological activity[5−7]. In addition, because the assay is generally performed in lysates, nonspecific adsorption of abundant or highly interactive background proteins may mask the true low-abundance targets, and washing conditions are often difficult to optimize without either increasing background or losing real binders[6,7]. Importantly, weak or transient interactions may dissociate during washing and elution, thereby increasing the risk of false-negative target assignment (Fig. 1a). Therefore, affinity chromatography remains highly useful when the chemistry is tractable. However, due to the contradiction between maintaining the native bioactivity and installing an enrichment handle, its general applicability to NPs is limited.
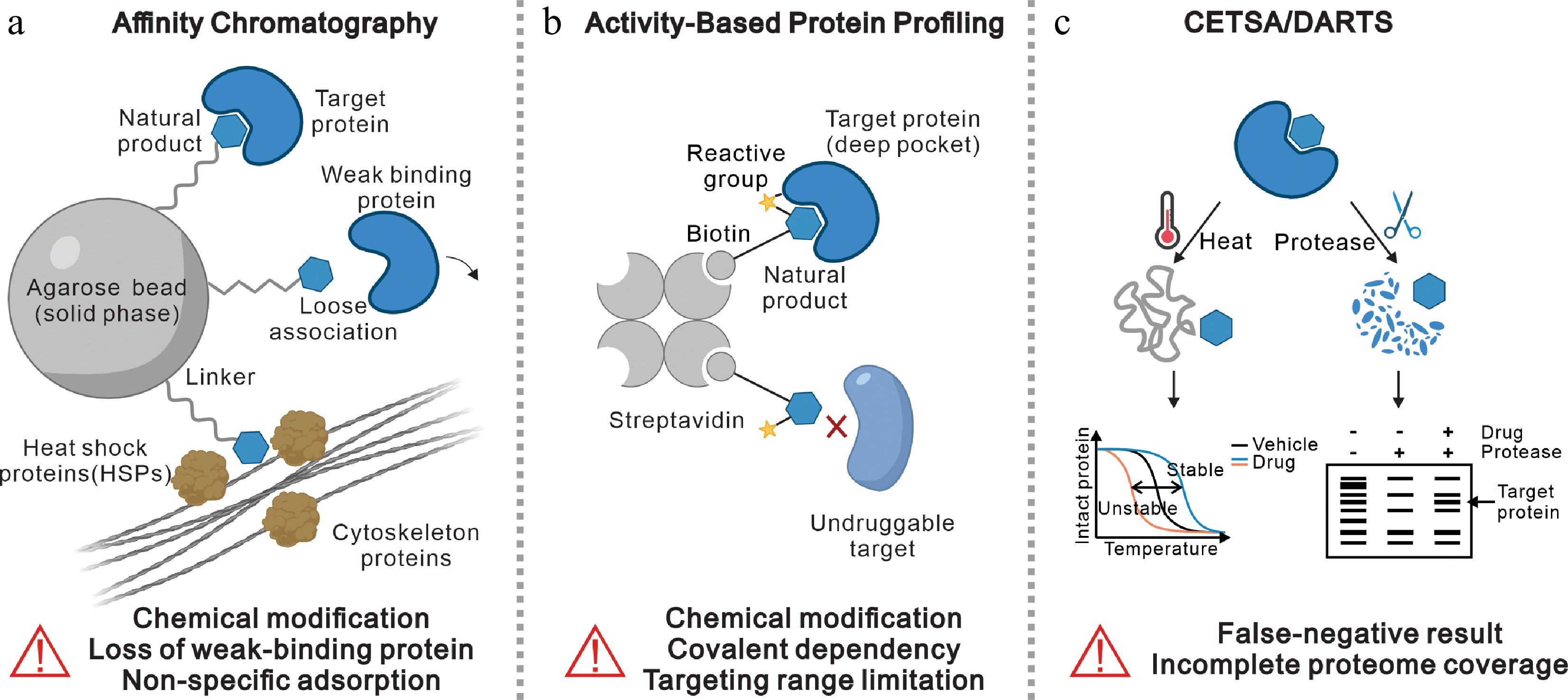
Figure 1.
Current target identification strategies and their limitations. (Contains elements from BioRender. Cheng, L. [2026]
https://BioRender.com/niqnoda ). (a) Affinity chromatography: NPs are immobilized on agarose beads via a linker to capture target proteins. Limitations include the requirement for chemical modification, loss of weak-binding proteins due to loose association, and non-specific adsorption of high-abundance proteins like HSPs and cytoskeleton proteins. (b) ABPP: uses probes containing a reactive group and a biotin tag to covalently bind and enrich target proteins via streptavidin. Limitations involve covalent dependency, chemical modification requirements, and limited targeting range (e.g., inability to capture undruggable targets). (c) CETSA/DARTS: label-free methods relying on ligand-induced changes in thermal stability (CETSA, left), or protease resistance (DARTS, right). Limitations include potential false-negative results and incomplete proteome coverage.ABPP
-
ABPP is a chemical proteomics strategy pioneered by Liu et al. in 1999. It utilizes small-molecule probes to directly profile protein function in complex biological systems[16,17]. In the context of NPs target identification, ABPP commonly encompasses both activity-based probes and affinity/photoaffinity probes, utilizing probe-directed chemoproteomic enrichment and proteomic readouts to capture and identify targets[16−19]. Compared with conventional affinity chromatography, ABPP offers a more direct approach for target engagement analysis, as it combines ligand recognition with covalent capture or photo-crosslinking, thereby enabling target identification in lysates, intact cells, or biological samples related to diseases. Recent NPs studies illustrate the importance of this strategy. For example, an ABPP workflow identified ACAT1 as a major target of chlorogenic acid in tumor mitochondria[20], while a 2025 ABPP study further showed that ENO1 is a direct target of chlorogenic acid in photoaging-related senescence models[21]. Likewise, chemoproteomic profiling of withaferin A mapped a broader covalent/non-covalent target landscape and validated previously unrecognized targets, including MVK, HNRNPF, and CKAP4[22]. More recently, ABPP-based studies have identified TRIM33 as a covalent target of parthenolide[23] and STAT3 as a key target of baicalin[24], further demonstrating the high universality of ABPP for NPs target deconvolution.
Despite these advantages, ABPP is fundamentally still limited by factors such as probe design, target reactivity, and interpretability. First, its successful application usually requires either an intrinsically reactive NP or a probe that can be synthesized without substantially perturbing bioactivity, permeability, or binding mode[5,6]. Second, classical covalent ABPP is inherently biased toward proteins containing suitably reactive residues, whereas photoaffinity-based variants may introduce nonspecific labeling and require additional filtering to distinguish true targets from background proteins[16,18]. Third, competitive ABPP signals are not always equivalent to direct active-site occupancy, because changes in probe labeling may also result from alterations in residue reactivity, allosteric effects, conformational perturbation, or protein-complex remodeling[5]. Finally, ABPP typically relies on binary small-molecule–protein interactions and is best suited for targets with well-defined, deep binding pockets; consequently, it often yields weak or undetectable signals for proteins with shallow binding sites or large, flat interaction interfaces, which are frequently engaged by NPs (Fig. 1b). Therefore, although ABPP is one of the most widely used chemical proteomics methods at present, its performance in NPs target identification still depends strongly on the chemical tractability of the scaffold, the compatibility of probe design, and the intrinsic ligandability of the target proteome.
CETSA and DARTS
-
CETSA, first introduced by Molina et al. in 2013, is a label-free biophysical method that detects ligand-induced changes in protein thermal stability in cells, lysates, or tissues[8,25−27]. When ligand binding stabilizes a protein, a larger fraction remains soluble after heating, and this shift can be monitored by Western blot or, at the proteome scale, by mass spectrometry. TPP, established soon thereafter by Savitski et al., extended CETSA to multiplexed quantitative proteomics and enabled proteome-wide identification of direct and indirect drug targets[28]. This approach has been applied successfully for many other NPs. For example, CETSA identified purine nucleoside phosphorylase (PfPNP) as a direct target of quinine in malaria parasites, thereby clarifying a long-standing mechanistic question[29]. Later, TPP revealed NOP14 as the key target of vioprolide A, linking this NP to impaired ribosome biogenesis[30]. More recently, Zhang et al. used the TPP strategy to identify CNPY3 as a cellular target of gambogic acid in prostate cancer pyroptosis, illustrating how proteome-wide thermal profiling can uncover previously unappreciated targets of NPs with complex structure[31].
DARTS was proposed by Lomenick et al. in 2009. It is another label-free target-identification method that infers ligand binding from increased resistance of the target protein to limited proteolysis[32,33]. Because it is simple, inexpensive, and does not require chemical derivatization of the NPs, DARTS is often used as an orthogonal validation method. Moreover, in certain specific cases, it can also serve as a discovery-oriented screening tool when integrated with mass spectrometry. Representative recent examples include the identification of KPNB1 as a candidate target of withaferin A[34], DCTPP1 as a candidate target of compound 2[35], and p75NTR as a primary binding target of astragaloside II[36].
Despite their clear advantages, CETSA/TPP and DARTS share important limitations that should be explicitly acknowledged. First, both methods assume that ligand binding generates a sufficiently measurable change in protein stability, thermal stability in CETSA/TPP, and protease susceptibility in DARTS, but this is not always the case. Weak binders, surface binders, allosteric modulators, low-abundance targets, or large multidomain proteins may fail to produce detectable shifts, thereby increasing the risk of false negatives[25−27]. Second, the magnitude of a CETSA or TPP shift is not necessarily proportional to ligand affinity, and TPP performed in intact cells may capture indirect downstream effects, protein complex remodeling, or post-translational changes in addition to direct binding events[5,6,27]. Third, DARTS is strongly influenced by protease choice, digestion conditions, and target abundance, and is generally less suitable than CETSA for assessing target engagement in intact cellular contexts[5,6,32] (Fig. 1c). For these reasons, CETSA/TPP and DARTS are best regarded as powerful complementary approaches rather than standalone solutions, and in NPs target identification they often require orthogonal validation to distinguish direct targets from indirect responders.
In this context, TPD provides a conceptually distinct and experimentally complementary strategy for TCM research. As an event-driven pharmacological modality, TPD converts target engagement into a more amplified and functionally informative outcome-selective protein depletion. Mechanistically, TPD is a form of chemically induced proximity: instead of depending solely on active-site occupancy, it uses chemical tools to induce the proximity between key biomacromolecules and thereby trigger downstream biological effects. TPD is therefore not intended to replace conventional occupancy- or stability-based methods, but to complement them, particularly when direct binding or ligand-induced stability changes are difficult to detect. These features make TPD particularly attractive for the target deconvolution of bioactive NPs.
-
Chemically induced proximity (CIP), or more broadly small-molecule-induced proximity, refers to a pharmacological strategy in which a ligand or engineered chemical modality brings two biomolecules into productive spatial proximity, thereby triggering a biological consequence that is often distinct from classical binary occupancy-driven inhibition[37]. Conceptually, the key event in CIP is not simply target binding, but the formation or stabilization of a new functional interaction. The origin of this viewpoint can be traced back to the 1990s, when it was discovered that simply bringing receptors or signaling proteins close together was sufficient to initiate the subsequent signaling process[38]. Recently, many reviews have summarized these early observations and constructed a broad induced-proximity framework, which now encompasses a variety of therapeutic and chemical-biological modalities[39,40].
As illustrated in Fig. 2, CIP can be broadly categorized according to the biological outcomes, including targeted protein inhibition and targeted protein post-translational modification (PTM). The former includes proximity-based inhibition or relocalization strategies such as regulated induced proximity targeting chimeras (RIPTAC)[41], whereas the latter includes induced phosphorylation, acetylation, ubiquitination, and related PTM-driven outcomes. Within this second category, TPD is currently the most mature and best validated consequence of induced proximity. However, TPD is not limited to a single degradation pathway: current TPD modalities include proteasome-based degraders, molecular glues (MGs), and lysosome- or autophagy-related systems. Among these, proteolysis targeting chimera (PROTAC) remains the most representative and mechanistically established class for intracellular degradation dependent on the ubiquitin-proteasome system (UPS)[42].
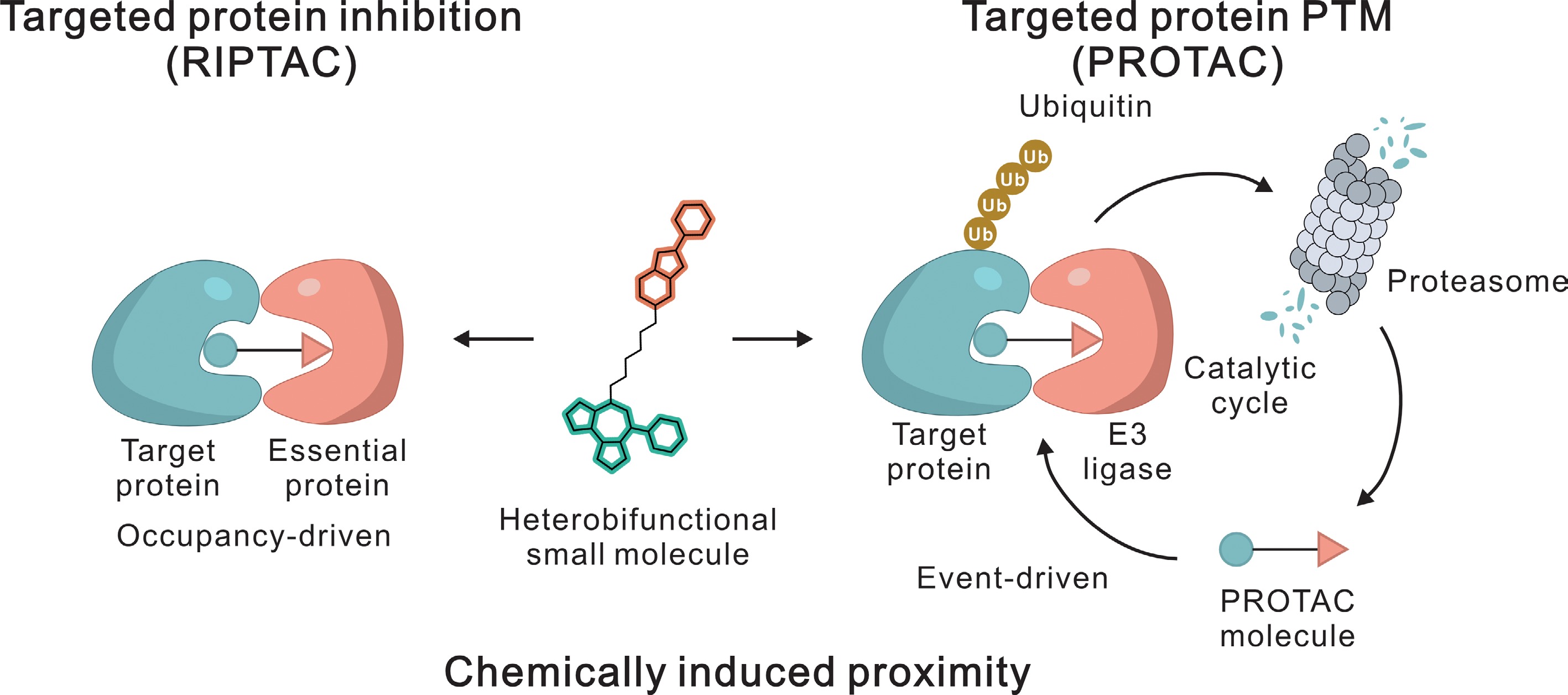
Figure 2.
Common modalities of chemically induced proximity. The schematic illustrates two distinct mechanisms mediated by heterobifunctional small molecules. Targeted protein inhibition (RIPTAC): This approach induces proximity between a target protein and an essential protein, resulting in functional inhibition. The mechanism acts via an occupancy-driven mode and requires stable binding to exert its effect. Targeted protein PTM (PROTAC): This approach recruits an E3 ligase to the target protein, facilitating the transfer of ubiquitin chains. The polyubiquitinated target is subsequently degraded by the proteasome. Subsequently, the PROTAC molecule was released and participated in the further reactions, thereby forming an event-driven and catalytic cyclic process.
PROTAC technology
-
PROTAC is a cutting-edge technology that hijacks the endogenous UPS to selectively eliminate a protein of interest (POI). In the canonical UPS cascade, ubiquitin is activated by the E1 enzyme, then transferred to the E2 conjugating enzyme, and finally installed onto a substrate under the control of the E3 ubiquitin ligase. PROTAC technology leverages this machinery by utilizing heterobifunctional molecules to simultaneously engage the POI and an E3 ligase. This artificial proximity promotes ternary-complex formation, enabling the transfer of E2-loaded ubiquitin onto the POI. Repeated ubiquitination eventually leads to the target protein being recognized by the 26S proteasome in a polyubiquitin-dependent manner and undergoing irreversible degradation. Unlike traditional inhibitors that require sustained target occupancy to maintain functional blockade, the PROTAC technology acts in an event-driven manner: transient but productive target engagement is sufficient to initiate a catalytic degradation cascade[43].
Structurally, a PROTAC molecule typically comprises three modular components: a POI-binding ligand, an E3 ligase-recruiting ligand, and a chemical linker that connects these two. This highly modularized architecture has greatly facilitated the wide application of this technology in both academic and industrial fields. Based on incomplete statistics, the PROTAC technology has been successfully applied to a wide range of internal cellular targets, which are continuously expanding. To date, over a hundred proteins have been reported as degradable in preclinical studies, and dozens of PROTAC molecules have advanced into the clinical trial stage. While initially concentrated in oncology, these clinical efforts are increasingly expanding into inflammation, autoimmune disorders, and neurodegenerative diseases[42,44,45]. Overall, these progressive refinements establish PROTAC as a robust and increasingly mature proximity-based technology, extending its utility beyond therapeutic interventions to serve as a powerful tool for target deconvolution.
PROTAC-based target identification: current reviews and the position of DBPP
-
The significance of NPs in PROTAC research is becoming increasingly prominent, as their structural complexity and unique interaction characteristics can provide useful ligands or starting scaffolds for proximity-based chemistry. Recent reviews have summarized NPs-derived PROTAC molecules and MG degraders mainly from the perspective of medicinal chemistry, including NPs used as POI ligands, E3 ligase ligands, or MGs[46,47]. Recently, there have been two reviews specifically discussing target identification based on PROTAC technology. Liu et al. proposed the concept of 'targeted degradomics'[48] and summarized early examples of using a single optimal PROTAC probe for target deconvolution. Leora Spitz et al. further discussed the general principles and precautions of applying PROTAC molecules in target validation, and emphasized the importance of orthogonal controls[49]. These works collectively recognized the potential of degradation-based strategies for target identification. However, neither of them provided a systematic methodological framework specifically designed for multi-target NPs, nor did they incorporate a probe-mixed strategy based on a PROTAC toolbox or a dual-path orthogonal validation integrating degradation proteomics with ternary-complex immunoprecipitation-mass spectrometry (IP-MS). The degradation-based protein profiling (DBPP) approach presented here directly addresses these gaps, offering a more flexible, cost-effective, and accurate framework tailored to the polypharmacological nature of NPs[50].
Importantly, DBPP should not be regarded as conceptually completely independent of the previous degradation-based approaches; instead, it incorporates these strategies into a more flexible and systematic framework, thereby enabling broader applicability and higher stability in target identification. Hence, the following sections focus on the conceptual basis, core workflow, methodological strengths and limitations, typical cases, summary, and future prospects of DBPP, rather than detailing the general medicinal chemistry of NP-derived degraders.
-
DBPP is a multidisciplinary target identification strategy that systematically integrates medicinal chemistry, proteomics, and molecular biology. The fundamental logic of DBPP is to chemically transform a bioactive NP into a PROTAC toolbox, thereby hijacking the UPS to induce the degradation of its potential targets. By transforming the detection mode from the 'physical binding' events that are traditionally difficult to capture to highly sensitive 'protein disappearance' phenotypes, DBPP is designed to reduce the impact of high background noise and minimize the loss of weak-binding targets that are common in conventional affinity-based strategies. The core workflow of DBPP comprises three key technical steps:
Construction and selection of a PROTAC toolbox
-
Probe design is the foundational step in DBPP. Because the efficient formation of the active ternary complex (Target-PROTAC-E3) strictly depends on the length, flexibility, and spatial orientation of the linker, relying on a single PROTAC molecule inevitably risks producing false negatives for targets that bind but lack a compatible spatial conformation for ubiquitination. To address this issue, DBPP introduces the concept of 'PROTAC Toolbox' and generates a PROTAC library from a chemically accessible modification site by utilizing varying linkers with different properties (e.g., rigid piperazine chains, alkyl chains, or PEG chains of different lengths) and different E3 ligase recruiters (e.g., cereblon [CRBN] or von Hippel-Lindau [VHL] ligands). Subsequently, based on the cell phenotypes and structural diversity, representative molecules were selected from the library, thereby forming a toolbox for subsequent proteomics studies.
Conceptually, this step distinguishes DBPP from conventional single-probe strategies. The goal of target identification is not to optimize one 'best' PROTAC molecule for a single target, but to construct a PROTAC toolbox that maximizes target coverage for multitarget NPs. In this framework, both single-probe and mixed-toolbox modes can be adopted. The former can more clearly attribute degradation events to specific degraders, while the latter can improve efficiency and reduce experimental cost when the purpose is rapid target identification.
Quantitative proteomics screening and data analysis
-
After treating cells with the PROTAC toolbox, we perform whole-proteome quantification via high-resolution mass spectrometry, typically using Tandem Mass Tag (TMT)-based quantitative proteomics. In order to capture primary degradation events without interference from secondary downstream remodeling of the proteome, the treatment time should typically be limited to a relatively short period, usually 8 to 12 h. The experimental design generally includes at least three comparison groups: a vehicle control group, the PROTAC toolbox group, and a negative control group. The negative control is usually constructed from PROTAC analogues that retain the NP-derived ligand and linker but contain an inactivated E3 ligase-binding moiety, such as an N-methylated glutarimide ring in the case of CRBN-recruiting degraders. This design is intended to preserve target binding affinity while abolishing effective E3 recruitment and subsequent degradation.
Subsequently, the candidate targets will be prioritized through a comprehensive comparative analysis rather than based on fold change alone. In practice, proteins are filtered by setting appropriate thresholds based on indicators such as abundance change, statistical significance, reproducibility, and identification confidence. The candidate targets with higher credibility refer to the proteins whose abundance significantly decreases in the active PROTAC group, while remaining basically unchanged in the DMSO group and the negative control group. When analyzing multiple probes in the toolbox separately, the individual data sets can be integrated. And those proteins that have an intersection in at least two separate sample groups can be given priority consideration. In the mixed-toolbox proteomics, degradation-centered results can be further integrated with IP-MS in the subsequent validation step. Thus, unlike ABPP, which enriches proteins by probe capture, DBPP identifies proteins by depletion and relies on comparative and integrative data analysis to distinguish likely direct degradation events from indirect proteome perturbations.
Orthogonal validation of DBPP candidates
-
One of the core features of DBPP is that candidate targets are not determined solely through proteomic degradation analysis. Because the apparent protein loss may be indirectly caused, and some true target proteins may form ternary complexes without undergoing effective degradation, DBPP requires orthogonal validation from both degradation and binding perspectives. In practice, the candidate proteins identified through quantitative proteomics are first confirmed at the degradation level. The common practice is to verify the dose- and time-dependent degradation through immunoblotting. Then, mechanism validation experiments are conducted, such as inhibiting the proteasome, inhibiting neddylation, performing E3 ligase competition experiments, or knocking out the recruited ligases, to determine whether the observed protein loss is indeed related to the degrader, ligase, and proteasome. Meanwhile, label-free assays (such as CETSA or DARTS) can further provide support for direct target validation. When the purified protein is available, biophysical measurement can also be introduced, including surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC).
In the meantime, DBPP also encompasses a second validation dimension based on the complex formation induced by PROTAC molecules. This process was accomplished through IP-MS, with the aim of capturing E3 ligase-centered ternary complexes and identifying associated proteins by mass spectrometry. This step holds significant importance. It can not only verify the real targets in degradation proteomics to enhance the efficiency of target identification, but also complement degradation proteomics by capturing those targets that are overlooked in degradation proteomics due to their inability to be effectively degraded. When feasible, structural approaches such as crystallography, cryo-EM, or computationally guided modeling may provide additional support, although these are not required for every target. Overall, an efficient DBPP validation strategy is to integrate degradation-centered analysis with binding/complex-centered analysis. Proteins supported by both types of evidence are generally the most reliable targets, which is consistent with the dual-path logic illustrated in Fig. 3.
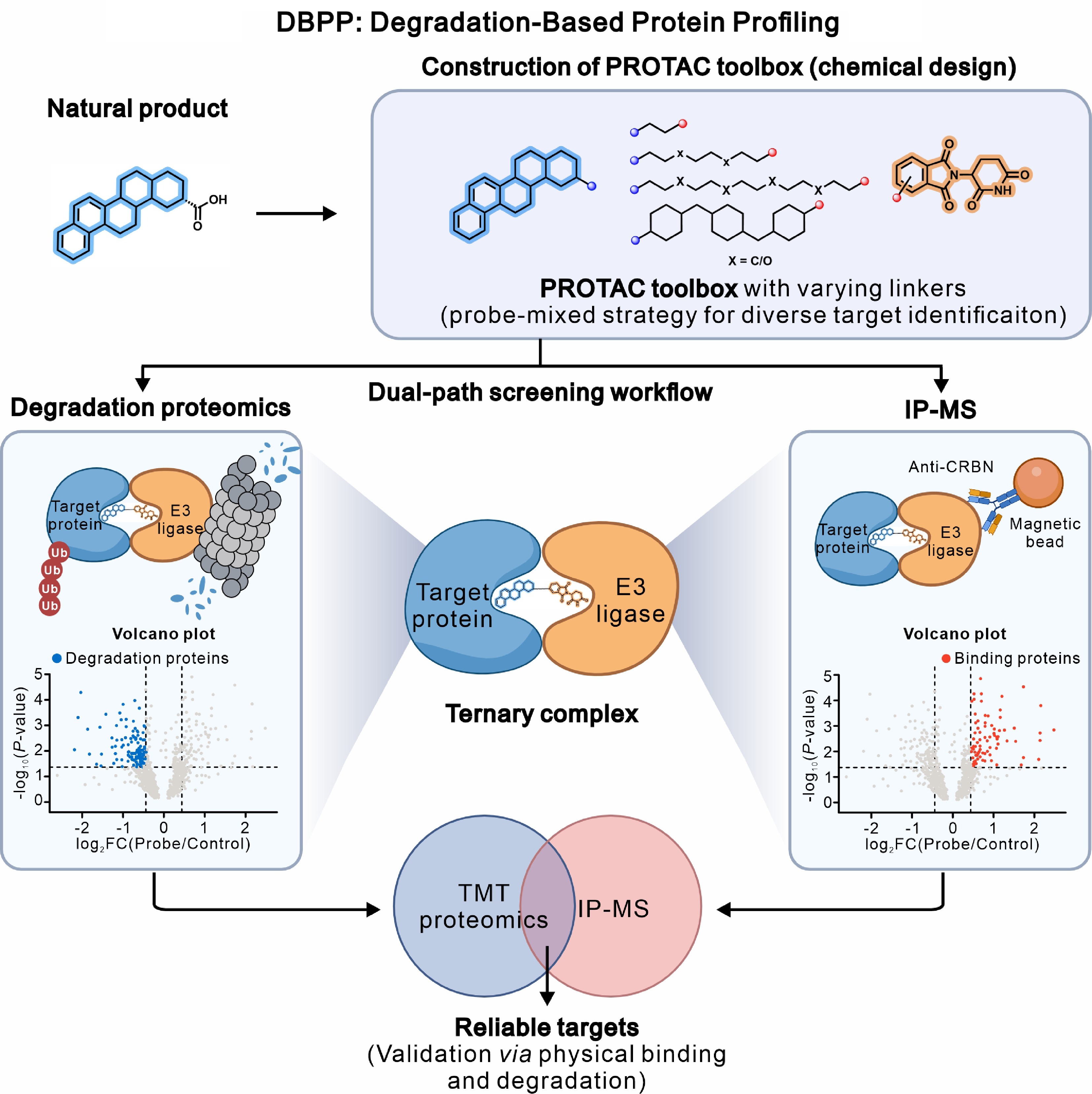
Figure 3.
Diagram of the DBPP strategy. The DBPP workflow integrates chemical design with a dual-path proteomics screening approach to systematically identify and validate NP targets. (Top) Construction of the PROTAC toolbox: A bioactive NP is chemically derivatized using a variety of linkers to connect with an E3 ligand. This generates a PROTAC library to accommodate the diverse spatial requirements necessary for ternary complex formation. (Bottom) Dual-path screening workflow: Upon treatment, the PROTACs induce the formation of target-PROTAC-E3 ligase ternary complexes, which are subsequently analyzed via two orthogonal parallel paths. (Left) Degradation proteomics: Quantitative TMT proteomics is employed to monitor global protein abundance. Significantly downregulated proteins are defined as potential degradation targets (blue dots in the volcano plot). (Right) IP-MS: Direct physical binding targets are captured by enriching the ternary complexes using E3 ligase-specific antibodies (e.g., Anti-CRBN magnetic beads) followed by mass spectrometry. Significantly enriched proteins are defined as potential binding targets (red dots in the volcano plot). (Center) Integration: The cross-validation of the functional 'degradation' dataset and the physical 'binding' dataset ultimately yields high-confidence, reliable targets for the NPs.
-
Compared with ABPP and earlier classical PROTAC-based proteomic strategies, DBPP has several methodological strengths. First, DBPP is toolbox-based rather than single-molecule-based. Since different degraders can exhibit different degradation spectra, using multiple representative PROTACs can reduce reliance on any single ternary complex geometry and enhance target coverage for multi-target NPs. The mixed-toolbox format further enhances screening efficiency and cost-effectiveness.
Second, DBPP employs a dual-pathway validation logic. The candidate proteins are not only supported by the proteomic degradation data, but also further evaluated by the complex evidence provided by IP-MS. This is important because some genuine targets may not be efficiently degraded, whereas some apparent degradation events may be indirect. Combining degradation-centered with complex-centered evidence can improve confidence in target assignment and reduce false positives.
Third, compared with conventional degradation-based strategies, the introduction of IP-MS provides a practical advantage. In intact cell degradation experiments, certain targets may be difficult to identify because of the improper subcellular localization and incompatible ternary-complex geometry. In contrast, IP-MS performed after cell lysis can still enrich proteins that are able to participate in PROTAC-induced ternary complexes, even if they are not efficiently degraded in intact cells. This feature allows DBPP to recover a broader class of target proteins, including non-degradable binders, and partially compensates for the E3 compartmentalization limitation of classical degradation-based strategies. For an overview of these comparative features, refer to Table 1.
Table 1. Comparison of current target identification strategies.
Label-free methods
(CETSA, TPP, DARTS)Activity-based protein
profiling (ABPP)Conventional single-probe
PROTAC profilingDegradation-based protein profiling (DBPP) Core mechanism Stability-based, label-free: ligand binding induces changes in protein thermal stability or protease susceptibility. Occupancy/activity-driven; probe captures target proteins through covalent or photoaffinity labeling. Event-driven discovery; a PROTAC induces ubiquitination and degradation, and downregulated proteins are screened by quantitative proteomics. Event-driven, dual-path discovery; combines degrader-induced depletion with ternary-complex/
binding-centered enrichment.Probe architecture No probe required; native compounds used directly. Warhead, linker, and reporter tag. Typically used as individual probes. Target ligand, linker, and E3 recruiter. Usually, one representative PROTAC molecule. A PROTAC toolbox with varied linker types/lengths; compatible with both single-toolbox and mixed-toolbox formats. Detection logic 'Indirect readout': targets inferred from ligand-induced stability shifts (thermal or proteolytic). 'Addition' readout: proteins are enriched by pull-down or probe capture. 'Subtraction' readout: proteins significantly decreased after degrader treatment are prioritized. Dual-path readout ('subtraction + addition'): candidate targets are prioritized by both degradation proteomics and IP-MS-based complex evidence. Target coverage Broad in principle (proteome-wide for TPP), but biased toward proteins with detectable stability changes. Best for ligandable/reactive proteins; biased toward proteins with suitable nucleophilic residues or photo-crosslinkable environments. Broad, but prone to false negatives due to strict spatial/conformational restrictions of a single ternary complex. Broadest among the three; can capture proteins that degrade and, through IP-MS, proteins that bind but do not degrade efficiently. Potential target number ~103 (thousands) ~102 (hundreds) 10–102 (tens to hundreds) Tens Main advantages 1. No derivatization required.
2. Preserves native binding properties.1. Direct target capture.
2. Strong mechanistic resolution for reactive ligands. 3. Mature chemoproteomic
framework.1. Converts binding into an amplified depletion signal. 2. Can reveal non-catalytic and weak-binding targets. 1. Toolbox strategy improves target coverage. 2. Dual-path validation reduces false positives/false negatives.
3. Suitable for multitarget NPs. 4. Compatible with the probe-mixed strategy to improve efficiency and reduce cost.Technical limitations 1. Requires measurable stability change (false negatives for weak/allosteric binding). 2. Limited sensitivity for low-abundance proteins.
3. Indirect readout may
include secondary effects.1. Probe synthesis is demanding. 2. Probe installation may perturb activity. 3. Coverage bias toward reactive or probe-compatible targets. 1. Strongly dependent on ternary complex geometry of one PROTAC molecule. 2. Indirect degradation effects may confound interpretation. 3. Coverage is further constrained by the subcellular accessibility and distribution of the recruited E3 ligase, making proteins in certain cellular compartments more difficult to identify. Requires construction and preliminary selection of a representative PROTAC toolbox, which may increase the entry barrier for groups without synthetic support. Despite these advantages, DBPP also has several limitations that should be considered. The strategy still relies on the successful construction of a representative PROTAC toolbox, which may require preliminary optimization of linker composition, E3 ligase selection, and overall molecular properties; this can present a practical barrier for groups without sufficient synthetic support. In addition, although the dual-path design reduces false positives, the degradation signals may still contain indirect or downstream effects, and IP-MS may capture proteins associated within complexes without direct binding to the parent NP. Furthermore, as with other proteomics-based methods, the detection sensitivity remains influenced by protein abundance, instrument performance, and data analysis thresholds. Overall, although DBPP has significant advantages, it also has inherent limitations. Therefore, it is best to apply it as part of an integrated target-identification framework. It is worth noting that no single strategy is universally applicable; instead, the synergistic integration of multiple orthogonal strategies is crucial for compensating for their respective limitations, thereby maximizing the accuracy and efficiency of target deconvolution.
-
Although the DBPP strategy innovatively integrates a rationally designed PROTAC toolbox, a probe-mixed strategy, and dual-path orthogonal validation, one of its core logics still involves leveraging protein degradation induced by PROTAC molecules as a functional readout of target engagement, which is essentially consistent with the current degradation-based target identification strategies. Therefore, classic degradation-based strategies are also included within the scope of the DBPP strategy. This section focuses on introducing several representative studies.
Celastrol
-
Celastrol, a pentacyclic triterpenoid derived from Tripterygium wilfordii, exhibits a wide range of pharmacological activities, including anti-inflammatory and anticancer effects. However, its clinical development has been hindered by unclear target profiles and potential toxicity. To systematically elucidate its molecular mechanism, Ni et al. established a DBPP framework by constructing a PROTAC toolbox incorporating diverse linker types and lengths[50] (Fig. 4a).
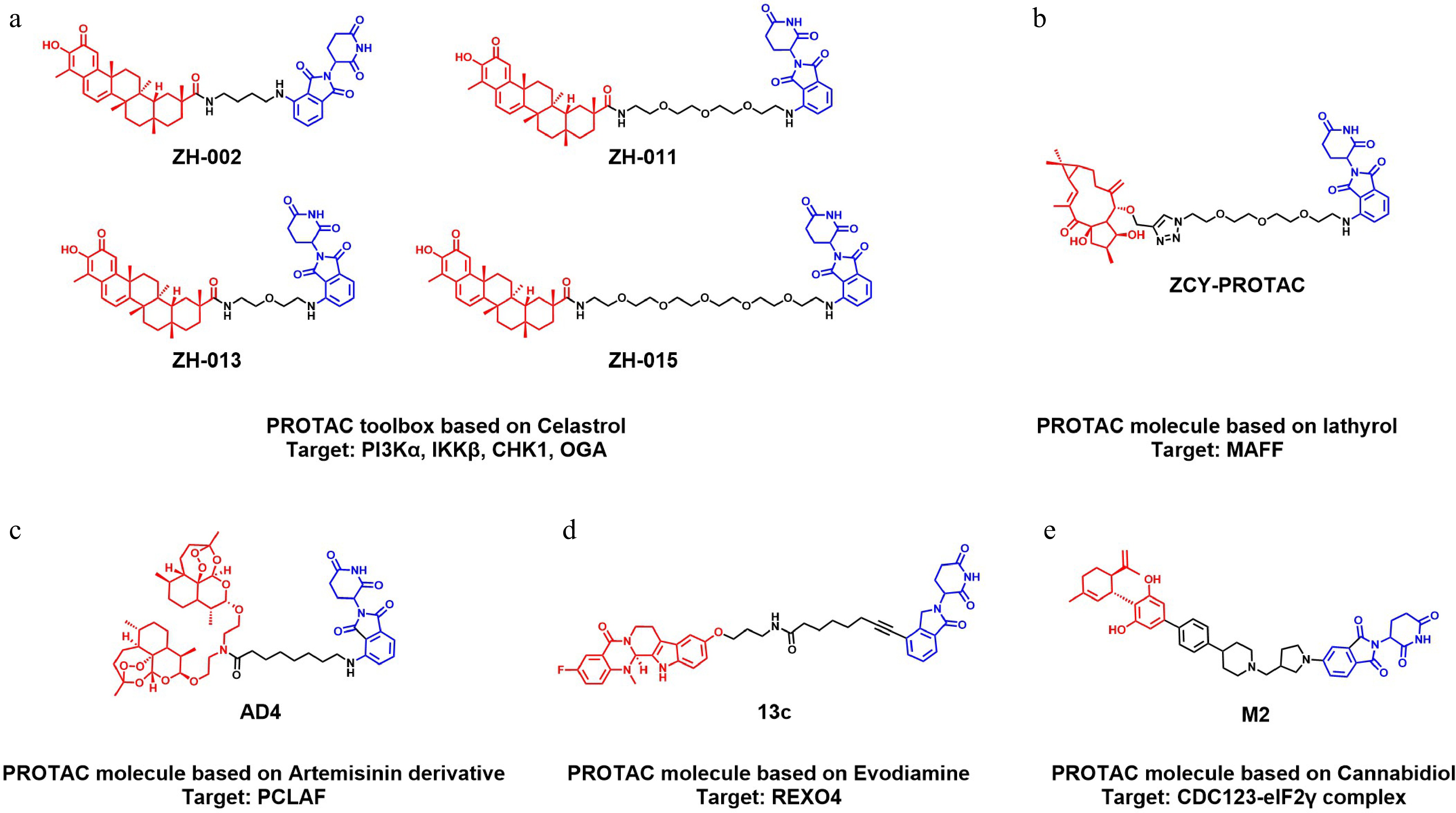
Figure 4.
Representative examples of NPs-derived PROTAC molecules. Chemical structures of various PROTAC molecules utilized in DBPP. The NP scaffolds are shown in red, the E3 ligands are shown in blue, and the linkers are shown in black. (a) A celastrol-based PROTAC toolbox (ZH-002, ZH-011, ZH-013, ZH-015) for multi-target identification (targets: PI3Kα, IKKβ, CHK1, OGA). (b) A PROTAC molecule derived from lathyrol (ZCY-PROTAC) (target: MAFF). (c) A PROTAC molecule derived from an artemisinin derivative (AD4) (target: PCLAF). (d) A PROTAC molecule derived from evodiamine (13c) (target: REXO4). (e) A PROTAC molecule derived from cannabidiol (M2) (target: CDC123-eIF2γ complex).
Through the integrated analysis of TMT-based degradation proteomics and IP-MS datasets, this study prioritized candidate targets supported by both degradation and ternary complex evidence. This strategy successfully reproduced previously reported targets, such as inhibitor of kappa B kinase β (IKKβ) and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PI3Kα). Additionally, other candidate targets were also discovered, including checkpoint kinase 1 (CHK1). It is notable that CHK1 exhibits relatively weak binding affinity and limited inhibition in conventional assays, illustrating that DBPP can transform weak or transient interactions into detectable degradation signals. In addition, O-GlcNAcase (OGA) was identified as a potential target, leading to the development of an OGA degrader. This study provides a representative example of how a toolbox-based DBPP strategy can improve target coverage and reduce false positives in multitarget NPs.
Lathyrane diterpenoids
-
Lathyrane diterpenoids, such as ZCY-001 derived from Euphorbia lathyris, display anti-inflammatory activity, although their direct targets have remained unclear. In this context, a PROTAC molecule (ZCY-PROTAC) was constructed by conjugating the lathyrol scaffold to a CRBN ligand[51] (Fig. 4b).
By using a TMT-based quantitative proteomic approach, V-maf musculoaponeurotic fibrosarcoma oncogene homolog F (MAFF) was identified as a candidate degradation target. Subsequent orthogonal validation, including microscale thermophoresis (MST), SPR, CETSA, and DARTS, supported direct binding between the compound and MAFF. Mechanistically, the interaction was reported to modulate MAFF dimerization dynamics, favoring MAFF-NRF2 heterodimer formation over MAFF homodimers, thereby activating the NRF2/HO-1 axis and suppressing the inflammatory response mediated by NF-κB. This study illustrated the applicability of target identification based on PROTAC technology in inflammation-related NPs, although it mainly reflected the single-molecule PROTAC strategy rather than the complete DBPP implementation.
Artemisinin
-
Artemisinin is widely used as an antimalarial agent and has been investigated for anticancer applications, although its direct targets in tumor cells have been debated. To address this issue, Li et al. developed a PROTAC molecule (AD4) based on artemisinin[52] (Fig. 4c).
The proteomic analysis of RS4;11 leukemia cells revealed that after treatment with AD4, the level of PCNA-associated factor (PCLAF) significantly decreased, and this phenotype was associated with the proteasome pathway. The degradation effect was dose-dependent and reversible after removal of the degrader. Functional studies suggested that degradation of PCLAF relieved its inhibitory effect on p21, leading to reduced Rb phosphorylation and subsequent G1 phase arrest. This work demonstrated how degradation-based strategies can link target identification with functional characterization. However, similar to the MAFF study, it employs a single-molecule PROTAC strategy rather than the complete DBPP framework.
Evodiamine
-
Evodiamine is a bioactive alkaloid that has been proven to have anti-cancer activity, but its molecular mechanism remains unclear. Chen et al. addressed this by designing a series of PROTAC molecules derived from evodiamine, alongside a negative control that retains target-binding capability but lacks E3 recruitment[53] (Fig. 4d).
Comparative proteomic analysis between active degrader and negative control led to the identification of RNA exonuclease 4 (REXO4) as a candidate target. Binding was further supported by CETSA and MST assays. Functional studies indicated that the degradation of REXO4 was associated with increased intracellular reactive oxygen species (ROS) and DNA damage. It is noteworthy that the addition of a negative control group can enhance the specificity of target identification by distinguishing degradation-dependent effects from ligand-induced effects. This design principle is conceptually aligned with DBPP, particularly in its emphasis on controlled comparison.
Cannabidiol (CBD)
-
Cannabidiol (CBD) exhibits anti-tumor activity, but its target is still unclear. In a recent study, Yu et al. developed a combined strategy termed stability- and degradation-based protein profiling (SDPP), which integrates the principles of TPP and DBPP[54] (Fig. 4e).
By using this dual-dimensional approach, CDC123 was identified as a candidate target for CBD. A variety of orthogonal validation methods, including CETSA, DARTS, ITC, and SPR, have confirmed the direct interaction between CBD and CDC123, as well as the disruption of the CDC123-eIF2γ complex. Mechanistic analysis suggested that this disruption activates the integrated stress response (ISR) and promotes apoptosis in colorectal cancer cells. This study highlights how DBPP can be integrated with label-free approaches to enhance target identification confidence and coverage, representing an extension of the DBPP concept.
-
DBPP provides a distinct framework for target identification of bioactive NPs by shifting the primary readout from binding affinity to functional protein depletion. Compared with conventional occupancy-driven or stability-based approaches, DBPP utilizes an event-driven mechanism that can transform transient or moderate interactions into a measurable degradation outcome. This feature is particularly important for NPs, which possess the features of multiple targets, multiple mechanisms, and relatively weak binding affinity. In this sense, DBPP complements rather than replaces existing strategies by providing an evidence dimension that is more directly linked to functional results at the proteome level.
Methodologically, DBPP integrates chemical design (PROTAC toolbox construction), quantitative proteomics, and orthogonal validation strategies, enabling a more comprehensive characterization of compound–target interactions. This dual-path logic that combines degradation data with complex formation data enhances the credibility of target identification and reduces the likelihood of misinterpreting indirect effects. The important point is that DBPP incorporates the early degradation-based target identification strategies and simultaneously extends them toward a more flexible, more accurate, and more suitable framework for NPs target identification.
Emerging opportunities through integration with cutting-edge technologies
-
To further expand the application scope of DBPP, we have combined DBPP with the cutting-edge technologies in biology and AI, and proposed several promising directions below:
Integrating DBPP with single-cell omics: resolving target dynamics within cellular heterogeneity
-
Conventional proteomics averages signals across cell populations, potentially obscuring cell state-specific degradation responses. The 'multi-target, low-affinity' characteristics of NPs suggest that different cellular subpopulations may respond heterogeneously to degrader treatment. Combining DBPP with single-cell proteomics or single-cell transcriptomics could enable profiling of degradation and downstream regulatory effects at single-cell resolution. In fact, treating heterogeneous tissues or organ-like tissues with PROTAC molecules derived from NPs, followed by the integration of emerging single-cell proteomics or single-cell RNA sequencing, may help identify rare cell subsets that are sensitive to specific degradation events and facilitate the study of the association between target degradation and phenotypes such as apoptosis or drug resistance. Such integration may provide mechanistic insight into the cellular heterogeneity underlying complex diseases such as cancer and neurodegeneration.
Integrating DBPP with spatial transcriptomics: reconstructing the 3D in-situ target network
-
The pharmacological effects of NPs are influenced by the spatial environment within tissues, yet current DBPP workflows lack spatial resolution. Spatial transcriptomics preserves native tissue architecture while measuring gene expression patterns, thereby achieving in-situ correlation between target degradation and transcriptional remodeling. Future workflows could extend DBPP to explants derived from patients or organoids. These models will be treated with NPs-derived PROTAC molecules, followed by spatial profiling. By correlating spatially resolved target loss (e.g., validated by immunofluorescence on adjacent sections) with local transcriptional changes, researchers can visually answer: In which cell niches (e.g., stroma vs tumor core) is the TCM ingredient most active? How does target degradation in one cell type spatially propagate signaling cascades to neighbors? This integration may extend DBPP from the molecular level investigation to the functional interpretation at the tissue level, providing a modern spatial annotation for traditional TCM formulation principles (e.g., the 'monarch, minister, assistant, and guide' synergy theory).
Synergizing DBPP with novel transgenic animal models: achieving real-time in vivo target visualization
-
To overcome the disconnect between in vitro DBPP results and in vivo validation, it is necessary to establish a validation system for DBPP in vivo. For example, transgenic animal models carrying bioluminescent or fluorescently labeled target proteins (e.g., Target-Luciferase or Target-GFP fusions) could be constructed. Upon administering optimized NP-derived PROTAC molecules into such models, the loss of signal (representing target degradation) could be monitored in real-time using in vivo imaging techniques like in vivo imaging systems (IVIS). Furthermore, by combining with Cre-LoxP systems, it is possible to construct tissue-specific conditional knockout animal models targeting specific E3 ligases. By observing the absence of target degradation induced by PROTAC in specific tissues, it is possible to infer whether this degradation process is dependent on the specific E3 enzyme in vivo. Such an in vivo DBPP platform can more accurately simulate the in vivo pharmacokinetic and pharmacodynamic behaviors, enabling the screening of PROTAC molecules with favorable in vivo activity and tissue selectivity, significantly enhancing the success rate of clinical translation for NPs-derived compounds.
Merging DBPP with artificial intelligence: target prioritization and mechanistic interpretation
-
Recent advances in artificial intelligence (AI) and machine learning provide useful computational frameworks for addressing one of the central challenges in DBPP: how to prioritize biologically meaningful targets from large degradation proteomics and IP-MS datasets. Because DBPP often yields multiple candidate proteins, especially for polypharmacological NPs, AI-assisted multi-omics integration and network pharmacology may help rank candidates by combining degradation degree, reproducibility across degrader groups, complex enrichment evidence, subcellular localization, disease relevance, and prior target knowledge[2]. Graph-based drug-target affinity models such as GraphDTA[55] and multimodal network frameworks such as Herb-CMap[56] illustrate how to integrate structure, interaction networks, and perturbational data to improve target ranking and mechanistic interpretation in complex NPs systems. For DBPP, this type of integration may be particularly valuable for distinguishing likely direct functional targets from indirect targets, and for aligning candidate targets with the known polypharmacological profile of the parent NP.
AI may also support mechanistic interpretation after DBPP has narrowed the candidate range. Degradability prediction models such as DeepPROTACs[57], MAPD[58], PrePROTAC[59], and DegradeMaster[60] indicates that degradation tendencies can be inferred from the features of the degrader, E3 ligase, and target proteins. This might help explain why some candidates can be effectively degraded, while others are mainly enriched by IP-MS or remain in a non-degradable state. In parallel, structure-based tools such as PRosettaC[61], AlphaFold3[62], and DeepTernary[63] may be used more cautiously at the candidate-refinement stage to generate testable hypotheses for ternary-complex compatibility, degrader geometry, or plausible ligand-binding regions on prioritized targets. Therefore, within the framework of DBPP, the most practical role of AI in the short term is to improve candidate prioritization, degradability interpretation, and structural hypothesis generation, thereby increasing the efficiency and interpretability of NPs target deconvolution rather than replacing orthogonal experimental validation.
In summary, DBPP is transforming from a single technique toward a systems platform through integration with single-cell and spatial omics, advanced animal models, and AI-assisted analysis. The integration of these technologies is expected to strengthen the connection from molecular perturbation to cellular and tissue-level function, and ultimately to systemic efficacy, thereby promoting the modernization of NPs research (especially in TCM).
This work was supported by the National Natural Science Foundation of China (Grant Nos 82125034, 82330115), the National Key R&D Program of China (Grant Nos 2021YFA1300200, 2021YFA1302100), the Beijing Outstanding Young Scientist Program (Grant No. JWZQ20240101007), and the Beijing Frontier Research Center for Biological Structure.
-
Not applicable.
-
The authors confirm their contributions to the work as follows: study conception: Rao Y; draft manuscript preparation: Rao Y, Ni Z, Shi Y. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing is not applicable to this review as no datasets were generated or analyzed.
-
The authors declare that they have no conflict of interest.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Ni Z, Shi Y, Rao Y. 2026. Degradation-based protein profiling in target identification and early-stage drug discovery of bioactive natural products. Targetome 2(3): e020 doi: 10.48130/targetome-0026-0019 

Degradation-based protein profiling in target identification and early-stage drug discovery of bioactive natural products
- Received: 05 March 2026
- Revised: 01 May 2026
- Accepted: 04 May 2026
- Published online: 19 May 2026
Abstract: Identifying the molecular targets of bioactive natural products (NPs) remains a critical bottleneck in drug discovery and the modernization of Traditional Chinese Medicine (TCM). Conventional target deconvolution strategies, such as affinity chromatography and activity-based protein profiling (ABPP), are often constrained by strict chemical modification requirements, incomplete proteome coverage, and difficulties in capturing weak or transient interactions. To overcome these limitations, this review highlights degradation-based protein profiling (DBPP), an emerging chemoproteomic strategy that applies proteolysis targeting chimera (PROTAC) technology to target identification. Unlike traditional occupancy-driven methods, DBPP employs an event-driven mechanism, converting complex physical binding events into amplified, detectable protein depletion signals via the ubiquitin-proteasome system. We systematically outline the core framework of DBPP, with particular emphasis on the rationally designed 'PROTAC toolbox', the probe-mixed strategy, and the dual-path orthogonal validation that integrates quantitative degradation proteomics with immunoprecipitation-mass spectrometry (IP-MS). Representative case studies involving NPs such as celastrol and artemisinin illustrate the potential of DBPP to identify elusive targets, including non-catalytic and weak-binding proteins, as well as reducing false positives through orthogonal validation. Finally, we discuss current methodological limitations and explore possible prospects for integrating DBPP with artificial intelligence (AI), single-cell omics, and spatial transcriptomics techniques as an effective way toward deciphering the polypharmacology of complex NPs.
-
Key words:
- Natural products /
- Target identification /
- DBPP /
- PROTAC /
- Chemical proteomics