






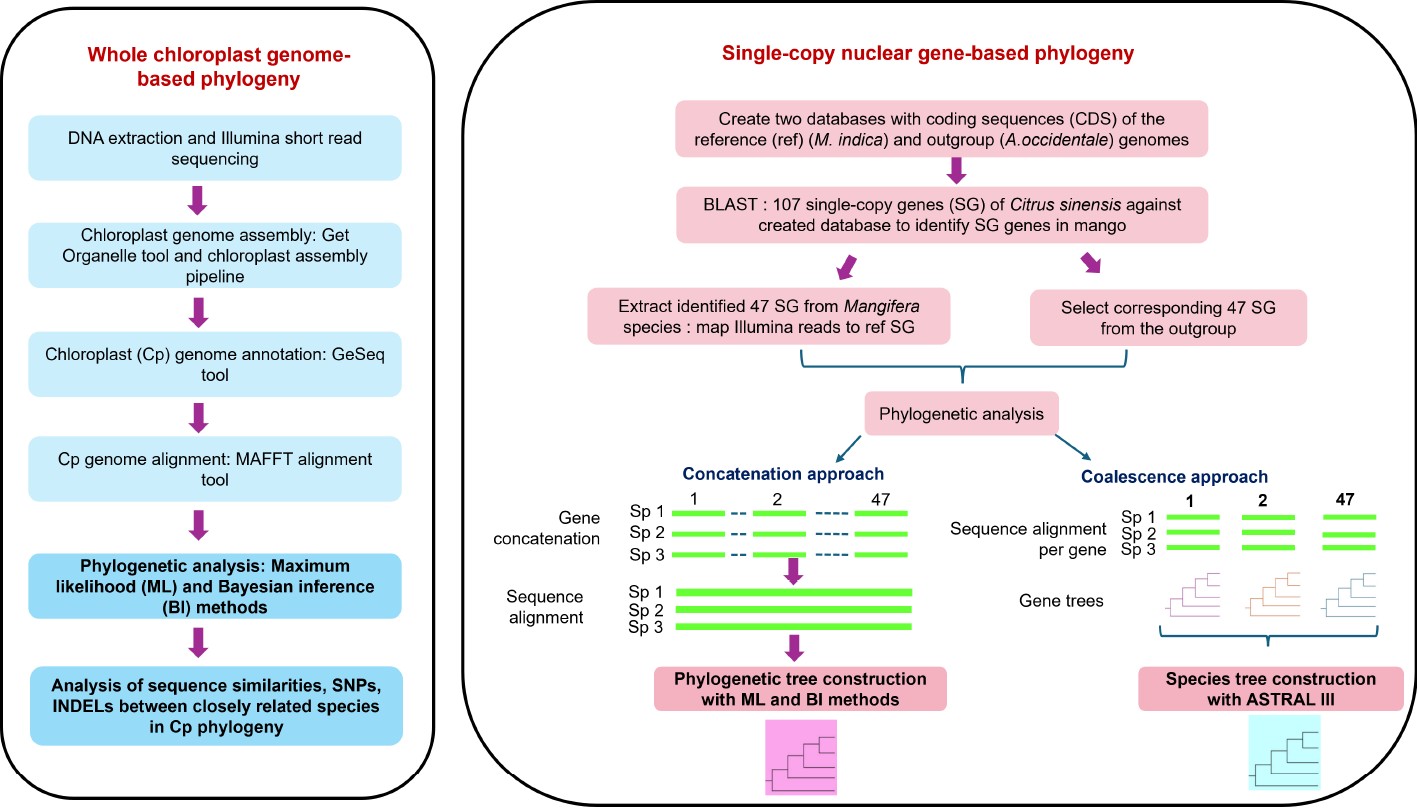



-
Mango ( Mangifera indica L.), an evergreen dicotyledonous angiosperm often referred to as the 'king of fruits' has adapted to grow in tropical and sub-tropical regions of the world [ 1− 3] . It is considered one of the most economically successful fresh fruits cultivated in more than 100 countries. India leads the global mango production producing approximately 24.7 million tonnes accounting for 45% of total mango production followed by Indonesia (6.6%), Mexico (4.3%), China (4.3%), and Pakistan (4.3%) [ 4] . Besides being consumed fresh, ripe, and unripe mangoes are used to produce pickles, chutney, juices, cereal flakes, sauce, and jam building high demand for mangoes on the international market.
The taxonomic lineage of the genus Mangifera ( Anacardiaceae) reveals consistent recognition of two major groups with the number of species reported varying between 45−69 [ 1, 5, 6] The most accepted classification [ 6] defines 69 species mainly based on morphological descriptors of reproductive tissues. Of the 69 species, 58 are divided into two subgenera, Mangifera and Limus, with the remaining 11 species placed in an uncertain position due to insufficient voucher material. The subgenus Mangifera includes 47 species further divided into four sections: Marchandora Pierre, Euantherae Pierre, Rawa Kosterm, and Mangifera Ding Hou. Mangifera Ding Hou is the largest section in the genus with more than 30 species including domesticated mango ( M. indica) [ 6, 7] . The 11 species in sub genus Limus are further divided into two sections: Deciduae and Perrennis.
Due to the high demand for mango globally, systemic breeding programs have been initiated recently to develop cultivars with high productivity, improved consumer, and transportability traits. However, breeding is time-consuming due to the long juvenile period, high heterozygosity, and polyembryony observed in mango. Currently, while M. indica stands as the primary cultivated species for commercial fruit production, with a set of selected commercial cultivars dominating the crop improvement programs, 26 other species have been reported to produce edible fruits [ 7− 9] . Many wild species exhibit potential significance in trait-specific breeding due to their favorable traits related to fruit quality, biotic and abiotic stress tolerance and potential as rootstocks [ 10− 12] . Effective exploitation of these species relies on a comprehensive understanding of their distinctive characteristics within a genetic framework, as they are primarily described based on morphological traits. Therefore, identifiying molecular evolutionary relationships within the genus Mangifera is vital to facilitate the efficient use of wild relatives in future breeding programs.
Recent studies have used molecular markers within the chloroplast genome [ 10, 13− 15] and a set of nuclear genes [ 16− 18] to analyze phylogenetic relationships in mango. However, the results are inconsistent, and many studies were unsuccessful in inferring evolutionary relationships with fully resolved phylogenies. Two studies have used whole chloroplast genome [ 19] and mitochondrial genome [ 17, 20] sequences alone with a small number of taxa. However, in most angiosperms, chloroplast, and mitochondrial genomes are maternally and paternally inherited, respectively [ 21] . Consequently, such studies prevent precise analysis of evolutionary relationships due to the use of uniparentally inherited genetic information for phylogenetic analysis.
The genus Mangifera is native to South and South-East Asia ranging from Indochina, Burma, Thailand, and the Malay Peninsula to Indonesia and the Philippines where some of the species are found only in the wild while others are locally grown in gardens and orchards [ 6] . With the introduction of the common mango to South-East Asia during the 4 th−5 th centuries [ 22] , M. indica and wild Mangifera species in the region might have come into contact with each other. Since both wild and domesticated mangoes are assumed to be self-incompatible, hybridization is expected among these outcrossing species when grown in close proximity. Among wild species, a hybrid origin has been reported for M. odorata [ 23] , and M. casturi [ 24, 25] . With molecular data suggesting the potential of cross-hybridization in the genus, more hybrids can be expected among these 69 Mangifera species that have been currently identified as distinct species. Comparative phylogenetic analysis based on both chloroplast genome and ideally, a set of single-copy nuclear genes, together representing maternal and biparental inheritance respectively will be a useful approach for the precise determination of evolutionary relationships [ 26] .
The availability of a suitable and precise reference genome is crucial in evolutionary studies to determine relationships among the species with higher accuracy. The first draft genome for M. indica was assembled for the Indian cultivar Amrapali [ 27, 28] and with the use of advanced sequencing platforms, a high-quality chromosome-level genome was developed for the cultivar 'Alphonso' [ 29] . The genetics and genomics of chloroplasts have progressed rapidly with the advent of high-throughput sequencing technologies. Chloroplast genomes in higher plants are typically double-stranded and organized into conserved quadripartite structure, consisting of a pair of inverted repeats (IR) separated by small single copy region (SSC) and a large single copy region (LSC). The chloroplast genome size, although far smaller than most of the plant nuclear genomes ranges from 120 to 160 kb [ 5] with 110 to 130 genes. Conflicts between the chloroplast and nuclear phylogenetic analysis provides valuable insights into speciation, hybridization and incomplete lineage sorting [ 30, 31] . So far, assembled chloroplast genomes of only six out of 69 species [ 19, 32] are available.
In this study, sequences of chloroplast genomes were compared [ 33] , and a selected set of common single-copy genes present in the nuclear genome of 14 Mangifera species used to analyze evolutionary relationships in the genus.
-
Nineteen samples belonging to 14 Mangifera species were selected ( Table 1). Leaf tissues of M. foetida, M. sylvatica, M. quadrifida, and one of the M. altissima and M. laurina species were sourced from The Botanical Ark, Mossman (16°22'21" S, 145°19'23" E), Queensland, Australia . M. caesia leaves were sourced from a tree at Fruit Forest Farm (
www.fruitforestfarm.com.au ), East Feluga, (17°53'46.0" S and 145°59'38.0" E), Queensland, Australia and leaves of two M. pajang species were sourced trees located at Treefarm, El Arish (−17°47'59.99" S and 146°00'0.00" E) and Durian Heaven Farm, Japoonvale (17°43'36" S, 145°54'35" E), Queensland, Australia. All M. indica varieties and other Mangifera species were sourced from trees grafted onto M. indica cv. Kensington Pride rootstock at the Walkamin Research Station, Mareeba, (17°08'02" S and 145°25'37'' E), North Queensland, Australia. DNA extraction was carried out from fine pulverized mango leaf tissue samples using a cetyltrimethylammonium bromide method [ 34] . The quality and quantity of DNA were assessed for acceptable absorbance ratios (ideal 1.8−2.0 at A260/280 and over 2.0 at 260/230) using a Nanodrop Spectrophotometer. DNA degradation and quantity were assessed by resolving sample and standard DNA by agarose gel electrophoresis. The isolated DNA was subjected to next-generation short read sequencing on an Illumina HiSeq 2000 platform at the Ramaciotti Centre for Genomics, University of New South Wales, Australia ( Supplemental Table S1).Table 1. Details of the 14 Mangifera species used in this study including country of origin, native distribution and important characteristics.
Sub genera Section Species/taxon Embryony Country of origin Geographical distribution of the species Ploidy level (2n) Prominent fruit/horticultural characteristics Mangifera Mangifera Ding Hou M. laurina Polyembryonic Indonesia Wild distribution: Myanmar, Vietnam, Malesia, Thailand. Cultivated in Borneo, Sumatra, Java, (Indonesia), and the Philippines. 40 Juicy, very acid and fibrous fruit: edible.
Resistance to anthracnose (fruit skin).
Used as a rootstock for M. indica in Malaysia.Limus Deciduae M. pajang Monoembryonic Indonesia Endemic to Borneo (Brunei Indonesia, Malaysia).
Common in cultivation.Unknown Fruit flesh: deep-orange-yellow, fibrous, acid to acid sweet, mildly fragrant: edible.
Largest fruit in the genus.Mangifera Euantherae Pierre M. caloneura (Xoài Quéo) Polyembryonic Vietnam Dry deciduous dipterocarp forests in Myanmar, Thailand, Laos, and Vietnam 40 Fruit with good aroma, sour taste: edible.
Drought tolerance / resistance to anthracnose (fruit skin) and gummosis.Limus Perrennis M. odorata Polyembryonic Malaysia Never been observed in the wild.
Origin is unknown, primarily cultivated in Guam, Philippines, Thailand, and Vietnam.
Introduced to Indonesia, Malaysia, and Singapore.40 Firm, fibrous, sweet to acid-sweet, juicy fruit with a strong smell and turpentine flavour: edible.
Resistance to anthracnose.Limus Perrennis M. foetida Monoembryonic Malesia Western part of Malesia (Sumatra, Java, Borneo, Malay Peninsula), wild and cultivates, introduced to Burma, Indochina. 40 Fruit with strong turpentine smell, sweet and pleasant, very fibrous: edible. Mangifera Mangifera Ding Hou M.altissima Polyembryonic Philippines Native to Philippines, Indonesia, Papua New Guinea, Solomon Islands. Unknown Smooth fibrous or non-fibrous fruit: edible. Limus Deciduae M. caesia Monoembryonic Indonesia Natural distribution: Peninsular Malaysia, Borneo (Brunei Darussalam, Indonesia, Malaysia) and Sumatra (Indonesia).
Cultivated from Peninsular Thailand, Bali, Java and to the Philippines.40 Sweet flesh with strong fragrance in the fruit: edible. Mangifera Mangifera Ding Hou M. zeylanica Monoembryonic Sri Lanka Endemic to Sri Lanka, not found under cultivation. Unknown Very juicy fruit, fibrous, pleasant, sweet taste pulp: edible. Mangifera Mangifera Ding Hou M. sylvatica Polyembryonic Myanmar Native to India (Sikkim), China, Myanmar, Thailand, Bangladesh, and Nepal. 40 Fruits almost fibreless, little pulp, sweet and sour taste: edible. Mangifera Mangifera Ding Hou M. quadrifida Unknown Malesia Sumatra, Malay Peninsula, Borneo, and Sunda Islands. Unknown Fruits with less fibres, sweet and pleasant smell. Mangifera Mangifera Ding Hou M. lalijiwa Polyembryonic Indonesia Indonesia (Java, Madura, Bali and probably in Sumatra). Very rare both in wild and in cultivation. Unknown Small glossy yellowish fruit with acid-sweet taste: edible. Mangifera Mangifera Ding Hou M. applanata Polyembryonic Malesia Native to Indonesia (Kalimantan and Sumatra) and Malaysia (Pahang, Sabah, and Sarawak).
Cultivated in some areas of Borneo.Unknown Juicy, very acid fruit with turpentine and lemon taste: edible. Mangifera Mangifera Ding Hou M. casturi Polyembryonic Indonesia Endemic to South Kalimantan, Indonesia.
Not known in the wild, only found in cultivation.Unknown Small fruits, with an attractive purple color and a distinctive aroma: edible. Mangifera Mangifera Ding Hou M. indica cv. 'Kensington Pride' Polyembryonic Australia First planted in Bowen, North Queensland, Australia.
Pre-Australian origin is unknown.
Grown throughout Australia.40 Soft and juicy flesh with moderate to little
fibre, sweet with a characteristic flavour excellent eating quality.Mangifera Mangifera Ding Hou M. i ndica cv. 'Alphonso' Monoembryonic India Prominently grown in India. 40 Firm to soft flesh, low in fibre content, sweet with characteristic aroma, very pleasant taste: edible. Mangifera Mangifera Ding Hou M. i ndica cv. 'Tommy Atkins' Monoembryonic USA (Florida) Originated and most widely grown commercial cultivar in USA. 40 Fruit with firm, medium juicy, medium amount of fibre of good eating quality.
Highly resistant to anthracnose disease.Chloroplast genome assembly
-
In addition to generating sequence data for the 14 Mangifera species, publicly available Illumina sequencing paired-end reads were downloaded from the National Centre for Biotechnology Information (NCBI) (
www.ncbi.nlm.nih.gov ) for five Mangifera species namely M. sylvatica, M. odorata, M. persiciformis, M. hiemalis and M. indica cv. Tommy Atkins ( Supplemental Table S2). Chloroplast genomes for each species were assembled using two methods: a chloroplast assembly pipeline (CAP) [ 35] in CLC Genomic Workbench (CLC-GWB) software (CLC Genomics Workbench 20.0,www.clcbio.com ) and 'Get Organelle' pipeline (http://github.com/Kinggerm/GetOrganelle ) [ 36] . Raw reads for all the species were imported to CLC-GWB and trimmed using the quality score limits of 0.01. The CAP processed two approaches to assemble the chloroplast genome, a reference-guided mapping approach and a de-novo assembly approach. For the reference guided mapping, M. indica cv. Tommy Atkins chloroplast genome (Accession: NC_035239.1) [ 37] was used as the reference. The two chloroplast sequences generated using the two approaches of the CAP for each species were aligned in Geneious 2022.2.2 software (www.geneious.com ) and Clone Manager Professional 9 to identify mismatches. Manual curation of mismatches involved observing the reads mappings at the position of the mismatch. De-novo assembled chloroplast genomes from Get Organelle pipeline were checked in Bandage v. 0.8.1 [ 38] to visualize the completeness of the assembled genomes. The final chloroplast genome assembled from CAP and Get Organelle pipeline were compared for mismatches and further manual curation, ensuring high-quality chloroplast genomes were assembled for all the species.Chloroplast genome annotation and identification of single nucleotide polymorphisms (SNPs), insertions, and deletions (INDELs)
-
Genome annotations were performed using GeSeq tool (
https://chlorobox.mpimp-golm.mpg.de/geseq.html ) and M. indica cv. Tommy Atkins (Accession: NC_035239.1) was used as the reference. Based on the phylogenetic relationships observed in chloroplast phylogeny, closely related species within the main clades and subclades were compared to determine their evolutionary relationships. The chloroplast genomes of the species were subjected to pairwise alignment in Geneious using the MAFFT alignment tool (MAFFT v7.490) [ 30] , and the number INDELs, substitutions and SNPs present between the sequences were identified.Nuclear gene sequence assembly
-
A set of single-copy nuclear genes was used to analyze phylogenetic relationships among the species. To analyze hybridization/introgression possibilities within the genus, it was necessary to develop individual gene trees to determine if they showed a discordance from the average phylogeny since different genes can have different evolutionary histories. The coalescence/ASTRAL approach, which employs multiple genes to develop gene trees to determine the degree to which they exhibit the same topology was applied.
Details of the genes were not available for Mangifera species. Therefore, Citrus sinensis, the closest relative of M. indica for which the details of single-copy nuclear genes were available [ 39] was used as the reference, to extract corresponding single-copy genes in mango. Single copy genes (107) in C. sinensis were mapped against the coding DNA sequences/gene models of M. indica cv. 'Alphonso' [ 29] in CLC-GWB. Out of 107, 47 were identified as single-copy genes in M. indica ( Supplemental Table S3). Then, trimmed paired-end illumina reads of each species were mapped against 47 single-copy genes of M. indica, and consensus gene sequences were extracted. The same 47 gene sequences were extracted from the outgroup ( A. occidentale) used in phylogenetic analysis.
Phylogenetic analysis
Chloroplast genome-based phylogenetic analysis
-
For chloroplast genomes-based phylogenetic analysis, sequences were imported to the Geneious and aligned using MAFFT alignment tool [ 30] . Two methods were used for phylogenetic analysis: Maximum likelihood (ML) method and Bayesian inference (BI) method. jMfodelTest v2.1.4 [ 40] was used to select the best-fitting nucleotide substitution model ( Supplemental Table S4). ML analysis was performed in RaXML GUI 2.0 (v 2.0.10) [ 41] with 1,000 bootstrap replicates. Bayesian analysis was carried out in Geneious software using MrBayes v. 3.2 [ 42] ( Supplemental Table S4). iTOL v.6 tool (
https://itol.embl.de ) [ 43] was used to visualize and edit the phylogenies. Using posterior probability (PP) and bootstrap support (BS) to evaluate the supports of the phylogenetic tree implemented under BI and ML methods respectively.Nuclear gene-based phylogenetic analysis
-
For nuclear gene sequences, phylogenetic trees were generated using two approaches: gene concatenation and coalescent approach to analyze any topological incongruence and for a better understanding of evolutionary relationships among species.
Gene concatenation approach
-
All 47 single-copy genes were concatenated in the same order to obtain one long sequence per species. Sequences for all the species were imported to the Geneious 2022.2.2 software and aligned by MAFFT alignment. Phylogenetic trees were constructed using ML method and BI methods after selecting the best-fitting nucleotide substitution model by running jModelTest v2.1.4 [ 40] . ML analysis was performed in RAxML (version 8) [ 41] with 1,000 bootstrap replicates, and Bayesian analysis was carried out in Geneious software using MrBayes v. 3.2 [ 42] ( Supplemental Table S4). iTOL v.6 tool (
https://itol.embl.de ) [ 43] was used to visualize and edit the phylogenetic trees.Coalescent approach
-
Single ML gene trees were constructed using RAxML (version 8) [ 41] . The best-scoring ML tree was searched using a GTR + GAMMA model with 1000 bootstrap replicates. Low support branches (BS < 10%) in gene trees were collapsed to minimize the potential impacts of gene tree error for species tree reconstruction. The gene trees were used to construct a coalescent-based species tree using ASTRAL-III [ 44] .
-
Illumina sequencing conducted for the 19 samples belonging to 14 Mangifera species in this study resulted in 55,999,560 to 181,601,786 raw reads with 150 bp mean read length. The number of trimmed paired-end reads trimmed at 0.01 quality limits (Phred score > 20) ranged between 52,547,125 and 171,303,402 reads. As the data size of the trimmed reads of all 19 samples corresponded to over 20 x of the genome size, all were selected for the chloroplast assembly ( Supplemental Table S1). Raw reads downloaded from NCBI for five Mangifera species ( M. odorata, M. sylvatica, M. persiciformis, M. hiemalis, and M. indica cv. Tommy Atkins) had a total of 99,649,506 to 127,708,722 reads which ranged from 94,450,606 to 117,445,811 after trimming at 0.01 quality limits. For all the species, the mean coverage was higher than 20x genome size, which enabled them to be included in the analysis ( Supplemental Table S2). The Get Organelle pipeline resulted in two output files for the chloroplast genome for each species/genotype, due to the possibility of the SSC occurring in both orientations in the chloroplast genomes in plants. Therefore, the two chloroplast sequences for each species were aligned with the reference ( M. indica; Accession: NC_035239.1) in Clone Manager Professional 9 to select the sequence with the widely accepted SSC orientation (5'LSC3':5'IR13':5SSC3':3'IR25').
The size of the chloroplast genomes of 15 wild Mangifera species and three cultivars of M. indica ranged from 151,752 to 158,965 bp of which the smallest and the largest genomes were recorded for M. caesia and two M. laurina respectively ( Table 2). The typical quadripartite structure of the chloroplast genome was recorded in all 16 Mangifera species and the lengths of LSC, SSC, and IR regions ranged between 86,507 to 98,334 bp, 18,319 to 19,064 bp, and 17,177 to 26,412 bp respectively where overall guanine–cytosine content (GC content) ranged from 37.6% to 37.9%. The chloroplast genomes for all species had the same number of total genes (115), rRNA (4) and tRNA (31) and protein encoding genes (80) ( Table 2). Although the size of the chloroplast genomes varies across the Mangifera species, three cultivars of M. indica, two M. laurina samples, and two M. altissima samples had identical chloroplast genomes. Two M. odorata samples had slightly different chloroplast genome sizes, where the accession we sequenced had a genome size of 158,889 bp while the sample for which the data was downloaded from NCBI ( M. odorata*) had a genome size of 158,883 bp, representing a 6 bp difference. The length difference was due to two deletions revealed in M. odorata*, one located in a non-coding region of LSC while the other located in the intron1 region of the PetD gene of LSC. The chloroplast genomes of two M. pajang samples collected from Treefarm, El Arish ( M. pajang) and Durian Heaven Farm ( M. pajang ‡ ) had 57 bp difference in length with 27 variants (insertions, deletions, SNPs, substitutions) between two genomes. However, only four SNPs were identified within coding regions of matK, atpA, rps2, and psbC genes in LSC, and none of these SNPs resulted in a change in amino acid sequence causing no effect on the produced protein. Of the remaining SNPs, four were in non-coding regions of LSC, one in the intron region of the pafI gene and two in the intron region of the NdhA gene. Except for one insertion and one deletion observed in intron regions of petD and trnK-UUU genes, respectively, all other insertions, deletions, and substitution occurred in non-coding regions of both the LSC and SSC. In addition, except of one deletion, all identified insertions, were characterized as tandem repeats, spanning lengths of 1-2 bp. Upon examination of two M. sylvatica samples, it was noted that they had different chloroplast genome sizes. Specifically, the accession sequenced in the present study was 245 bp larger than the accession data downloaded from NCBI ( M. sylvatica*). Comparative analysis of annotated chloroplast genomes revealed 183 variants, including 119 SNPs, 29 insertions, 27 deletions, and eight substitutions in M. sylvatica*. However, only 47 variants were in the coding region (46 SNPs, one insertion) resulting in codon change in 13 genes. The chloroplast sequence of M. indica cv. 'Kensington Pride' ( Fig. 1) is a representation of the chloroplast sequence of the 16 Mangifera species, which have the same number of genes. Despite this consistency, differences exist in total chloroplast size, as well as the sizes of the LSC, IR1, and IR2, and the SSC regions.
Table 2. Annotation of the chloroplast genomes of Mangifera species.
Species/
genotypeGenome
size (bp)Overall GC content LSC (bp) SSC (bp) IR (bp) Total no.
of genesTotal no.
of protein coding genesTotal no.
of tRNAsTotal no.
of rRNAsM. laurina 158,965 37.8% 87,714 18,427 26,412 115 80 31 4 M. laurina † 158,965 37.8% 87,714 18,427 26,412 115 80 31 4 M. pajang 158,830 37.8% 87,654 18,424 26,376 115 80 31 4 M. pajang ‡ 158,887 37.8% 87,709 18,426 26,376 115 80 31 4 M. caloneura 157,780 37.9% 86,672 18,350 26,379 115 80 31 4 M. odorata 158,889 37.8% 87,708 18,427 26,377 115 80 31 4 M. foetida 158,882 37.8% 87,706 18,422 26,377 115 80 31 4 M. altissima 157,780 37.9% 86,673 18,349 26,379 115 80 31 4 M. altissima † 157,780 37.9% 86,673 18,349 26,379 115 80 31 4 M. caesia 151,752 37.6% 98,334 19,064 17,177 115 80 31 4 M. zeylanica 157,604 37.9% 86,507 18,319 26,389 115 80 31 4 M. lalijiwa 157,779 37.9% 86,672 18,349 26,379 115 80 31 4 M. applanata 157,779 37.9% 86,672 18,349 26,379 115 80 31 4 M. casturi 158,942 37.8% 87,733 18,425 26,392 115 80 31 4 M. quadrifida † 158,889 37.8% 87,679 18,424 26,393 115 80 31 4 M. sylvatica † 158,025 37.9% 86,856 18,387 26,391 115 80 31 4 M. sylvatica* 157,781 37.9% 86,712 18,347 26,361 115 80 31 4 M. odorata* 158,883 37.8% 87,702 18,427 26,377 115 80 31 4 M. persiciformis* 158,952 37.8% 87,566 18,536 26,368 115 80 31 4 M. hiemalis* 158,838 37.8% 87,681 18,535 26,368 115 80 31 4 M. i ndica cv. 'Kensington Pride' 157,780 37.9% 86,673 18,349 26,379 115 80 31 4 M. indica cv. 'Alphonso' 157,780 37.9% 86,673 18,349 26,379 115 80 31 4 M. indica cv. 'Tommy Atkins'* 157,780 37.9% 86,673 18,349 26,379 115 80 31 4 GC, guanine or cytosine; IR, inverted repeats; LSC, large single copy; SSC, small single copy. * Mangifera species for which chloroplast genomes were assembled using raw data downloaded from NCBI. † Mangifera species collected from the Botanical Ark, Queensland, Australia. ‡ Mangifera species collected from the Durian Heavan Farm, Queensland, Australia. M. pajang is collected from TreeFarm, Queensland, Australia. 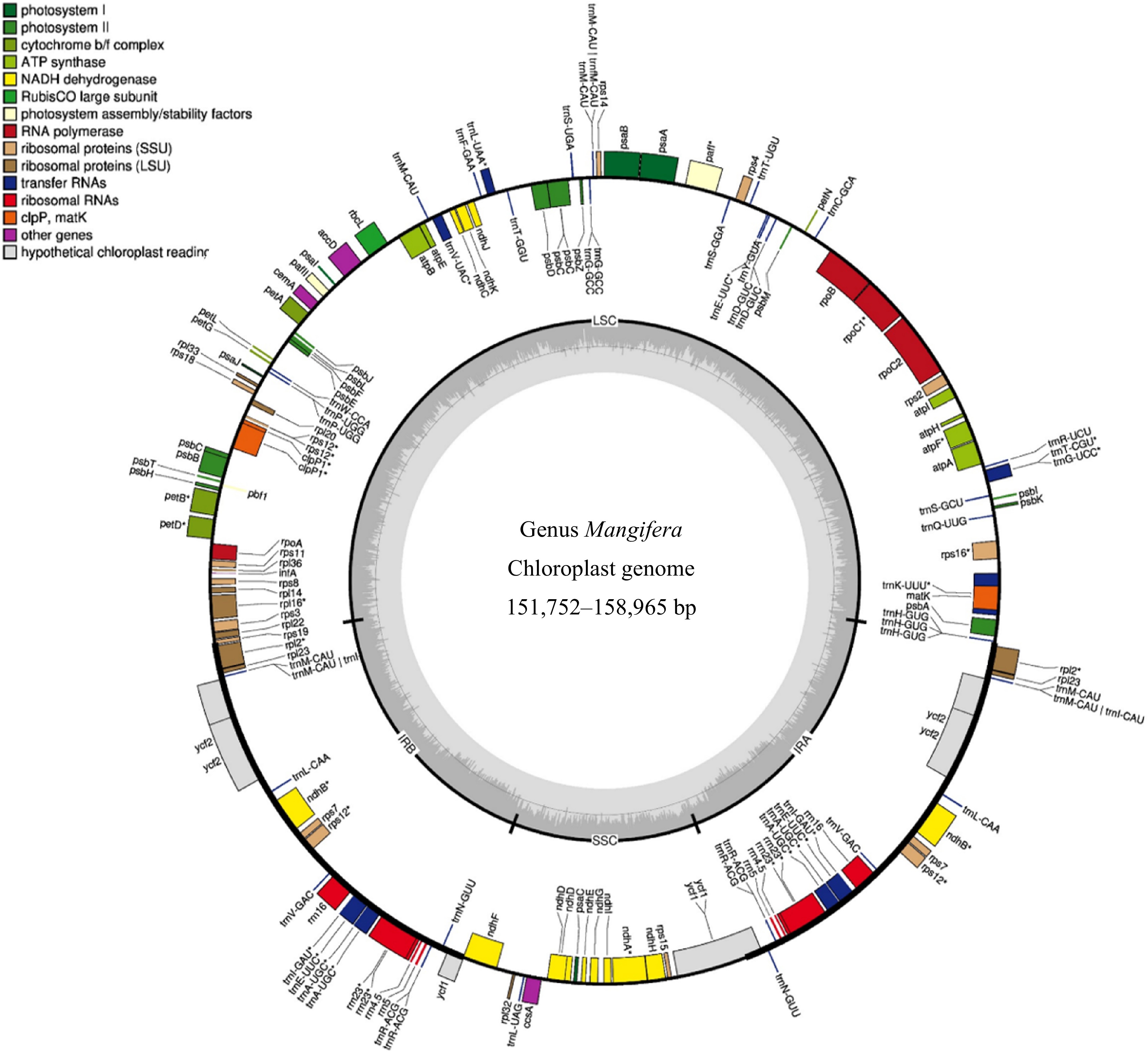
Figure 1.
Genome map of the chloroplasts in the genus Mangifera. The genome size of the 16 Mangifera species ranges from 151,752 to 158,965 bp for M. caesia and M. laurina, respectively. In the most outer circle, the thick black border/line indicates Inverted Repeat Regions (IR) whereas the thin lines indicate Large Single Copy (LSC) and the Small Single Copy (SSC). Genes inside the circle are transcribed in the clockwise direction whereas the genes outside the circle are transcribed in the counter-clockwise direction. Different colours are given for the genes with respect to their functions. The darker grey in the inner circle corresponds to GC content, whereas the lighter grey corresponds to AT content.
Chloroplast phylogeny and identification of SNPs and INDELs
-
A multiple chloroplast sequence alignment conducted using A. occidentale as the outgroup followed by phylogenetic tree construction resulted in an ML tree and a Bayesian tree with same tree topology. BS and PP values of the final tree are presented in Fig. 2. The model of nucleotide substitutions for ML analysis was GTR + G, whereas, for the Bayesian analysis, it was TPM1uf + G. The tree developed with the ML approach showed a BS of 100 at most of the nodes and a PP of one in all the nodes. In the whole plastome tree, three main clades were identified. First, 16 Mangifera species were clustered into two distinct clades in which only M. caesia belonging to section Dissidue in the subgenus Limus was placed in the first clade (Clade A). The other 15 species were grouped into a separate clade indicating their evolutionary distinct relationship to M. caesia, which were then clustered into two subclades (Clade B and Clade C). Clade B included a total of eight species that belong to different categories in the classification. M. pajang, M. foetida, and M. odorata belong to the sub genus Limus while M. casturi, M. quadrifida, and M. laurina belong to the subgenus Mangifera. M. persiciformis, and M. hiemalis are two species placed under uncertain position in the classification. Within Clade B, species in subgenera Mangifera (Clade BI), Limus (Clade BII), and species being classified in an uncertain position (Clade BIII) have localized into well-supported distinct clades (BS = 100, PP = 1). The species belong to subgenera Mangifera and Limus and were sisters to each other and both together have become a sister clade to species placed in uncertain positions in the classification. Clade C had species belonging only to the subgenus Mangifera. Interestingly, four wild species ( M. lalijiwa, M. applanata, M. altissima, and M. caloneura) were clustered with three cultivars of domesticated mango ( M. indica) (Clade CI). Although species belonging to sections Mangifera and Euantherae are characterized by the presence of one and multiple fertile stamens respectively, M. caloneura in section Euantherae was clustered with species belonging to section Mangifera. Furthermore, M. zeylanica, and M. sylvatica (both samples); two species within Clade C were separately clustered into distinct clades (Clades CII and III). Therefore, the phylogeny based on whole chloroplast genome clustered species belong to different groups, inferring the close genetic and evolutionary relationships of their chloroplast genomes ( Fig. 2).
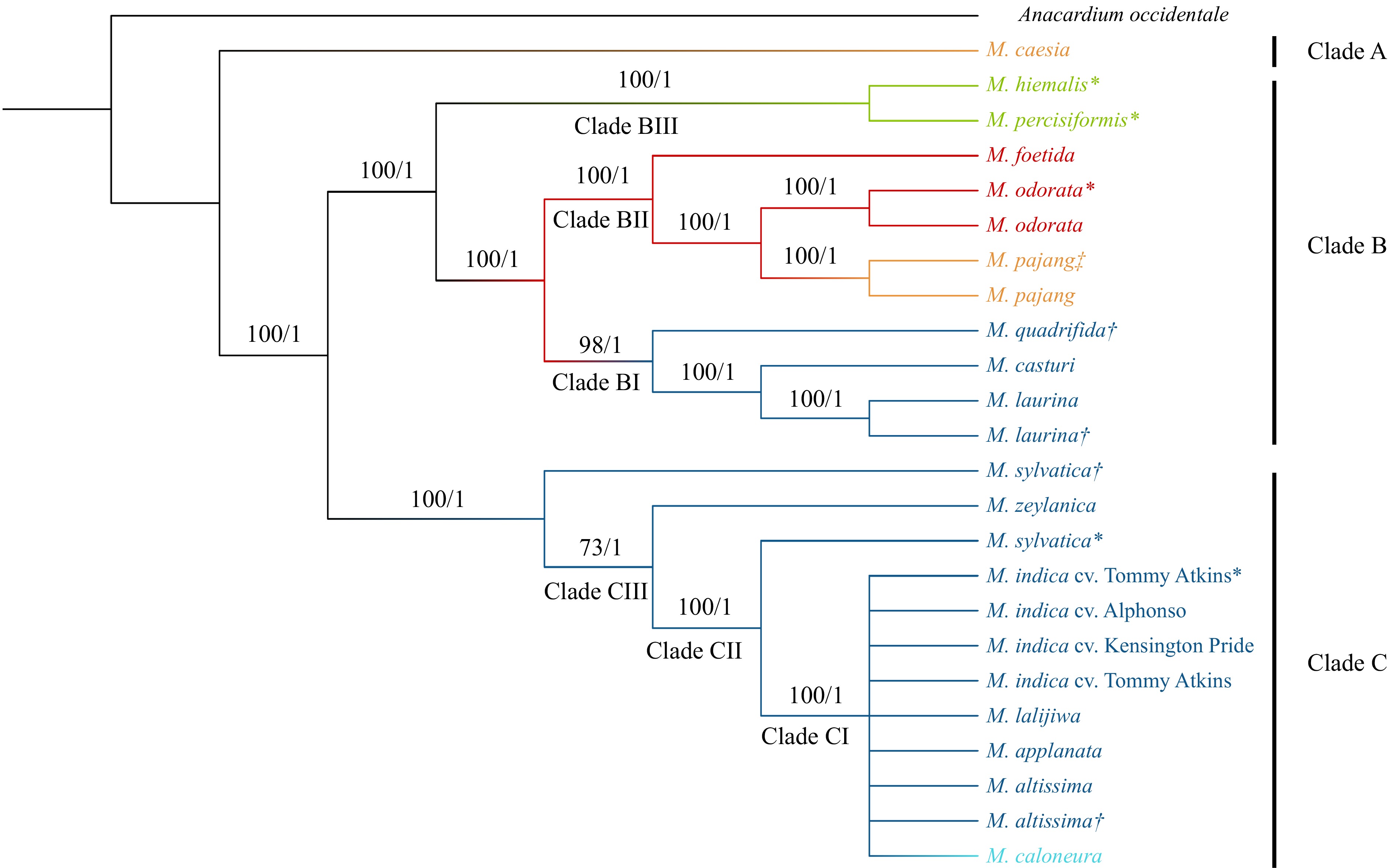
Figure 2.
Phylogenetic tree developed for Mangifera species based on whole chloroplast genomes. The phylogenetic tree of 24 accessions belongs to 16 species with A. occidentale used as the outgroup. Trees were generated using Maximum Likelihood (ML) and Bayesian inference (BI) method. Numbers associated with the branches are ML bootstrap value (/100) and BI posterior probabilities (/1). Dark Blue: Sub genus Mangifera, Section Mangifera, Light blue: Sub genus Mangifera, Section Euantherae, Red: Sub genus: Limus, Section Perrennis, Yellow: Sub genus: Limus, Section: Deciduae, Light Green: species placed in uncertain position in the classification. * Mangifera species for which chloroplast genomes were assembled using raw data downloaded from NCBI. † Mangifera species collected from the Botanical Ark, Queensland, Australia. ‡ Mangifera species collected from the Durian Heaven Farm, Queensland, Australia.
The assembled chloroplast genomes were imported to Geneious software to conduct pairwise alignment to identify the number and types of variants present between the species clustered as sister taxa in the two main clades (Clade B and C) of the chloroplast phylogeny. In Clade B, two M. laurina samples had identical chloroplast genomes whereas two M. odorata samples had two structural variants (deletions). The chloroplast genome of M. pajang had a total of 27 structural variants including one substitution, 11 SNPs, seven insertions, and eight deletions compared to M. pajang. A total of 116 variants were observed between M. laurina and M. casturi while 119 variants were found between M. casturi and M. quadrifida. Furthermore, a total of 75 variants were found between M. odorata and M. pajang while there were 108 variants between M. odorata and M. foetida ( Table 3). In addition, the two species M. persiciformis and M. hiemalis, for which the chloroplast genomes were assembled from raw read data available in NCBI, differed by 45 variants, overall revealing the close evolutionary relationships among the taxa in Clade B. In Clade CI, pairwise comparison of wild species with M. indica cv Kensington Pride showed that, M. altissima and M. indica had identical chloroplast sequences. Moreover, M. lalijiwa and M. applanata also had an identical chloroplast genome which differed from M. indica only by having a single nucleotide deletion located in a non-coding region in LSC. Furthermore, despite reporting distinct morphological characteristics from M. indica, M. caloneura only had one single nucleotide insertion and one single nucleotide deletion in non-coding regions compared to M. indica. Diversity within the chloroplast genomes clustered in Clade CI was very low.
Table 3. INDELs, SNPs and substitutions identified with respect to clustering pattern in chloroplast phylogeny.
Clade name in the phylogenetic tree Species/
genotypes in comparisonSpecies/
genotype ASpecies/
genotype BTotal no. of
variations between A vs BTypes of variation Insertions Deletions SNPs Substitutions BI M. laurina
M. laurina †
M. casturi
M. quadrifida †M. laurina M. laurina † 0 − − − − M. laurina M. casturi 116 30 30 51 5 M. casturi M. quadrifida 191 32 25 119 15 BII M.odorata
M. odorata*
M. pajang
M. pajang ‡
M. foetida †M. odorata M. odorata* 2 − 2 − − M. pajang M. pajang ‡ 27 7 8 11 1 M. odorata M. pajang 75 17 16 39 3 M. odorata M. foetida 108 21 16 68 3 BIII M. persiciformis*
M. hiemalis*M. persiciformis* M. hiemalis* 45 9 9 27 − CI M. altissima
M. altissima †
M. lalijiwa
M. applanata
M. caloneura
M. indica cv. 'Kensington Pride'
M. indica cv. 'Tommy Atkins'
M. indica cv. 'Alphonso'M. indica cv.
'Tommy Atkins'M. indica cv.
'Kensington Pride'0 − − − − M. indica cv.
'Tommy Atkins'M. indica cv.
'Tommy Atkins'*0 − − − − M. indica cv. 'Alphonso' M. indica cv. 'Kensington Pride' 0 − − − − M. indica cv. 'Alphonso' M. indica cv.
'Tommy Atkins'0 − − − − M. altissima M. altissima † 0 − − − − M. altissima M. indica cv. 'Kensington Pride' 0 − − − − M. lalijiwa M. indica cv. 'Kensington Pride' 1 − 1 − − M. applanata M. indica cv. 'Kensington Pride' 1 − 1 − − M. caloneura M. indica cv. 'Kensington Pride' 2 1 1 M. lalijiwa M. applanata 0 − − − − M. caloneura (Xoài Quéo) M. lalijiwa 3 2 1 − − CII M. sylvatica*
M. indica cv. 'Kensington Pride'M. sylvatica* M. indica cv. 'Kensington Pride' 96 22 14 57 3 CIII M. zeylanica
M. indica cv. 'Kensington Pride'M. zeylanica M. indica cv. 'Kensington Pride' 175 18 26 122 9 * Species for which chloroplast genomes were assembled using raw data downloaded from NCBI. † Mangifera species collected from the Botanical Ark, Queensland, Australia. ‡ Mangifera species collected from the Durian Heavan Farm, Queensland, Australia. Nuclear gene phylogeny
Concatenation-based nuclear phylogeny
-
The same approach was used to construct a nuclear phylogeny with a concatenation-based approach as was applied in constructing the chloroplast phylogeny. A. occidentale was used as the outgroup. A total of 47 common single-copy nuclear genes out of 107 [ 39] were identified and selected for Mangifera species. The multiple-sequence alignment was 71,881 bp in length and ML and Bayesian trees resulted in almost the same tree topology. The final tree with BS values and PP values is presented in Fig. 3a. Although some of the nodes showed less BS support values, all the nodes were supported with high PP values. The model of nucleotide substitutions for ML analysis was GTR + I + G whereas TPM1 + I + G was used for the Bayesian analysis.
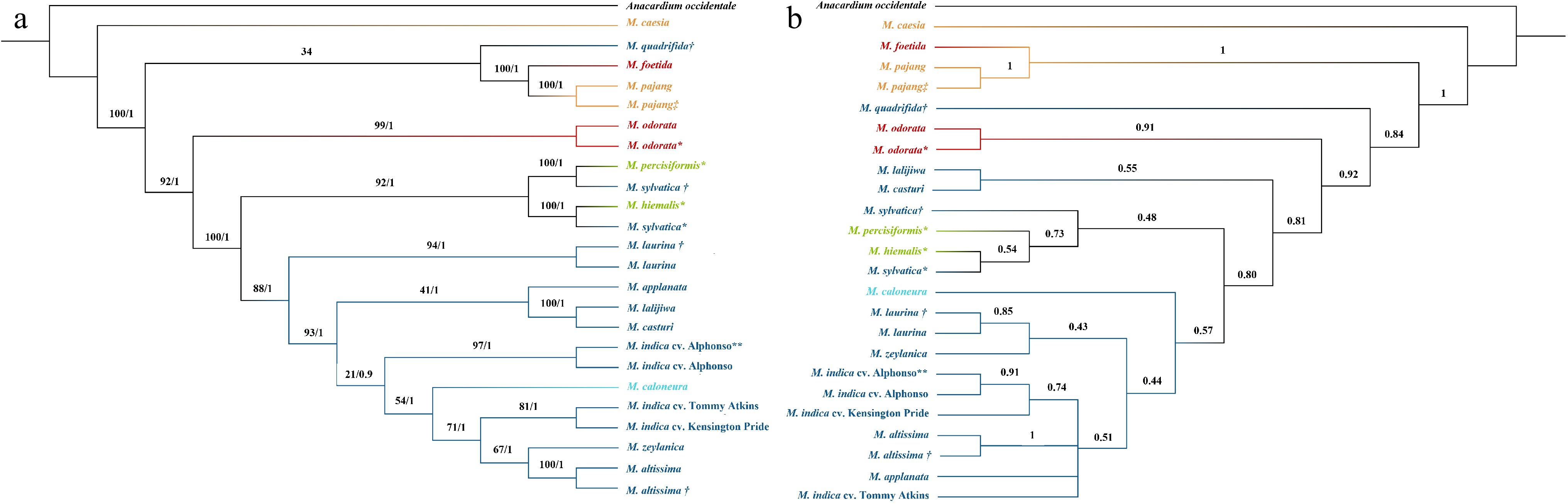
Figure 3.
Phylogenetic tree developed for Mangifera species based on a selected set of nuclear genes using (a) concatenation and (b) coalescence-based methods. Phylogenetic tree of 24 accessions with A. occidentale used as the outgroup. Concatenation-based trees were generated using Maximum Likelihood (ML) and Bayesian inference (BI) methods and consensus tree is shown in the figure. Numbers associated with the branches are ML bootstrap value (/100) and BI posterior probabilities (/1). In the coalescence-based tree (ASTRAL tree), numbers associated with branches are local posterior probability values (/1). Dark Blue: Sub genus Mangifera, Section Mangifera, Light blue: Sub genus Mangifera, Section Euantherae, Red: Sub genus: Limus, Section Perrennis, Yellow: sub genus: Limus, Section: Deciduae. * Species for which nuclear genes were extracted using raw data downloaded from NCBI. ** M. indica cultivar from which gene models were downloaded from NCBI and used to create local database in CLC-GWB for the selection of single copy nuclear genes in M. indica. † Mangifera species collected from the Botanical Ark, Queensland, Australia. ‡ Mangifera species collected from the Durian Heaven Farm, Queensland, Australia.
Except for M. sylvatica and M. quadrifida, the other eight species belonging to subgenus Mangifera were clustered into one main distinct clade. Among the eight clustered species, two M. laurina samples clustered as sister taxa and is the most distinct clade from the others. Of the seven remaining species, M. lalijiwa, M. applanata with M. casturi were clustered into one clade while M. altissima, M. caloneura, M. zeylanica, and the M. indica cultivars were clustered into another clade within the main clade. Two M. altissima samples were clustered into one clade and M. casturi and M. lalijiwa were sister taxa to each other. Furthermore, showing the close genetic relationship of the two M. indica cultivars (Kensington Pride and Tommy Atkins) to M. zeylanica and M. altissima revealed a close evolutionary relationship of the two wild species to domesticated mango. Moreover, although two M. sylvatica species closely related to species in the domesticated clades in chloroplast phylogeny, they were clustered with the two species placed in an uncertain position in the classification in the nuclear phylogeny. In addition, M. quadrifida was clustered with M. foetida and M. pajang; two species belong to the subgenus Limus. Both chloroplast and concatenation-based nuclear phylogenies revealed that M. caesia is evolutionarily distant from the rest of the Mangifera species ( Figs 2 & 3a). Grouping of species in both chloroplast genome and nuclear genes-based analysis does not completely concur with the accepted classification [ 6] for genus Mangifera. Incongruence in tree topologies could be seen between the phylogenies developed based on the whole plastome genome and the nuclear genes.
Coalescence-based nuclear phylogeny
-
Previously proposed hybrids and their proposed parents were included in the dataset and the possibility of hybridization events also were observed for some species when compared chloroplast and concatenation-based nuclear phylogenies. Therefore, to further analyze the phylogenetic relationships among Mangifera species with respect to nuclear genes, a coalescence approach was utilised to develop individual nuclear gene trees thereby developing a species tree. Individual gene trees were analyzed to see close evolutionary relationships among species.
In coalescence-based species tree, local posterior probability support (LPP) values are indicated in the branches (/1). In both concatenation and coalescence based nuclear phylogenies, except M. quadrifida, species belonging to subgenus Mangifera, and species placed in uncertain positions in the classification were clustered in one clade with high support values (BS = 100, PP = 1, LPP = 0.81) ( Fig. 3). Within this clade, the pattern of clustering into sub-clades was different for some species between the two nuclear phylogenies but BS and LPP support values also were low for some sub-clades. In both concatenation and coalescence-based phylogenies, M. casturi and M. lalijiwa are clustered together as sister taxa (BS = 100, PP1, LPP = 0.55) and M. hiemalis, M. persiciformis, and two M. sylvatica samples were clustered into one sub-clade (BS = 100, PP = 1, LPP = 0.48). In the coalescence-based tree, M. hiemalis, M. sylvatica, and M. persiciformis were closely related to six species belonging to the subgenus Mangifera ( M. altissima, M. applanata, M. indica, M. caloneura, M. zeylanica, and M. laurina). However, in the concatenation-based tree, M. casturi, and M. lalijiwa were more closely related to the six species than M. sylvatica, M. persiciformis, and M. hiemalis.
M. odorata is a proposed hybrid between M. indica and M. foetida, and M. casturi is a proposed hybrid between M. indica, and M. quadrifida. Within the present dataset, there are both parental and hybrid species. Therefore, 47 nuclear gene trees were analyzed to support the hybridity by recording the number of gene trees where the hybrids were clustered with their parents. Out of 47 gene trees, M. odorata was clustered with M. indica as sister taxa in only four gene trees and they were not supported with high BS values ( Table 4, Supplemental Fig. S1). Similarly, M. odorata was clustered with M. foetida as sister taxa only in four gene trees and one of the trees showed less BS value ( Table 4, Supplemental Fig. S1). M. casturi, was clustered with M. indica as a sister taxa in only two gene trees where BS values were weak in one of them ( Table 4, Supplemental Fig. S1). Furthermore, only one gene tree had M. casturi and M. quadrifida clustered into one clade that had less BS. Therefore, for both the proposed hybrid species, the number of gene trees in which the proposed hybrids clustered with their parental species was low and showed low BS values.
Table 4. Gene trees indicating clustering of suggested hybrids/ wild relatives with their proposed parents as sister taxa.
Gene name Gene
tree no.Species proposed to be hybrid Species supposed to be undergone domestication/hybridization Trees in which M. sylvatica clusters with M. persiciformis
in the same clade as sister taxaM. odorata M. casturi M. zeylanica M. sylvatica Trees in which M. odorata clusters with M. indica in the same clade as sister taxa Trees in which M. odorata clusters with M. foetida in the same clade as sister taxa Trees in which M. casturi clusters with M. indica in the same clade as sister taxa Trees in which M. casturi clusters with M. quadrifida in the same clade as sister taxa Trees in which M. zeylanica clusters with M. indica in the same clade as sister taxa Trees in which M. sylvatica clusters with M. hiemalis in the same clade as sister taxa 4-alpha-glucanotransferase 1 × × × × √ × × A49-like RNA polymerase I associated factor 2 × × × × √ × × Acyl-CoA dehydrogenase,
C-terminal domain3 × × × × × √ × Alphabeta hydrolase family 4 √ × × × √ × √ Aminomethyltransferase folate-binding domain 5 × × × × × × × Aminopeptidase I zinc metalloprotease (M18) 6 × × × × × × × ArgJ family 7 × × × × × × × Armadillobeta-catenin-like repeat 8 × × × × × × × Brix domain 9 × × × × × × √ Cactus-binding C-terminus of cactin protein 10 × × × × × × × Carbon-nitrogen hydrolase 11 √ × × × × × × CobWHypBUreG, nucleotide-binding domain 12 × × × × × × × Cohesin loading factor 13 × × × × × × × Creatinase Prolidase N-terminal domain 14 × × × × × × × Cyclophilin type peptidyl-prolyl cis-trans isomeraseCLD 15 × × × × × × × Cytochrome b5-like HemeSteroid binding domain 16 × × × × × × × DDRGK domain 17 × √ × × × × × Dienelactone hydrolase family 18 × √ × × × √ × Divergent CRALTRIO domain 19 × × × × × √ × Dual specificity phosphatase, catalytic domain 20 × × × × √ √ × Dynamin family (2) 21 × × × × × × × ER membrane protein complex subunit 1, C-terminal 22 × × × × × × × Eukaryotic protein of unknown function (DUF866) 23 × × √ × × × × FAD binding domain 24 × × × × √ × × Glucose-6-phosphate isomerase 25 × × × × × × √ Glucosidase II beta subunit-like protein 26 × × × × × √ × Glycosyl hydrolases family 31 27 × × × × × √ × Glyoxalase bleomycin resistance protein dioxygenase superfamily 28 × × × × × × × Hydroxyacylglutathione hydrolase C-terminus 29 × × × × × × × Methyltransferase domain 30 × √ × × × × × NmrA-like family 31 × × × × √ √ × N-terminal domain of lipoyl synthase of Radical_SAM family 32 × × × × × × × PAP2 superfamily C-terminal 33 × × × × × × × Prolyl oligopeptidase, N-terminal beta-propeller domain 34 × × × × × × × Putative tRNA binding domain 35 × × × × √ √ × Pyruvate phosphate dikinase, AMPATP-binding domain 36 × × × × × √ √ Redoxin 37 √ × × × × × × Ribosomal protein L13 38 × × × √ √ √ × RNA recognition motif.
(a.k.a. RRM, RBD, or RNP domain)39 × × × × × √ × Rubrerythrin 40 × × × × × × √ snoRNA binding domain, fibrillarin 41 × × × × √ × × Sodium Bile acid symporter family 42 × √ × × √ × × TFIIS helical bundle-like domain 43 × × × × √ × × Transcription factor TFIIB repeat 44 √ × × × × √ √ tRNA synthetase class II core domain (G, H, P, S and T) 45 × × × × × × √ Uncharacterized ACR, YdiUUPF0061 family 46 × × √ × × × √ WLM domain 47 × × × × × × √ According to nuclear gene phylogeny, close evolutionary relationship between M. zeylanica and M. indica was observed where M. zeylanica was clustered with the species in domesticated clade. Analyzing individual gene trees revealed that M. zeylanica was clustered with M. indica as sister taxa in 11 gene trees and with M. altissima in eight gene trees while in some of the other gene trees, M. zeylanica clustered with species in domesticated clade. In addition, it was also observed that there is a close evolutionary relationship between M. hiemalis, M. persiciformis, and M. sylvatica in nuclear phylogenies. M. sylvatica species were clustered with M. hiemalis as sister taxa in 12 gene trees and with M. persiciformis in nine gene trees. Furthermore, all three species ( M. sylvatica, M. persiciformis, and M. hiemalis) were clustered into one subclade in four gene trees. However, individual gene trees for M. zeylanica, and M. sylvatica showed less BS support when clustering with M. indica and M. hiemalis/M. persiciformis respectively ( Table 4, Supplemental Fig. S1). Therefore, it was assumed that M. zeylanica might have undergone domestication and there is also a possibility that M. sylvatica may have cross-hybridised with M. hiemalis or M. persiciformis during the evolution of these species.
-
Determination of phylogenetic relationships among crop species provides basic information for predicting their evolutionary history, taxonomical classification, and evaluating their diversity and importance in plant breeding [ 45] . Although genetic analysis of plants has improved rapidly with advanced sequencing technology, many phylogenetic studies in the genus Mangifera have relied on a set of molecular markers such as amplified fragment length polymorphisms (AFLP), rapid amplified polymorphic DNA and simple sequence repeats and the sequencing of limited numbers of targeted regions in the chloroplast genome and nuclear ribosomal DNA [ 10, 13− 18] .
Chloroplast genomes for seven species were assembled for the first time in this study for M. pajang, M. altissima, M. caesia, M. lalijiwa, M. z eylanica, M. appalanta, and M. casturi. Different pipelines and programs are available to assemble organelle genomes. Here, CAP and Get Organelle pipeline have been used. The two approaches used in CAP (reference-guided mapping and de-novo assembly) eliminate many errors in genomes developed from each approach giving a highly accurate final chloroplast genome. The Get Organelle pipeline is also capable of generating all possible arrangements of the chloroplast genome present [ 36] . Therefore, a comparison of chloroplast genomes generated from CAP and the Get Organelle pipeline validated the development of highly accurate final chloroplast genomes for all the species. More genes have been annotated in our analysis compared to previous studies, which reported a total of 112 genes (78 protein-coding genes, 30 tRNA genes, four rRNA genes) [ 46] and 113 genes (79 protein-coding genes, 30 tRNA genes, four rRNA genes) [ 19] .
Phylogenetic relationships within the genus Mangifera showed topological incongruence for some species with respect to whole chloroplast and nuclear genes trees which maybe caused by introgressive hybridization, allopolyploidy or incomplete lineage sorting. Reproductive compatibility between different species allows the native cytoplasm of a species to be easily replaced by another through hybridization which has been detected both in animals (mitochondrial capture) [ 47] and plants (chloroplast capture) [ 48] . In plants, chloroplast capture events have been reported in many plant families [ 49− 52] . Hybridization followed by recurrent backcrossing have explained discrepancies between chloroplast and nuclear gene-based phylogenies in diverse families of plants [ 53− 56] . In mango, evidence for inter-specific reproductive compatibility was reported for M. indica and M. laurina. A cross between M. indica, and M. laurina have produced 60 successful hybrids [ 57] . Hybrid origins were reported for M. odorata and M. casturi.
Close genetic relationship between M. applanata, and M. altissima has been reported in a phylogenetic analysis conducted based on Maturase K gene [ 15] . In the present study, M. laijiwa, M. applanata, M. altissima, and M. caloneura were clustered with M. indica in the chloroplast phylogeny sharing 99.9% sequence similarity. These four wild relatives clustered with domesticated mango into a distinct clade even in concatenation-based nuclear phylogeny showing their close evolutionary relationships whereas only M. laijiwa out of the above four species clustered separately in the coalescent approach . Furthermore, M. indica cultivars Kensington Pride and Tommy Atkins were more closely related to M. altissima than M. indica cv. 'Alphonso' in concatenation-based nuclear phylogeny failing to resolve M. indica from M. altissima. A close evolutionary relationship between M. altissima, and M. indica was also confirmed in the coalescence approach. Due to remarkably close evolutionary relationships observed in chloroplast and nuclear phylogenies, we suggest these four wild relatives and cultivated mango are very closely related and might have shared or descended from a common ancestor.
Although a single domestication event has been reported for M. indica based on historical records [ 2] , two independent domestication events have been proposed in India and Indochina [ 7] . A population genomics study [ 58] suggested that mango domestication is a complex process and it may involve multiple domestication events and interspecific hybridization; two common phenomena observed in the domestication of perennial fruit crops. Their results indicated a high genetic diversity among M. indica cultivars distributed outside of the region where the mango originated and a unique genetic diversity in Southeast Asian cultivars compared to other populations. Furthermore, they suggest that the origin and initial cultivation of mango may have taken place in Southeast Asia and further improvement and domestication may have occurred in India. In addition, cross-hybridization was highly likely to occur between wild relatives and M. indica at the early stages of domestication due to the presence of a high number of species which is supported by evidence for crossbreeding. Thus, apart from descending from a common ancestor, cross-hybridization between M. indica and the four wild relatives is also a possible phenomenon that may have further contributed to the close evolutionary relationships observed in our study. However, this could be further supported by including multiple replicates for the species which is a limitation in this study.
M. zeylanica, is an endemic species to Sri Lanka. A close evolutionary relationship was observed in the concatenation-based nuclear phylogeny between M. zeylanica, and M. indica despite having a distinct chloroplast genome. Therefore, it was hypothesized that cross-hybridization might have occurred between an early lineage of M. zeylanica, and M. indica or its close wild relative. Since the species have a distinct chloroplast genome, it was assumed that M. indica may have most likely acted as the paternal parent, resulting in hybrids that carry the chloroplast genome of M. zeylanica and nuclear genes of both M. zeylanica, and M. indica or its close relative. The nuclear phylogeny/species tree based on the coalescence approach also showed a close relationship between M. indica, and M. zeylanica. Clustering of M. zeylanica with M. indica and with M. altissima which is a close relative to M. indica suggested the possibility of M. zeylanica having a hybrid origin. But as the BS/PP and LPP values are relatively low for this clade in gene trees as well as in both the consensus trees, it is also possible that the set of genes is not sufficiently variable to give a better resolution in the phylogeny. Therefore, it is difficult to conclude the cross-hybridization of M. zeylanica.
M. casturi is a cultivated species in Indonesia. This endemic species is only found in cultivation [ 59] and was proposed to be a natural hybrid between M. indica, and M. quadrifida according to a SNP analysis [ 25] . Since M. casturi has shown a higher affinity to M. indica than to M. quadrifida instead of being direct intermediate between two species, it was further suggested that M. casturi is most likely a result of an F1 hybrid backcrossed with M. indica [ 25] . Microsatellite marker-based analysis showed broad genetic variation among four M. casturi samples and DNA barcoding-based phylogenetic analysis suggested several species as ancestors for M. casturi [ 24] . Genetic variation has also been confirmed between 16 accessions of M. casturi using SNP markers (N. Dillon, pers. comm.). Therefore a combination of microsatellite and DNA barcoding data support that M. quadrifida and M. indica have hybridised to result in M . casturi and F1 hybrids may have further hybridized with the ancestors of the parental species or multiple other Mangifera species to generate hybrids with high genetic diversity [ 24] . In the present study, M. casturi has a distinct chloroplast genome and it is closely related to M. quadrifida in chloroplast phylogeny. However, a close evolutionary relationship was observed between M. casturi and species in the domesticated clade in the concatenation-based nuclear phylogeny where it clusters with M. lalijiwa as sister taxa and distinctly related to M. quadrifida. In contrast, in the coalescence approach, M. casturi showed a relatively distant evolutionary relationship with M. indica, and M. quadrifida both in species trees and in individual gene trees . Therefore, according to our results, coalescence-based nuclear phylogeny and gene trees don't strongly support the parentage of M. indica, and M. quadrifida for M. casturi. Since a very low number of genes are shared between M. indica/M. quadrifida, and M. casturi, is not possible that M. casturi is a first-generation hybrid if the two species are the parents. The F1 of M. casturi may have cross-hybridized with other wild relatives as previous study suggested [ 24] . Also, the absence of replicates for M. casturi, and M. quadrifida and other wild relatives limit analysis of any other species for the hybrid origin of M. casturi.
M. laurina is a cultivated species in Indonesia where its wild distribution ranges from Myanmar, Cambodia, Vietnam and Malesia, Thailand to New Guinea. Analysis of ITS genomic region [ 18] have revealed close evolutionary relationship between M. laurina, and M. indica. Analysis of Maturase K chloroplast genomic region has differentiated Indonesia and Thailand specimens collected for M. laurina. Since common interspecific hybridization has been suggested for this species [ 14] , it is possible that M. laurina may have cross-hybridized with other species after introduction to the regions where it is widely cultivated. Due to the relatively close evolutionary relationship observed between M. laurina, and M. indica in nuclear gene analysis despite the chloroplast genome being distinct, it might be possible to occur hybridization between the early lineage of M. laurina and M. indica its close relative. Current data and results only support the close evolutionary relationship between the two species, but further analysis should be conducted with multiple samples for both species.
Among M. pajang, M. foetida, M. odorata, M. persiciformis, and M. hiemalis clustered within the same main clade in chloroplast phylogeny, M. pajang is an endemic species originating from and cultivated in Borneo, Indonesia. Based on the AFLP marker analysis, M. odorata is proposed as a hybrid between M. indica, and M. foetida and it has shown more affinity to M. foetida than to M. indica [ 18, 23] . The present results also confirm that M. odorata is closely related to M. foetida than to M. indica according to both chloroplast and nuclear phylogenies. Since chloroplast genomes are relatively conserved, have less rate of evolution and in general, shows maternal inheritance (in angiosperms), hybrids share the chloroplast genomes of maternal parents. A study conducted on the inheritance of Solanum chloroplast genomes in four known interspecific hybrids revealed that two hybrids had identical chloroplast genomes while the other two showed only 2 bp difference with respect to their maternal parents [ 31, 60] . Furthermore, it was also revealed that only one hybrid had two substitutions in the coding sequence and in intergenic region while the other three were consistent with the maternal parent. In the present study, although the chloroplast genomes of M. foetida and M. odorata differ in 7 bp, there are 108 variants between the two species including 27 SNPs and nine insertions. In both concatenation and coalescence approaches for nuclear genes, M. odorata showed a relatively distant evolutionary relationship with M. indica. Individual gene trees clustered the M. odorata with each proposed parent in four gene trees only and some clades showed weak BS. Although the whole chloroplast genome and multiple nuclear genes provide more information compared to molecular markers, evidence for the hybrid origin of M. odorata is not strong enough and the parentage of M. odorata is inconclusive according to the results. Therefore, further analysis is required with populations for proposed parents and the hybrid to confirm the hybridity.
Another discrepancy observed from the chloroplast and nuclear trees is related to the position of M. sylvatica. Previous studies have revealed a close evolutionary relationship between M. indica and M. sylvatica based on restriction fragment length polymorphism (RFLP) [ 10] , ITS [ 18] marker analysis and whole chloroplast genome analysis [ 19] . Here, two M. sylvatica samples had different chloroplast genome sizes and had some structural differences. Therefore, M. sylvatica samples didn't cluster as sister taxa in chloroplast phylogeny. Another study [ 61] also reported that the M. sylvatica chloroplast genome assembled has a different length (157,368 bp) compared to the one available in NCBI and clustering them into two separate subclades. Since two M. sylvatica samples have been collected from different countries, the suggestion is that the regional separation might have mediated these evolutionary differences. Furthermore, another assembled M. sylvatica chloroplast genome is 158,063 bp in size [ 46] . Therefore, different chloroplast genome sizes and their structural variations of M. sylvatica might have occurred due to their different geographical distribution. Despite having structural variations, two M. sylvatica samples in our study were also closely related to M. indica. However, in nuclear phylogenies, they were nested with M. hiemalis and M. persiciformis suggesting that M. sylvatica might have a hybrid origin which has been occurred a long time ago, but the low BS values in individual gene trees do not provide strong support for this hypothesis.
Topological incongruence observed by the chloroplast genome and single-copy nuclear gene-based phylogenies reveal that there is a potential for inter-specific hybridization in the genus. But less BS values and weak resolution in gene trees of coalescence approach and low BS/PP/LPP support values in some of the branches of concatenation-based nuclear phylogeny and species tree are clear evidence that the nuclear genes are not well distinguished/ might not vary across the group of species studied. Less variability of nuclear genes and absence of multiple replicates for proposed hybrids limited conclusions about possible hybridization event/s and hybrid origin of M. odorata, and M. casturi. Although both the proposed parents are present, phylogenies will show their close evolutionary relationships if it is a recent generation hybrid. Therefore, the results of this study suggests that the whole group is sufficiently closely related with each other, so we needed a large amount of data to get well-resolved and highly supported phylogenetic trees. The history of evolution of the species and hybridization is complex in the genus and requires more species to get a better understanding. However, it is possible that out of 69 distinct species identified in the genus, some or many of them may have either domestication input or cross-hybridized with other wild relatives.
-
The analysis of determining evolutionary relationships within the genus Mangifera revealed a close genetic relationship among species and discrepancies between whole plastome and nuclear gene-based phylogenies. We suggest that the five species including M. indica, M. altissima, M. applanata, M. caloneura, and M. lalijiwa are very closely related and might have descended from the same common ancestor. It was difficult to validate the hybrid origin of M. odorata, and M. casturi as suggested previously due to the absence of multiple replicates for the proposed parents within our dataset, clustering of the proposed parent in only a few number of gene trees, and due to weak support obtained in gene trees. Relatively high numbers of gene trees showed a close evolutionary relationship between M. zeylanica, and M. indica, and M. sylvatica and M. hiemalis/M. persiciformis. However, the evidence did not strongly support the possible hybridization due to weak BS/PP and LPP supports. Moreover, it was observed that geographical proximity might have facilitated possible hybridization events. Despite limited number of species used in the study, it seems that evolution and hybridization in the genus Mangifera is a complex process. This is the first comparative analysis of evolutionary relationships within the genus with whole chloroplast genome and multiple nuclear genes. These findings provide an understanding about the nature of hybridization within the genus between wild and domesticated mangoes revealing potential domestication input into some species. Validation of hybridity and accuracy of evolutionary relationships within the genus can be highly supported and improved by adding more species including multiple replicates for the potential parents and sampling species from different geographical locations.
-
The authors confirm contribution to the paper as follows: Study conception and design: Henry RJ, Furtado A, Dillon NL; data collection: Wijesundara UK, Furtado A, Dillon NL, Masouleh AK; analysis and interpretation of results: Wijesundara UK, Furtado A, Dillon NL, Masouleh AK, Henry RJ; draft manuscript preparation: Wijesundara UK. All authors reviewed the results and approved the final version of the manuscript.
-
All data supporting the findings of this study are available within the paper and its supplementary information. Raw Illumina sequence read data were submitted to NCBI's Sequence Read Archive (SRA) database under Bio project ID PRJNA940204 and under Bio sample ID's SAMN33621737-SAMN33621749 and SAMN40922882-SAMN40922886.
Authors acknowledge The University of Queensland Research Computing Centre (UQ-RCC) for providing all the computation resources. David K Chandlee, Peter Salleras, Kerry McAvoy and Alan Carle for providing leaf materials for the species M. pajang, M. caesia, M. foetida, and M. sylvatica to use in the project. This project was funded by the Department of Agriculture and Fisheries-Queensland Alliance for Agriculture and Food Innovation (QAAFI) Collaboration Fund (Genomics of Mangifera species – HF11422) and the Hort Frontiers Advanced Production Systems Fund (National Tree Genomics – AS17000) as part of the Hort Frontiers strategic partnership initiative developed by Hort Innovation, with co-investment from the Queensland Government and contributions from the Australian Government.
-
The authors declare that they have no conflict of interest.
-
accompanies this paper at ( https://www.maxapress.com/article/doi/10.48130/tp-0024-0031)
-
Received 12 April 2024; Accepted 7 June 2024; Published online 12 October 2024
-
All Mangifera species had same gene number and order in the chloroplast genome.
Phylogenies based on nuclear genes and chloroplast genomes were discordant.
Topological incongruences suggest possible inter-specific hybridization in mango.
M. indica and four wild relatives were closely related.
Evidence of gene flow between species suggests hybrids to be common within the genus.
- Supplemental Table S1 Details of Illumina sequencing data of the Mangifera species.
- Supplemental Table S2 Details of Illumina sequencing data of the Mangifera samples downloaded from NCBI SRA database.
- Supplemental Table S3 Details of single copy genes used for nuclear phylogeny.
- Supplemental Table S4 Best substitutional model for generating phylogenetic trees.
- Supplemental Fig. S1 Individual gene trees constructed for 47 single-copy nuclear genes. Maximum likelihood trees developed are shown here and the numbers associated with the branches of the trees are ML bootstrap alues (/100).
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wijesundara UK, Furtado A, Dillon NL, Masouleh AK, Henry RJ. 2024. Phylogenetic relationships in the genus Mangifera based on whole chloroplast genome and nuclear genome sequences. Tropical Plants 3: e034 doi: 10.48130/tp-0024-0031 

Phylogenetic relationships in the genus Mangifera based on whole chloroplast genome and nuclear genome sequences
- Received: 12 April 2024
- Revised: 01 June 2024
- Accepted: 07 June 2024
- Published online: 12 October 2024
Abstract: The genus Mangifera ( Anacardiaceae) includes 69 species with Mangifera indica L. being the most important and predominantly cultivated species for commercial mango production. Although the species are classified based on morphological descriptors, molecular evidence has proposed the hybrid origin of two species suggesting the possibility that more of the species may be of hybrid origin. To analyze evolutionary relationships within the genus, 19 samples representing 14 Mangifera species were sequenced. Whole chloroplast genomes and 47 common single-copy nuclear gene sequences were assembled and used for phylogenetic analysis using concatenation and coalescence-based methods. The chloroplast genome size varied from 151,752 to 158,965 bp with M. caesia and M. laurina having the smallest and largest genomes, respectively. Annotation revealed 80 protein-coding genes, 31 tRNA, and four rRNA genes across all the species. Comparative analysis of whole chloroplast genome sequence and nuclear gene-based phylogenies revealed topological conflicts suggesting chloroplast capture or cross-hybridization. The chloroplast genomes of M. altissima, M. applanata, M. caloneura, and M. lalijiwa were similar to those of M. indica (99.9% sequence similarity). Their close sequence relationship suggests a common ancestry and likely cross-hybridization between wild relatives and M. indica. This study provides improved knowledge of phylogenetic relationships in the genus Mangifera, indicating extensive gene flow among the different species, suggesting that hybridization may be common within the genu s.