









-
Huanglumu (derived from Berberis amurensis), a traditional Chinese herb with a millennium-long medicinal history, is used to treat gastrointestinal inflammation (enteritis, dysentery), cardiovascular disorders (hypertension), and hepatic conditions (jaundice)[1,2]. The medicinal value of B. amurensis primarily stems from its production of benzylisoquinoline alkaloids (BIAs)[2−4]. BIAs constitute a structurally diverse class of nitrogen-containing plant metabolites, comprising over 2,500 identified variants. These compounds are phylogenetically widespread in Ranunculales, especially in the Berberidaceae, Ranunculaceae, Papaveraceae, and Menispermaceae families[5−7]. Nine bioactive BIAs have been isolated from B. amurensis, of which berberine is common in Berberis species[8,9]. Recent pharmacological research has indicated that berberine, a main active ingredient in B. amurensis (with a fresh weight content of 2.6%)[3,10,11], has a series of important functions such as antibacterial, anti-inflammatory, antioxidant, hypoglycemic, lipid-lowering, neuroprotective, and antitumor activities[12−25]. These multifaceted bioactivities underscore the significance of berberine in traditional and modern pharmacopeias.
Extensive research has elucidated the biosynthetic pathways of structurally diverse BIAs in various plant species, including berberine in Coptis chinensis (Ranunculaceae) as well as sanguinarine, papaverine, and morphine in Papaver somniferum (Papaveraceae), with the key catalytic enzymes responsible for their structural diversification being well-characterized[26−31]. Despite their significant structural diversity, the biosynthesis of the benzylisoquinoline alkaloid (BIA) core skeleton initiates from L-tyrosine[6]. L-tyrosine undergoes decarboxylation by tyrosine decarboxylase (TYDC) and hydroxylation by tyramine 3-hydroxylase (3OHase) to form dopamine, a crucial precursor for the tetrahydroisoquinoline moiety[32]. Concurrently, tyrosine aminotransferase (TyrAT) and 4-hydroxyphenylpyruvate decarboxylase (HPPDC) convert L-tyrosine to 4-hydroxyphenylacetaldehyde (4-HPAA), which provides the benzyl moiety[32]. Through Pictet–Spengler condensation, dopamine, and 4-HPAA form the tetrahydroisoquinoline and benzyl moieties of (S)-norcoclaurine, respectively[33,34]. These two key intermediates then undergo Pictet-Spengler condensation, catalyzed by pathogenesis-related (PR)10/Bet v1-type (S)-norcoclaurine synthase (NCS), to form (S)-norcoclaurine - the universal precursor of all BIAs[35]. Notably, this NCS-mediated condensation represents the first committed and rate-limiting step in BIA biosynthesis, exerting strict control over the metabolic flux toward downstream derivatives. Based on the basic skeleton formed by this enzymatic reaction, structural diversification of BIAs is achieved through subsequent enzymatic modifications, including methylation and oxidation mediated by methyltransferases and oxidases, respectively[36,37]. (S)-norcoclaurine is converted to the pivotal intermediate (S)-reticuline through four enzymatic steps: 6-O-methylation, N-methylation, 3'-hydoxylation, and 4'-O-methylation. These steps are catalyzed by (S)-norcoclaurine 6-O-methyltransferase (6OMT), (S)-coclaurine N-methyltransferase (CNMT), (S)-N-methylcoclaurine 3'-hydroxylase (NMCH, CYP80B), and (S)-3'-hydroxy-N-methylcoclaurine 4'-O-methyltransferase (4'OMT), respectively[38−42]. Notably, O-methyltransferases (OMTs) are crucial in producing structural diversity and enhancing the biological properties of BIAs. For instance, OMTs in Coptis species catalyze position-specific methylations that are critical for protoberberine alkaloid formation[37]. Genome-guided mining in Corydalis tomentella identified novel methyltransferases involved in cavidine biosynthesis[43]. The catalytic versatility and substrate promiscuity exhibited by O-methyltransferases from diverse medicinal plants—including CtOMTs (Coptis teeta), CyOMTs (Corydalis yanhusuo), PsOMTs (Papaver somniferum), and StOMT2 (Stephania tetrandra)—significantly contribute to the structural diversification of benzylisoquinoline alkaloids (BIAs). The enzymatic flexibility enables the rational exploitation of their known modification capabilities, facilitating the directed biosynthesis of novel BIA derivatives with potential pharmacological activities[27,44−48].
Recent advances in omics have accelerated the discovery of active BIA pathway genes in non-model species[49−51]. Despite the notable accumulation of berberine in B. amurensis, the molecular mechanisms underlying its biosynthesis remain poorly characterized due to limited genetic information, hindering the identification of key pathway enzymes. In this study, transcriptome sequencing, assembly, and functional annotation were performed on tissues derived from the roots, stems, and leaves of B. amurensis. Through systematic analysis, five key biosynthetic genes were cloned and characterized: One NCS, one 6OMT, two 4'OMT, and one SOMT gene. Functional characterization revealed that: (1) BaNCS4 facilitates the condensation reaction between dopamine and 4-HPAA, resulting in the formation of (S)-norcoclaurine; (2) Ba6OMT4 subsequently converts (S)-norcoclaurine to (S)-coclaurine; (3) Ba4'OMT2 converts (S)-3'-hydroxy-N-methylcoclaurine to (S)-reticuline; and (4) BaSOMT1 successively methylates (S)-scoulerine at the C9 and C2 position, generating (S)-tetrahydrocolumbamine and (S)-tetrahydropalmatine as products. These findings provide molecular foundation for future metabolic engineering efforts aimed at optimizing benzylisoquinoline alkaloid production, with particular relevance to berberine biosynthesis.
-
Plant materials of B. amurensis were collected in July 2022 from their natural habitat in Yichun City, Jiangxi Province, China. Species identification was confirmed through DNA barcoding analysis. Fresh tissues, including leaves (L), stems (S), and roots (R), were immediately frozen in liquid nitrogen and stored at -80°C until RNA extraction. Authentic standards—(S)-norcoclaurine, N-methylcoclaurine, (S)-reticuline, and (S)-scoulerine—were obtained from BioBioPha Co., Ltd. (Kunming, Yunnan, China). While (S)-tetrahydrocolumbamine, (S)-tetrahydropalmatine and (S)-coclaurine were obtained from Shanghai Yuanye Bio-Technology Co., Ltd. (Shanghai, China). The suppliers of 4-HPAA and 3'-hydroxy-N-methylcoclaurine were Toronto Research Chemicals Inc. (Toronto, Canada), and S-adenosyl-L-methionine (SAM) was procured from Sigma-Aldrich (St. Louis, MO, USA). These commercial compounds had a purity level of at least 98%. Chromatographically pure reagents were employed in the compound detection tests.
Transcriptome sequencing and de novo assembly
-
In Berberis amurensis, transcriptome sequencing was performed on the roots, leaves, and stems using Illumina HiSeq 2000 sequencing technology. Frozen leaf, root, and stem samples were used to extract total cellular RNAs (Supplementary Method S1). The Illumina TruSeq RNA Sample Prep Kit v3-cBot-HS was utilized to create a cDNA library, and the Illumina HiSeq platform form Novogene (Beijing, China) was utilized for paired-end sequencing (Supplementary Method S2). Following RNA-seq, adaptors, low-quality nucleotides, and unknown nucleotides were eliminated from the raw reads to create clean reads. Raw sequencing reads were quality-filtered using Trimmomatic (v0.39)[52] with the following parameters: removal of bases with Phred quality scores < 20, exclusion of reads shorter than 40 bp after trimming, elimination of reads containing terminal 'N' nucleotides, and adapter sequence clipping using the ILLUMINACLIP algorithm with a 2 bp seed mismatch allowance. FastQC software[53] was used for quality control both before and after cutting. The transcriptome sequences were constructed using the de novo RNA-Seq Assembly Pipeline (
https://github.com/abims-sbr/drap ) with Trinity (v2.13.2)[54], employing the default settings. The CD-HIT-EST program[55] eliminated duplicate contigs. TransDecoder (v3.0.1) was finally used to predict the coding region and the read counts. The completeness of the B.amurensis transcriptome assembly was evaluated using the Embryophyta_odb10 dataset, which contains 1,614 evolutionarily conserved embryophyte genes, through Benchmarking Universal Single-Copy Orthologs (BUSCO) tool (v5.7.1)[56].Functional annotation, CDS prediction, and phylogenetic analysis
-
Transcriptomic data from B. amurensis leaves, stems, and roots were compared against publicly available databases, including the nonredundant protein (NR), SwissProt, Pfam, the Kyoto Encyclopedia of Genes and Genomes (KEGG) database and Gene Ontology (GO)[57,58]. After GO annotations were obtained, GO functional classification was performed using WEGO software[59]. The transcriptome sequences were blasted with an E-value cutoff of 1 × 10−5 to identify the sequences encoding the berberine biosynthetic enzymes. ClustalW[60] was employed for aligning the protein sequences, followed by phylogenetic analysis. Using MEGA X (v10.2.4)[61], a maximum likelihood phylogenetic tree was constructed to identify NCSs and OMTs from B. amurensis.
Identification of differentially expressed genes
-
For transcriptomic analysis, gene expression levels were quantified as FPKM (fragments per kilobase of exon per million fragments mapped) values using R packages[62,63]. The resulting expression matrix was visualized as a heatmap using OmicStudio tools (
www.omicstudio.cn ). To ensure data stability and comparability, normalization was performed by adding a pseudo-count of 1 to each FPKM value, followed by a log10 transformation. Differential expression analysis was conducted using edgeR (v4.6.1) with p value ≤ 0.05 and |log2FC| > 1. Functional annotation of DEGs was performed through GO and KEGG pathway enrichment analysis, with statistical significance set at p ≤ 0.05.Recombinant protein expression and purification
-
Utilizing an RNAprep pure plant kit from TIANGEN (Beijing, China), total RNA was extracted from the leaf, stem, and root tissues of B. amurensis. The TransScripte One-Step gDNA Removal and cDNA Synthesis SuperMix from TransGen Biotech (Beijing, China) was used to synthesize first-strand cDNA.
Total RNA was isolated from B. amurensis leaf, stem, and root tissues using the RNAprep Pure Plant Kit (TIANGEN, Beijing, China). First-strand cDNA was synthesized from 1 μg total RNA using TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China) according to the manufacturer's protocol. Full-length open reading frames (ORFs) of putative BaNCS and BaOMT genes were amplified from cDNA templates using 2× Phanta Flash Master Mix (Vazyme, Nanjing, China) with gene-specific primers containing overlapping sequences for subsequent cloning (Supplementary Table S1). PCR products were purified and cloned into the pET-28a(+) prokaryotic expression vector using E.Z.N.A.® Gel Extraction Kit (Omega) and the Basic Seamless Cloning and Assembly Kit (TransGen Biotech). Recombinant plasmids were transformed into E. coli DH5α competent cells for propagation and subsequently purified using the TIANprep Mini Plasmid Kit (TIANGEN). The verified recombinant plasmids were transformed into E. coli BL21(DE3) for heterologous protein expression. Cultures were grown to OD600 ≈ 0.6 at 37 °C, then induced with 0.5 mM IPTG at 15 °C for 20 h with shaking (140 rpm). The cells were harvested by centrifugation (8,000 × g, 10 min, 4 °C) and resuspended in lysis buffer (100 mM Tris-HCl, pH 7.5) supplemented with 1 mM PMSF. The cell disruption was achieved by sonication (10 min, 5 s pulse/5 s rest) on ice, followed by centrifugation (12,000 × g, 20 min, 4 °C) to remove cell debris. The supernatant containing soluble proteins was incubated with Ni-NTA resin (CWBIO, Beijing, China) for 1 h at 4 °C (Supplementary Method S3). After washing with 20 mM imidazole in the binding buffer, target proteins were eluted with 250 mM imidazole, as previously optimized[48]. Target protein purity was assessed by 12% SDS-PAGE with Coomassie Brilliant Blue staining, and concentrations were determined by A280 measurement using a Spark multimode microplate reader (Tecan, Switzerland).
In vitro enzyme assays and functional characterization
-
The enzyme assay was conducted on the activity of the genes encoding NCS, 6OMT, 4'OMT, and SOMT using a reaction mixture containing 100 µl of 100 mM Tris-HCl (pH 7.4), 25 mM sodium ascorbate, 10% (v/v) glycerol, 1 mM β-mercaptoethanol, 2 mM SAM, 0.5 mM potential substrate, and crude enzyme (or 10 µg of purified recombinant enzyme). The CcNCS, Cc6OMT, Cc4'OMT, and CcSMT were used as the positive control, whereas the empty vector was used as the negative control. Enzymatic assays were performed at 37 °C and 200 rpm for 2 h. The reaction was terminated by adding 50% (v/v) methanol, followed by centrifugation at 12,000 rpm for 20 min. The resulting supernatant was collected, spin-dried, and reconstituted in 150 μL of methanol for Liquid chromatography with high performance (HPLC) and Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) analysis.
Identification of enzymatic products using HPLC and LC-MS/MS
-
Chromatographic separation was performed on a Shimadzu LC-2050C 3D system (Shenyang, China) using an Ultimate® UHPLC XB-C18 column (2.1 mm × 100 mm, 1.8 μm) maintained at 40 °C. The mobile phase consisted of 0.1% (v/v) formic acid in water (A) and acetonitrile (B), with the following gradient program at 0.5 mL/min: 5% B (0−1 min), linear increase to 30% B (1−3 min), 60% B (3−15 min), 100% B (15−20 min), returning to 5% B (20−25 min), and held for 5 min (25−30 min). Detection was performed at 282 nm with a 10 µL injection volume. For LC-MS/MS analysis, the Shimadzu LCMS-9030 Q-TOF system employed identical mobile phases with an optimized gradient: 15% B (0−1 min), linear increase to 45% B (1−8 min), 80% B (8−10 min), returning to 15% B (10−11 min) and held for 4 min (11−15 min) at 0.2 mL/min (column temperature, 35 °C). Mass spectrometric analysis was performed in data-dependent acquisition (DDA) mode with the following parameters: full MS scans (m/z 100−800), followed by top-10 MS/MS scans. MS detection used positive electrospray ionization (ESI+) with nitrogen nebulizing gas (3.0 L/min) and drying gas (10 L/min at 300 °C), scanning m/z 100−1,000. Each sample was injected at a volume of 1 μL.
-
To systematically characterize the B. amurensis transcriptome, three strand-specific cDNA libraries (BaL, BaS, and BaR) were constructed from leaf, stem, and root tissues, respectively. Following quality control (removal of adapter sequences and low-quality reads), 21,665,463 (3.2 Gb), 25,527,459 (3.8 Gb), and 18,494,454 (2.8 Gb) clean reads were obtained for BaL, BaS, and BaR libraries, respectively (Supplementary Table S2). The GC content of clean reads of BaL, BaS, and BaR were 45%, 44%, and 44%, respectively. De novo assembly using a standardized short-read pipeline generated 55,016 coding genes with an N50 of 1,170 bp. Benchmarking against the Embryophyta_odb10 dataset revealed exceptional assembly completeness, with 90.8% of the 1,614 conserved single-copy orthologs being fully represented (Table 1, Supplementary Table S3). This high-quality transcriptome provides a robust foundation for downstream gene discovery and expression analyses.
Table 1. Overview of de novo assembly statistics.
Item Transcriptome assembly Unigenes (the longest isoform) Coding
genesTotal sequences 325,523 262,191 55,016 Complete BUSCOs 92.40% 92.00% 90.80% Total bases 229,930,964 111,153,438 48,237,144 Min sequence length (bp) 178 201 255 Max sequence length (bp) 18,516 15,698 12,783 Average sequence length (bp) 706.34 423.94 876.78 Median sequence length (bp) 385 294 618 N50 length (bp) 1,186 435 1,170 GC content 41.10% 40.02% 43.83% The coding genes were annotated by searching the sequences against the following five public protein databases: NR, Swiss-Prot, GO, KEGG, and Pfam. In B. amurensis, these databases contained 53,926 genes (98.02%) that were annotated overall. In addition, 48,464 (88.09%), 35,005 (63.63%), 14,735 (27.32%), 9,376 (17.04%), and 25,133 (57.76%) genes were annotated that were annotated in the NR, Swiss-Prot, GO, KEGG, and Pfam databases, respectively (Table 2, Supplementary Data S1). A total of 14,735 genes from B. amurensis were categorized into three classes in the GO analyses (Supplementary Fig. S1). Using the KEGG database for annotation, 265 biological pathways were ascribed to 10,811 gene from B. amurensis, with 3,443 genes representing the largest annotated set for metabolic pathways. This included 1,523 genes involved in the biosynthesis of secondary metabolites (ko01110), including BIA biosynthesis (Supplementary Fig. S2).
Table 2. Statistics on the number of gene annotated for function by KEGG, NR, GO, PFAM, and Swissprot databases.
Database Number of genes Percentage KEGG 9,376 17% NR 48,464 88% GO 14,735 34% Pfam 25,133 58% Swiss-Prot 35,005 64% Total 53,926 98% Identification of differentially expressed genes
-
The differential expression analysis (p value ≥ 0.05 and |log2FC| ≥ 1) identified 23,794 DEGs across three tissue comparisons (BaL, BaS, BaR). The BaR vs BaL comparison showed the highest number of DEGs (18,095), with 10,564 up-regulated and 7,531 down-regulated genes. Similarly, BaS vs BaL revealed 14,837 DEGs (9,714 up/5,123 down), while BaR vs BaS had 12,315 DEGs (4,761 up/7,554 down) (Supplementary Table S4). Further analysis of the up-regulated DEGs exhibited significant differences in the number of tissue-specific DEGs across comparisons, with BaS vs BaL showing the highest number of tissue-specific DEGs (4,057), followed by BaR vs BaL (2,039) and BaR vs BaS (836). Intersection analysis identified substantial overlap between comparisons, with 4,600 DEGs co-upregulated in both BaR vs BaL and BaS vs BaL contrasts, and 2,868 genes shared between BaL-BaR and BaS-BaR (Supplementary Fig. S3a). These results facilitate the identification of candidate genes potentially involved in berberine biosynthesis.
To further characterize the DEGs, KEGG and GO enrichment analyses were performed. KEGG analysis identified 7,336 (BaR vs BaL), 5,856 (BaS vs BaL), and 5,929 (BaR vs BaS) DEGs, with 'biosynthesis of secondary metabolites' being the most significantly enriched pathway across all comparisons. Notably, the isoquinoline alkaloid biosynthesis (ko00950) pathway was consistently enriched and ranked among the top 20 pathways in all three comparisons. Specifically, BaR vs BaL showed 91 DEGs (75 up-regulated/16 down-regulated) in ko00950 pathway, along with 61 DEGs (32 up-regulated/29 down-regulated) in the tyrosine metabolism (ko00350) pathway and 44 DEGs (19 up-regulated/33 down-regulated) in nitrogen metabolism (ko00910) pathway (Fig. 1a). Similar patterns were observed in BaS vs BaL (79 DEGs: 71 up-regulated/8 down-regulated in ko00950 pathway; 51 DEGs: 34 up-regulated/17 down-regulated in ko00350 pathway; 38 DEGs: 10 up-regulated/28 down-regulated in ko00910 pathway) and BaR vs BaS (69 DEGs: 39 up-regulated/30 down-regulated in ko00950 pathway; 40 DEGs: 18 up-regulated/32 down-regulated in ko00350 pathway; 33 DEGs: 14 up-regulated/19 down-regulated in ko00910 pathway), all potentially related to berberine biosynthesis (Fig. 1b−c). Functional characterization through GO enrichment identified oxidoreductase activity (GO:0016491) as the most significantly represented molecular function across BaR vs BaL, BaS vs BaL, and BaR vs BaS comparisons (Supplementary Fig. S3b−S3d), highlighting the importance of redox reactions in the observed differential gene expression patterns.
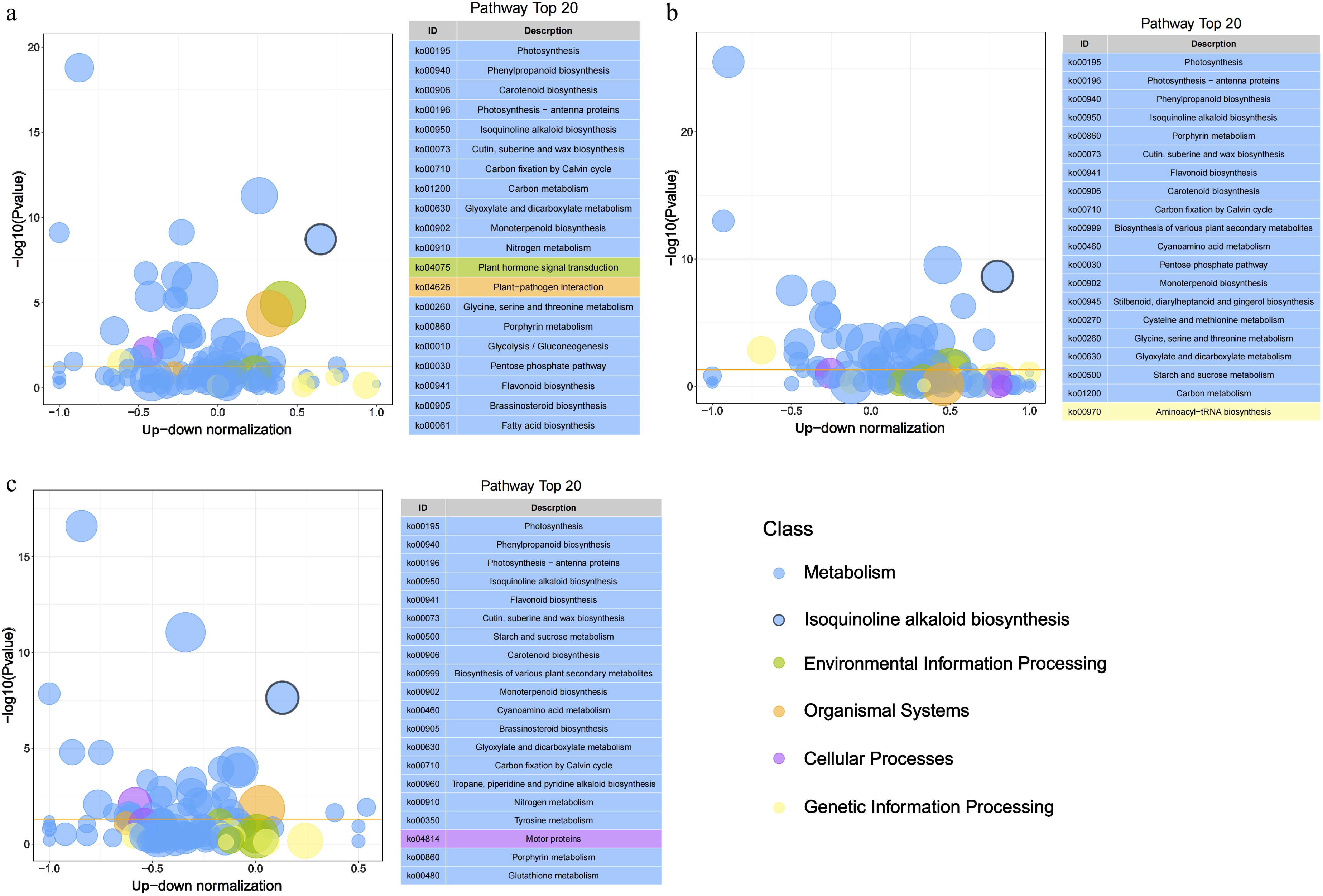
Figure 1.
Bubble plot of KEGG pathway enrichment analysis based on DEGs across tissues. The x-axis represents the up-down normalization value calculated as (Number of up-regulated genes − Number of down-regulated genes)/(Number of up-regulated genes + Number of down-regulated genes), reflecting the directional bias of pathway regulation. Bubble size indicates the number of DEGs per pathway, while color denotes different pathway classes. The yellow line represents the threshold value of p value = 0.05. On the right is the path list of the top 20 with p values.
Identification and expression pattern analysis of berberine biosynthetic genes
-
The berberine biosynthetic pathway initiates with the stereospecific condensation of dopamine and 4-HPAA, catalyzed by NCS to form the pivotal intermediate (S)-norcoclaurine. This tyrosine-derived scaffold then undergoes sequential modifications through a series of reactions involving multiple methyltransferases (OMTs and NMTs) and oxidases (CYP450, BBE, and STOX), ultimately yielding berberine (Fig. 2). Comprehensive transcriptome analysis revealed distinct expression profiles of 37 genes encoding nine key enzymatic components of the berberine pathway in B. amurensis, including four NCSs, six 6OMTs, twelve CNMTs, two CYP80Bs, two 4'OMTs, two BBEs, three SOMTs, three CYP719A21s, and three STOXs (Fig. 2). Tissue-specific expression patterns demonstrated clear partitioning of the biosynthetic active enzymes. The early pathway genes (NCS, 4'OMT, and CYP80B) showed predominant expression in stems, suggesting this organ as a major site for intermediate production (Fig. 2, Supplementary Table S5). In contrast, downstream enzymes (BBE and STOX) exhibited root-enriched expression, indicating roots as the primary site for late-stage modifications and berberine accumulation. Notably, BaCYP719A2, as the only leaf-specific isoform, may contribute to organ-specific alkaloid profiles, given the established role of CYP719A in methylenedioxy bridge formation (Fig. 2). These transcriptional patterns strongly correlated with the tissue distribution of BIAs, where roots and stems showed the highest berberine accumulation.
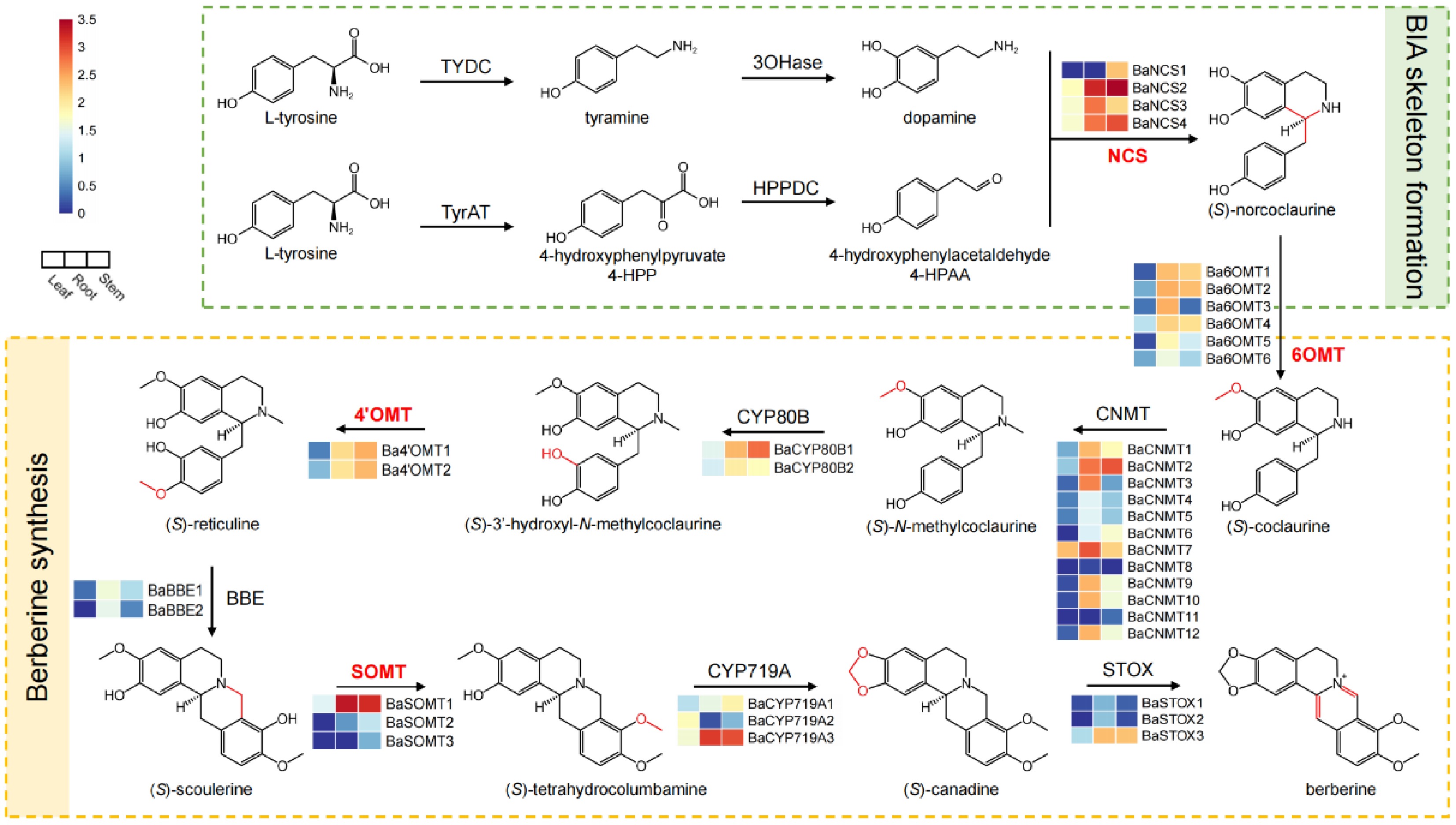
Figure 2.
Gene expression in the biosynthetic pathway of berberine in B. amurensis. The columns from left to right in each heat map display the expression levels of the leaf, root, and stem of B. amurensis. The log10(FPKM+1) values were used for normalization.
Phylogenetic analysis of NCSs and OMTs involved in berberine biosynthesis
-
In this study, four BaNCSs and 11 BaOMTs (6 Ba6OMTs, 2 Ba4'OMTs, 3 BaSOMTs) from the transcriptome of B. amurensis (Fig. 3). The NCS-derived phylogenetic tree revealed a close relationship between the NCSs of Berberidaceae (four BaNCSs from B. amurensis and ShNCS from Sinopodophyllum hexandrum) and Ranunculaceae (CjPR10A from Coptis japonica and TfNCS form Thalictrum flavum), while the NCS of Papaveraceae (PsNCS1 and PsNCS2 from P. somniferum, PbNCS from Papaver bracteatum, CyNCS from Corydalis yanhusuo, and AmNCS from Argemone mexicana) is a separate branch of divergence, basically consistent with the phylogenetic relationships between species (Fig. 3a)[64]. Therefore, these BaNCSs in this clade might be specifically related to the NCS activity in B. amurensis. Notably, the sequence identity among the four genes was more than 90%. The OMT-derived phylogenetic tree indicated distinct functional clades among BaOMTs, including 6OMT, 4'OMT, and SOMT. It showed significant gene expansion, particularly in the 6OMT branch (Fig. 3b). Ct7OMT (norcoclaurine 7-O-methyltransferase) is a separate branch involved in jatrorrhizine biosynthesis that contains an unusual 7-O-methylation pattern[27]; however, 7OMT was not annotated in our transcriptome dataset. These findings indicate gene expansion and evolutionary conservation of NCS and OMT within the lineage.
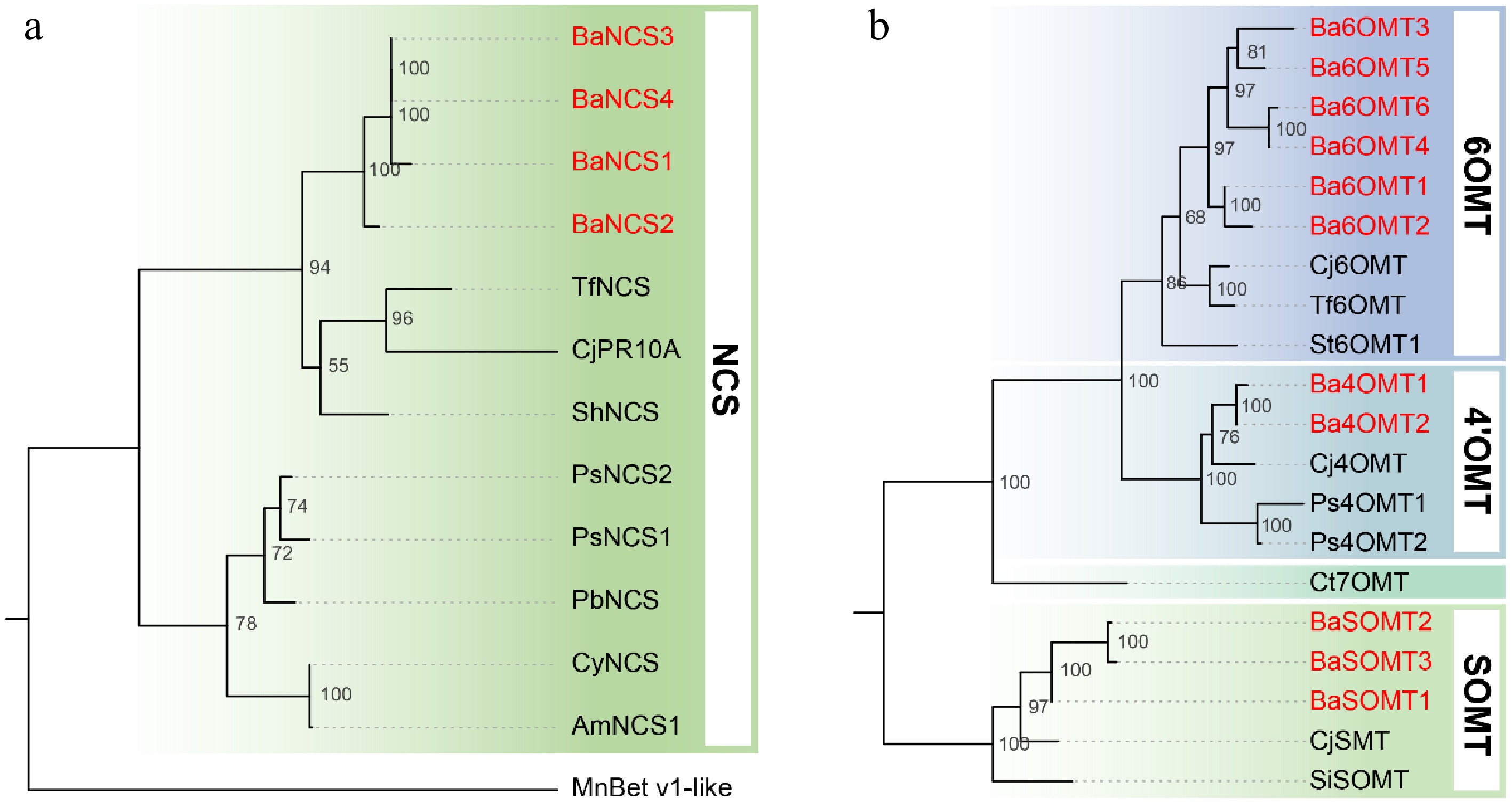
Figure 3.
Phylogenetic analysis of (a) NCS, and (b) OMT protein sequences. The maximum-likelihood tree was constructed using aligned amino acid sequences from B. amurensis (highlighted in red) and homologous enzymes from other plant species. Bootstrap values (1000 replicates) are shown at branch nodes. A higher value indicates that the branch is more reliable. Abbreviations and GenBank accession numbers for the sequences are listed as follows: TfNCS (ACO90248.1), CjPR10A (BAF45338.2), ShNCS (AIT42265.1), PsNCS2 (AAX56304.1), PsNCS1 (AAX56303.1), PbNCS (ACO90258.1), CyNCS (WDS97777.1), AmNCS1 (ACJ76785.1), MnBet_v1-like (EXB29146.1), Cj6OMT (BAB08004.1), Tf6OMT (AAU20765.1), St6OMT1 (6OMT from S. tetrandra)[48], Cj4'OMT (BAB08005.1), Ps4'OMT1 (AAP45313.1), Ps4'OMT2 (AAP45314.1), CjSMT (BAA06192.1), and SiSOMT (MK749415).
To elucidate the molecular basis of berberine biosynthesis in B. amurensis, five candidate genes (BaNCS4, Ba6OMT4, Ba4'OMT1, Ba4'OMT2, and BaSOMT1) were cloned and heterogenously expressed in E. coli. Both crude extracts and purified recombinant proteins were subsequently used for in vitro enzymatic assays to characterize their catalytic activities (Supplementary Fig. S4).
Functional characterization of NCS in vitro
-
In vitro enzymatic assays were conducted to characterize the catalytic activity of BaNCS4 with dopamine and 4-HPAA as substrates. Using CcNCS as the positive control, the results showed the same product peak under the catalysis of CcNCS and BaNCS4 (Supplementary Fig. S5). LC-MS analysis revealed a reaction product with mass-to-charge ratio (m/z) 272 [M+H]+, whose retention time (8.42 min) and MS/MS fragmentation pattern matched authentic (S)-norcoclaurine standards (Fig. 4). These results demonstrate that BaNCS4 catalyzes the stereospecific condensation of dopamine and 4-HPAA to form (S)-norcoclaurine, the universal precursor of benzylisoquinoline alkaloids.
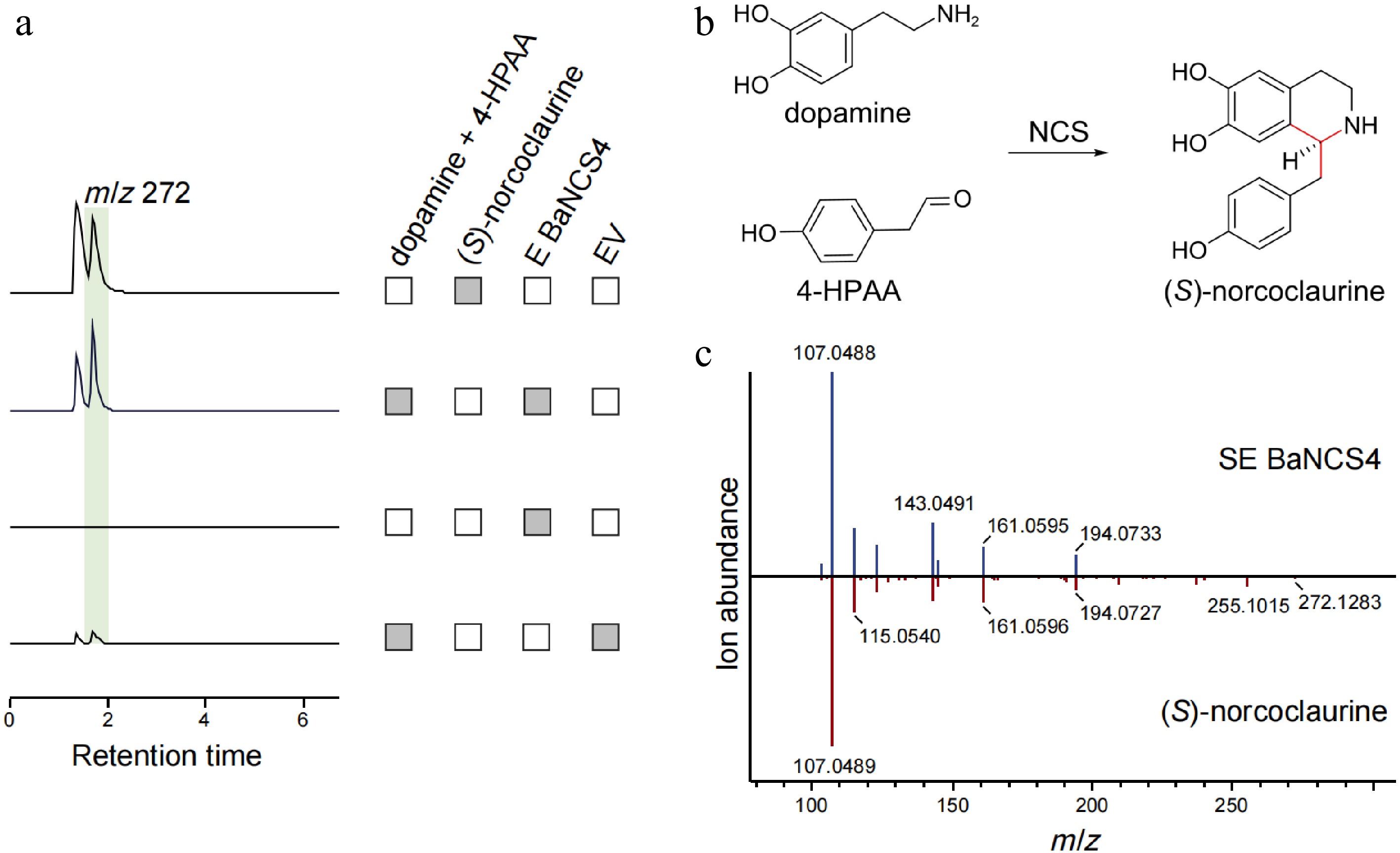
Figure 4.
Functional characterization of BaNCS related to berberine biosynthesis in B. amurensis. (a) The LC-MS extracted ion chromatogram at m/z 272 represents the catalytic product generated by BaNCS. The reaction system without substrate and pET-28a (+) empty vector (EV) was used as the negative control. (b) NCS mediates the Pictet-Spengler condensation of dopamine and 4-HPAA, yielding (S)-norcoclaurine. (c) Comparison of MS/MS fragments between the product from BaNCS (above) and authentic (S)-norcoclaurine (below).
Functional characterization of 6OMT in vitro
-
The in vitro O-methyltransferase activity of the candidate 6OMT protein, the first modifying enzyme after the formation of the BIA basic skeleton, was assessed using (S)-norcoclaurine as substrate, with Cc6OMT from C. chinensis as the positive control (Supplementary Fig. S5). LC-MS analysis showed that a product peak with a m/z of 286 appeared at the same position under the catalysis of Ba6OMT4 and Cc6OMT purified protein (Fig. 5a). After comparing the retention time and mass spectrum information of the product with the standard, the product was identified as (S)-coclaurine (Fig. 5b and c). The result indicated that Ba6OMT has (S)-norcoclaurine 6OMT activity and could catalyze the O-methylation of the C6 hydroxyl group to convert (S)-norcoclaurine to (S)-coclaurine. This modification represents the first committed step in the diversification of benzylisoquinoline alkaloid scaffolds.
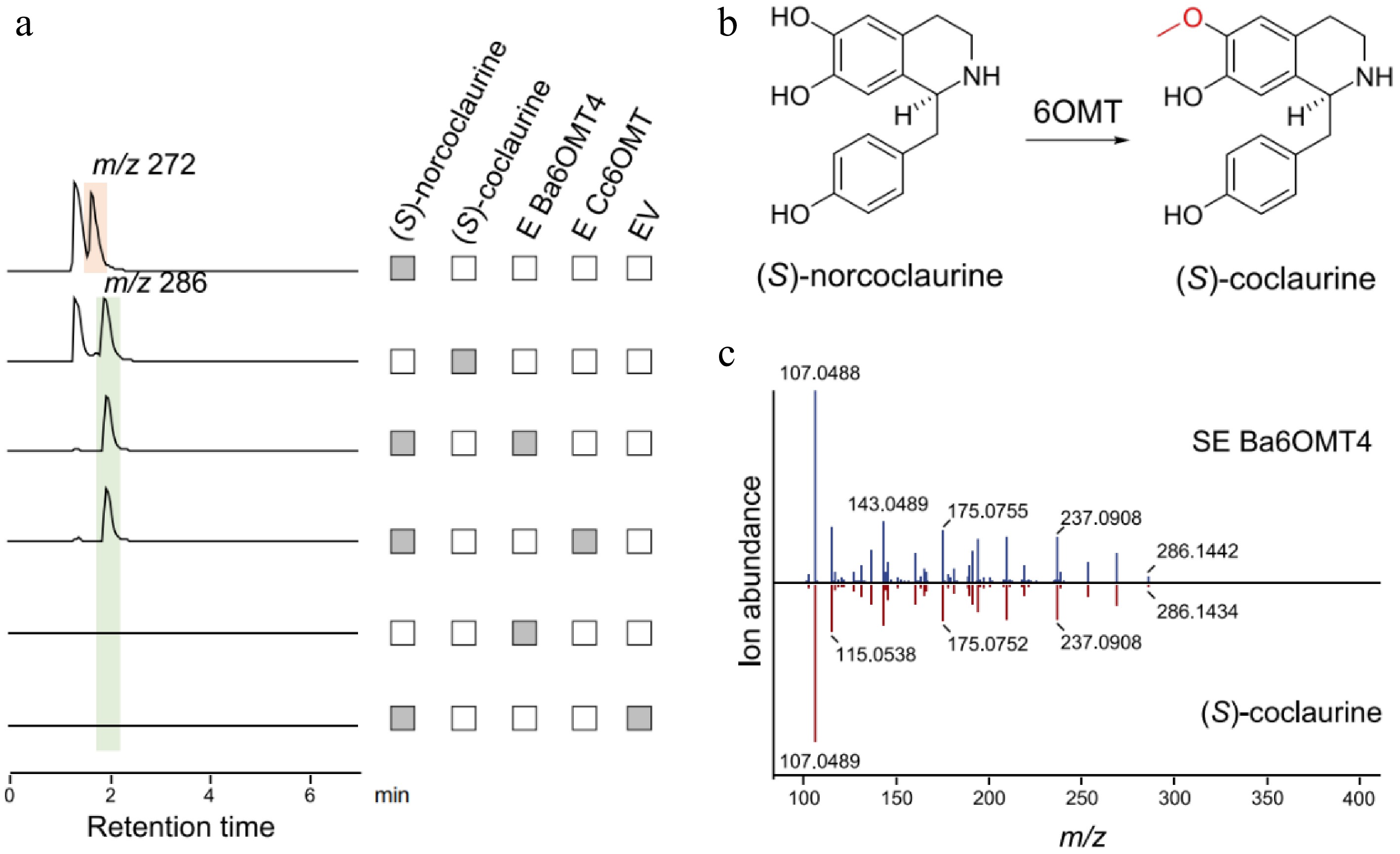
Figure 5.
Functional characterization of Ba6OMT related to berberine synthesis in B. amurensis. (a) The LC-MS extracted ion chromatogram at m/z 272 and 286 represent the substrate (S)-norcoclaurine and the catalytic product generated by Ba6OMT, respectively. The pET-28a (+) empty vector (EV) was used as the negative control. (b) The synthesis of (S)-coclaurine catalyzed by 6OMT. (c) Comparison of MS/MS fragments between the product from Ba6OMT (above) and authentic (S)-coclaurine standard (below).
Functional characterization of 4'OMT in vitro
-
In vitro catalytic activity of Ba4'OMT1 and Ba4'OMT2 was assessed using (S)-3'-hydroxy-N-methylcoclaurine as substrate, with the Cc4'OMT from C. chinensis as the positive control, and it was found that only Ba4'OMT2 exhibited activity (Supplementary Fig. S5). The LC-MS results showed that under the catalysis of Ba4'OMT2 and Cc4'OMT, a product peak with the m/z of 330 appeared at the same position (Fig. 6a). After comparing the retention time and mass spectrum information of the product with the standard, the product was identified as (S)-reticuline (Fig. 6b & c). The result demonstrated that Ba4'OMT2 could catalyze the methylation of the C4' hydroxyl group in (S)-3'-hydroxy-N-methylcoclaurine to form (S)-reticuline and has higher catalytic activity than Cc4'OMT.
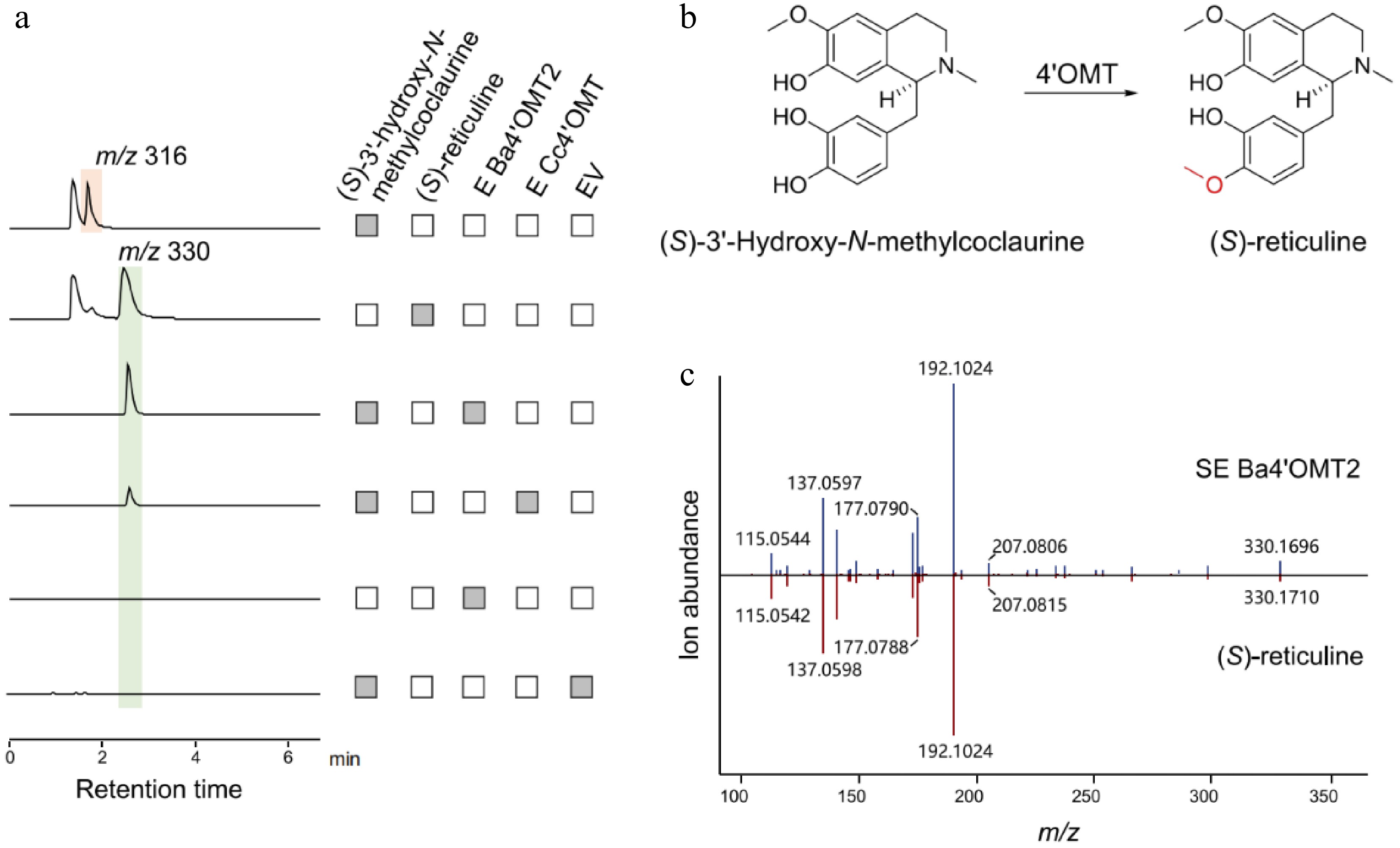
Figure 6.
Functional characterization of Ba4'OMT2 related to berberine biosynthesis in B. amurensis. (a) The LC-MS extracted ion chromatogram at m/z 316 and m/z 330 represent the substrate (S)-3'-hydroxy-N-methylcoclaurine and the catalytic product generated by Ba4'OMT2 enzymes, respectively. (b) The synthesis of (S)-reticuline catalyzed by 4'OMT. (c) Comparison of MS/MS fragments between the product from Ba4'OMT2 (above) and authentic (S)-reticuline (below).
Functional characterization of SOMT in vitro
-
Furthermore, HPLC analysis showed that, using (S)-scoulerine as the substrate, BaSOMT had the same catalytic function as CcSOMT (Supplementary Fig. S5). The LC-MS results showed that both enzymes could catalyze (S)-scoulerine to produce product peaks with m/z of 342 and 356 (Fig. 7a). The two products were identified as (S)-tetrahydrocolumbamine and (S)-tetrahydropalmatine, respectively, based on comparison with the mass spectrometry information of the standard sample (Fig. 7b & c). This shows that BaSOMT can catalyze the successive methylation of the C9 and C2 hydroxyl groups to convert (S)-scoulerine to (S)-tetrahydrocolumbamine and (S)-tetrahydropalmatine. The identical retention times and mass spectra of the methylated derivatives confirmed conserved functional characteristics between these orthologous enzymes.
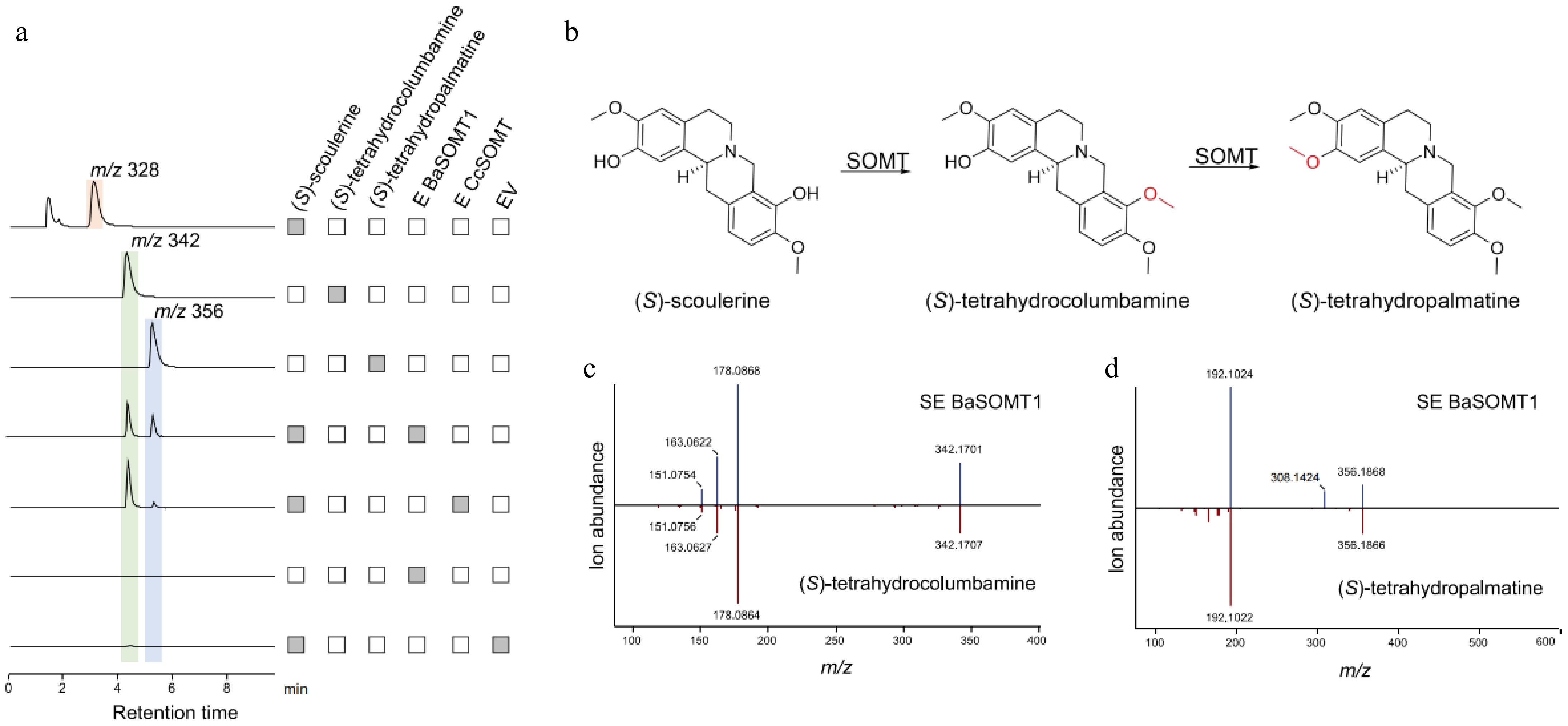
Figure 7.
Functional characterization of BaSOMT related to berberine synthesis in B. amurensis. (a) The LC-MS extracted ion chromatogram at m/z 328, 342, and 356 represent the substrate (S)-scoulerine and two continuous catalytic products generated by BaSOMT, respectively. (b) The synthesis of (S)-tetrahydrocolumbamine and (S)-tetrahydropamatine catalyzed by SOMT. (c), (d) Comparison of MS/MS fragments between the product from BaSOMT (above) and their corresponding standards (below).
-
The roots and stems of B. amurensis accumulates high levels of berberine—a prominent BIA with broad pharmacological activities and a key bioactive component in traditional medicine[10]. The transcriptomic analysis revealed significant differential expression of all identified genes involved in berberine biosynthesis in various tissues. The enrichment of 'oxidoreductase activity' (GO:0016491) and the prominence of the isoquinoline alkaloid biosynthesis pathway (ko00950) in DEGs support the tissue-specific pattern of BIA metabolism in B. amurensis. Notably, the spatial separation of early and late biosynthetic genes suggests potential inter-organ transport of pathway intermediates in B. amurensis, a phenomenon well-documented in other berberine-producing plants[65].
Given that the NCS enzyme catalyzes the first rate-limiting step of the berberine pathway, and methyltransferases catalyze the biochemical reactions involved in the biosynthesis and modification of bioactive BIAs in plants, the NCS and OMT functional enzymes were identified in B. amurensis. Based on the assembled transcriptome, the gene expression profile of 37 candidate genes was observed. In addition to BaNCS4, which showed high expression levels in the stem, Ba6OMT4, Ba4'OMT2, and BaSOMT1 showed high expression in the roots, which may play a direct role in the substantial accumulation of BIAs observed in both the roots and stems of B. amurensis. The biosynthetic pathways for diverse arrays of BIAs with significant pharmacological activity have been established in several plants, such as noscapine and morphine in opium poppy[28,66], berberine in C. chinensis[26], nelumboferine in sacred lotus[67], berberine in Phellodendron amurense[49], etc. Among them, the upstream pathway of BIA is highly conserved in Ranunculales. In the same way, these findings confirm the evolutionary conservation of NCS and OMT in Berberis.
The growing demand for bioactive natural products has outpaced traditional plant extraction methods, driving increasing interest in heterologous biosynthesis approaches. To establish sustainable microbial production platforms, a critical focus must be placed on engineering high-performance enzymes capable of supporting efficient biomanufacturing processes. For instance, due to the low enzymatic conversion efficiency of (S)-N-methylcoclaurine to (S)-reticuline, Rao et al.[68] streamlined biosynthesis by eliminating NMCH and 4'OMT-mediated methylation steps in traditional protoberberine pathways, achieving high yields of rotundine at the gram level. In this study, in vitro functional verification revealed Ba4'OMT2 with significantly higher activity than Cc4'OMT, providing a potentially efficient biosynthetic element. This discovery holds significant promise for applications in synthetic biology. Integration of Ba4'OMT2 into microbial hosts (e.g., Saccharomyces cerevisiae or E. coli) may enhance titers of protoberberine alkaloids, as evidenced by recent successes with plant-derived OMTs in heterologous systems[68−70]. The superior activity of Ba4'OMT2 could increase the flow to the target alkaloid without changing the natural substrate and eliminating the methylation step.
In summary, based on transcriptome analysis, this study systematically revealed high conservation with homologs from other berberine-producing plants. Four functional enzymes were identified in vitro: BaNCS4 for fundamental skeleton formation, along with three methyltransferases (Ba6OMT4, Ba4'OMT2, and BaSOMT1) for subsequent modifications. In the first committed step of berberine biosynthesis in B. amurensis, dopamine and 4-HPAA condense to produce (S)-norcoclaurine, catalyzed by BaNCS4. Likewise, these OMTs are vital for the diversification of BIA. Notably, a highly catalytically active Ba4'OMT2 enzyme was discovered. These findings clarify the enzymatic framework for berberine biosynthesis in B. amurensis and provide functional genetic elements to address current yield limitations in microbial production of these medicinally important alkaloids. These results advance the understanding of specialized metabolism in Berberis species and offer practical resources for metabolic engineering applications.
This work was supported by the National Natural Science Foundation of China (Grant Nos 82274037, 32460117), and the Beijing Life Science Academy (BLSA) (Grant No. 2024500CD0120), and (Grant Nos YNDG202402ZY02, YNDG202302ZY02, CNTC-110202201016 (JY-16), and YNTC-2023530000241009). The author sincerely thanks the China Association for Science and Technology (CAST) for the academic support through the Youth Talent Lift Project (PhD Student Special Support Program).
-
The authors confirm their contributions to the paper as follows: study design and coordination: Xu Z, Kong L, An Z; sample preparation of plant materials : Kong L; bioinformatic analysis: Shah SBA, Kong L, Fu Y; experiments conduction and data analysis: Shah SBA, Dong Y, Tian Y; draft manuscript preparation: Bai G, An Z, Shah SBA, Dong Y, Xu Z, Du Y. All authors reviewed the results and approved the final version of the manuscript.
-
Raw sequencing reads have been deposited in the Genome Sequence Archive in the Beijing Institute of Genomics Data Center, Chinese Academy of Sciences, under accession number CRA024471, which are publicly accessible at https://ngdc.cncb.ac.cn/gsa/s/vYxYc7xZ. The primer sequences and the data on gene expression levels are available in the supplementary information.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Syed Basit Ali Shah, Lingzhe Kong
- Supplementary Method S1 Step-by-step RNA extraction protocol for plant tissues.
- Supplementary Method S2 Library Construction Parameters.
- Supplementary Method S3 Induce expression and purification of target protein.
- Supplementary Data S1 The coding genes were annotated by searching the sequences against the following five public protein databases: NR, Swiss-Prot, GO, KEGG, and Pfam.
- Supplementary Table S1 The gene specific primers with homologous arms were used in this study.
- Supplementary Table S2 Raw data summary and statistics of transcriptome data in B. amurensis.
- Supplementary Table S3 The results of BUSCO assessment of de novo assemblies.
- Supplementary Table S4 The results of BUSCO assessment of de novo assemblies.
- Supplementary Table S5 The expression level of gene involved in the berberine biosynthetic pathway in B. amurensis.
- Supplementary Fig. S1 Gene ontology (GO) classification and statistical analysis of gene in B. amurensis. 14,735 gene were functionally categorized into three main domains: biological process, molecular function, and cellular component, based on their annotated roles in cellular systems.
- Supplementary Fig. S2 There are three panels in the KEGG pathway analysis plot: (a) A bar graph shows gene counts of varies pathway. (b) A dot plot shows pathway percentages. (c) A depiction of network interactions that shows possible linkages between pathways. The visualization offers a thorough summary of biological processes and functional annotations by highlighting differences in gene counts and percentages across various pathways using a viridis color gradient.
- Supplementary Fig. S3 GO enrichment analysis of DEGs in different comparisons.
- Supplementary Fig. S4 SDS-PAGE analysis of heterologous expression of proteins in E. coli. Lane M indicates protein marker; lane EV indicates empty vector (negative control); lanes 1−9 represent BaNCS4, CcNCS, Ba4'OMT2, Cc4'OMT, Ba4'OMT1, CcSOMT, Ba6OMT4, BaSOMT1, Cc6OMT, respectively.
- Supplementary Fig. S5 HPLC analysis of in vitro catalytic assays of crude enzyme.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Shah SBA, Kong L, Dong Y, Fu Y, Tian Y, et al. 2025. Selection and functional identification of berberine biosynthetic genes in Berberis amurensis. Medicinal Plant Biology 4: e024 doi: 10.48130/mpb-0025-0022 

Selection and functional identification of berberine biosynthetic genes in Berberis amurensis
- Received: 27 April 2025
- Revised: 17 May 2025
- Accepted: 20 May 2025
- Published online: 23 July 2025
Abstract: Berberine, a pharmacologically important benzylisoquinoline alkaloid (BIA) exhibiting antimicrobial and anti-inflammatory properties, accumulates abundantly in Berberis amurensis. However, there is a lack of reports concerning the berberine biosynthesis pathway in B. amurensis. In this study, berberine biosynthetic genes were systematically identified in B. amurensis, and key functional enzymes (BaNCS4, Ba6OMT4, Ba4'OMT2, and BaSOMT1) were characterized through transcriptomic and biochemical analyses. Notably, Ba4'OMT2 was found to exhibit superior catalytic efficiency compared to its ortholog Cc4'OMT, highlighting its potential as a valuable molecular tool for heterologous biosynthesis platforms. These findings not only elucidate the key enzymes of BIA skeleton formation and berberine O-methylation modification, as well as their evolutionary conservation but also offer potential molecular elements for achieving the biosynthesis of berberine through synthetic biology approaches.
-
Key words:
- Berberis amurensis /
- Berberine /
- Biosynthesis /
- Norcoclaurine synthase /
- O-methyltransferases