








-
Plastic waste valorization remains a central challenge for sustainable chemical engineering because polymers are energy-dense carbon resources, but are difficult to recycle into high-value chemicals without property loss. Catalytic thermochemical conversion—particularly catalytic pyrolysis and downstream upgrading—offers a scalable route to transform polymer-derived hydrocarbons into lighter olefins and aromatic chemicals[1,2]. Recent reviews comparing microwave-assisted pyrolysis/catalytic pyrolysis with conventional heating indicate that microwave operation can provide rapid volumetric heating and distinct heating-rate profiles, which may influence product distributions and energy efficiency, although outcomes remain system-dependent[3,4]. However, achieving both high selectivity and long catalyst lifetime is non-trivial, since polymer cracking generates bulky and highly reactive intermediates that readily form carbonaceous deposits and rapidly deactivate acid catalysts.
Among solid acids, ZSM-5 (MFI topology) is frequently adopted as a benchmark shape-selective catalyst for hydrocarbon cracking and aromatization reactions, owing to its well-defined micropore network and strong Brønsted acidity[5,6]. Yet the same microporous architecture that provides shape selectivity also imposes diffusion constraints, especially when the reacting pool contains large, multi-step intermediates[7]. This transport limitation can increase residence time and secondary reactions, accelerate the formation of coke precursors, and ultimately shorten catalyst lifetime. Introducing hierarchical porosity (micro-mesoporous coupling) has therefore become a widely used strategy to mitigate diffusion limitations, improve accessibility of active sites, and extend catalytic stability[8,9]. In particular, hierarchical ZSM-5 architectures can be created by controlling crystallization and growth processes, offering a synthesis-led route to stability enhancement without fundamentally changing the zeolite framework[10,11].
A critical yet underutilized synthesis parameter for hierarchical zeolites is the crystallization temperature, which directly controls nucleation-growth kinetics, crystal size development, and the probability of generating intercrystalline or intracrystalline mesopores[12]. In an illustrative study on ZSM-5, adjusting crystallization temperature was shown to tune hierarchical structure formation and even influence framework Al distribution, leading to measurable changes in catalytic behavior and stability in hydrocarbon conversion[13,14]. More broadly, studies on other zeolite families (e.g., Beta) demonstrate that crystallization temperature, time, and gel composition jointly govern texture (mesopore volume, micropore fraction), Si/Al, and acidity descriptors, which in turn control selectivity in refinery-scale hydrocarbon transformations[15,16]. These insights motivate a temperature-controlled, one-pot synthesis strategy as a rational handle to co-optimize transport and acidity, two parameters that often trade off when stability is targeted.
Catalyst lifetime, however, is ultimately dictated by deactivation pathways, of which coke formation is the most prevalent for acidic zeolites in hydrocarbon conversions[17]. Mechanistic evidence across MFI systems indicates that the location and chemical nature of coke critically determine the severity of deactivation. In methanol-to-hydrocarbons chemistry, detailed analysis of MFI zeolites showed that internal coke formed within micropores can be substantially more detrimental than external coke deposited on the crystal surface, because internal deposits directly restrict access to the shape-selective channel system and mask catalytically relevant acid sites[18]. Complementary investigations in n-hexane cracking over H-ZSM-5 further reinforce that coke growth is closely tied to diffusion constraints and secondary reactions, linking transport limitations to accelerated deactivation[19]. In reactions where surface processes dominate, carbonaceous layers at the external surface can also severely reduce accessibility to Brønsted acid sites, thereby suppressing catalytic turnover even when the framework remains intact. Recent studies on hierarchical ZSM-5 for plastic catalytic pyrolysis have highlighted that introducing secondary porosity can mitigate diffusion limitations and delay coke-induced deactivation, thereby improving catalyst stability and time-on-stream performance[20]. Collectively, these findings highlight that lifetime is governed not only by how much coke forms, but also where it forms, and how effectively reactants/products can access and leave the active zeolite domain.
From a catalyst-design perspective, both acid-site distribution and pore architecture must be tuned to suppress rapid coking without sacrificing desired selectivity[21,22]. Recent work demonstrates that optimizing acidity and pore structure can simultaneously enhance aromatics selectivity and catalyst lifetime in ZSM-5-based systems, emphasizing the importance of balancing acid strength/density with diffusion pathways[23]. In addition, modifying the external surface, such as reducing surface aluminum via controlled dealumination has been reported to improve lifetime by suppressing surface coking pathways associated with external acid sites, again underscoring that 'accessible acidity' and 'where acidity resides' are as important as total acidity[24,25]. These concepts are especially relevant for polymer-derived feeds, where bulky intermediates and strong secondary reactions make diffusion-coke coupling particularly pronounced[26].
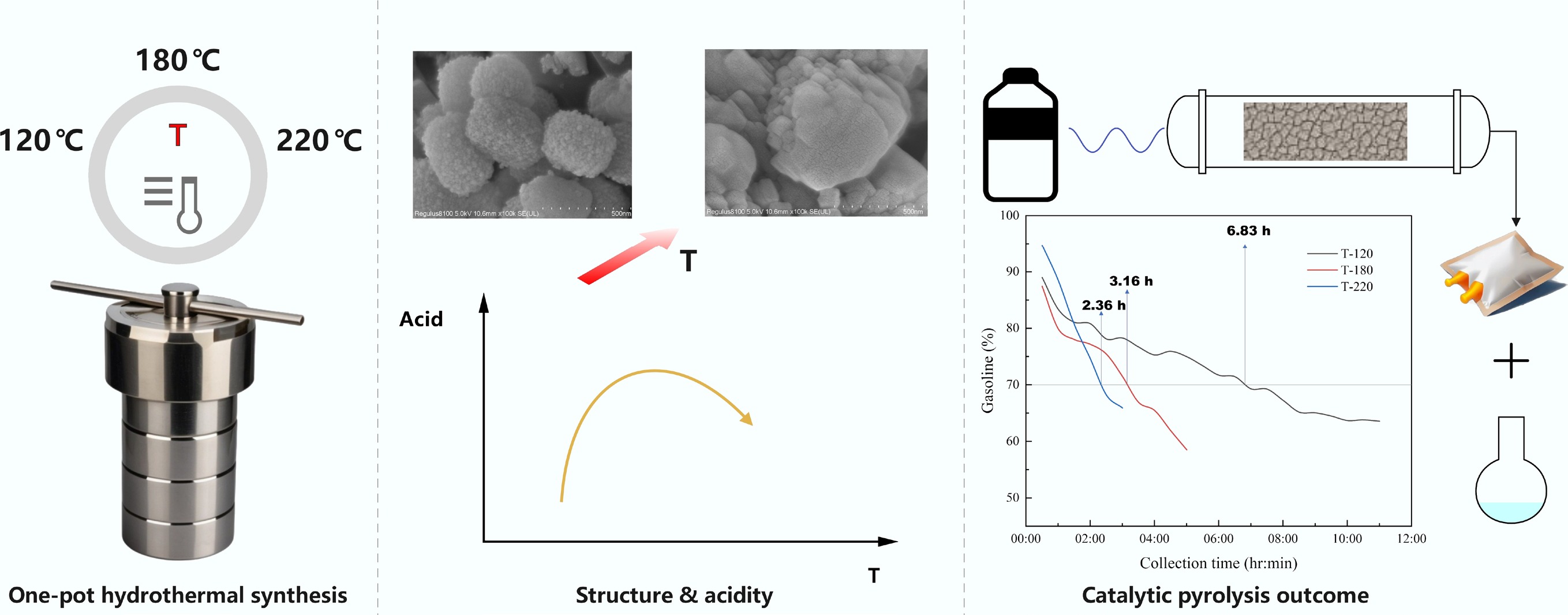
Despite the above progress, a consistent structure performance deactivation framework that links crystallization temperature (as a single, controllable synthesis knob) to hierarchical structure/acidity, and further to time-resolved product distributions and lifetime in plastic catalytic conversion remains insufficiently established. Here, we address this gap by synthesizing hierarchical ZSM-5 in a one-pot manner at crystallization temperatures of 120, 140, 180, 200, and 220 °C, followed by systematic characterization (XRD, N2 physisorption/BET, SEM, and NH3-TPD) and catalytic evaluation in plastic catalytic pyrolysis/upgrading. By combining time-on-stream gas composition, liquid yield metrics, and GC-MS-resolved liquid families, we construct a mechanistic interpretation of catalyst lifetime in terms of transport-acidity balance and coke-location-controlled deactivation. The resulting design rules provide practical guidance for selecting crystallization temperature windows that maximize catalyst lifetime while maintaining target selectivity in plastic waste valorization.
-
Tetrapropylammonium hydroxide (TPAOH, structure-directing agent), tetraethyl orthosilicate (TEOS, silica source), sodium aluminate (alumina source), ammonium chloride (NH4Cl), and deionized water were used as received. High-density polyethylene (HDPE) granules (2–4 mm) were provided by Goodfellow and used without further treatment in this study.
One-pot synthesis of hierarchical ZSM-5
-
Hierarchical ZSM-5 zeolites were synthesized via a one-pot hydrothermal route by varying the crystallization temperature. In a typical synthesis, an aqueous precursor mixture containing TPAOH, sodium aluminate, and TEOS was formulated to obtain an overall gel molar composition of 32 TPAOH : 80 SiO2 : 1 Al2O3 : 4,800 H2O. The mixture was vigorously stirred at room temperature for 8 h to ensure complete homogenization and sufficient hydrolysis/condensation of TEOS[27]. The resulting precursor solution was then transferred into a Teflon-lined stainless-steel autoclave, sealed, and crystallized at 120, 140, 180, 200, or 220 °C for 24 h. After hydrothermal crystallization, the solid product was recovered by filtration, thoroughly washed with deionized water, and dried at 100 °C overnight. Template removal was performed by calcination in air at 600 °C for 4 h. The calcined zeolite was subsequently converted to the protonic form via ion exchange: the powder was treated with a 1 M NH4Cl aqueous solution at 80 °C, and the exchange procedure was repeated three times. The exchanged sample was filtered, washed, dried, and calcined again in air at 600 °C for 4 h, yielding the H-form hierarchical ZSM-5 catalysts. For clarity, the catalysts are denoted as HZSM-5-T, where T represents the crystallization temperature (120–220 °C).
Catalytic pyrolysis process
-
The hierarchical ZSM-5 catalysts were immobilized on silicon carbide (SiC) foam supports with a pore density of 20 pores per inch (PPI). Briefly, zeolite slurries were prepared by dispersing the as-synthesized ZSM-5 powder in deionized water. In a typical procedure, 20 g of zeolite slurry was obtained by mixing the zeolite powder with 150 g of water under vigorous stirring until a homogeneous suspension was formed. Six pieces of SiC foam were then fully immersed in the slurry and repeatedly impregnated to ensure uniform penetration of the zeolite into the macroporous framework. After successive immersion and stirring steps, approximately 6 g of zeolite powder was deposited onto the SiC foams. The resulting zeolite-coated SiC foams were subsequently dried and used directly as structured catalysts in the catalytic microwave-assisted pyrolysis experiments. Figure 1 shows the experimental system diagram used in this study; catalytic performance was evaluated in the pyrolysis of a model plastic waste at 500 °C in a fixed-bed quartz reactor, using N2 as carrier gas at 50 L min−1. Catalyst loading on each SiC foam piece was determined by weighing the foams before and after coating/drying. In each run, 3 g of catalyst was loaded and continuously contacted with the plastic feed at a rate of 60 g h−1, corresponding to a plastic feed rate of 20 g h−1 g−1 catalyst (WHSV = 20 h−1).
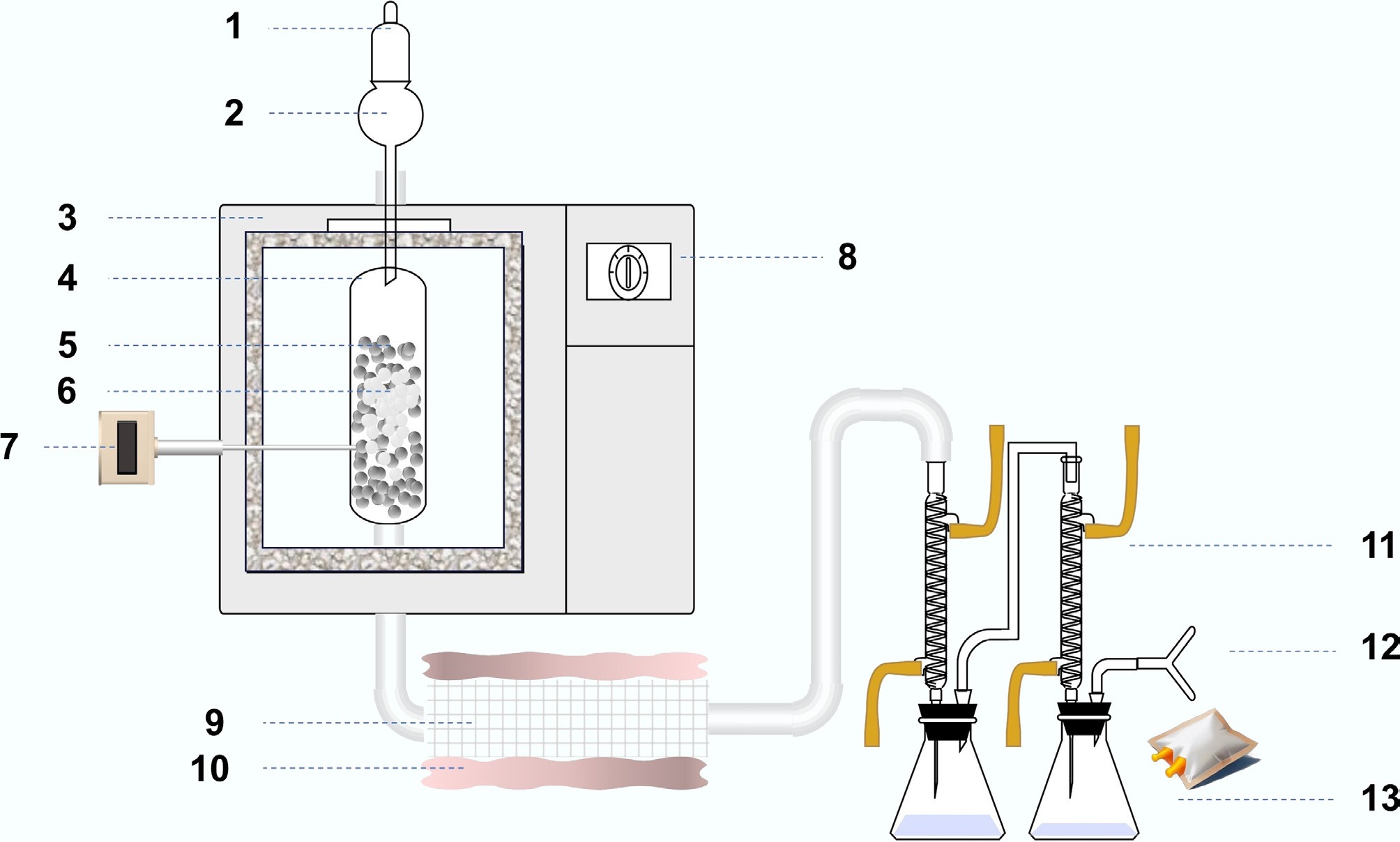
Figure 1.
Schematic of the catalytic pyrolysis system and product collection setup. (1) N2 carrier gas; (2) feed hopper; (3) microwave pyrolysis furnace; (4) quartz reactor; (5) SiC bed zone; (6) plastic particles; (7) temperature sensor; (8) system controller; (9) catalyst; (10) thermal insulation belt; (11) condenser; (12) gas collection (sampling bag); (13) liquid collection.
The bed temperature was continuously monitored by the temperature sensor located at/near the catalyst zone and maintained at the setpoint by adjusting microwave power through the controller. The temperature fluctuation during continuous operation was typically within ± 10 °C. Under these conditions, all hierarchical ZSM-5 catalysts produced a mixture of gas and liquid products together with a carbonaceous residue deposited on the catalyst. The hot effluent from the reactor was first passed through a condenser to recover the condensable fraction as the liquid product. The non-condensable fraction was subsequently collected in a gas sampling bag connected downstream of the condenser. Condensed liquids were collected at predefined time-on-stream intervals; each liquid sample represents the cumulative condensate collected over the preceding interval, and the collection vessel was replaced immediately at each sampling point. To minimize condenser hold-up effects, the condenser temperature and cooling conditions were kept constant throughout each run, and the condensate receiver was exchanged promptly at each sampling point. Any residual holdup is expected to mainly influence the earliest collection window; subsequent samples reflect the defined time intervals with stable condensation conditions. Liquid products were collected in pre-weighed receivers. Before weighing, the receivers were equilibrated to room temperature to ensure consistent mass determination. The liquid products were analyzed by gas chromatography-mass spectrometry (GC-MS) using a gas chromatograph (Agilent GC 7890) coupled to a mass spectrometer (Agilent MSD 5975C). Gas-phase products collected in the sampling bag were analyzed by the same GC-MS platform. In the following, we focus on five representative catalysts, T-120, T-140, T-180, T-200, and T-220, to illustrate the effects of crystallization temperature on product distribution and apparent lifetime. Coke deposited on the structured catalyst was quantified by mass change of the dried structured catalyst before and after reaction.
Catalyst characterization
X-ray diffraction (XRD)
-
Powder XRD patterns were recorded on a Rigaku SmartLab SE diffractometer using Cu Kα radiation (λ = 1.54060 Å) to verify the crystalline phase and evaluate relative crystallinity.
N2 physisorption (BET)
-
N2 adsorption–desorption isotherms were measured at 77.3 K using an automatic surface area and porosimetry analyzer (Micromeritics ASAP 2460) with N2 as the adsorptive. The specific surface area was calculated by the BET method. Micropore parameters were evaluated by the t-plot method, while mesopore size distributions were derived from the BJH method (adsorption/desorption branches; mesopore range reported in the test output).
Scanning electron microscopy (SEM)
-
SEM images were collected on a Hitachi Regulus/SU8100 field-emission SEM using the secondary-electron signal (SE). The accelerating voltage was 5.0 kV, with a typical working distance of 10.6 mm. Representative micrographs were acquired at 2,000×, 20,000×, and 100,000× magnifications.
NH3 temperature-programmed desorption (NH3-TPD)
-
NH3-TPD was performed following a standard procedure to quantify total acidity and acid strength distribution (weak/medium/strong sites), including catalyst pretreatment, NH3 adsorption to saturation, inert-gas purge to remove physisorbed NH3, and programmed heating with continuous detection of desorbed NH3.
-
Powder XRD confirmed that all catalysts synthesized at different crystallization temperatures (T-120 to T-220) exhibited the MFI framework characteristic of ZSM-5. Figure 2 shows the normalized XRD patterns of ZSM-5 catalysts synthesized at different crystallization temperatures.
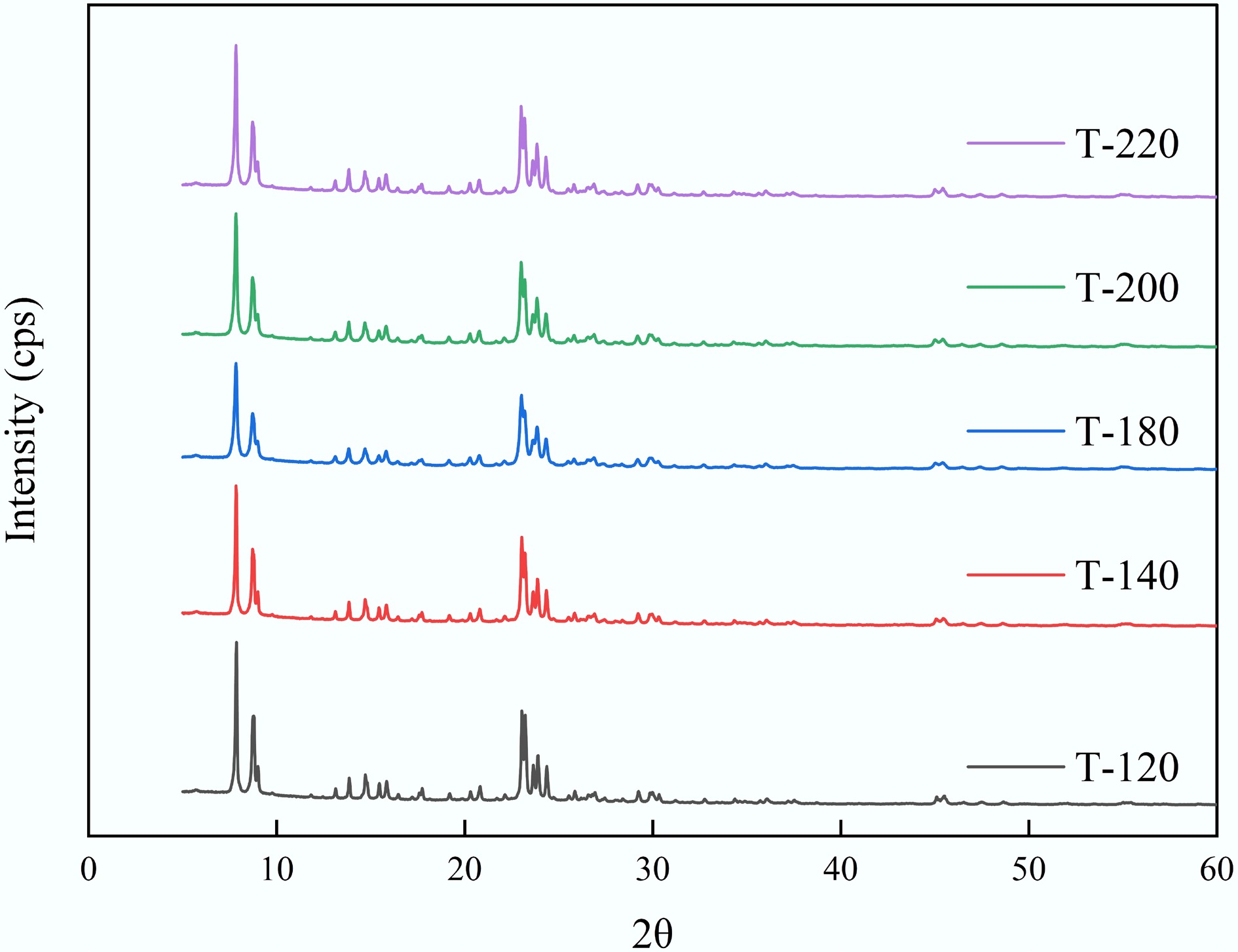
Figure 2.
Normalized XRD patterns of ZSM-5 catalysts synthesized at different crystallization temperatures (T-120, T-140, T-180, T-200, and T-220).
The diffraction patterns displayed the representative reflections at 2θ = 7.82°–7.84°, 14.70°–14.74°, 20.76°–20.80°, 23.02°–23.12°, and 29.88°–29.92°, with nearly identical d-spacings across the series (e.g., d = 1.127–1.130 nm at 7.8°, and d = 0.384–0.386 nm at 23°), indicating that varying crystallization temperature did not alter the zeolite framework or unit-cell metrics in a detectable manner. No additional intense reflections attributable to secondary crystalline phases were observed within the detection limit, suggesting phase-pure ZSM-5 under all synthesis conditions. Peak broadening analysis further indicated nanosized coherent domains: Scherrer sizes were typically 36–40 nm for the low-angle reflection (7.8°) and 20–21 nm for the main reflection near 23°[28]. Only minor variations in FWHM (e.g., 0.38° to 0.40° at 23°) were detected with increasing temperature, implying that the crystallization temperature exerted a relatively modest influence on the coherent crystallite size compared with the pronounced morphology evolution observed by SEM.
SEM analysis: morphology evolution at multi-scales
-
SEM revealed a systematic, crystallization-temperature-driven evolution of ZSM-5 morphology from nanocrystal-assembled hierarchical aggregates to coarsened, densely intergrown crystallites. As shown in Fig. 3, T-120 was dominated by fine particles and cauliflower-like agglomerates; at high magnification, the prevalent 'raspberry-like' secondary particles were constructed from tens-of-nanometer primary units, consistent with hierarchy arising largely from intercrystalline voids between assembled nanograins[29]. Increasing the temperature to T-140 promoted crystal maturation: well-faceted, smooth micrometer-scale crystals (1–2 μm) appeared while a fraction of nanograin-assembled aggregates persisted, indicating concurrent crystal growth and retention of hierarchical domains. At T-180, discrete raspberry-like spheres largely disappeared, and the microstructure shifted to compact, intergrown clusters composed of closely packed cuboidal subcrystallites (50–150 nm), with visibly reduced open void space. Further heating to T-200 and T-220 drove continued coarsening and densification, yielding predominantly blocky/plate-like intergrowths; notably, T-220 also exhibited abundant elongated/rod-like crystallites and the most compact surfaces. Collectively, the progressive loss of loosely assembled nanograin texture with increasing temperature suggested reduced interparticle porosity and transport accessibility at higher crystallization temperatures, providing a structural basis for interpreting temperature-dependent activity-stability trends.

Figure 3.
SEM images of hierarchical ZSM-5 synthesized at different crystallization temperatures (T-120, T-140, T-180, T-200, and T-220). For each sample, images were acquired at 2,000×, 20,000×, and 100,000× to illustrate the morphology evolution from nanocrystal-assembled aggregates to coarsened, densely intergrown crystallites with increasing crystallization temperature.
BET and porosity analysis: hierarchical vs microporous structure
-
Figure 4a compares the N2 adsorption–desorption isotherms of the hierarchical ZSM-5 series. All samples showed a pronounced uptake at low relative pressure (P/P0 < 0.1), consistent with a dominant microporous contribution from the MFI framework, followed by an obvious high-pressure hysteresis that evidenced additional meso-/interparticle porosity. The extent of high- P/P0 uptake followed T-180 > T-120 ≈ T-200 > T-140 > T-220, with the adsorbed amount at P/P0 = 0.99 reaching 197.5 cm3 g−1 (T-180), but only 146.0 cm3 g−1 (T-220). Correspondingly, the total pore volume was maximized for T-180 (0.306 cm3 g−1) and minimized for T-220 (0.226 cm3 g−1), indicating that crystallization temperature predominantly tuned pore volume rather than simply shifting the isotherm shape.
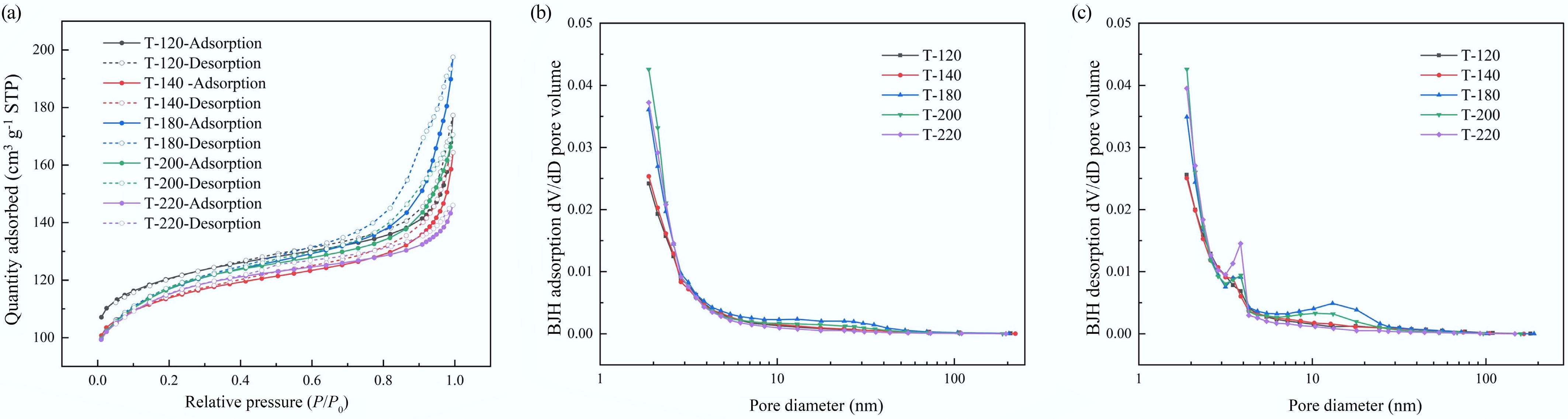
Figure 4.
N2 physisorption characterization of hierarchical ZSM-5 catalysts synthesized at different crystallization temperatures: (a) BET isotherm (Isotherm linear plot); (b) BJH adsorption pore size distribution (dV/dD pore volume); (c) BJH desorption pore size distribution (dV/dD pore volume).
Figure 4b (BJH adsorption) and Fig. 4c (BJH desorption) show that all catalysts were dominated by a sharp pore-size feature centered at 1.88–1.89 nm, while the intensity and breadth of the distributions varied with crystallization temperature. Notably, T-200 exhibited the highest dV/dD peak intensity at 1.88–1.89 nm (reflecting a strong contribution from small mesopores), whereas T-180 delivered the largest BJH cumulative pore volume (0.157–0.158 cm3 g−1), together with a more evident tail at larger diameters (10–30 nm), suggesting a broader hierarchical pore network. In contrast, T-220 showed the smallest BJH cumulative pore volume (0.075–0.077 cm3 g−1) and a narrower desorption-derived pore structure (BJH average pore diameter 4.25 nm), consistent with a less-developed mesoporous component. Overall, while the BET surface areas remained in a relatively narrow range (433–466 m2 g−1), the accessible pore volume and mesoporous contribution were strongly modulated by crystallization temperature, which was expected to impact diffusion and coke tolerance during catalytic pyrolysis.
N2 physisorption evidenced a clear crystallization temperature-dependent redistribution between microporosity and secondary (meso-/interparticle) porosity. Table 1 shows the textural properties of ZSM-5 catalysts synthesized at different crystallization temperatures.
Table 1. Textural properties of ZSM-5 catalysts synthesized at different crystallization temperatures derived from N2 physisorption (BET), t-plot, and BJH analyses
Sample SBET (m2 g−1) Vtotal (cm3 g−1) Vmicro (cm3 g−1) Sext (m2 g−1) Vmeso (cm3 g−1) DBJH (nm) T-120 467.3 0.274 0.147 88.7 0.111 7.67 T-140 439.0 0.254 0.136 91.1 0.102 6.89 T-180 437.1 0.306 0.125 126.5 0.157 7.87 T-200 435.7 0.264 0.122 132.7 0.115 5.85 T-220 434.5 0.226 0.127 116.5 0.075 4.47 SBET from BET method; Vtotal from single-point adsorption at P/P0 = 0.995; Vmicro and Sext from t-plot; Vmeso and average pore diameter DBJH from BJH adsorption branch (1.7–300 nm). All samples exhibited high BET surface areas (434–467 m2 g−1), consistent with well-developed ZSM-5 frameworks. The t-plot micropore volume showed a moderate decrease from T-120 (0.147 cm3 g−1) to T-200 (0.122 cm3 g−1), indicating partial densification/structural reorganization at elevated temperatures without loss of intrinsic microporosity. In contrast, the external surface area increased markedly with temperature, from 89–91 m2 g−1 (T-120/T-140) to 126–133 m2 g−1 (T-180/T-200), suggesting enhanced non-microporous surface contribution associated with hierarchical textures. Notably, T-180 displayed the highest total pore volume (0.306 cm3 g−1) and the largest BJH mesopore volume (1.7–300 nm; 0.157 cm3 g−1), indicating maximized secondary porosity. At the highest temperature, T-220 showed suppressed mesoporosity (0.075 cm3 g−1) and reduced total pore volume (0.226 cm3 g−1) together with a smaller BJH average pore diameter, consistent with a more compact morphology. These trends provided a quantitative basis for correlating transport accessibility with catalytic stability across the series.
NH3-TPD: acid strength and density distribution
-
NH3-TPD profiles of the ZSM-5 series could be described by two desorption features: a low-temperature peak centered at 111–120 °C and a high-temperature peak at 360–420 °C (Fig. 5). As shown in Table 2, while the low-temperature maximum varied only slightly across samples, the high-temperature maximum shifted markedly to higher temperatures from T-120 (360 °C) to T-180/T-200 (420 °C), indicating an increase in NH3 binding strength and thus stronger acid-site character at elevated crystallization temperatures. Quantitatively, the total acidity decreased sharply from T-120 (0.559 mmol g−1) to T-140 (0.197 mmol g−1), and then partially recovered for T-180–T-220 (0.289–0.327 mmol g−1). More importantly, the contribution of the high-temperature (strong-acid) component increased for intermediate-to-high temperatures, reaching a maximum strong-acid fraction at T-180 (40%). This evolution suggested that crystallization temperature not only tuned the amount of accessible acid sites, but also redistributed acid strength, which was expected to impact aromatization/cracking intensity and deactivation propensity in plastic catalytic conversion[30].
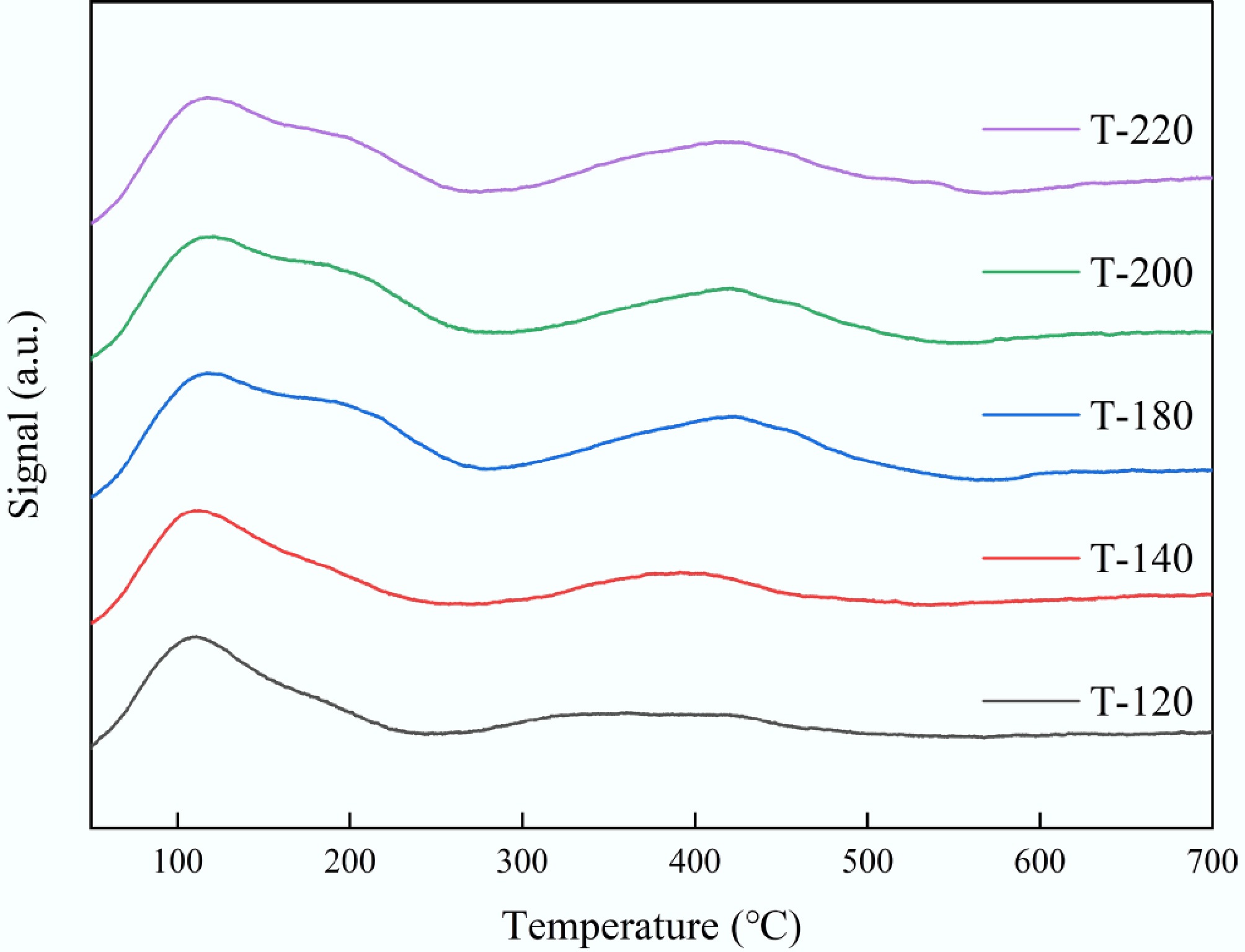
Figure 5.
NH3-TPD profiles of ZSM-5 synthesized at different crystallization temperatures (T-120, T-140, T-180, T-200, and T-220).
Table 2. NH3-TPD-derived acidity of ZSM-5 catalysts synthesized at different crystallization temperatures
Sample Peak 1 Tmax (°C) Peak 1 NH3 (mmol g−1) Peak 2 Tmax (°C) Peak 2 NH3 (mmol g−1) Total acidity (mmol g−1) Strong-acid fraction (%) T-120 111.06 0.40 360.15 0.16 0.56 28.79 T-140 112.27 0.14 391.26 0.06 0.20 29.77 T-180 116.88 0.20 419.65 0.13 0.32 40.15 T-200 120.38 0.19 420.05 0.10 0.29 34.05 T-220 116.81 0.19 412.20 0.12 0.32 38.72 Peak 1 (low-temperature) is typically assigned to weak/medium acid sites and/or weakly bound NH3; Peak 2 (high-temperature) corresponds to stronger acid sites (more strongly bound NH3). Reported acidity is the integrated NH3 quantity (mmol g−1). Strong-acid fraction = Peak 2/(Peak 1 + Peak 2). Catalytic pyrolysis of plastic waste: product distribution and apparent lifetime
Overall behaviour: liquid fuels and gasoline-range fraction
-
In this work, the gasoline fraction in the condensed liquid is defined based on thermogravimetric analysis (TGA) using a simulated boiling cut at 200 °C. The mass fraction volatilized below 200 °C is denoted as gasoline, and a higher gasoline fraction indicates more extensive cracking/upgrading and thus a healthier catalyst state.
The gasoline fraction in the liquid at a given time-on-stream (TOS) is defined as:
$ G(t)=\dfrac{m_{ \lt 200\;^\circ {\rm C}}(t)}{m_{\rm{liq}}(t)}\times 100{\text{%}} $ (1) where, m< 200 °C(t) is the mass loss of the collected liquid sample below 200 °C in TGA, and mliq(t) is the total collected liquid mass.
To quantitatively track activity retention, the normalized gasoline retention is defined as:
$ A(t)=\dfrac{G(t)}{G({t}_{0})} $ (2) where, t0 is the first sampling time (here, 0.5 h).
A practical lifetime descriptor is the time required for the gasoline fraction to drop to a selected threshold x (e.g., 70% or 60%):
$ {t}_{G=x}\;\text{such that}\;G({t}_{G=x})=x $ (3) In this study, tG=x was obtained by linear interpolation between two adjacent sampling points (t1, G1) and (t2, G2), where G1 > x > G1:
$ {t}_{G=x}={t}_{1}+\dfrac{x-{G}_{1}}{{G}_{2}-{G}_{1}}({t}_{2}-{t}_{1}) $ (4) An overall gasoline decay rate (in percentage points per hour) is reported as:
$ {r}_{G}=\dfrac{G({t}_{\text{end}})-G({t}_{0})}{{t}_{\text{end}}-{t}_{0}} $ (5) where, tend is the last sampling time of each run.
Figure 6 summarizes the evolution of the condensed liquid composition during catalytic pyrolysis as a function of time-on-stream (TOS), using the TGA-derived gasoline cut as an operational indicator of cracking severity and catalyst health. All catalysts initially produced liquids dominated by gasoline-range compounds, with G(t0) in the range of 87.4%–94.7% at t0 = 0.5 h (Table 3).
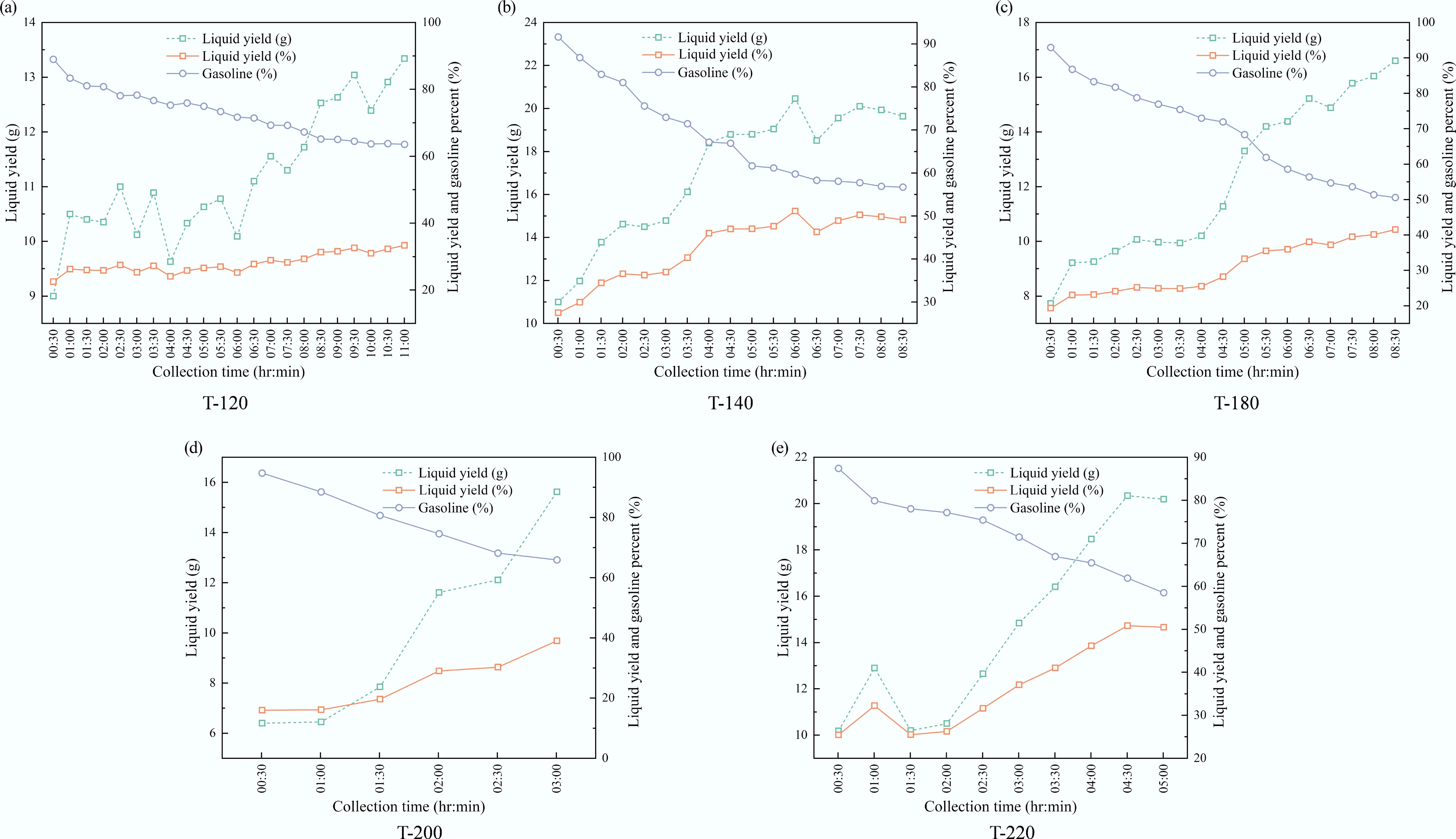
Figure 6.
Time-resolved composition of condensed liquid products over ZSM-5 catalysts synthesized at different crystallization temperatures: (a) 120 °C, (b) 140 °C, (c) 180 °C, (d) 200 °C, and (e) 220 °C.
Table 3. Gasoline indicators and deactivation metrics derived from TGA of condensed liquids
Catalyst G(t0) at 0.5 h (%) G at 3 h (%) tG=70% (h) tG=60% (h) G(tend) (%) tend (h) rG (%-pt h−1) T-120 88.98 78.28 6.83 - 63.55 11.0 −2.42 T-140 91.61 72.94 3.67 5.92 56.71 8.5 −4.36 T-180 92.92 76.92 4.77 5.78 50.58 8.5 −5.29 T-200 94.73 65.90 2.36 - 65.90 3.0 −11.53 T-220 87.41 71.45 3.16 4.78 58.50 5.0 −6.42 Gasoline is defined as the liquid fraction volatilized below 200 °C in TGA. tG=x values were obtained by linear interpolation (Eq. [4]). rG is calculated using Eq. (5). With continued operation, G(t) decreased for every catalyst, indicating a progressive shift of the condensed liquid toward heavier fractions, consistent with catalyst deactivation (reduced secondary cracking/upgrading). The decay kinetics, however, strongly depended on crystallization temperature. T-120 exhibited the most stable gasoline selectivity, maintaining G > 70% until 6.83 h and still delivering 63.6% at 11 h, corresponding to the smallest magnitude of gasoline-decay rate (rG = –2.42 pt h−1). In contrast, T-140 and T-220 crossed G = 70% much earlier (3.67 and 3.16 h, respectively), while T-180 displayed the deepest long-term decline, reaching 50.6% at 8.5 h. The short T-200 run showed a rapid early loss of gasoline selectivity, dropping below 70% by 2.36 h. Collectively, the gasoline fraction provided a simple, visually interpretable proxy that directly linked product distribution evolution to catalyst lifetime.
Gas-phase product distribution
-
Time-resolved gas analysis (Fig. 7) showed that the C1–C4 slate evolved markedly with catalyst crystallization temperature, and that this evolution became more pronounced as the run proceeded.
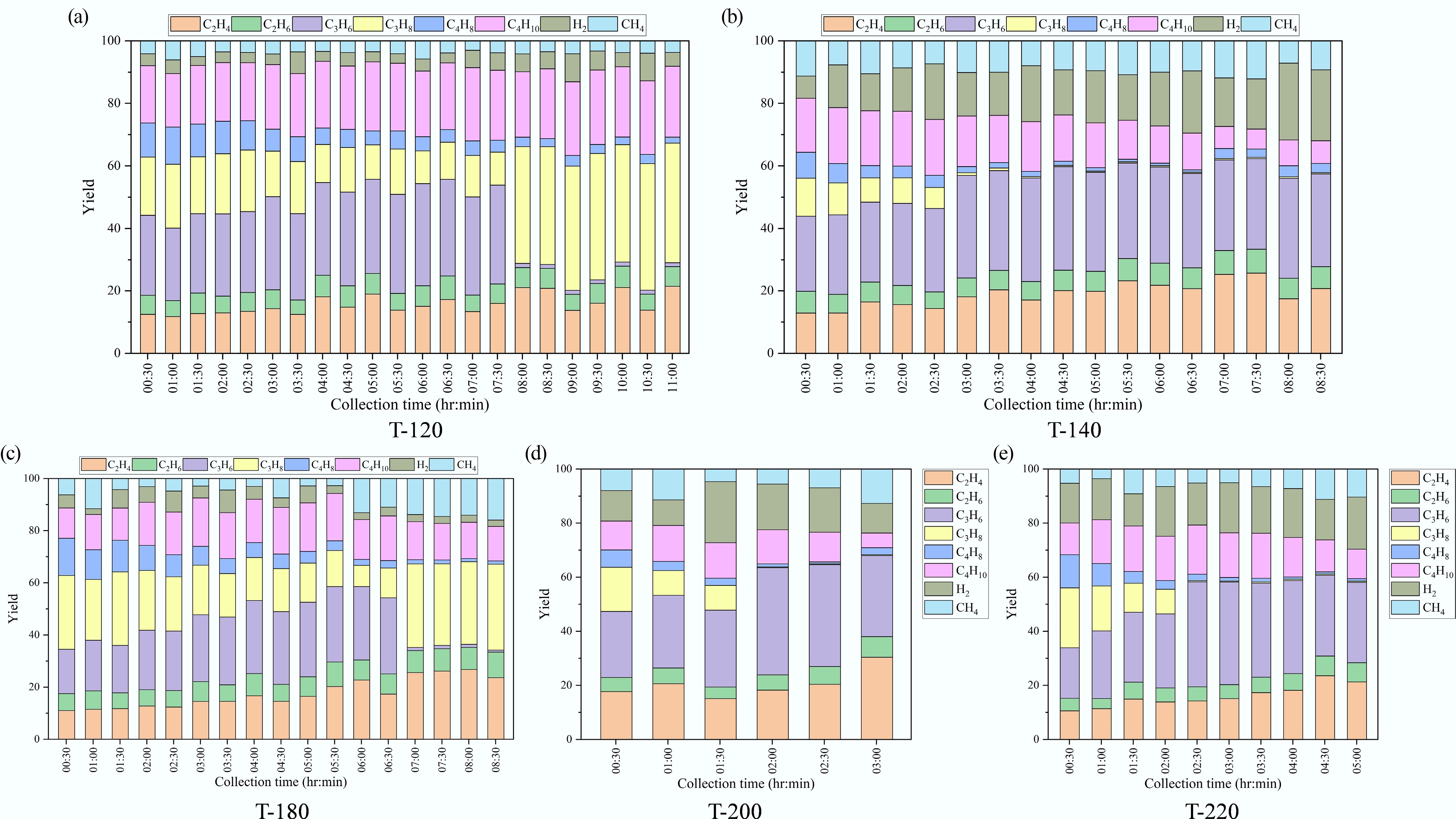
Figure 7.
Time-resolved composition of gas products over ZSM-5 catalysts synthesized at different crystallization temperatures: (a) 120 °C, (b) 140 °C, (c) 180 °C, (d) 200 °C, and (e) 220 °C.
For T-120, the early gas was distributed across C2–C4 species (e.g., at 0:30, C3H6 25.7%, C3H8 18.6%, C4H8 10.9%, C4H10 18.3%), but with time the composition shifted strongly toward more saturated C3–C4 products, with C3H6 collapsing to 1.2% and C3H8 rising to 38.3% by 11:00; C4H8 also dropped to 1.9% while C4H10 remained high (22.7%). T-180 exhibited a similar shift: C3H6 decreased from 17.0% (0:30) to 0.8% (8:30), while C3H8 stayed high (28.3% to 32.9%) and CH4 increased substantially (6.3% to 15.9%). In contrast, higher-temperature samples maintained larger olefin fractions later in the run. For T-140, C3H6 increased from 24.1% to 29.6% and C3H8 dropped to 0.4% by 8:30, accompanied by a strong rise in H2 (7.1% to 22.7%). T-200 (0:30–3:00) and T-220 (0:30–5:00) also showed very low terminal C3H8 (0.3%–0.5%) with sustained/high C2H4 and C3H6 (e.g., T-220 at 5:00: C2H4 21.2%, C3H6 29.7%, H2 19.3%). Overall, low/intermediate crystallization temperatures (T-120, T-180) drifted toward propane-butane-rich gases over time, whereas higher-temperature catalysts (T-140, T-200, T-220) retained higher ethylene/propylene and H2 fractions at later collection times.
Liquid-phase composition from GC-MS
-
Figure 8 summarizes the time-resolved mass fraction of major liquid families (paraffins, olefins, monoaromatics, BTX, and others) over ZSM-5 catalysts synthesized at different crystallization temperatures.
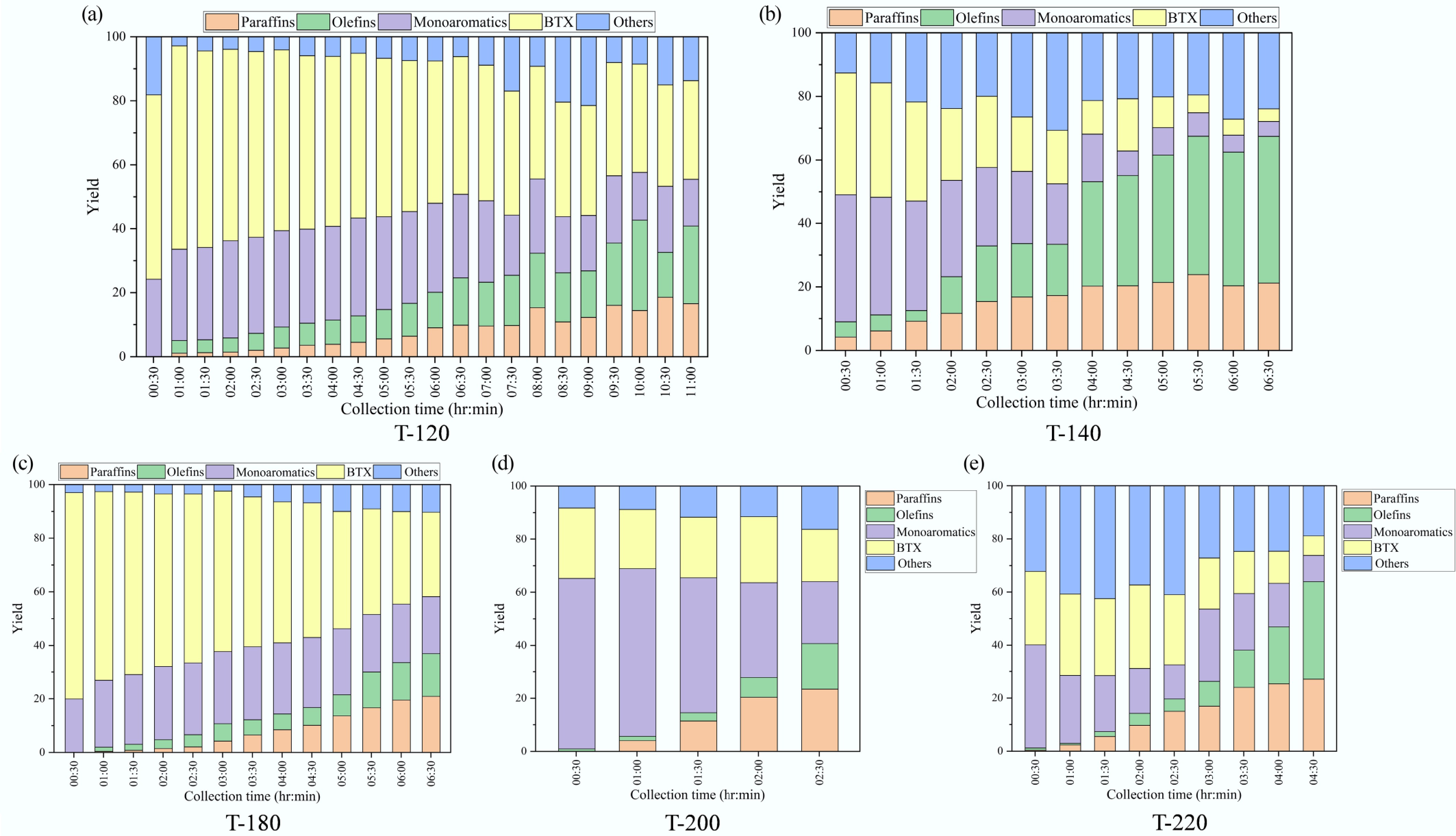
Figure 8.
Time-resolved liquid product speciation during catalytic pyrolysis over ZSM-5 catalysts synthesized at different crystallization temperatures (paraffins, olefins, monoaromatics, BTX, and others; mass fraction): (a) 120 °C, (b) 140 °C, (c) 180 °C, (d) 200 °C, and (e) 220 °C.
Overall, the liquid pool evolved from an aromatic-rich composition at early collection times toward more aliphatic (paraffin/olefin) products as the run proceeded, with the extent and rate of this shift strongly depending on the catalyst. For T-120, the early liquid was dominated by aromatics (BTX 57.6 wt% and monoaromatics 24.2 wt% at 0.5 h). With continued operation, BTX decreased gradually (to 44.4 wt% at 6 h and 30.8 wt% at 11 h), while olefins and paraffins increased steadily, reaching 24.2 wt% and 16.6 wt% at 11 h, respectively; monoaromatics also declined to 14.7 wt%. T-180 showed an even stronger initial BTX enrichment (77.1 wt% at 0.5 h), followed by a pronounced decrease to 31.5 wt% at 6.5 h, accompanied by growth of paraffins (0 wt% to 20.9 wt%) and olefins (0.1 wt% to 16.0 wt%); monoaromatics increased to 27.0 wt% at 3 h and then moderated to 21.2 wt% at 6.5 h. In contrast, T-140 underwent a rapid transition from aromatics to aliphatics: BTX dropped from 38.3 wt% (0.5 h) to 17.1 wt% (3 h) and further to 4.0 wt% (6.5 h), while olefins rose sharply to 46.2 wt% and paraffins to 21.2 wt% by 6.5 h. T-200 exhibited a similarly fast compositional drift within a short time window, with monoaromatics decreasing from 64.3 wt% (0.5 h) to 23.3 wt% (2.5 h) and concurrent increases in paraffins (0 wt% to 23.4 wt%) and olefins (0.9 wt% to 17.2 wt%). T-220 started with a large other fraction (32.2 wt% at 0.5 h), reached a local maximum of BTX at 2 h (31.6 wt%), and then shifted strongly toward aliphatics at 4.5 h (olefins 36.8 wt% and paraffins 27.1 wt%) with BTX and monoaromatics decreasing to 7.3 wt% and 9.9 wt%, respectively. These results demonstrate that crystallization temperature not only influenced the initial aromatic distribution, but also governed how rapidly the liquid composition migrated toward paraffin/olefin-rich products during extended operation.
Deactivation mechanism
-
Powder XRD confirmed that all samples retained the phase-pure MFI framework across the entire crystallization-temperature window (T-120 to T-220), with nearly identical characteristic reflections and no detectable secondary phases. Therefore, the time-on-stream (TOS) performance decay was not attributable to framework collapse, but instead to progressive loss of effective accessibility of acid sites and shape-selective channels during operation. The crystallization temperature primarily rewired two control knobs that governed this accessibility: (1) the hierarchical morphology/porosit; and (2) the density and strength distribution of acid sites. SEM revealed a systematic evolution from nanocrystal-assembled aggregates with abundant intercrystalline voids (T-120) toward more compact, densely intergrown crystallites at higher temperatures, especially for T-200/T-220. In parallel, N2 physisorption showed that while intrinsic microporosity was preserved within a narrow range (Vmicro = 0.122–0.147 cm3 g−1), the secondary porosity (Vmeso), and total pore volume varied strongly: T-180 maximized Vtotal and Vmeso, whereas T-220 exhibited the lowest Vmeso and a smaller BJH pore diameter, consistent with a more diffusion-restricted architecture. Acidity evolved just as decisively: NH3-TPD indicated a sharp drop in total acidity from T-120 (0.559 mmol g−1) to T-140 (0.197 mmol g−1), followed by partial recovery at higher temperatures, while the strong-acid fraction increased and reached a maximum at T-180 (40%).
These structure acidity differences manifested directly in the time-resolved product fingerprints and the apparent lifetime metrics. Using the TGA-defined gasoline cut (volatile below 200 °C) as an operational indicator of cracking/upgrading severity, all catalysts showed a monotonic decline in gasoline fraction with TOS, but with highly catalyst-dependent kinetics. The stability spread was large: T-120 retained G > 70% until 6.83 h and showed the slowest decay rate (rG = −2.42 %-pt h−1), whereas T-220 and T-140 crossed G = 70% much earlier (3.16 and 3.67 h), and T-200 exhibited the fastest early loss (tG=70% = 2.36 h; rG = −11.53 %-pt h−1). Importantly, the gasoline-fraction loss was not simply a family shift from aromatics to aliphatics: GC-MS family analysis showed that, as catalysts deactivated, the condensed pool could become simultaneously more aliphatic yet less gasoline-range (i.e., heavier olefin/paraffin oligomers increasingly dominate), consistent with weakened secondary cracking/upgrading rather than a single-path selectivity switch.
A mechanistic linkage emerged when the liquid-family evolution (Fig. 8) and gas-phase slate (Fig. 7) were interpreted as in situ reporters of acid-function retention. At early TOS, aromatic-rich liquids indicated that polymer-derived intermediates underwent sufficient secondary chemistry on the zeolite to form BTX/monoaromatics. As deactivation proceeded, suppression of these secondary steps was accompanied by increased prevalence of paraffins/olefins in the condensed phase and persistent light olefins (C2H4/C3H6) in the gas phase for the less stable catalysts[31]. For example, T-140 exhibited a rapid collapse of BTX from 38.3 wt% (0.5 h) to 4.0 wt% (6.5 h), while olefins rose to 46.2 wt% and paraffins to 21.2 wt%, demonstrating a fast loss of the aromatic-forming function. Consistently, its gas-phase evolution retained high propylene and strongly rising H2 (H2: 7.1% to 22.7%; C3H6 increased to 29.6%, while C3H8 dropped to 0.4% by 8:30), indicating that olefin-rich cracking/dehydrogenation signatures persisted while hydrogen-transfer-associated saturation became weak at later times. This behavior aligned with its low total acidity (0.197 mmol g−1), which limited the number of active turnovers available for sustained secondary conversion as the catalyst surface became progressively covered/blocked. At the opposite end, T-120 sustained aromatic production for much longer, despite having the highest total acidity (0.559 mmol g−1). Its morphology was dominated by nanograin-assembled aggregates with abundant intercrystalline voids (Fig. 3), which could preserve pathway continuity between external surfaces and the micropore network under coking conditions. Accordingly, the aromatic-to-aliphatic drift was gradual: BTX decreased from 57.6 wt% (0.5 h) to 30.8 wt% (11 h), while olefins and paraffins rose steadily to 24.2 wt% and 16.6 wt%, respectively. Meanwhile, the gas composition evolved toward more saturated C3–C4 products over time (C3H6: 25.7% to 1.2%; C3H8: 18.6% to 38.3% by 11:00), consistent with continued secondary conversion capacity and a slower loss of the gasoline-range fraction (tG=70% = 6.83 h; rG = −2.42 %-pt h−1). The key implication was that lifetime was not dictated by total acidity alone, but by whether the pore-morphology network could keep a meaningful fraction of acid sites accessible as deposits accumulated.
T-180 occupied a distinct regime: it delivered the most BTX-enriched initial liquids (77.1 wt% at 0.5 h) and exhibited the highest strong-acid fraction (40%) together with the largest secondary porosity (Vmeso = 0.157 cm3 g−1; Vtotal = 0.306 cm3 g−1). This combination intensified the aromatic-forming chemistry early on, but also accelerated the decline of gasoline fraction at longer TOS (G[tend] = 50.58% at 8.5 h; rG = −5.29 %-pt h−1). The gas slate shifted toward propane/butane enrichment as the run proceeded (C3H6: 17.0% to 0.8%; C3H8 remained high, 28.3% to 32.9%), suggesting that the catalyst maintained substantial secondary-conversion character even while the gasoline fraction decayed.
Together, these signatures are most consistent with a scenario in which strong acid sites drive rapid secondary chemistry (supporting high BTX), but progressive pore blocking within the shape-selective domain increasingly suppresses further cracking to gasoline-range products, causing the condensed pool to drift heavier despite continued formation of aromatics/monoaromatics in the remaining accessible domain[32]. Finally, the high-temperature end (T-220) illustrated the combined penalty of stronger acidity and reduced mesoporosity. T-220 had a high strong-acid fraction (38.72%) but the lowest mesopore volume (0.075 cm3 g−1) and a more compact morphology with rod-like crystallites, pointing to more severe transport restriction. Correspondingly, deactivation was rapid (tG=70% = 3.16 h; rG = −6.42 %-pt h−1). Its gas phase retained high ethylene/propylene and elevated H2 at later times (e.g., at 5:00: C2H4 21.2%, C3H6 29.7%, H2 19.3%) with very low propane, while its liquid pool shifted strongly toward paraffins/olefins at 4.5 h (olefins 36.8 wt%, paraffins 27.1 wt%) with BTX and monoaromatics dropping to 7.3 wt% and 9.9 wt%. This pattern indicated that once access to the catalytic domain was curtailed, the reactor increasingly expressed primary cracking signatures (olefin-rich light gases and aliphatic liquids), while the aromatic-upgrading function collapsed early.
Overall, the combined evidence supports a deactivation picture governed by crystallization temperature-controlled balancing between (1) hierarchical accessibility that buffers the zeolite domain against progressive blockage; and (2) acid strength/density that controls the intensity of secondary conversion and the propensity for rapid loss of effective functionality. The longest-lived catalyst (T-120) coupled a highly open, nanograin-assembled architecture with sufficient acidity to sustain secondary chemistry for extended times, yielding the slowest gasoline-fraction decay. In contrast, catalysts with more compact morphologies and/or stronger-acid enrichment (notably T-220 and the fast-decaying T-200 run) lost gasoline-range selectivity much earlier, consistent with faster loss of accessible catalytic pathways under continuous plastic conversion.
-
Hierarchical ZSM-5 catalysts synthesized at different crystallization temperatures (120–220 °C) were evaluated for microwave-assisted catalytic pyrolysis of plastic waste. All catalysts retained the MFI framework, while crystallization temperature strongly regulated secondary porosity and acidity without altering phase purity. Among them, T-180 exhibited the largest mesopore volume (0.157 cm3 g−1) and strongest acid-site contribution (40.15%), whereas T-220 showed restricted mesoporosity and diffusion limitations. Catalyst lifetime was quantitatively assessed using a TGA-defined gasoline fraction (≤ 200 °C). T-120 showed the highest stability, maintaining gasoline yields above 70% for 6.83 h and 63.55% after 11 h, corresponding to the slowest deactivation rate (−2.42 %-pt h−1). In contrast, catalysts synthesized at higher temperatures deactivated more rapidly, with T-200 reaching 70% gasoline within 2.36 h. Product distribution analysis revealed that deactivation was associated with suppressed secondary upgrading, reflected by declining BTX yields and increased light hydrocarbon fractions. These results demonstrate that balancing hierarchical porosity and acidity via crystallization-temperature control is critical for extending catalyst lifetime in plastic waste valorization.
The authors gratefully acknowledge the Analytical and Testing Center of Southeast University for providing instrument access and technical support.
-
The authors confirm their contributions to the paper as follows: Cunfeng Ke: writing – review & editing, writing – original draft, visualization, validation, investigation, formal analysis, data curation, conceptualization; Yunlong Li: data curation, investigation; Leilei Dai: writing – review & editing, methodology, formal analysis, data curation, investigation, supervision, resources; Zhaoyang Liu: data curation; Lata Suman: data curation; Roger Ruan: methodology, investigation; Yugang Wang: formal analysis, data curation; Yaming Gao: data curation; Chunfeng Chen: formal analysis, data curation; Huiyan Zhang: writing – review & editing, visualization, supervision, resources, project administration, investigation, formal analysis, conceptualization. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
-
This work was supported by the National Natural Science Fund for Distinguished Young Scholars of China (Grant No. 52425607), and the Natural Science Foundation of Jiangsu Province (BK20240010).
-
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
-
Full list of author information is available at the end of the article.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Ke C, Li Y, Dai L, Liu Z, Lata S, et al. 2026. One-pot synthesis of hierarchical ZSM-5 for lifetime improvement in catalytic conversion of plastic waste. Sustainable Carbon Materials 2: e018 doi: 10.48130/scm-0026-0013 

One-pot synthesis of hierarchical ZSM-5 for lifetime improvement in catalytic conversion of plastic waste
- Received: 17 February 2026
- Revised: 02 March 2026
- Accepted: 17 March 2026
- Published online: 08 April 2026
Abstract: Hierarchical ZSM-5 catalysts were synthesized via a one-pot route at crystallization temperatures between 120 and 220 °C and applied to microwave-assisted catalytic pyrolysis of plastic waste at 500 °C under continuous operation. All materials preserved the MFI framework, while crystallization temperature strongly tuned secondary porosity and acidity; for example, the mesopore volume increased from 0.075 cm3 g−1 (T-220) to 0.157 cm3 g−1 (T-180). Catalyst lifetime was quantified using a TGA-defined gasoline fraction (≤ 200 °C) of the condensed liquid as a product-relevant activity descriptor. The gasoline fraction decreased monotonically with time-on-stream for all catalysts, but with markedly different decay rates. T-120 showed the highest stability, maintaining gasoline yields above 70% for 6.83 h and still achieving 63.55% after 11 h, whereas catalysts synthesized at higher crystallization temperatures deactivated much faster, with T-200 and T-220 crossing the 70% threshold after only 2.36 and 3.16 h, respectively. Time-resolved product analysis further revealed that deactivation was accompanied by a strong decline in aromatic upgrading (e.g., BTX dropping from 38.3 wt% to 4.0 wt% for T-140) and an increase in less-upgraded aliphatic fractions. These results demonstrate that controlling crystallization temperature offers a simple, scalable means to balance hierarchical porosity and acidity, thereby extending catalyst lifetime and improving liquid-fuel quality in plastic waste valorization.