








-
Bacterial wilt, caused by the Ralstonia solanacearum (R. solanacearum) species complex, is a devastating disease affecting peanuts (Arachis hypogaea L.) worldwide[1,2]. The pathogen invades vascular tissues, causing sudden wilting, stunted growth, and plant death, often leading to complete yield loss in severely affected fields[3,4]. Its prevalence spans major peanut-growing regions in Africa, Asia, and the Americas, where conducive environmental conditions, particularly warm and humid climates, favor pathogen proliferation[5].
In peanut-growing regions such as South and Southeast Asia, sub-Saharan Africa, and Latin America, outbreaks frequently occur despite resistant cultivars and standard management practices[6]. The pathogen's persistence in soil, water, and alternate hosts, combined with reliance on symptom-based diagnostics, often leads to undetected strain variation and inconsistent control outcomes.
Economically, bacterial wilt represents a substantial threat, with estimated losses reaching millions of dollars annually, compromising both smallholder livelihoods and commercial production systems[7]. Despite decades of research, the disease continues to challenge peanut production, reflecting the complexity of pathogen-host-environment interactions.
Current management strategies for bacterial wilt in peanuts encompass a combination of approaches, including the deployment of resistant cultivars, chemical treatments, cultural practices such as crop rotation and soil amendments, and biological control agents[8]. While these measures have provided varying levels of success, their effectiveness is often inconsistent across regions and seasons. Resistant cultivars sometimes fail under high pathogen pressure, chemicals offer temporary suppression with environmental concerns, and biological control approaches, although promising, frequently show variable field performance[9,10]. This inconsistency emphasizes the need for a deeper understanding of underlying factors that may influence disease outbreaks beyond traditional epidemiological assessments.
In the R. solanacearum species complex, cryptic diversity refers to genetically distinct lineages that are difficult to distinguish with routine diagnostics[11]. These lineages, classified by phylotypes and sequevars, can differ in virulence, host range, and response to control measures, explaining why resistant cultivars or biological and chemical treatments may fail in certain peanut fields.
One factor that has received relatively little attention is cryptic diversity within bacterial wilt pathogens. Cryptic diversity denotes biologically meaningful variation within pathogen populations that escapes detection by routine diagnostic, phenotypic, or taxonomic approaches[12]. In bacterial wilt systems, it encompasses concealed genetic, epigenetic, phenotypic, and ecological differences that collectively shape virulence, adaptability, and disease dynamics[13]. This hidden variation can drive unexpected outbreaks, cultivar-specific susceptibility, and variable disease severity. Cryptic diversity differs from general genetic variability by focusing on hidden, functionally consequential variation that eludes standard typing and requires high-resolution genomic or ecological analyses for detection[14]. It extends beyond phenotypic plasticity by encompassing stable lineage differences that persist across environments, explaining why routine diagnostics often underestimate pathogen complexity.
Phylogenetic analyses, particularly using multilocus sequence typing (MLST), whole-genome sequencing, and selected marker loci, have become essential for resolving diversity within the R. solanacearum species complex[15]. Genome-based classification has reshaped the understanding of bacterial wilt pathogens, as traditional host-range and biochemical systems offered limited resolution and obscured evolutionary relationships[16]. Phylogenomics now reveals a phenotypically similar, yet deeply divergent species complex, where cryptic diversity within and across lineages emerges as a defining feature rather than a fine-scale anomaly.
Routine diagnostics often detect pathogens only at broad taxonomic levels, while single-marker and even multilocus assays lack the resolution to capture accessory genomic and regulatory variation linked to virulence[17]. Although whole-genome methods provide superior resolution, their adoption is constrained by cost, infrastructure, and inconsistent protocols, underscoring the need for harmonized sampling, standardized workflows, and open data reporting to improve detection of cryptic pathogen populations.
These approaches reveal cryptic lineages that are often indistinguishable phenotypically but differ in virulence, host specificity, and environmental adaptation, including those infecting peanuts[18]. In peanut-growing regions, molecular phylogenetics has identified lineage-specific patterns that explain variable disease severity, resistance breakdown, and inconsistent responses to chemical or biological controls[19]. Despite these advances, the extent to which cryptic genomic and ecological diversity drives recurrent outbreaks in peanut fields remains underexplored. Therefore, this review aims to synthesize current knowledge of R. solanacearum cryptic diversity, highlighting its adaptive potential, implications for host-pathogen interactions, and relevance for designing targeted, region-specific management strategies.
-
Hidden genomic diversity and complex evolutionary mechanisms underpin the remarkable adaptability and persistence of plant pathogenic bacteria[20]. Traditional classification based on biovars, races, or phylotypes has provided a coarse understanding of pathogen diversity to capture subtle, functionally significant differences[21]. Genetic variation within R. solanacearum and closely related species infecting peanuts is increasingly recognized as a key factor in pathogen adaptability and disease persistence[22,23].
It harbors exceptional genomic variability resulting from an expansive and dynamic pan-genome. Comparative genomic analyses show that the core genome constitutes only about 17% of the species' total gene pool, while the majority of genes belong to the accessory genome[24]. This accessory fraction includes many genes involved in pathogenicity, host specificity, and ecological adaptation, and ongoing horizontal gene transfer and recombination continually reshape this genomic landscape, enabling rapid evolutionary change[25].
Recent evidence suggests that pathogen populations harbor extensive cryptic variation, which may manifest as differences in virulence, host specificity, or environmental tolerance[26,27]. Such hidden diversity challenges assumptions that a single resistant cultivar or management practice can effectively control all strains, potentially underpinning unexplained disease outbreaks.
Advances in molecular and genomic tools have revolutionized the ability to uncover this hidden diversity[28,29]. Single-nucleotide polymorphism (SNP)-based phylogenomics, whole-genome sequencing (WGS), and pan-genome analyses allow researchers to resolve genetic differences at unprecedented resolution, revealing lineage-specific adaptations and novel gene clusters[30,31]. These approaches not only differentiate closely related strains, but also identify genes associated with virulence, stress tolerance, and metabolic flexibility. In peanut-infecting Ralstonia, applying these techniques has uncovered previously unrecognized subpopulations that may escape detection by conventional diagnostic or phenotypic assays[32].
Evolutionary mechanisms driving this cryptic diversity are multifaceted. Horizontal gene transfer (HGT) facilitates the acquisition of virulence factors and metabolic genes from unrelated strains or species, while plasmids and prophages serve as mobile genetic elements that can rapidly reshape pathogen genomes[33,34]. Spontaneous mutations, including point mutations, insertions, and deletions, further contribute to intra-population variability[35]. Together, these mechanisms generate a dynamic landscape of genomic diversity, enabling the emergence of cryptic lineages (a pathogen species that look identical but differ in traits) capable of evading host resistance, surviving environmental stress, or exploiting novel ecological niches.
Regional populations of R. solanacearum have undergone allopatric divergence driven by long-term geographic isolation and adaptation to distinct climatic conditions and host assemblages, resulting in structured phylogenetic lineages[36,37]. At the same time, frequent intra-phylotype recombination reshuffles genetic loci among closely related strains, generating novel allelic combinations and continuously amplifying genomic diversity within regional populations.
Pathogen populations show pronounced geographic structuring driven by environmental adaptation and historical isolation, with distinct lineages linked to specific climatic and agroecological zones[38]. Human-mediated dispersal via contaminated materials, water, soil, and equipment can spread lineages across regions, introducing novel variants that trigger disease emergence and resistance breakdown[39].
Critically, the hidden genomic diversity offers a plausible explanation for unexpected bacterial wilt outbreaks even in fields planted with resistant cultivars. Resistant lines may be highly effective against dominant pathogen strains, but ineffective against rare or cryptic variants that harbor unique virulence determinants[40]. This observation emphasizes the limitations of current resistance-based strategies and highlights the need for integrative approaches combining genomic surveillance, predictive modeling, and dynamic breeding strategies. Hence, understanding the evolutionary drivers and functional consequences of cryptic diversity is therefore essential for developing sustainable and long-term control measures for bacterial wilt in peanuts.
Hidden genomic diversity continuously drives evolutionary processes horizontal gene transfer, mutations, recombination, and mobile elements producing cryptic lineages that evade resistance, tolerate stress, and persist in agroecosystems (Fig. 1). This dynamic genomic turnover underlies unexpected bacterial wilt outbreaks, highlighting the need for continuous surveillance and adaptive disease management strategies.
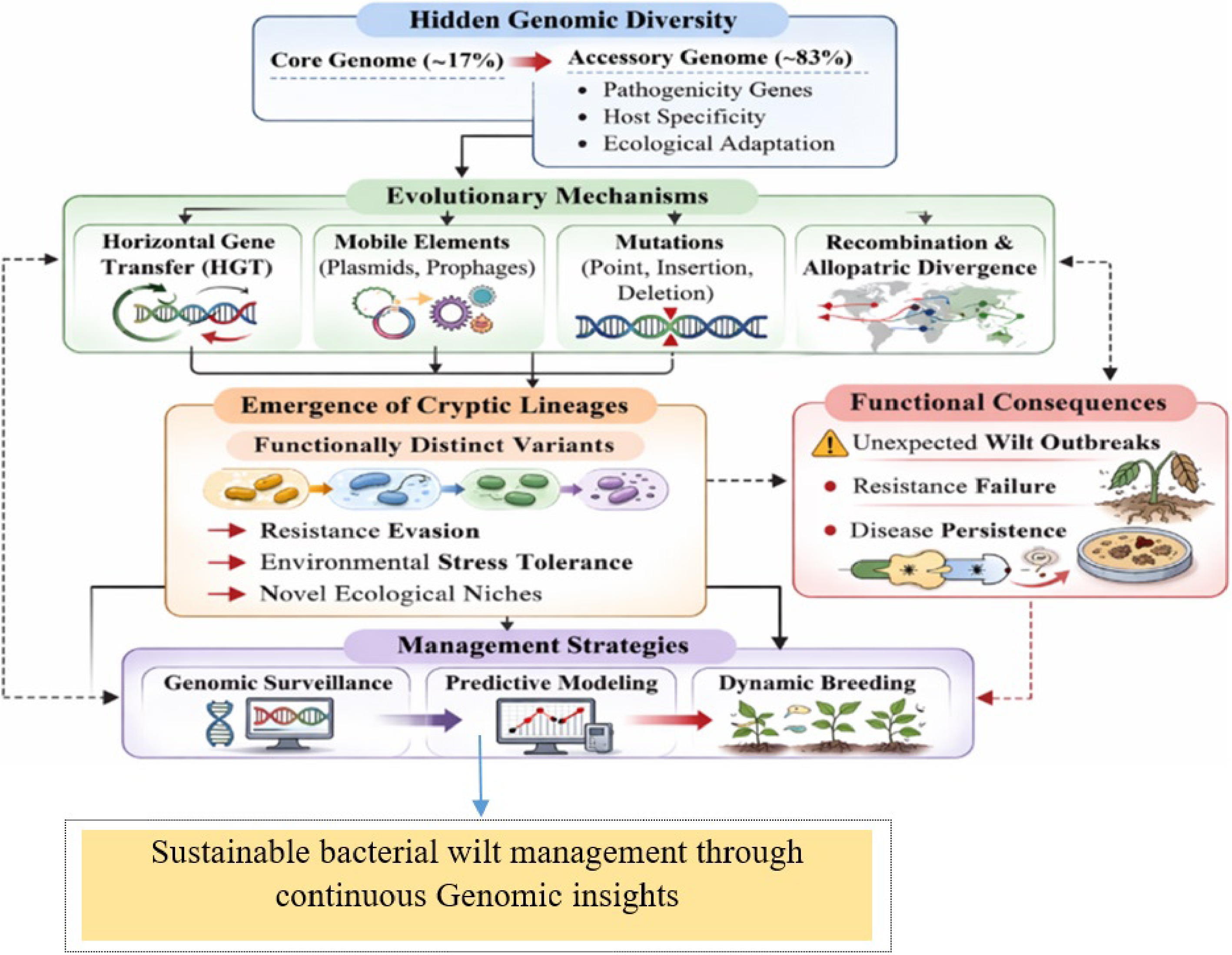
Figure 1.
Cryptic genomic diversity and evolutionary mechanisms in R. solanacearum.
-
Epigenetics, traditionally studied in eukaryotes, is increasingly recognized as a significant factor in bacterial biology, including plant pathogens such as R. solanacearum[41,42]. In bacteria, epigenetic mechanisms involve DNA methylation, modifications mediated by histone-like proteins, and the regulatory functions of small RNAs[43,44]. Unlike genetic mutations, these modifications do not alter the nucleotide sequence but can profoundly influence gene expression, enabling pathogens to fine-tune their physiological and pathogenic responses to changing environments[45,46]. This regulatory layer adds complexity to the understanding of bacterial adaptation and disease dynamics in crop systems.
DNA methylation, one of the best-characterized bacterial epigenetic processes, can regulate the timing and intensity of virulence gene expression[44,47]. Similarly, histone-like proteins influence chromosomal organization and accessibility, modulating transcriptional responses to environmental cues[48]. Small RNAs serve as rapid, reversible regulators, adjusting mRNA stability or translation to optimize pathogen fitness[49,50]. Together, these mechanisms allow bacteria to swiftly respond to stress, evade host defenses, or adjust metabolic pathways without permanent genomic changes, providing a flexible system for survival and infection.
Despite its recognized importance in bacterial physiology and virulence in other crops, epigenetic regulation remains largely unexplored in the context of bacterial wilt in peanuts. There is a striking paucity of studies examining how methylation patterns, nucleoid-associated proteins, or small RNAs influence Ralstonia pathogenicity in this host[41,51]. This knowledge gap limits the ability to fully explain instances where outbreaks occur, despite the deployment of resistant cultivars or rigorous cultural practices, suggesting that epigenetic mechanisms may be silently shaping disease outcomes.
Epigenetic plasticity provides a compelling explanation for rapid pathogen adaptation to environmental stressors or host immune responses[52,53]. Thus, by modulating virulence gene expression in response to fluctuating soil conditions, temperature, or host-derived signals, cryptic epigenetic changes could enable certain Ralstonia lineages to overcome control measures that would otherwise be effective. In addition, incorporating epigenetic insights into bacterial wilt research may reveal previously overlooked drivers of control failure, offering novel avenues for more precise disease management and durable crop protection strategies. While DNA methylation, histone-like proteins, and small RNAs often act synergistically to enhance bacterial virulence, their interactions can also show inverse relationships depending on environmental or host conditions. For instance, excessive DNA methylation may suppress virulence gene activation when host resistance is strong, counteracting the effects of histone-like proteins that promote transcriptional accessibility[54,55]. Similarly, small RNAs can downregulate transcripts activated by methylation or chromatin remodeling, balancing gene expression to prevent metabolic stress[56]. In some cases, reduced histone-like protein binding can increase genomic accessibility but destabilize regulatory precision, leading to lower pathogen fitness[57]. mRNAs also serve as key intermediates in this network, where their stability and translation are finely regulated by DNA methylation patterns, and chromatin structure[58]. Thus, the interplay among these epigenetic mechanisms is both cooperative and antagonistic, maintaining a fine-tuned equilibrium between virulence expression, environmental adaptation, and survival efficiency.
Figure 2 summarizes the key epigenetic mechanisms: DNA methylation, histone-like proteins, and regulatory RNAs that collectively modulate pathogen virulence. It highlights dynamic feedback between these molecular layers and gene expression outputs, enabling pathogens to rapidly adjust to environmental and host-derived cues. This framework emphasizes epigenetic regulation as a flexible, non-genetic driver of pathogenicity with important implications for disease persistence and management.
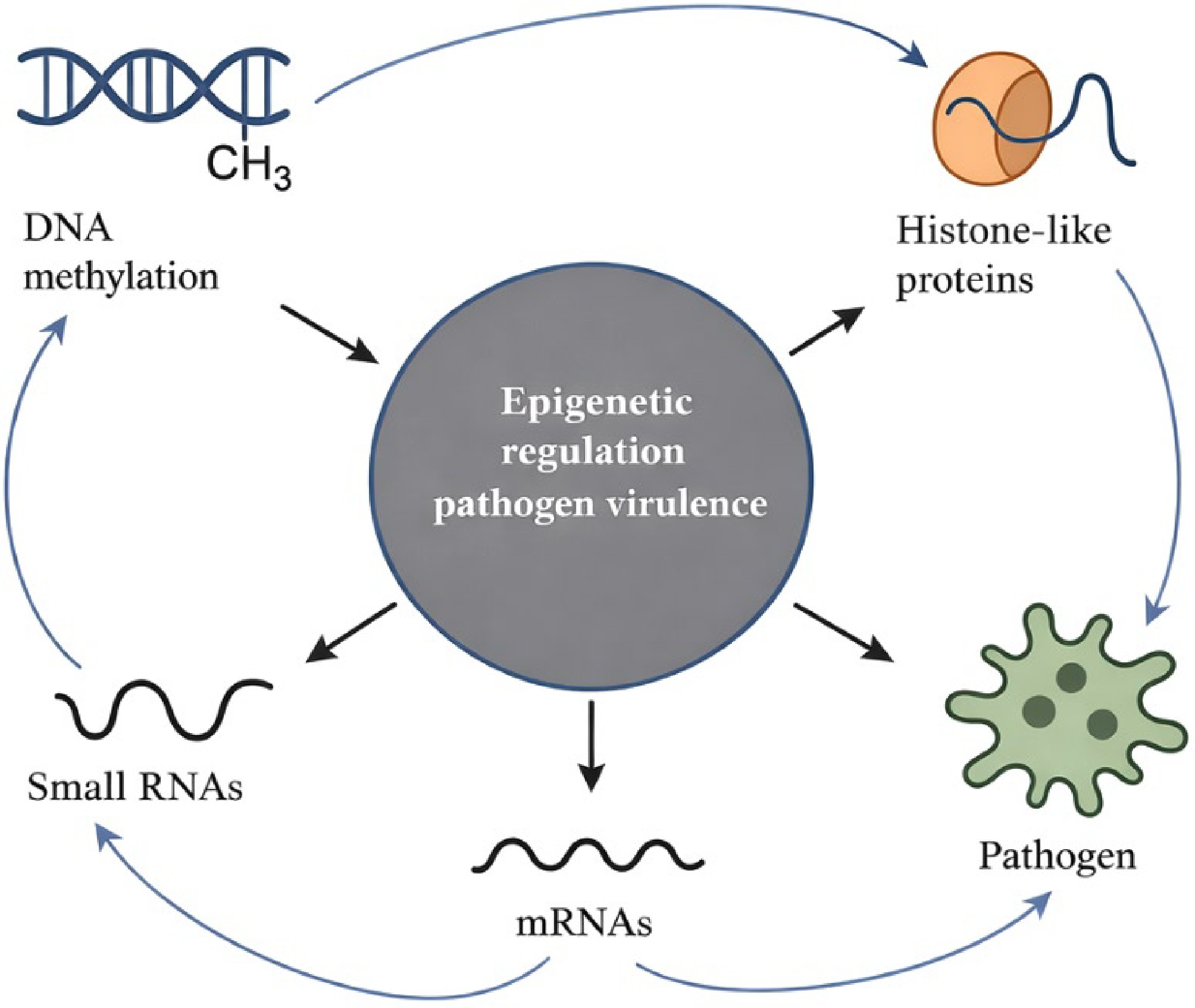
Figure 2.
Epigenetic regulation of pathogen virulence mechanisms.
-
Cryptic diversity among R. solanacearum populations can significantly influence host-pathogen interactions, even within a single peanut cultivar[20]. Different cryptic lineages may exhibit subtle variations in virulence, tissue colonization patterns, and environmental adaptability, leading to differential disease outcomes[59,60]. These hidden differences often remain undetected through conventional diagnostics, but can result in some plants succumbing while others remain asymptomatic, creating heterogeneous disease patterns in the field.
Hidden genetic variation frequently results in measurable biological differences, with cryptic lineages varying in aggressiveness, infection efficiency, host range, and environmental tolerance[61]. Some drive rapid disease progression, whereas others persist as latent infections, and differences in soil and water survival further sustain inoculum and complicate epidemiology. The coexistence of multiple lineages within hosts or environments promotes genetic exchange through recombination and horizontal gene transfer, enabling rapid emergence of more adaptable variants[62]. Mobile genetic elements facilitate the spread of virulence- and stress-tolerance traits, accelerating evolution and sustaining cryptic diversity. Environmental factors such as soil moisture, temperature, nutrient status, and cropping practices shape pathogen population dynamics by selectively favoring particular lineages[63]. Agronomic interventions, including irrigation, soil amendments, and crop rotation, may inadvertently enhance the persistence of highly adaptable cryptic populations. Cryptic lineages vary in their responses to chemical and biological control, with some showing tolerance to antimicrobials or evading beneficial microbes[64]. These differences can shift population structures, and reduce the long-term effectiveness of management strategies. Fragmented surveillance and management systems hinder effective responses to emerging lineages, as inconsistent reporting and limited real-time monitoring delay detection[65]. Integrating genomic surveillance with field management remains a critical gap in disease control. Managing cryptic diversity requires shifting from uniform interventions to adaptive and diversity-aware strategies. Continuous monitoring, multi-lineage resistance screening, and ecological integration are key for sustainable disease control[66]. Immediate priorities include using high-resolution surveillance, multi-lineage screening in breeding, and region-specific monitoring, alongside strengthened biosecurity to limit pathogen spread. Management should combine genetically diverse resistant cultivars, targeted microbial consortia, and agronomic practices that limit pathogen survival, integrating environmental, genomic, and ecological data for durable disease control[67].
The existence of lineage-specific variation challenges the assumptions underpinning many resistance breeding programs[68]. Traditional strategies often rely on the notion that a cultivar resistant to one strain will be broadly resistant to all pathogen populations[69]. However, cryptic lineages with distinct virulence determinants may overcome cultivar-specific defenses, producing partial resistance or complete susceptibility in unexpected scenarios[70]. This mismatch between breeding assumptions and pathogen reality may explain why resistant cultivars sometimes fail under field conditions despite rigorous selection and screening. Resistance failure often arises from hidden lineage diversity, as breeding programs focus on dominant strains while overlooking rare variants capable of overcoming resistance. When resistant cultivars suppress major populations, minor lineages may rapidly expand, and environmental effects can further destabilize resistance expression in the field[71].
Emerging evidence from genomic and phenotypic studies indicates the potential for lineage-dependent virulence profiles. For example, subtle genetic differences among strains can lead to variable expression of effector proteins, toxins, or enzymes that influence host recognition and disease progression[72]. In peanuts, while comprehensive studies are limited, preliminary observations suggest that certain Ralstonia lineages preferentially infect specific cultivars, causing sporadic or cultivar-dependent outbreaks[73]. Such specificity emphasizes the need for more refined screening tools capable of assessing resistance against a spectrum of cryptic pathogen variants[74].
The implications of cryptic diversity for host-pathogen specificity are profound, and hence, breeding programs must consider not only the dominant pathogen strains but also minor, cryptic lineages that could bypass resistance mechanisms. Integrating genomic surveillance, multi-lineage pathogenicity assays, and predictive modeling into breeding pipelines may enhance the durability of resistance[75]. Recognizing the role of hidden diversity in shaping host-pathogen interactions is therefore essential for designing effective and long-term management strategies for bacterial wilt in peanuts.
Cryptic diversity in R. solanacearum shapes host pathogen specificity, with each determinant linked to its biological basis and breeding implications (Table 1).
Table 1. Cryptic diversity and interaction specificity in R. solanacearum.
Key determinant Defining characteristic Biological basis Translational significance Ref. Cryptic diversity Hidden pathogen variation Undetected genetic Leads to unexpected disease patterns [76] Lineage-specific virulence Strain-dependent aggressiveness Distinct virulence genes or effectors Requires lineage-targeted screening [77] Host interaction specificity Cultivar-dependent infection Certain lineages prefer specific hosts Explains cultivar-specific outbreaks [78] Diagnostic limitations Conventional tools miss variants Hidden lineages escape detection Necessitates genomic surveillance tools [79] Resistance breakdown Resistance fails in field Cryptic strains bypass defenses Breeding must include multi-lineage testing [74] Effector variability Variable toxin/enzyme expression Alters host recognition and response Complicates durable resistance breeding [80] Environmental adaptability Lineage adapts to niche Enhanced survival in diverse conditions Promotes regional disease variability [81] Genomic and phenotypic integration Use multi-source data Correlates lineage traits with virulence Enables predictive resistance models [82] Refined breeding strategy Diversity-informed selection Incorporates cryptic pathogen profiles Ensures long-term resistance durability [83] -
The soil and rhizosphere microbiome plays a crucial role in shaping pathogen populations and influencing disease outcomes in peanut fields. Beneficial microbes can suppress pathogen growth through competition, antibiosis, or induction of plant systemic resistance, while complex microbial networks contribute to the soil health and ecosystem stability[84,85]. The contribution of beneficial taxa can be conceptualized at two complementary scales. At the community level, highly diverse microbiomes confer resistance to pathogen invasion through niche pre-emption, functional redundancy, and enhanced network stability[86]. At the individual level, specific microbial taxa suppress pathogens via direct competition, antibiosis, or the induction of systemic resistance in the host plant[87]. Thus, together, these multiscale mechanisms underpin microbiome-mediated resilience against disease.
However, the presence of cryptic pathogen lineages complicates this dynamic, as subtle genetic or phenotypic differences among Ralstonia strains may determine how they interact with the surrounding microbial community[88]. Such interactions can ultimately influence colonization success, virulence, and disease severity.
Microbiome–pathogen interactions ultimately determine colonization success, virulence, and disease severity by regulating niche access, resource use, and microbial antagonism[89]. Pathogen entry commonly disrupts the native community, but the magnitude of this disturbance is governed by strain-specific invasion strategies rooted in underlying genomic diversity[90]. For instance, highly virulent strains can restructure microbial networks to facilitate establishment, whereas less aggressive strains are constrained by resident competitors, leading to attenuated disease[91].
Beyond taxonomic profiling, the functional attributes of the rhizosphere microbiome, such as antimicrobial compound production, resource competition, and disruption of pathogen signaling, play decisive roles in regulating pathogen establishment[92]. The influence of microbial community architecture, including interaction networks and keystone taxa on the suppression or persistence of cryptic pathogen populations remains insufficiently addressed. Horizontal gene transfer within the rhizosphere further contributes to pathogen adaptation by enhancing virulence and environmental resilience[93]. Plant genotype microbiome interactions critically shape pathogen diversity and should be systematically incorporated into microbiome-informed disease management frameworks.
Cryptic lineages of Ralstonia may engage differently with rhizosphere microbiota[94]. Certain lineages could be more tolerant of antagonistic microbes, evade microbial suppression, or exploit specific microbial metabolites to enhance survival and infection[95]. These lineage-specific interactions remain largely unexplored in peanut systems, yet they may be a key factor in the heterogeneous disease patterns observed in the field. Therefore, by understanding these relationships, researchers can begin to elucidate why some pathogen variants thrive while others are naturally constrained by microbial competition[96].
Biocontrol agents that are effective against dominant pathogen strains may fail against rare or cryptic lineages that interact differently with the soil microbiome[97]. Such failures are often attributed solely to environmental variability or insufficient inoculum density, overlooking the potential for cryptic diversity to shape these responses. Consequently, this gap reveals the need for a more nuanced approach that integrates pathogen genomics with microbial ecology to predict and enhance biocontrol performance.
Incorporating microbiome-informed strategies offers a promising avenue for managing bacterial wilt in peanuts[98,99]. Accordingly, by targeting specific cryptic lineages with compatible biocontrol consortia or microbiome-modifying practices, it may be possible to suppress pathogen populations more effectively and sustainably. Integrating genomic surveillance of cryptic lineages with detailed rhizosphere microbiome analyses could guide the development of precision interventions, ensuring that management strategies account for both pathogen diversity and the ecological context in which disease develops[100,101].
Figure 3 presents hidden pathogen diversity and rhizosphere microbiome interactions as an integrated, circuit-like system centered on peanut roots, emphasizing that disease development is an emergent property of interconnected biological processes. It visually indicates cryptic Ralstonia lineages and shows how subtle genomic differences can alter microbial competition, metabolite utilization, and evasion of antagonistic microbes. The pathways illustrate how microbial antagonism, virulence adaptation, and metabolite exchange feed into pathogen survival and colonization success. By depicting failures of conventional biocontrol alongside genomic surveillance and microbiome analysis, the figure features why single-strain or one-size-fits-all approaches are often ineffective. The graphic conveys a precision disease-management framework in which pathogen genomics and rhizosphere ecology are jointly used to design targeted and microbiome-informed interventions.
Figure 3.
Microbiome–pathogen interaction cycle in disease emergence.
-
Cryptic diversity among R. solanacearum populations presents a significant challenge to conventional disease management strategies in peanuts. Recognition of cryptic diversity challenges the assumption of pathogen uniformity underlying conventional control strategies, explaining failures of single-genotype resistance and standardized treatments[102]. A diversity-informed framework instead integrates genomic surveillance, lineage-resolved diagnostics, and ecological monitoring to enable dynamic, population-based management.
Resistant cultivars, long considered a cornerstone of control, may fail when minor or cryptic pathogen lineages harbor unique virulence traits that bypass host defenses[74,103]. Similarly, chemical treatments often provide only temporary suppression, as pathogen populations can rapidly adapt or persist in niches less affected by agrochemicals[104]. Biocontrol agents, while environmentally friendly and increasingly popular, also exhibit inconsistent effectiveness due to differential interactions with diverse cryptic lineages and the surrounding microbiome[105,106]. Collectively, these observations indicate that current approaches often underestimate the complexity and adaptability of pathogen populations.
Thus, addressing this challenge requires innovative and diversity-aware strategies that consider the full spectrum of pathogen variation. One approach is the deployment of multi-line or multi-genotype cultivars, combining complementary resistance traits to buffer against lineage-specific virulence[107,108]. Genome-informed pathogen surveillance can further guide the selection of cultivars and interventions, providing early warning of emergent lineages that threaten resistance durability[66]. By integrating high-resolution genomic data with field observations, breeders and pathologists can make more targeted decisions, reducing the risk of unexpected outbreaks.
Customized microbial consortia represent another promising avenue for biocontrol. Recognizing that cryptic lineages may interact differently with rhizosphere communities, designing microbial assemblages specifically adapted to suppress multiple pathogen variants could enhance efficacy[109,110]. Thus, such strategies may include combinations of antagonistic bacteria, fungi, or bacteriophages, selected based on their ability to inhibit or outcompete cryptic lineages. These approaches require a deep understanding of both pathogen diversity and ecological interactions, emphasizing the need for integrative research bridging genomics, microbiology, and soil ecology.
In general, adaptive management strategies are essential for addressing evolving pathogen populations[111,112]. Rather than relying on static control measures, dynamic approaches that continuously monitor pathogen diversity, adjust cultivar deployment, and modify biocontrol interventions can mitigate the risks posed by cryptic lineages[113,114].
In general, integrating genomic, epigenetic, and ecological insights into practical disease management provides a holistic framework capable of anticipating pathogen evolution, enhancing the resilience of peanut production systems, and reducing reliance on single-strategy interventions.
An integrative framework links pathogen diversity, multi-genotype cultivars, microbial consortia, and genomic surveillance to improve peanut disease management. Cyber-physical systems are also emerging tools for monitoring and adapting control strategies in response to evolving pathogen dynamics.
Figure 4 illustrates how cryptic diversity within R. solanacearum populations complicates disease management in peanut, necessitating more nuanced and flexible control strategies. It emphasizes integrative approaches that combine genome-informed pathogen surveillance, deployment of multi-genotype cultivars, and customized microbial consortia to suppress diverse pathogen variants. Collectively, the framework highlights adaptive, data-driven disease management enabled by interdisciplinary research and cyber-physical systems to monitor and respond to dynamic disease pressures.
Figure 4.
Integrative framework for managing cryptic diversity of R. solanacearum in peanut production.
-
Bacterial wilt caused by R. solanacearum and related species remains a major constraint to peanut production worldwide. While resistant cultivars, chemical treatments, and biocontrol strategies are widely used, their inconsistent success points to underlying factors beyond conventional pathogen assessments. One such factor is cryptic diversity—hidden genetic, epigenetic, and phenotypic variation within pathogen populations that can drive unexpected outbreaks, cultivar-specific susceptibility, and variable disease severity.
Genomic analyses have revealed extensive hidden variation among Ralstonia lineages, with mechanisms such as horizontal gene transfer, plasmids, prophages, and mutations generating cryptic subpopulations. Epigenetic regulation, including DNA methylation, histone-like proteins, and small RNAs, further modulates virulence gene expression, allowing rapid adaptation to host defenses or environmental stress without altering the genome. These hidden variations, together with lineage-specific interactions with the soil and rhizosphere microbiome, can compromise the effectiveness of traditional control measures and partially explain the inconsistent outcomes of breeding and biocontrol programs.
The implications for disease management are profound. Multi-line or multi-genotype cultivars, genome-informed pathogen surveillance, and microbiome-configured biocontrol consortia represent promising strategies to counter the effects of cryptic diversity. Integrating genomic, epigenetic, and ecological knowledge into adaptive management frameworks can enhance resistance durability, predict lineage emergence, and optimize biocontrol efficacy, offering a more resilient approach to peanut bacterial wilt.
In conclusion, acknowledging and addressing cryptic diversity is essential for sustainable disease management. Future research should focus on epigenetic regulation, lineage-specific microbiome interactions, and precision breeding and biocontrol strategies informed by pathogen genomics. Hence, by embracing this complexity, peanut production systems can move toward more predictable, durable, and effective bacterial wilt control, turning hidden pathogen variation from a threat into an opportunity for informed intervention.
-
The author confirms sole responsibility for the following: study conception and design, data collection, analysis and interpretation of results, manuscript preparation, and approval for the final version of the manuscript.
-
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
-
No funding was received for this review work. Figures and diagrams were created using BioRender.com under a free trial; no publication license number is available.
-
The authors declare that they have no conflict of interest.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Gelaye Y. 2026. Cryptic diversity in bacterial wilt pathogens infecting peanut: an overlooked driver of control failure. Circular Agricultural Systems 6: e009 doi: 10.48130/cas-0026-0009 

Cryptic diversity in bacterial wilt pathogens infecting peanut: an overlooked driver of control failure
- Received: 03 November 2025
- Revised: 15 February 2026
- Accepted: 28 February 2026
- Published online: 28 April 2026
Abstract: Bacterial wilt caused by the R. solanacearum species complex remains a major constraint to global peanut (Arachis hypogaea L.) production. Despite decades of control efforts, inconsistent management outcomes suggest the influence of hidden biological variability beyond conventional pathogen characterization. This review critically synthesizes current knowledge on how cryptic genomic, epigenetic, and ecological diversity within R. solanacearum drives its adaptability, host specificity, and management failures in peanut bacterial wilt. Recent findings reveal that hidden genomic variation, including single-nucleotide polymorphisms, horizontal gene transfer, and mobile genetic elements, underpins the adaptability of Ralstonia lineages. Epigenetic mechanisms such as DNA methylation and small RNAs further fine-tune virulence gene expression, while lineage-specific interactions with the rhizosphere microbiome modulate pathogen persistence and disease severity. Additionally, evidence of stress-induced epigenomic reprogramming suggest that environmental fluctuations can trigger reversible virulence states and enhance outbreak unpredictability. Newly identified communication between cryptic lineages and beneficial microbes indicate that pathogen adaptation can restructure the rhizosphere network, and indirectly compromise biocontrol stability. Collectively, these insights explain the recurrent breakdown of host resistance, the inconsistent field performance of control measures, and the evolutionary resilience of pathogen populations. The review concludes that sustainable management of bacterial wilt requires diversity-informed, adaptive strategies integrating genomic surveillance, multi-genotype resistant cultivars, and microbiome-optimized biocontrol systems. Future research should prioritize elucidating epigenetic regulation, stress-responsive virulence plasticity, and lineage microbiome feedback to guide predictive and evolution-resilient disease control frameworks.