






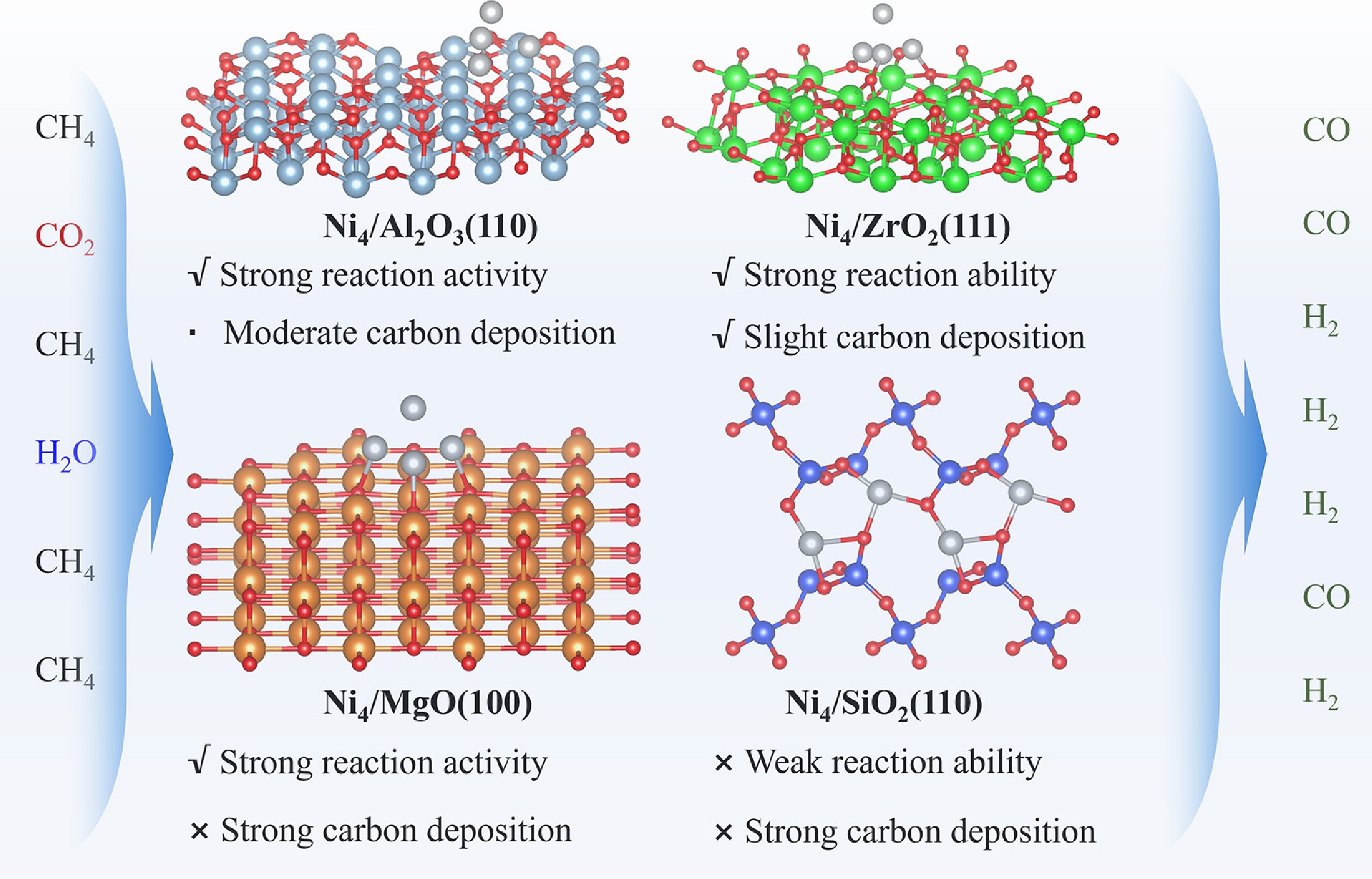



-
Biomass stands as a pivotal sustainable carbon source that offers a renewable pathway to platform chemicals such as methane via gasification[1]. The reforming of biomass-derived methane presents an efficient route to produce green syngas (CO + H2), a fundamental feedstock for industrial chemical synthesis. Catalytic conversion by adopting Ni-based catalysts is the main route for methane reforming, and the support is essential for the optimized activity and stability[2]. Concurrently, it is widely recognized that electronic structure modification by different supports plays a decisive role in determining catalytic activity and stability[3−5], for instance, via the surface acidity/basicity[6] and strong metal-support interaction (SMSI)[7]. They can refine the adsorption of reactants and intermediates, while SMSI can also inhibit metal particle agglomeration to enhance catalyst stability[8,9].
The predominant supports for Ni-based catalysts are oxide substrates, encompassing Al2O3, ZrO2, MgO, and SiO2[10−13]. Among these, Al2O3 exhibits exceptional mechanical strength, and the intrinsic pore architecture facilitates effective dispersion of Ni particles[14,15], while surface acidic sites promote methane adsorption and activation through enhanced C–H bond cleavage. Nevertheless, the phase transition can induce sintering of active sites, compromising catalytic performance. ZrO2 demonstrates remarkable crystal phase stability at elevated temperatures (> 800 °C), effectively suppressing Ni particle aggregation while simultaneously inhibiting carbon deposition[16,17]. The basic sites of MgO enhance chemical adsorption and activation of CO2 during methane dry reforming. Furthermore, SMSI facilitates electron transfer from the support to the active components, thereby activating the active species and reducing the energy barrier for methane reforming[18−20]. SiO2 presents a well-developed pore structure coupled with relatively low chemical reactivity, primarily providing a chemical environment conducive to Ni component dispersion. Notably, mesoporous zeolites can encapsulate active Ni species, making them ideal supports suitable for harsh operating conditions. However, their inherent weak metal-support interactions may also compromise active site stability[21,22].
Given that different supports can fundamentally alter catalytic activity, a comprehensive understanding of how various supports influence methane reforming is imperative. This study focuses on four common support surfaces—Al2O3(110), ZrO2(111), MgO(100), and SiO2(110)—that have been extensively observed in experiments. Employing density functional theory (DFT) calculations, we systematically investigated the influence of supports on the active Ni component and the adsorption behaviors of key reaction intermediates. The investigation provides microscopic insights into SMSI phenomena and establishes a theoretical foundation for the rational design of high-performance methane reforming catalysts.
-
The optimized lattice parameters for the investigated materials are as follows: α-Al2O3 exhibits hexagonal symmetry with a = b = 4.81 Å and c = 13.12 Å; monoclinic ZrO2 (m-ZrO2) displays lattice constants of a = 5.15 Å, b = 5.23 Å, c = 5.33 Å, and β = 99.2°; face-centered cubic MgO presents a lattice parameter of a = b = c = 4.19 Å; and trigonal SiO2 shows lattice parameters of a = b = 4.91 Å, c = 5.43 Å, and γ = 120°. Based on experimental observations demonstrating their stability under methane reforming conditions[23,24], the Al2O3(110), m-ZrO2(111), MgO(100), and SiO2(110) surfaces were selected for theoretical model construction. A Ni4 cluster was employed as the active catalytic component, positioned on the respective support surfaces.
Spin-polarized DFT calculations were performed using the Vienna ab initio simulation package (VASP)[25]. The Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional was employed in conjunction with projected-augmented wave (PAW) pseudopotentials, utilizing a plane-wave cutoff energy of 400 eV. The k-point mesh resolution value was set to 0.03 (2π/Å) for all calculations. The convergence accuracies of electron self-consistent field and force during geometry optimization were 1 × 10−6 eV and −0.04 eV/Å, respectively. The Grimme D3 correction was considered in all calculations[26]. A vacuum layer of 15 Å was implemented to eliminate spurious interactions between periodic images. Structural visualization and computational output analysis were conducted using VESTA[27] and VASPKIT[28] packages, respectively. The adsorption energy was determined according to Eq. (1).
$E_{\rm{ads}}=E_{\rm{total}}-E_{\rm{slab}}-E_{\rm{gas}}$ (1) where, the Eads, Etotal, Eslab, and Egas are the adsorption energy, the total energy of the adsorbed slab system, the energy of the pristine slab, and the energy of the isolated gaseous molecule.
The electron density difference (EDD, ∆ρ) is calculated as follows:
$ \Delta \rho =\rho _{a+b}-\rho_{a}-\rho_{b} $ (2) where, the ρa+b, ρa, and ρb are the electron density distributions of complex a and b, single a, and single b. Furthermore, density of states (DOS) and crystal orbital Hamilton population (COHP) analyses were utilized to elucidate the electronic interactions between adsorbate species and adsorbent surfaces.
-
The optimized structures and EDD diagrams for the Ni4 cluster supported on Al2O3(110), ZrO2(111), MgO(100), and SiO2(110) are shown in Fig. 1, together with the corresponding binding energies between the active clusters and their respective substrates.
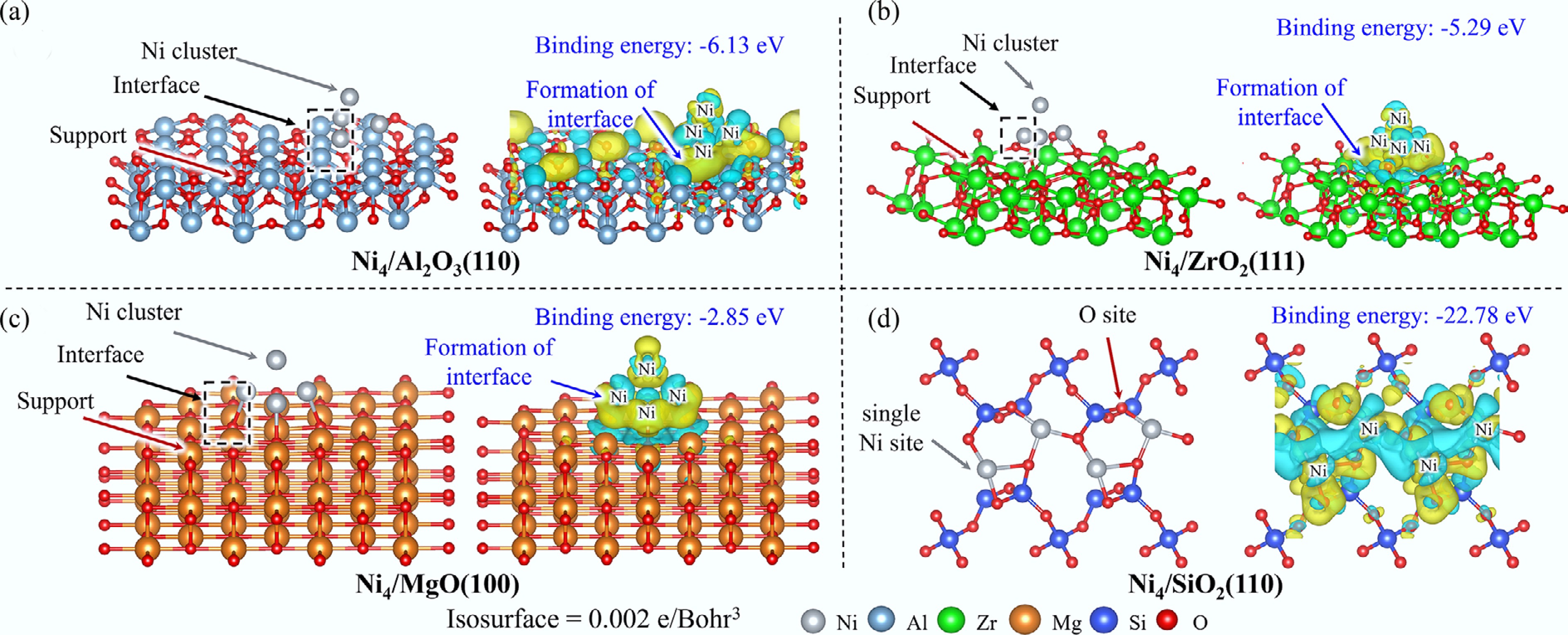
Figure 1.
Theoretical models and the electron transfer between the Ni4 cluster and substrates.
On the Al2O3(110) surface, the Ni4 cluster adopts a tetrahedral configuration, wherein two Ni atoms coordinate with surface Al atoms and a third Ni atom binds to surface O atoms. EDD analysis reveals electron accumulation in the interfacial region, originating from both the Ni4 cluster and adjacent surface atoms, which signifies a robust metal-support interaction corroborated by a substantial binding energy of −6.13 eV. Similar interaction patterns are observed for Ni4 clusters on ZrO2(111) and MgO(100) surfaces, exhibiting binding energies of −5.29 and −2.85 eV, respectively, with slight electron transfer between the Ni4 cluster and the substrate. Bader charge analysis reveals distinct electron transfer behaviors between the Ni4 cluster and oxide substrates. The calculated Bader charges of the Ni4 fragment are −1.201 |e|, +0.078 |e|, and –0.368 |e| for the Ni4/Al2O3(110), Ni4/ZrO2(111), and Ni4/MgO(100) systems, respectively. These small charge perturbations indicate that the Ni4 cluster retains its intrinsic metallic character across all three supports, with the main charge redistribution at the cluster-substrate interface. In contrast, the Ni4/SiO2(110) system displays a fundamentally different interaction mechanism: the four Ni atoms become individually dispersed and embedded within the support surface, with each Ni atom coordinating to three O atoms. This configuration yields an exceptionally high binding energy of −22.78 eV. Consistently, EDD analysis reveals substantial charge redistribution across the entire surface. The pronounced positive charge accumulation on the Ni4 cluster (+4.021 |e|) indicates a markedly ionic character, arising from the individual coordination of all Ni atoms with surface O and Si sites. This substantial electron depletion elucidates the exceptionally strong interfacial binding between Ni4 and the SiO2(110) surface.
The adsorption of methane reforming-related species on Ni4/MOn
-
Guided by the mechanistic framework of methane steam and dry reforming reactions, we systematically investigated the adsorption behaviors of key surface species, including CH4, CO2, H2O, CO, H2, CH, C, O, OH, and H across various catalyst surfaces. Multiple adsorption sites were comprehensively examined, as illustrated in Fig. 1. For the Ni4/Al2O3(110), Ni4/ZrO2(111), and Ni4/MgO(100) systems, distinct Ni4 cluster sites, interfacial sites, and support sites were identified and evaluated. In the case of Ni4/SiO2(110), a unique set of adsorption sites was specifically characterized, encompassing single Ni, Si, and O sites, as well as their bridge sites. The adsorption energies of all investigated species on these catalyst surfaces, as well as on the reference Ni(111) surface, are compiled in Supplementary Tables S1–S5 and Supplementary Figs S1–S4. Meanwhile, Fig. 2 and Supplementary Table S6 summarizes the highest adsorption energy of all reaction species on various catalysts, with detailed analysis in the subsequent sections.
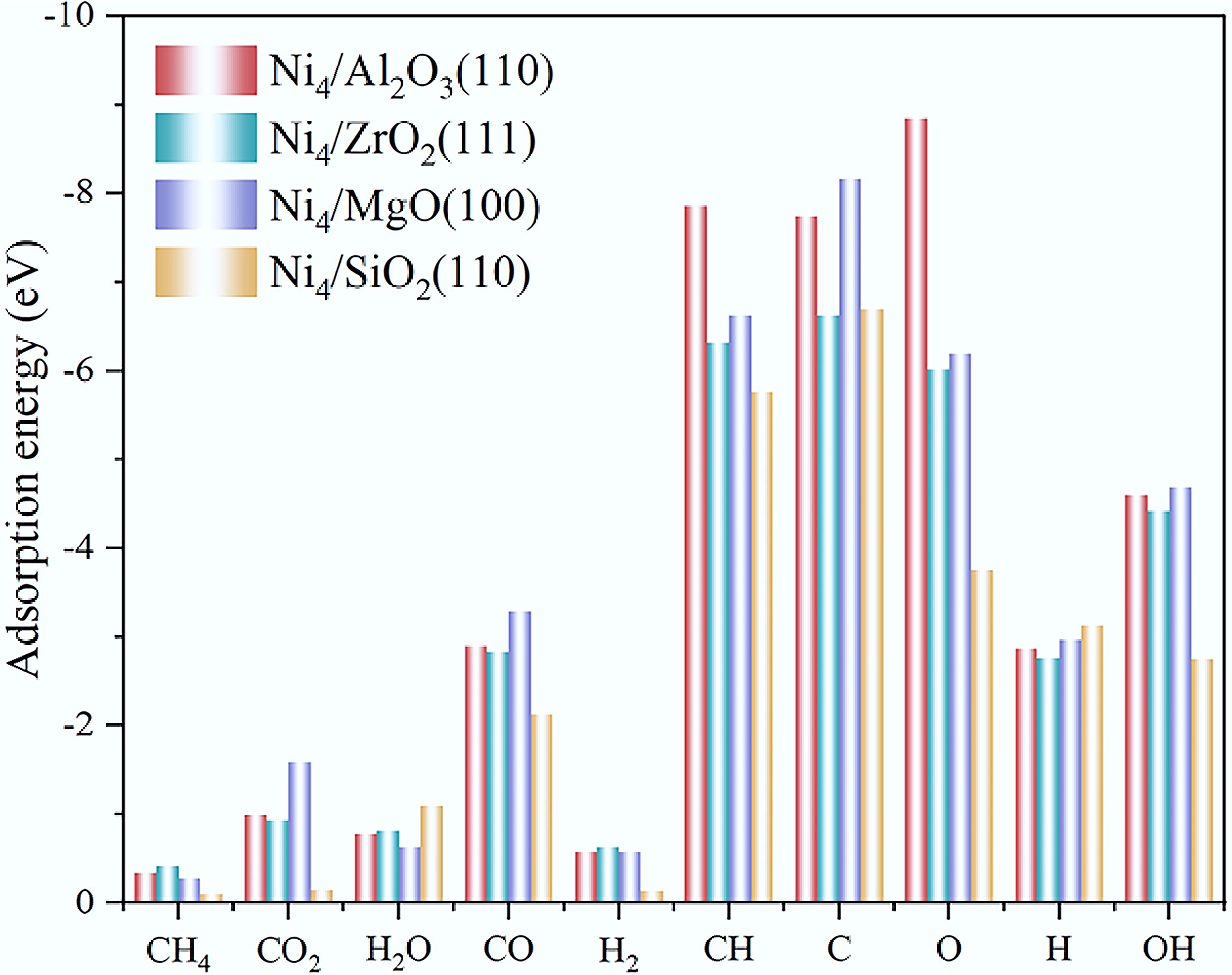
Figure 2.
The adsorption energies of all reaction species on different catalysts.
The adsorption of reactants (CH4, CO2, and H2O)
-
The adsorption of the reactant CH4 constitutes a critical initial step in the reforming process, and CO2 and H2O serve as the fundamental feedstocks for methane dry and steam reforming. All reactants exhibit distinct adsorption behaviors across various supported catalysts.
CH4 preferentially adsorbs atop the Ni4 cluster on Ni4/Al2O3(110) (Fig. 3), with an Eads of –0.34 eV. EDD analysis reveals electron depletion from the H atoms of CH4 toward the interfacial region between the Ni cluster and C atom, corroborated by DOS mapping, which demonstrates significant orbital overlap between the C 2p orbitals of CH4 and the Ni 3d orbitals (Supplementary Fig. S5). Similar adsorption behavior is observed for CH4 on Ni4/ZrO2(111) and Ni4/MgO(100), where CH4 stably adsorbs on the Ni4 cluster with adsorption energies of −0.41 and −0.27 eV, respectively. Notably, in the Ni4/MgO(100) system, substrate adsorption is also energetically favorable, exhibiting a comparable adsorption energy of −0.28 eV. Consistent with the Al2O3 system, electron transfer occurs from the hydrogen atoms of CH4 to the Ni4 cluster, with the C 2p and Ni 3d orbitals primarily governing the interfacial interaction. In marked contrast, CH4 adsorption on Ni4/SiO2(110) follows a fundamentally different mechanism, with CH4 preferentially occupying the Ni site. EDD analysis indicates electron flow from both the Ni atom and the hydrogen atoms of CH4 into the interfacial region between CH4 and Ni. However, DOS and COHP analyses reveal negligible interaction between Ni and CH4, with only weak bonding between the C 2p and Ni 3d orbitals, consistent with the substantially reduced Eads of −0.10 eV.
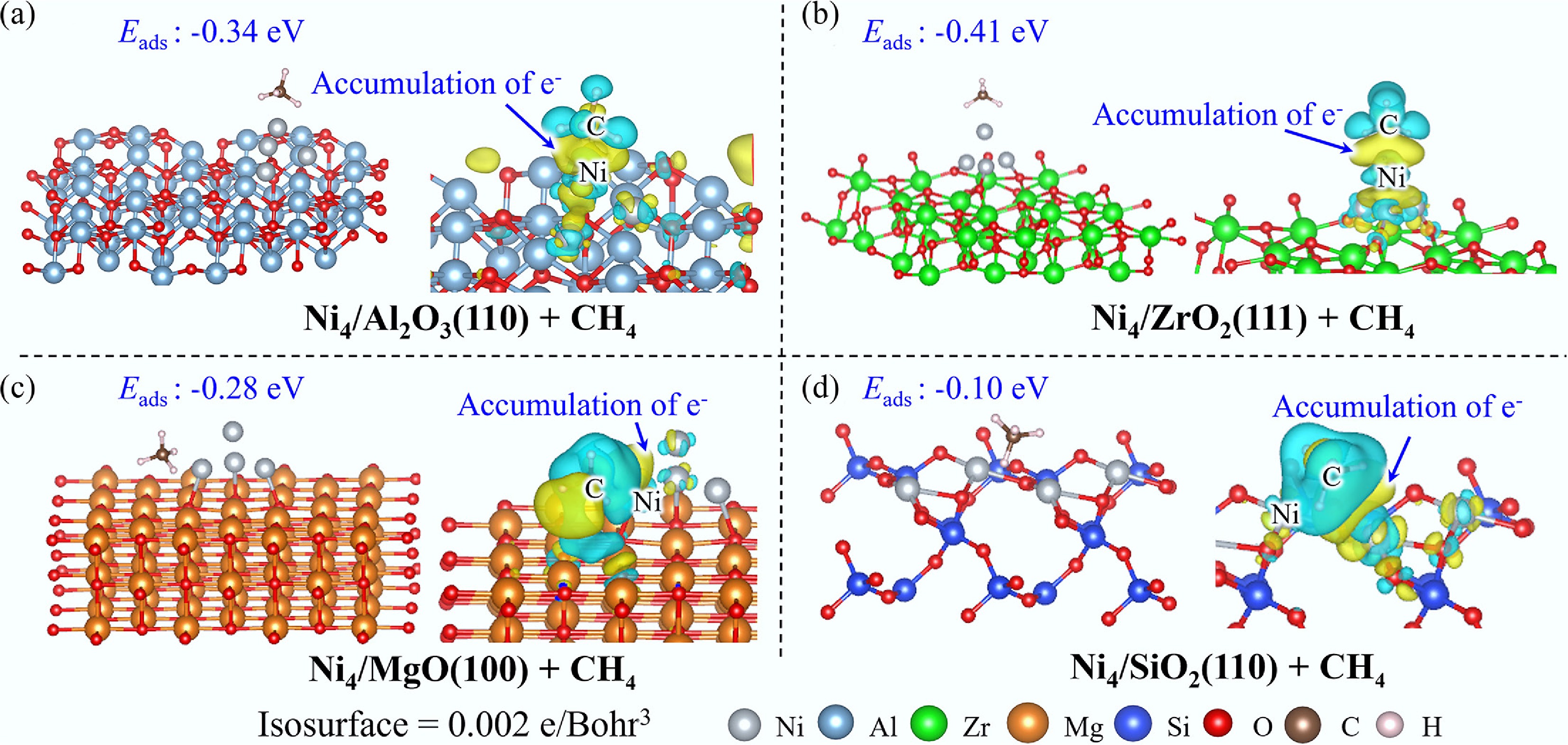
Figure 3.
The most stable adsorption configurations and EDD diagrams of CH4.
The adsorption of CH4 on Ni4/ZrO2(111) exhibits the most exothermic behavior among the investigated systems (Fig. 3), with an Eads of −0.41 eV, surpassing those observed for Ni4/Al2O3(110), Ni4/MgO(100), and Ni4/SiO2(110) surfaces. Notably, this adsorption strength also exceeds that on the pristine Ni(111) surface (Eads = −0.33 eV, Supplementary Table S5), indicating that the Ni4 cluster configuration provides enhanced advantages for CH4 adsorption processes. Conversely, the markedly weak adsorption of CH4 on Ni4/SiO2(110) (Eads = −0.10 eV) suggests that isolated single Ni atoms do not constitute optimal active sites for methane activation.
On Ni4/Al2O3(110), CO2 adopts a side-on configuration on the Ni4 cluster with a substantial Eads of −0.99 eV, wherein two oxygen atoms coordinate with two Ni atoms, resulting in a pronounced bending of the CO2 molecule to 141°. EDD analysis reveals electron transfer from both the Ni4 cluster and C atom of CO2 into the interfacial region between O and Ni, indicative of Ni–O bond formation mediated by hybridization between O 2p and Ni 3d orbitals, as confirmed by DOS analysis (Supplementary Fig. S6). Analogous adsorption phenomena are observed for Ni4/ZrO2(111) and Ni4/MgO(100) systems, where CO2 similarly adsorbs on the Ni4 clusters with adsorption energies of −0.93 and −1.59 eV, respectively. Notably, the Ni4/MgO(100) system demonstrates the most robust CO2 adsorption, reflected in the most severe CO2 bending to 134°. EDD mapping corroborates electron redistribution from the Ni4 cluster and CO2 toward Ni–O bonds, while DOS and COHP analyses collectively confirm the significant contribution of O 2p and Ni 3d electrons to Ni–O bond formation. In marked contrast, CO2 interaction with Ni4/SiO2(110) is characterized by weak physisorption (Eads = −0.15 eV), wherein CO2 remains spatially separated from the catalyst surface. Consistent with this minimal interaction, DOS and COHP analyses reveal negligible electronic coupling between Si and CO2, confirming the absence of significant chemical bonding.
The adsorption behavior of CO2 demonstrates remarkable consistency across Ni4/Al2O3(110), Ni4/ZrO2(111), and Ni4/MgO(100) systems, wherein CO2 preferentially adsorbs on the Ni4 cluster (Fig. 4). Among these systems, Ni4/MgO(100) exhibits the most thermodynamically favorable adsorption configuration with an Eads of −1.59 eV. Notably, CO2 undergoes weak physisorption on Ni4/SiO2(110) (Eads = −0.15 eV), highlighting the superior efficacy of Ni clusters over isolated Ni atoms for CO2 adsorption. When compared with the pristine Ni(111) surface (Eads = −0.36 eV), all Ni4 cluster configurations demonstrate enhanced affinity for CO2 adsorption. These findings collectively establish Ni clusters as the primary reactive sites for CO2 adsorption and subsequent activation processes.
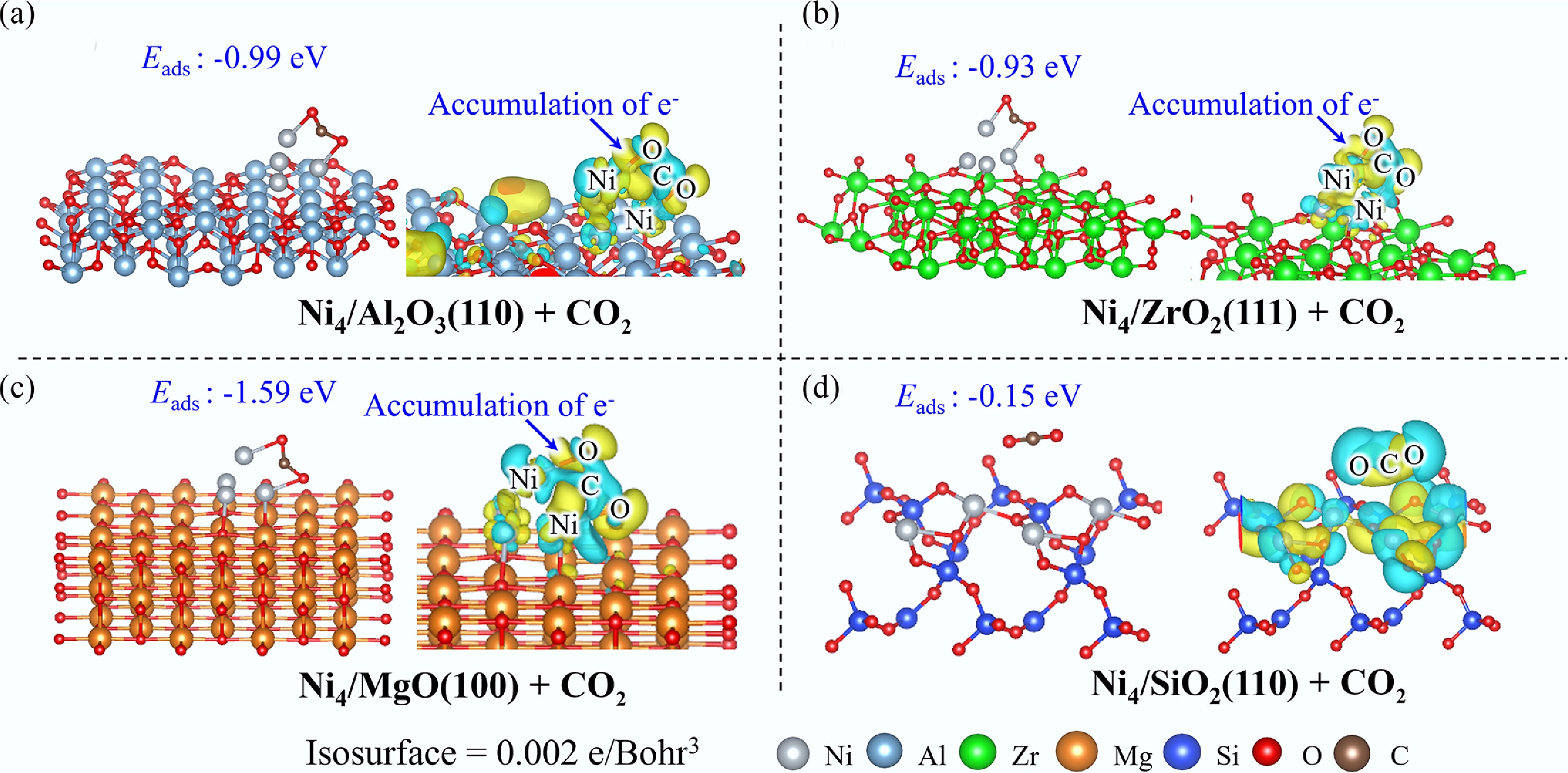
Figure 4.
The most stable adsorption configurations and EDD diagrams of CO2.
As illustrated in Fig. 5, on Ni4/Al2O3(110), H2O preferentially adsorbs atop the Ni4 cluster with a moderate Eads of −0.78 eV, establishing a Ni–O bond with an interatomic distance of 2.01 Å. EDD analysis reveals electron depletion from both H2O and the Ni4 cluster toward the Ni–O interfacial region, corroborated by DOS and COHP analyses, which demonstrate significant orbital hybridization between O 2p and Ni 3d states contributing to bond formation (Supplementary Fig. S7). In contrast, on Ni4/ZrO2(111) and Ni4/MgO(100) systems, H2O preferentially adsorbs on the substrate surfaces, forming robust Zr–O and Mg–O bonds with adsorption energies of −0.81 and −0.63 eV, respectively. Consistent with this substrate-mediated adsorption, electron density accumulation predominantly occurs around the Zr–O/Mg–O bond regions, arising from the hybridization of O 2p electrons with Zr 5s or Mg 3s electrons (Supplementary Fig. S7). Notably, Ni4/SiO2(110) exhibits the most thermodynamically favorable H2O adsorption (Eads = −1.10 eV), wherein H2O coordinates with Ni atoms to form strong Ni–O bonds.
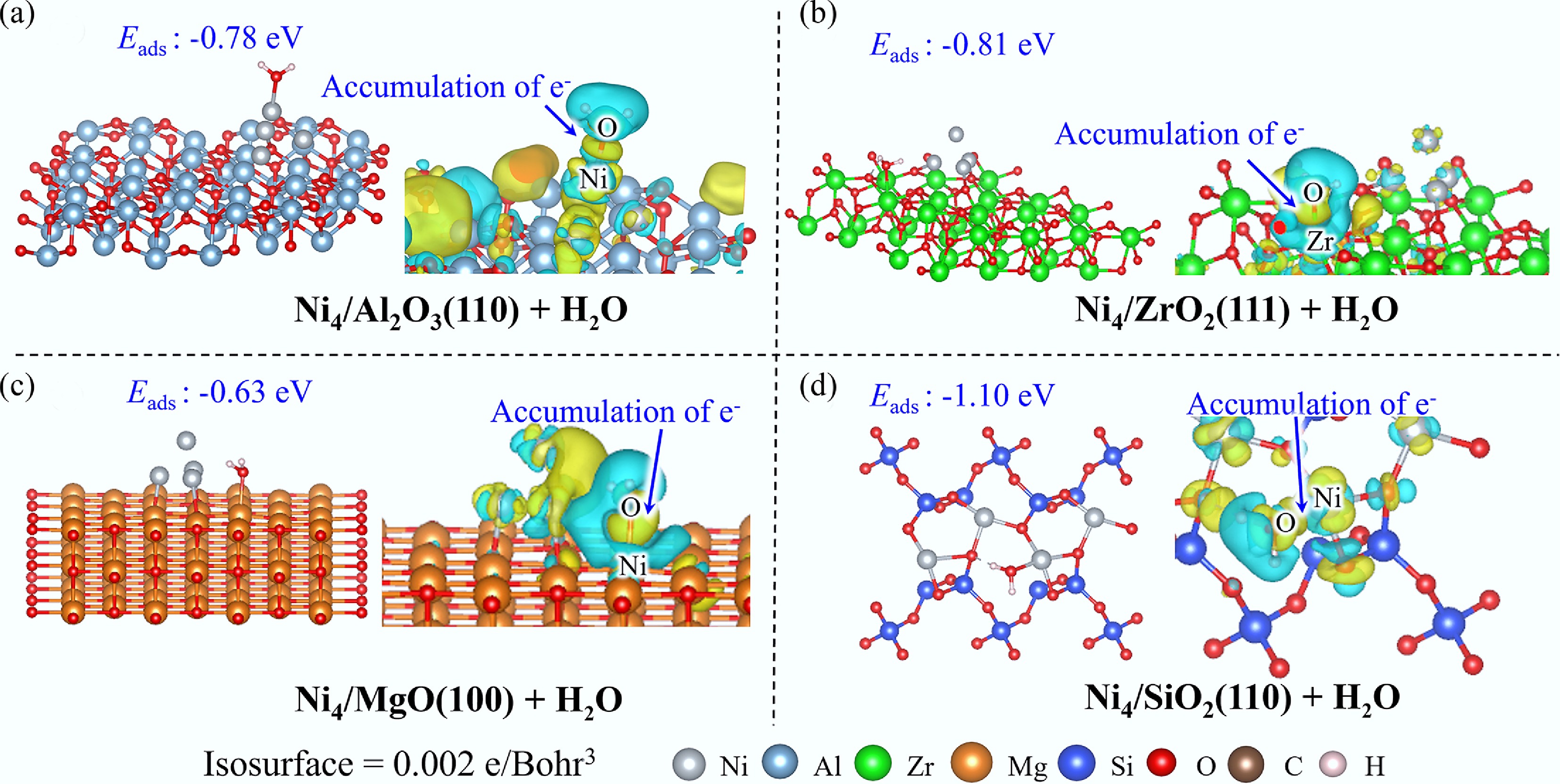
Figure 5.
The most stable adsorption configurations and EDD diagrams of H2O.
The adsorption energy hierarchy follows the order Ni4/SiO2(110) > Ni4/Al2O3(110) > Ni(111) (Eads = −0.66 eV), indicating a clear preference for H2O adsorption on isolated atoms and small clusters rather than extended metallic surfaces. Furthermore, the substrate-mediated adsorption behavior observed for ZrO2(111) and MgO(100) supports suggest a strategic advantage in mitigating H2O-induced active center sintering, thereby potentially enhancing catalyst stability under steam reforming conditions.
The adsorption of products (CO and H2)
-
CO and H2 are the principal products of methane reforming processes, requiring rapid desorption for sustained catalytic activity to avoid active site poisoning[29].
The adsorption behavior of CO across various catalysts reveals distinct electronic and structural characteristics. On Ni4/Al2O3(110), Ni4/ZrO2(111), and Ni4/MgO(100) surfaces, CO exhibits remarkably similar adsorption configurations, preferentially coordinating to the Ni4 cluster (Fig. 6). EDD analysis demonstrates significant electron accumulation in the interfacial region between the Ni cluster and CO molecule, indicative of strong metal-adsorbate interactions. Complementary DOS and COHP analyses reveal substantial orbital overlap near the Fermi level between Ni and CO, with bonding contributions predominantly arising from the hybridization of C 2p orbitals with Ni 3d/4s orbitals (Supplementary Fig. S8). In contrast, CO adsorption on Ni4/SiO2(110) occurs preferentially at the Ni site, where the Ni–C bond formation is mediated through C 2p and Ni 3d electronic interactions.
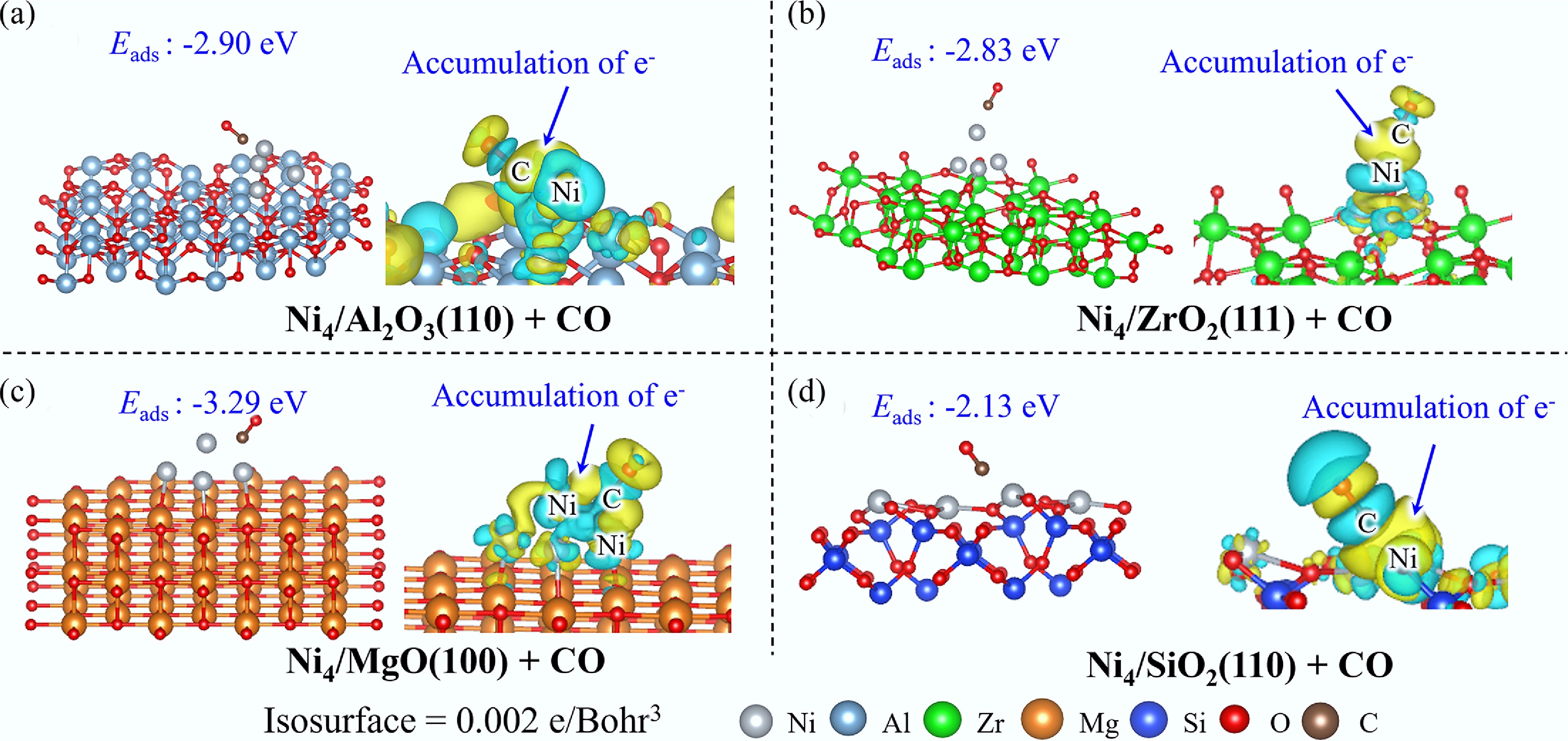
Figure 6.
The most stable adsorption configurations and EDD diagrams of CO.
The thermodynamic hierarchy of CO adsorption energies follows the order: Ni4/MgO(100) (Eads = −3.29 eV) > Ni4/ZrO2(111) (Eads = −2.90 eV) > Ni4/Al2O3(110) (Eads = −2.83 eV) > Ni(111) (Eads = −2.17 eV) > Ni4/SiO2(110) (Eads = −2.13 eV). Notably, CO demonstrates a pronounced preference for adsorption on Ni4 clusters rather than on extended metallic surfaces or isolated atomic sites. Given the exceptionally strong CO adsorption energies observed, particularly on Ni4/MgO(100), there exists a significant risk of catalyst poisoning through CO over-adsorption, which could potentially compromise the catalytic activity and operational stability of these systems.
The adsorption behavior of H2 exhibits remarkable uniformity across the investigated supported Ni4 catalysts (Fig. 7), wherein H2 preferentially adsorbs atop the Ni4 cluster in Ni4/Al2O3(110), Ni4/ZrO2(111), and Ni4/MgO(100) configurations, while coordinating to isolated Ni sites in Ni4/SiO2(110). EDD analysis reveals significant electron accumulation in the interfacial region between H2 and the Ni cluster, arising from the hybridization of H 1s and Ni 3d electronic orbitals (Supplementary Fig. S9). Notably, COHP analysis demonstrates that the H2–Ni interaction in Ni4/SiO2(110) is substantially weaker compared with the other catalyst systems.
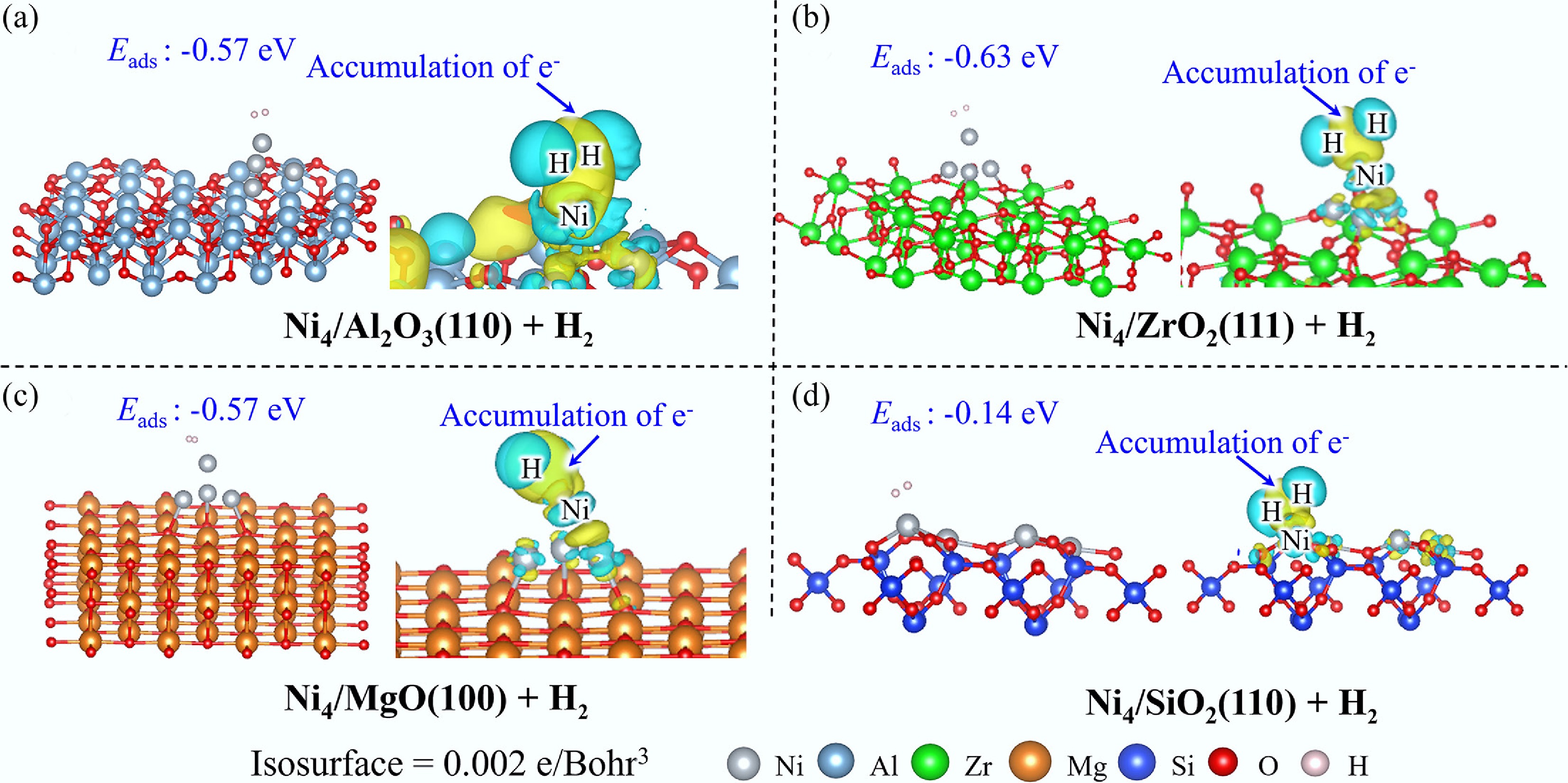
Figure 7.
The most stable adsorption configurations and EDD diagrams of H2.
Consistent with these electronic structure findings, the adsorption energy of H2 on SiO2(110) is markedly lower (Eads = −0.14 eV), reflecting its weak interaction strength. For the remaining three supported catalysts, H2 adsorption energies are comparable (Eads = −0.57 to −0.63 eV) and significantly stronger than that observed on the pristine Ni(111) surface (Eads = −0.25 eV). These findings collectively establish that H2 exhibits a pronounced preference for adsorption on Ni clusters rather than on extended metallic surfaces or isolated atomic sites, highlighting the enhanced catalytic potential of cluster-based architectures.
The adsorption of key intermediates (CH, C, O, H, and OH)
-
CH and C intermediates represent critical cracking species in methane reforming, whose adsorption is also a key indicator for carbon deposition. In addition, O, H, and OH groups represent critical secondary surface intermediates throughout the reaction pathway.
Across Ni4/Al2O3(110), Ni4/ZrO2(111), and Ni4/MgO(100) systems, CH intermediates demonstrate preferential interaction with Ni4 clusters rather than with the substrates (Fig. 8). EDD mapping reveals significant electron accumulation in the interfacial region between Ni clusters and the C atom, indicative of robust Ni–CH interactions (Supplementary Fig. S10). Complementary DOS and COHP analyses demonstrate substantial orbital overlap near the Fermi level between Ni4 clusters and CH species, with bonding contributions predominantly arising from C 2p and Ni 3d orbital hybridization. In contrast, CH exhibits dual-site interaction with Ni4/SiO2(110), simultaneously coordinating with both the Ni atom and surface oxygen atoms. Electronic structure analysis indicates electron depletion from Ni and O atoms toward the Ni–CH interfacial region, mediated through Ni 3d and C 2p orbital overlap (Supplementary Fig. S10).
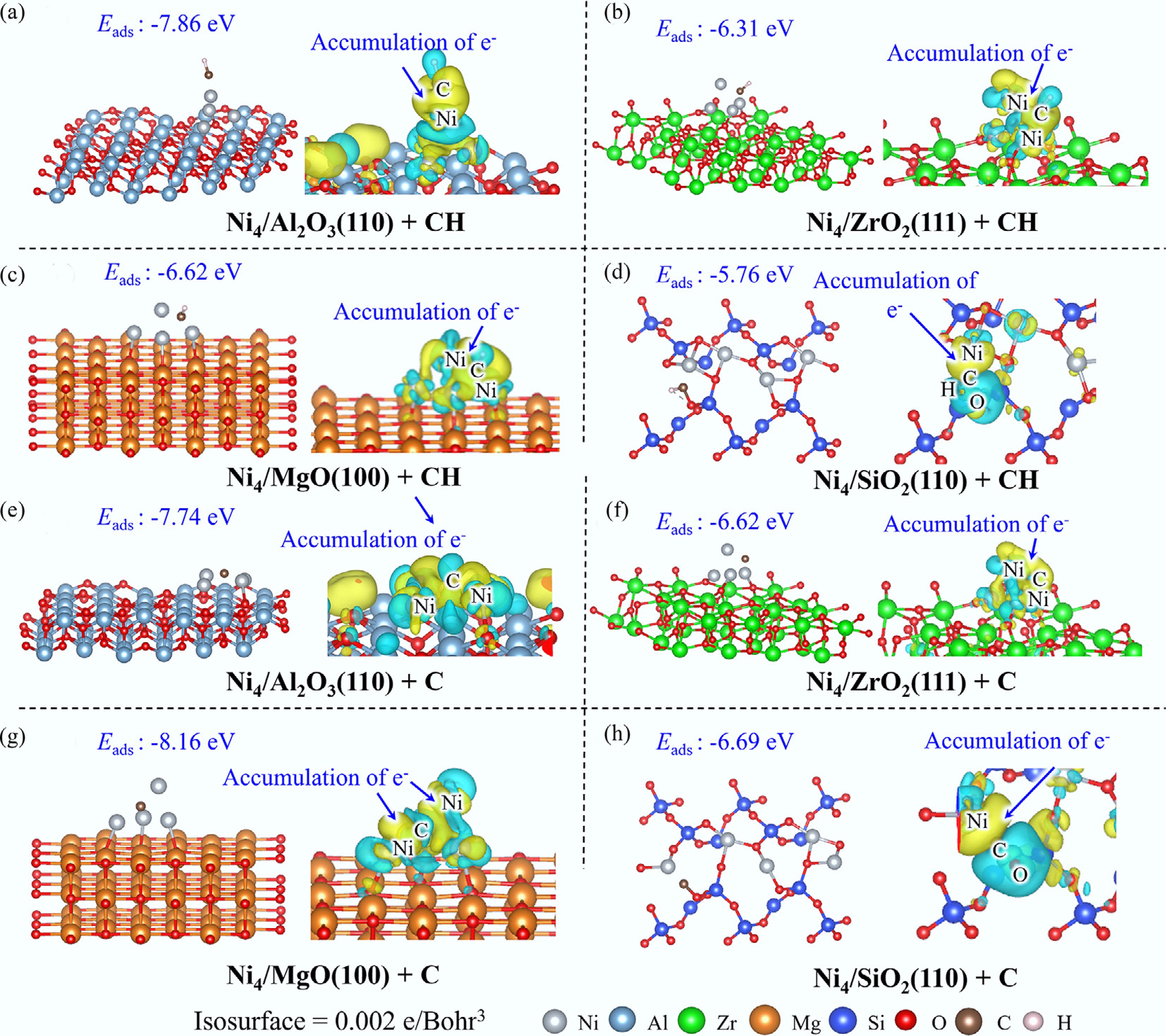
Figure 8.
The most stable adsorption configurations and EDD diagrams of (a)–(d) CH, and (e)–(h) C.
Thermodynamic analysis of adsorption strengths reveals that the interaction between CH and Ni4/Al2O3(110) exhibits the highest binding affinity (Eads = −7.86 eV), consistent with the strong orbital interactions evidenced by COHP analysis. This adsorption energy substantially exceeds that of CH on the pristine Ni(111) surface (Eads = −6.76 eV), while other catalyst systems demonstrate moderate binding strengths (Eads = −5.76 to −6.62 eV). Notably, the adsorption energy of CH on Ni4/SiO2(110) is significantly diminished compared with other catalysts, highlighting the superior affinity of Ni clusters and bulk metallic surfaces for CH intermediates relative to isolated Ni sites.
The adsorption behavior of C atoms across different catalyst systems reveals distinct structural and electronic characteristics that govern catalyst–carbon interactions. On Ni4/Al2O3(110), C adsorption induces significant reconstruction of the Ni4 cluster, wherein the C atom simultaneously coordinates with four Ni atoms (Fig. 8). EDD analysis demonstrates substantial electron accumulation in the interfacial region between the C atom and Ni4 cluster, while DOS and COHP analyses reveal pronounced orbital overlap between Ni 3d and C 2p orbitals, indicating a robust interaction (Supplementary Fig. S11). Analogous adsorption phenomena are observed for Ni4/ZrO2(111) and Ni4/MgO(100) systems, where the C atom interacts with Ni4 clusters that maintain their structural integrity without significant distortion. Similar electron accumulation patterns emerge in the Ni–C interfacial regions, mediated through C 2p and Ni 3d orbital hybridization. In contrast, the Ni4/SiO2(110) system exhibits distinctive adsorption behavior, wherein the C atom simultaneously coordinates with both surface Ni and O atoms, forming Ni–C and C–O bonds with interatomic distances of 1.25 and 1.65 Å, respectively. The Ni–C interaction predominates in this system, with electronic contributions arising from the C 2p and Ni 3d orbital overlap.
Thermodynamic analysis of adsorption strengths reveals a clear hierarchy: Ni4/MgO(100) demonstrates the most stable carbon adsorption (Eads = −8.16 eV), followed by Ni4/Al2O3(110) (Eads = −7.74 eV). Both systems exhibit substantially stronger C affinity compared with the pristine Ni(111) surface (Eads = −7.11 eV). Conversely, Ni4/ZrO2(111) (Eads = −6.62 eV) and Ni4/SiO2(110) (Eads = −6.69 eV) demonstrate relatively weaker carbon binding capabilities, highlighting the critical role of support materials in modulating the catalyst–carbon interaction.
On Ni4/Al2O3(110) and Ni4/ZrO2(111) surfaces, oxygen species preferentially adsorb on the substrate, forming robust Al–O or Zr–O bonds that induce significant surface electronic structure reconstruction (Fig. 9). DOS and COHP analyses elucidate that these interactions predominantly arise from orbital hybridization between O 2p and Al 3p or Zr 4d orbitals (Supplementary Fig. S12). In contrast, on Ni4/MgO(100), O adsorption occurs at the sublayer of the Ni4 cluster, with electrons flowing from the Ni4 cluster into the interfacial region between O and the metallic cluster. Both DOS and COHP analyses confirm the substantial contribution of Ni 3d and O 2p orbitals to these metal–oxygen interactions. Distinctively, oxygen adsorption on Ni4/SiO2(110) results in the formation of a Ni–O–Ni bridge structure, wherein both Ni–O interactions are mediated through Ni 3d and O 2p orbital hybridization.
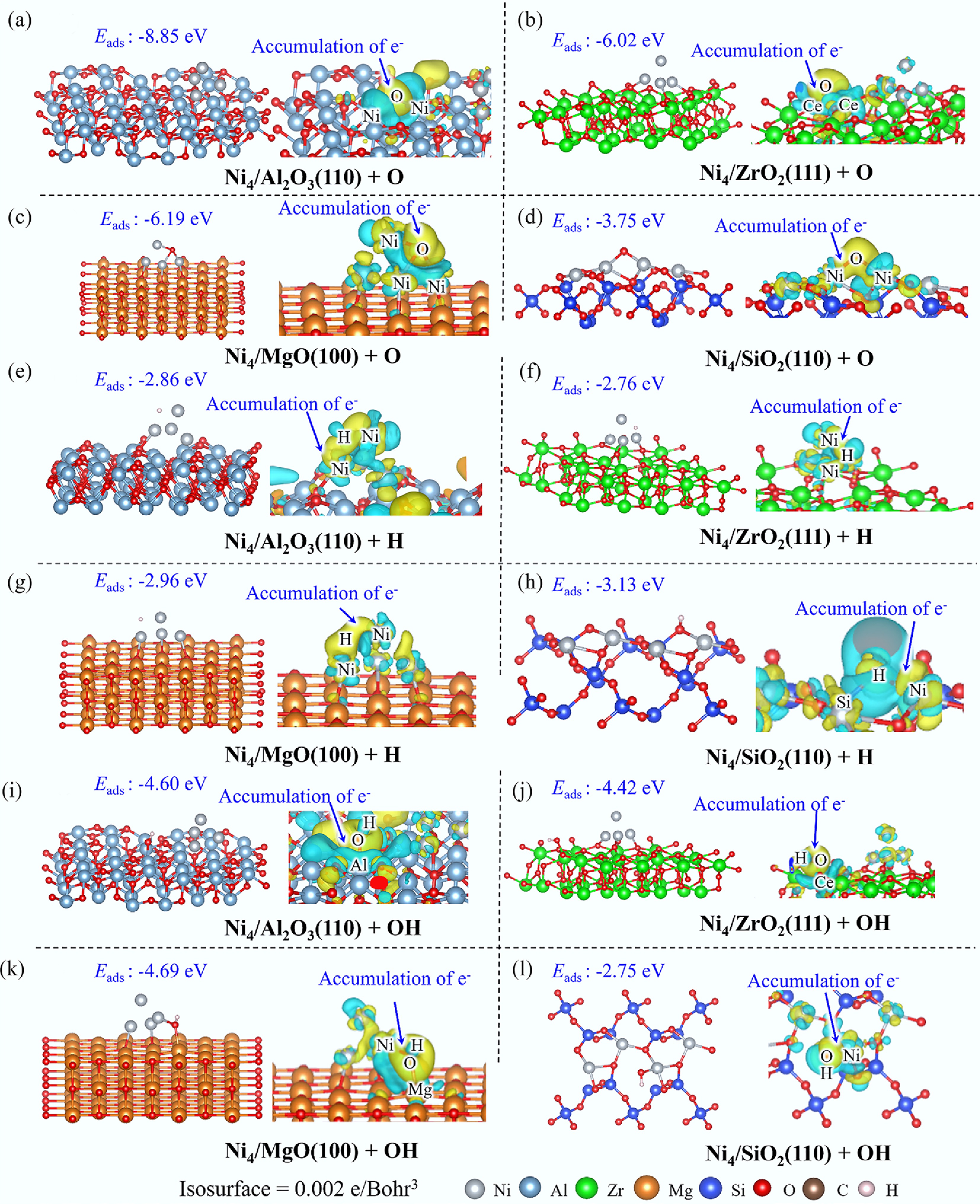
Figure 9.
The most stable adsorption configurations and EDD diagrams of (a)–(d) O, (e)–(h) H, and (i)–(l) OH.
Thermodynamic analysis reveals that O adsorption on Ni4/Al2O3(110) exhibits the strongest binding (Eads = −8.85 eV), reflecting the pronounced oxygen affinity of aluminum species. The O adsorption on Ni4/MgO(100) and Ni4/ZrO2(111) demonstrates moderate binding strengths with adsorption energies of −6.19 and −6.02 eV, respectively. Notably, all these systems display superior O affinity compared with the pristine Ni(111) surface (Eads = −5.95 eV), with the exception of Ni4/SiO2(110), which exhibits significantly weaker O adsorption (Eads = −3.75 eV). These findings collectively demonstrate that O adsorption is highly sensitive to the supports, with a clear tendency for oxygen to preferentially adsorb on hydrophilic substrates such as Al2O3 rather than on the metallic active sites.
On Ni4/Al2O3(110), Ni4/ZrO2(111), and Ni4/MgO(100), H preferentially adsorbs on the Ni4 cluster driven by nickel's high hydrogen affinity (Fig. 9). EDD, DOS, and COHP analyses collectively reveal significant electron accumulation in the interfacial region between the H atom and the Ni4 cluster, with interactions predominantly arising from H 1s and Ni 3d orbital hybridization (Fig. 9 and Supplementary Fig. S13). In contrast, on Ni4/SiO2(110), H preferentially adsorbs on substrate oxygen atoms, resulting in O–H bond formation with interactions primarily mediated through O 2p and H 1s orbital overlap.
Thermodynamic analysis demonstrates that H adsorption on Ni4/SiO2(110) exhibits the highest stability (Eads = −3.13 eV), marginally exceeding that on the pristine Ni(111) surface (Eads = −3.07 eV). Conversely, H adsorption on Ni4/Al2O3(110), Ni4/MgO(100), and Ni4/ZrO2(111) demonstrates reduced binding strength compared with Ni(111), with adsorption energies of −2.89, −2.96, and −2.76 eV, respectively. These findings indicate a slight preference for H interacting with bulk Ni(111) surfaces over dispersed clusters, albeit the energetic differences are minimal.
The adsorption behavior of OH also exhibits distinct substrate-dependent interactions. On Ni4/Al2O3(110) and Ni4/ZrO2(111), OH species preferentially adsorb on the substrate surfaces, establishing interactions with metal atoms through electron transfer from OH and Al/Zr species into Al/Zr–O bonds (Fig. 9). These interactions are predominantly mediated through orbital hybridization between O 2p and Al 3p or Zr 4d states (Supplementary Fig. S14). In contrast, OH adsorption on Ni4/MgO(100) occurs preferentially at the interfacial region, where significant electron accumulation is observed, and O 2p and Ni 3d orbitals contribute substantially to these metal-adsorbate interactions. Distinctively, on Ni4/SiO2(110), OH coordinates directly with Ni atoms, forming Ni–O bonds through the hybridization of O 2p and Ni 3d orbitals, as evidenced by EDD, DOS, and COHP analyses.
Thermodynamic analysis of adsorption strengths reveals that OH adsorption on Ni4/MgO(100) exhibits the most exothermic (Eads = −4.69 eV), reflecting the exceptionally strong interaction intensity between the interfacial site and OH. Concurrently, OH demonstrates robust adsorption on Al2O3(110) and ZrO2(111), with adsorption energies of −4.60 and −4.42 eV, respectively; both substantially exceeding that observed on the pristine Ni(111) surface (Eads = −3.74 eV). Exceptionally, OH interaction with the Ni atom in Ni4/SiO2(110) releases only 2.75 eV of energy, indicating the relatively weak affinity of isolated Ni atomic sites for OH species compared with cluster-based configurations.
Discussion
The reactivity of different supported catalysts
-
Different supports profoundly influence the catalytic performance of Ni4 clusters for methane reforming. Different supports influence both adsorption sites and energies, and the adsorption strength order is summarized in Supplementary Table S7.
Carbon-containing species (CH4, CO2, CO, CH, and C) and H species (H/H2) preferentially adsorb on the Ni4 cluster of Ni4/Al2O3(110), Ni4/ZrO2(111), and Ni4/MgO(100), indicating that the complete methane dry reforming process (CH4 + CO2 → 2CO + 2H2) can proceed entirely on the active Ni4 cluster. Conversely, O-containing species (O, OH, and H2O) exhibit a strong affinity for the substrate due to the superior oxygen-binding capability of support metals compared to Ni, enabling substrates to participate in steam reforming as H2O adsorption sites. Ni4/SiO2(110) represents a distinctive case, where most reaction species adsorb on isolated Ni sites.
Compared to the Ni(111) surface, supported Ni4 clusters on Al2O3(110), ZrO2(111), and MgO(100) carriers exhibit significantly enhanced adsorption energies for all species. In particular, Ni4/Al2O3(110) exhibits substantially increased adsorption energies for C, CH, and O species by about 0.6, 1.1, and 2.8 eV, respectively, leading to the promoted deep cracking activity. Ni4/ZrO2(111) also demonstrates generally elevated adsorption energies for all species, especially for reactants (CH4, CO2, and H2O), indicating that ZrO2 can facilitate the reaction by enhancing the interaction between the Ni4 cluster and reactants. Similarly, Ni4/MgO(100) displays markedly increased adsorption energies for most species, with OH, C, and CO2 exhibiting increases exceeding 1 eV. In contrast, the Ni4 cluster disperses into isolated single-atom sites on SiO2(110), resulting in diminished adsorption energies, except for H2O adsorption. Consequently, SiO2 support fails to improve Ni activity and may promote H2O-induced sintering due to the strengthened Ni–H2O interactions[30].
Quantitatively, the adsorption strength of CH4, CO2, and H2O on Ni4/Al2O3(110), Ni4/ZrO2(111), and Ni4/MgO(100) is comparable (Eads = −0.28 to −0.41 eV, and −0.93 to −1.59 eV), substantially higher than those on the Ni(111) surface (−0.33 eV and −0.36 eV) and Ni4/SiO2(110) (−0.10 eV and −0.15 eV). Notably, MgO exhibits exceptional CO2 adsorption capabilities, which substantially enhances dry reforming activity. This promotional effect is corroborated by experimental evidence where Jin et al.[31] reported a 26% increase in methane dry reforming conversion at 850 °C upon MgO doping into Ni/Al2O3 catalysts, underscoring the critical role of basic oxide supports in facilitating CO2 activation. For deep CH4 dissociation intermediates (C/CH), the sequence is MgO (−8.16/−6.62 eV) > Al2O3 (−7.74/−7.86 eV) > ZrO2 (−6.62/−6.31 eV) > SiO2 (−6.69/−5.76 eV). The exceptionally strong adsorption on MgO and Al2O3 facilitates complete methane cracking, consistent with prior studies where MgO doping enhanced dry reforming via strengthened SMSI[32]. However, for the adsorption of CO, the sequence is MgO (−3.29 eV) > Al2O3 (−2.90 eV) > ZrO2 (−2.83 eV) > SiO2 (−2.13 eV). Notably, MgO exhibits the strongest affinity toward both CO and CO2 among the investigated supports, which may also lead to active-site poisoning. This observation aligns with the Sabatier principle that excessively strong adsorption of reactants can impede product desorption, thereby compromising catalytic performance. This also highlights a critical trade-off: while stronger adsorption of reactants/intermediates is beneficial, excessive CO adsorption can lead to active site poisoning and reaction inhibition[29].
Furthermore, reaction kinetics govern the overall catalytic performance. Reported literature identifies C–H bond activation as the rate-determining step for methane reforming, particularly under high-temperature conditions[33]. Accordingly, we evaluated the activation barriers for CH4 dissociation across the four supported Ni4 clusters (Supplementary Fig. S15). The calculated energy barriers follow the sequence: Ni4/Al2O3(110) (0.397 eV) ≈ Ni4/MgO(100) (0.400 eV) < Ni4/ZrO2(111) (0.588 eV) < Ni4/SiO2(110) (1.170 eV). This kinetic hierarchy indicates superior catalytic activity for Al2O3- and MgO-supported systems, corroborating both the thermodynamic predictions and experimental observations that Ni/Al2O3 outperforms Ni/ZrO2 and Ni/SiO2[34], while MgO doping further enhances catalytic turnover[31].
In summary, Ni4 supported on MgO, Al2O3, and ZrO2 supports generally exhibits promoted intrinsic reactivity by strengthening the adsorption of reactants and cracking intermediates.
Carbon deposition and elimination
-
The Sabatier principle also provides a fundamental framework for understanding carbon deposition resistance. Carbon adsorption energy serves as the fundamental descriptor governing carbon deposition propensity on supported Ni catalysts. According to the foregoing analysis, atomic C exhibits exceptionally strong binding on Ni4/MgO(100), with an adsorption energy of −8.16 eV. According to the Sabatier principle, such excessive adsorption strength implies a substantial kinetic barrier for carbon removal or further conversion to gaseous products. Hence, this pronounced binding renders deposited carbon thermodynamically and kinetically trapped. Consistent with this, Zhang et al.[35] reported that deep cracking products of CH4 on MgO-supported catalysts exhibit limited removability, indicating inferior resistance to carbon deposition. This suggests that, despite the promotional effect of MgO on CO2 activation, its inadequate capability for carbon elimination may compromise long-term catalytic stability. The availability of active oxygen species represents another crucial factor governing carbon elimination through the C* + O* → CO* pathway. Although C is also strongly adsorbed on Al2O3(110) support surface (Eads = −7.74 eV), Al2O3(110) also demonstrates remarkable capability to stably adsorb oxygen atoms with exceptionally high binding energy (Eads = −8.85 eV), effectively creating a rich 'oxygen reservoir' that facilitates carbon removal. In contrast, ZrO2(111) exhibits a more balanced profile with moderate carbon adsorption strength (Eads = −6.62 eV) coupled with strong oxygen affinity (Eads = −6.02 eV), resulting in superior resistance to carbon deposition, in agreement with experimental findings[36−38]. Ni4/SiO2(110) presents an interesting case study, displaying carbon adsorption intensity comparable to ZrO2(111) (Eads = −6.69 eV), yet significantly weaker oxygen affinity (Eads = −3.75 eV), exhibiting a stronger tendency toward carbon deposition. These theoretical predictions are corroborated by experimental studies. Xu et al.[39] demonstrated the inferior stability of Ni/SiO2 relative to Ni/Al2O3, attributing this to the enhanced carbon deposition propensity. Meanwhile, Pizzolitto et al.[34] established a stability hierarchy of Ni/Al2O3 > Ni/ZrO2/Ni/SiO2 under reaction conditions. Furthermore, Zhang et al.[40] revealed that carbonaceous deposits on Ni/ZrO2 exhibit superior reactivity toward CO2 oxidation at lower temperatures compared with those on Ni/SBA-15, indicating the enhanced carbon resistance of ZrO2 supports. Collectively, these findings establish the following trend in anti-coking performance: Al2O3 ≈ ZrO2 > SiO2. This comparative analysis reveals the delicate balance between carbon binding strength and oxygen availability in determining overall catalyst stability.
From the perspective of carbon deposition resistance, ZrO2 emerges as the optimal support, offering an ideal compromise between moderate carbon adsorption and sufficient oxygen supply for effective carbon removal. This balanced approach ensures sustained catalytic activity while minimizing carbon accumulation.
Summary
-
Figure 10 presents a comprehensive comparison of oxide support effects on Ni-catalyzed methane reforming. Ni4/Al2O3(110) exhibits superior catalytic performance, attributable to its optimal adsorption of reaction intermediates, facile C–H bond activation, and strong oxygen affinity that promotes carbon removal via CO formation. Ni4/ZrO2(111) similarly demonstrates excellent activity, with balanced carbon and oxygen adsorption energetics that effectively suppress coking. In contrast, Ni4/MgO(100) displays high intrinsic activity, but suffers from excessive binding of atomic C and CO, leading to severe carbon deposition and active site poisoning that compromises operational stability. Finally, Ni4/SiO2(110) exhibits both low catalytic activity and pronounced carbon deposition propensity.
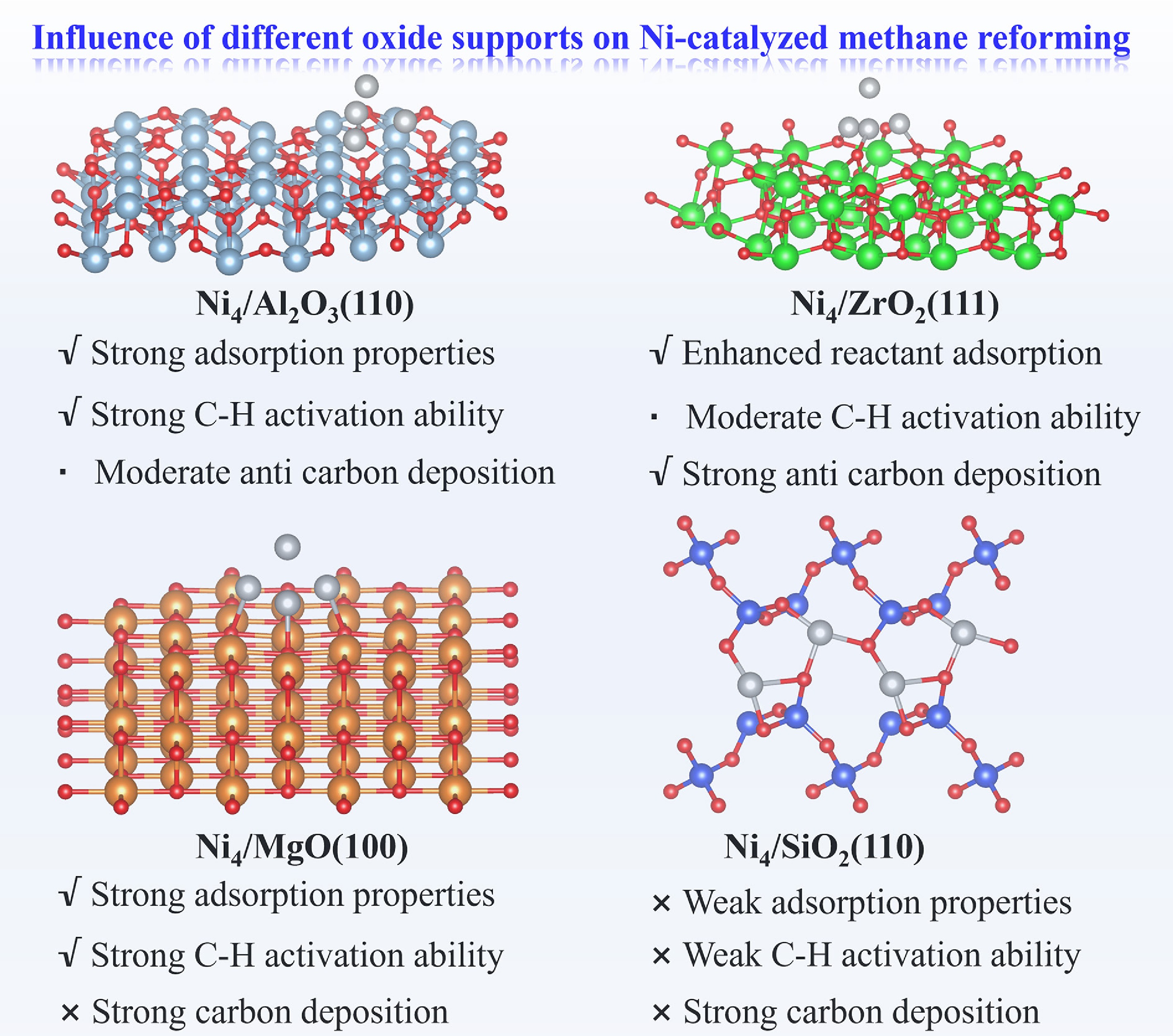
Figure 10.
The influence of different substrates on Ni-catalyzed methane reforming.
-
The present study elucidates the profound influence of different oxide supports on the catalytic performance of Ni-based catalysts in methane reforming, through systematic DFT calculations. By examining the adsorption behavior of reactants, key intermediates, and products across various supported catalyst systems, the fundamental structure-activity relationships that guide catalyst design optimization are established.
Comparative analysis reveals that supported Ni catalysts generally exhibit superior reactivity for methane reforming compared with the bulk Ni(111) surface. Cluster-based catalysts, including Ni4/Al2O3(110), Ni4/ZrO2(111), and Ni4/MgO(100), demonstrate highly consistent adsorption behavior for reactants (CH4, CO2, and H2O), indicating universal enhancement mechanisms across different oxide supports.
Distinctively, Al2O3(110) and MgO(100) supports significantly enhance reaction thermodynamics and exhibit strong reaction activity, as evidenced by the increased adsorption energies of C/CH and CO species, as well as their low C–H activation energy barrier. However, the strong adsorption of carbon-containing species also increases the risk of carbon deposition and potential active site poisoning by CO, especially for MgO(100), representing a classic activity-stability dilemma. ZrO2 emerges as a particularly promising support, by offering moderate enhancement of Ni4 cluster activity while simultaneously reducing carbon deposition tendencies. This balanced approach provides an effective strategy for addressing the fundamental activity-stability trade-off that plagues conventional Ni-based catalysts. In addition, Ni4/SiO2(110) exhibits relatively weak reactivity, although it is important to note that this system cannot represent the full spectrum of Si-based supported catalysts.
The development of composite catalysts that precisely control the distribution of active components while optimizing the anti-carbon deposition properties of substrates remains a crucial research priority. Such rational design strategies hold the key to breaking the long-standing activity-stability trade-off and enabling next-generation methane reforming catalysts with unprecedented performance and durability.
-
It accompanies this paper at: https://doi.org/10.48130/scm-0026-0018.
-
The authors confirm their contributions to the paper as follows: Yuangu Xia: investigation, methodology, formal analysis, writing – original draft; Haoyu Wang: investigation, methodology, formal analysis; Bin Hu: conceptualization, writing – revised draft, funding acquisition; Huaide Sun: investigation, review – revised draft; Tahir Iqbal: investigation, methodology; Ji Liu: conceptualization, writing – revised draft, funding acquisition; Qiang Lu: supervision, writing – revised draft, funding acquisition. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and analyzed during the current study are available from the corresponding author on reasonable request.
-
This work was supported by the National Natural Science Foundation of China (Grant Nos 52376182 and 52436009) and the Fundamental Research Funds for the Central Universities (Grant No. 2024JG003).
-
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
-
Full list of author information is available at the end of the article.
- The supplementary files can be downloaded from here.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Xia Y, Wang H, Hu B, Sun H, Iqbal T, et al. 2026. An atom-level insight into the oxide support effect of Ni-based catalysts on the syngas production in methane reforming. Sustainable Carbon Materials 2: e023 doi: 10.48130/scm-0026-0018 

An atom-level insight into the oxide support effect of Ni-based catalysts on the syngas production in methane reforming
- Received: 08 January 2026
- Revised: 02 March 2026
- Accepted: 07 April 2026
- Published online: 11 May 2026
Abstract: The rational design of highly efficient and stable catalysts is pivotal for the sustainable carbon source-derived methane reforming. The support material governs both the catalytic activity and long-term stability directly, by providing the surface acid/base sites and regulating strong metal-support interaction (SMSI). However, a comprehensive atomic-level understanding of how different oxide supports influence catalytic performance remains lacking. In the present study, a series of catalyst models that consist of common oxide supports and a Ni4 cluster were employed to elucidate support effects, using bulk Ni(111) as a reference. The catalytic properties, such as reactivity and carbon deposition tendency, were evaluated by mainly examining the adsorption of reactants (CH4, CO2, H2O), key intermediates (CH, C, O, H, OH), and products (CO, H2) on Ni4/MOn models. Notably, the Ni4 cluster supported on Al2O3(110), ZrO2(111), and MgO(100) generally enhances the reaction by strengthening the adsorption of reactants and cracking intermediates. However, excessively strong adsorption of carbon-containing intermediates on MgO also increases the risks of carbon deposition and CO poisoning. ZrO2 emerges as a more balanced support, providing moderate activity enhancement while inherently suppressing carbon deposition. In contrast, Ni4 clusters are redispersed into single atoms on SiO2(110), resulting in significantly weaker reactivity. This work elucidates the tailoring effect of supports on adsorption properties and activity-stability trade-off, offering a theoretical basis for the rational design of methane reforming catalysts.
-
Key words:
- Methane reforming /
- Support influence /
- Adsorption /
- DFT