










-
The endosperm of cereal crops is a main source of human food and animal feed, and is also used as raw materials for industry and biofuel. Endosperm functions as an absorptive structure that supports embryo development and later germinated seedling in angiosperms[1,2]. In the process from the double fertilization to seed maturity, early endosperm development only represents a relatively short duration, but it plays a decisive role in establishment of the endosperm function. Many cases showed that mutation of key factors in this process led to kernel abortion[3−7].
Maize kernel development originates from double fertilization in the embryo sac, where one sperm fertilizes the egg to form a diploid zygote that develops into the embryo; the other genetically identical sperm fuses with the two polar nuclei in the central cell and grows into the triploid endosperm. After double fertilization, early endosperm development undergoes four distinct cytological stages: coenocyte, cellularization, cell proliferation, and differentiation[6]. Early endosperm development usually spans from 0 to 144 h after pollination (HAP). Generally, from 0 to 48 HAP, the fertilized central cell undergoes several rounds of nuclear division without cytokinesis, thereby resulting in formation of a coenocyte. Afterward, cellularization occurs till 96 HAP, creating a fully cellularized structure. From 96 to 144 HAP, the endosperm rapidly proliferates and begins to differentiate from 120 HAP. Although the four representative cell types (the embryo surrounding region (ESR), starchy endosperm (SE), basal endosperm transfer layer (BETL), and aleurone (AL)) are not morphologically distinguishable at 120 HAP, the marker genes for a specific cell type begin to express in the corresponding regions. The time point at 144 HAP is generally considered as the end of early endosperm development, after which the endosperm activity is characterized by rapid cell proliferation and cell differentiation. From 192 HAP, the endosperm begins to synthesize storage reserves, which is called endosperm filling[1,6,8−10].
Over the years, several transcriptome profiling studies have advanced our understanding of the gene regulatory networks and cellular processes that control maize kernel development. Several endosperm cDNA libraries were constructed to identify gene expression at early-to-middle growth stages (4−6 days after pollination (DAP) and 7−23 DAP by using expressed-sequence-tag (EST) sequencing)[11]. A microarray with approximately 58,000 probes was used to study dynamic gene expression from 1 to 35 DAP (five days as an interval)[12]. By using high-throughput RNA sequencing (RNA-seq), a transcriptome atlas was generated using samples including embryo, endosperm and intact kernel, covering a long-time span from fertilization to maturity (0-38 DAP, two days as an interval), which revealed the extensive genetic control over the seed development[13]. A coupled laser-capture microdissection (LCM) and RNA-seq approach was used to capture different cell types at 8 DAP, illustrating the gene spatial expression pattern and correlation of gene regulation between each cell types during cell differentiation[14].
Recent studies reported a high-resolution transcriptome data for 31 consecutive time points by manual dissection of the nucellus within the first 144 HAP of kernel development (four or six hours as an interval)[10]. This provides a useful data source for functional research of maize early endosperm development. However, the early embryo sac (including the endosperm and the embryo) is small in size and is surrounded by the maternal tissue nucellus, which contributes a large portion to the whole kernel for transcriptome sequencing in that study, especially at the first 48-HAP stage. Therefore, it is possible that some genes expressing in a specific pattern but at a low level failed to be identified, even they might be important for early endosperm development.
Dissection of intact endosperms for RNA-seq is challenging, especially for 0−48 HAP endosperms. Here, we isolated early endosperm samples at five time points (24 h as an interval) by free hand and used them for RNA-seq. To compare the transcriptome data created from the free-hand samples, we generated another set of 96-HAP endosperm transcriptome data, where the endosperm samples were made by paraffin embedding and LCM[15]. In total, 22,853 unique genes were detected to express in at least one of the 22 samples ([FPKM] ≥ 1). Through WGCNA analysis, we identified nine distinct modules of co-expressed gene sets, of which Module 7 was composed of 5,555 genes that showed the highest expression level than genes in other modules at the coenocytic stage. In Module 7, a total of 391 genes were not detected for expression in nucellus, and therefore were regarded as the newly identified coenocyte-expressed (CE) gene set. We verified the reliability of the transcriptome data by in situ hybridization. Our work provides a valuable resource for studying early endosperm development in maize.
-
The maize (Zea mays) inbred line B73 was grown in the Songjiang experimental field in Shanghai (China) in 2021. All individual plants were self-pollinated at the same time. The embryo sac and endosperm were collected by manual dissection under a stereo microscope (Leica, Cat: DM2500) or a magnifying glass, and then were frozen immediately in liquid nitrogen in a 2 ml sterile tube. The materials were stored at −80°C before further processing. Each time point contains 3−4 replicates, and each replicate has at least 100 endosperms except for the one at 144 HAP, as 30−50 endosperms at 144 HAP are enough to extract 50 μg RNA.
Laser capture microdissection (LCM)
-
The self-pollinated B73 ears at 96 HAP were harvested for LCM. The uniform kernels in the middle region of the cob were dug out with the pedicle using sharp tweezers. To promote fixative penetration, 1-mm-thin sections in the middle of a kernel were longitudinally cut using a double-edge blade, and put immediately into a glass vial with prechilled Farmer's fixative (ethanol and glacial acetic acid in 3:1 ratio)[16], and kept at 4 oC overnight. The fixed kernel sections were then dehydrated in a graded ethanol series, cleared in graded n-butanol, embedded in paraffin wax (McCormick Scientific, Leica Biosystems, Cat: 39503002) using microwave[17]. Sections with 8−10 μm thickness were made as ribbons using a manual rotary microtome (Leica, Cat: RM2235), and mounted on PSA 1× White Slides (Leica, Cat: 39475275). Shortly before capture, sections were deparaffinized in Xylenes and air-dried[17]. Individual cell types were selected and captured using a laser microdissection system (Leica, Cat: LMD6500), with an optimized set of conditions (cutting speed, width, and laser energy).
RNA extraction, cDNA amplification, RNA-seq library construction and sequencing
-
High-quality total RNA of LCM samples and manually dissected samples was extracted with Arcturus® PicoPure® Frozen RNA Isolation Kit (ThermoFisher, Cat: KIT0214). High fidelity linear amplification of total RNA extracted from LCM samples was performed using Arcturus™ RiboAmp™ HS PLUS Kit (ThermoFisher, Cat: KIT0525) following the manufacturer’s protocol. The quality and quantity of the selected RNA samples were checked on an Agilent 2100 Bioanalyzer (Agilent Technologies). A total of 22 RNA samples used for preparing RNA-seq libraries and sequencing on a NovaSeq 6000 platform (Illumina, Inc., San Diego, CA, USA) by OE Biotech (Shanghai, China).
Quality control and mapping
-
Raw data (raw reads) were processed using Trimmomatic[18]. Clean reads were obtained by removing low-quality reads with ambiguous nucleotides, and adapter sequences were filtered from raw reads. An index of the reference genome was built using Bowtie v.2.0.6 and clean reads were aligned to the reference genome (
ftp://ftp.ensemblgenomes.org/pub/plants/release-45/fasta/zea_mays/dna/ ) using Hisat2[19]. Read counts of genes were acquired by HTSeq-count[20], and expression levels were calculated using the fragments per kilobase per million reads (FPKM) method[21].The relative expression level of each transcript was calculated by the statistical package DEGseq2 (version 3.12), and the resulting p-values were adjusted by controlling for the false discovery rate (FDR). Genes with |log2-fold change| > 1 and FDR < 0.05 were considered as differentially expressed genes (DEGs)[22].
WGCNA
-
The highly co-expressed gene modules were identified from the DEGs using WGCNA[23,24]. Genes with low FPKM ([FPKM] < 1) or low coefficient of variation of FPKM (SD ≤ 0.5) were filtered out, and 9,322 genes were obtained as the input gene set for WGCNA. WGCNA network construction and module detection was conducted using an unsigned type of topological overlap matrix (TOM), a minimal module size of 30, and a branch merge cut height of 0.25. WGCNA was performed using online tools (
https://cloud.oebiotech.com/task/ ). The intramodular connectivity value was calculated and used to evaluate the association of genes with modules. The top 50 hub genes in the CE Gene Set were filtered by their higher intramodular connectivity value. The connection weight to each gene pair was calculated, and 3,279 gene pairs were screened with a threshold of 0.5 in the CE Gene Set. The degree of the top 48 hub genes was greater than 70, the other is less than 40. So, these 48 genes are the real hub genes. The regulatory network of six hub TFs in the CE Gene Set was represented using Cytoscape 3.1[25]. The FPKM values of each gene were convert to Z-score and then used to draw clustered heatmaps by pheatmap in R.Functional enrichment analysis
-
To analyze the biological functions or pathways that are overrepresented in WGCNA modules. GO (Gene Ontology) analysis was performed using the OmicShare tools, a free online platform for data analysis (
www.omicshare.com/tools ). The significant threshold of p-value less than 0.05 would be identified for each GO category.Histocytochemical analysis
-
To confirm the developmental stages of endosperms that were sampled for RNA-seq samples, the same batch of each sample was used for the histocytochemical analysis. Kernels were fixed in Farmer’s fixative (ethanol and glacial acetic acid in 3:1 ratio) as processed for LCM. After dehydration using a gradient concentration of ethanol, the sections were embedded in epoxide resin for semi-thin sectioning. The sections were stained with 0.5% toluidine blue solution for 10 min and photographed under bright field using a Leica DM2500 for photos.
Kernel fixation and in situ hybridization
-
The spatial expression pattern of selected specific genes was validated by RNA in situ hybridization. kernels at 48 HAP were fixed in 4% paraformaldehyde solution (Sigma) with 0.1% TritonX-100 (Sigma) and 0.1% Tween-20 in PBS (Solarbio, Cat: 9005-64-5) for 16 h. After dehydration using graded ethanol and vitrification by xylene, the samples were embedded in paraffin. Kernels were longitudinally cut into 7-μm sections using a Leica manual microtome (Leica, Cat: FM2235). The paraffin sections with visible coenocyte were intercepted and affixed to glass slides. In situ hybridization was carried out according to the protocol in previous studies[5]. The fragment of each cDNA sequence was cloned and inserted into the pEasy-blunt-zero cloning vector (TransGen, Cat: CB501-01). The vectors used for the synthesis of antisense and sense RNA probes were transcribed in vitro by T7 and SP6 RNA polymerase, labeled by digoxigenin (DIG) according to the company instructions for the DIG RNA labeling kit (Roche, Cat: 11175025910).
-
To identify the genes expressing in early endosperm, we used the free-hand method to isolate early endosperm samples of the maize inbred line B73 from 48 to 144 HAP with a stereomicroscope or magnifier. The width of a 48-HAP endosperm is only about 250 μm, which is the most difficult to isolate than samples at later stages. However, there is a clear boundary between the endosperm and the nucellus tissue, and therefore the endosperm could be dug out along the boundary with tweezers. We successfully separated the endosperm samples at 48, 72, 96, 120 and 144 HAP, and collected about 30 to 100 endosperms at each time point for RNA-seq (Fig. 1a−e). We also collected the 96-HAP endosperms by LCM (Fig. 1f & g). We captured about 32,000 cells from 160 endosperm sections for each replicate.

Figure 1.
Isolation of early endosperms from 48 to 144 HAP by free hand and LCM. (a) Longitudinal free-hand sectioning of a developing B73 kernel at 48 HAP. (b) The enlarged area containing the endosperm as shown in (a) by an arrow. (c) Longitudinal free-hand sectioning of a developing B73 kernel at 72 HAP. (d) The enlarged area containing the endosperm as shown in (c) by an arrow. (e) Isolated endosperms from 48 to 144 HAP by free hand. (f) The endosperm at 96 HAP before LCM. (g) The endosperm at 96 HAP after LCM. (h) Light microscopy of semi-thin sections of developing B73 endosperm at 48 HAP. (i) Light microscopy of semi-thin sections of developing B73 endosperm at 96 HAP. The endosperms are indicated by white arrows. The embryo is indicated by a red asterisk. Bars = 500 μm in (a)−(d), (f), and (g). Bar = 1 mm in (e). Bars = 200 μm in (h) - (i).
Due to the difficulty in isolating early endosperm samples, we used all the samples obtained for RNA extraction and RNA-seq, with 3−4 biological replicates at each time point (Supplemental Data Set 1). The RNA extracted from free-hand samples had a higher RNA integrity compared with that from LCM samples (RIN: 9-10, Supplemental Fig. S1). Overall, the transcripts of different biological replicates were highly correlated (r ≥ 0.92; Supplemental Fig. S2). We did not eliminate biological replicates and directly used all samples for further analysis.
To confirm the corresponding endosperm developmental stage at each time point, we fixed, embedded and sectioned kernels from the same batch of samples. Observation of semi-thin 48-HAP kernels showed that the free nuclei shared a common cytoplasm and were arranged along the central cell membrane (Fig. 1h), while that of 96-HAP kernels displayed a fully cellular endosperm (Fig. 1i), confirming that our samples were collected correctly.
Maize endosperm develops at different rates in spring and autumn in Shanghai (China). Some studies showed that the coenocytic stage continued to 44 HAP[10], and other studies reported that the coenocytic stage could continue to 60 HAP. The development rate was most likely affected by the temperature or growing degree-days (GDD)[6]. However, the most accurate approach to evaluate the endosperm development process is cytological observation. In our results, En48 corresponded to the coenocytic stage (Fig. 1a & h), En72 to En96 corresponded to the cellularization stage, and En120-144 corresponded to the cell proliferation stage.
Global analyses of transcriptome data of early endosperm
-
We collected and captured 22 samples and each sample was sequenced at a depth of over 50 million read pairs to achieve a sufficient sequencing depth (Supplemental Data Set 1). Mapping of clean reads onto the B73 genome (RefGen_V4) revealed that 22,853 unique genes were detected for expression in at least one of the 22 samples ([FPKM] ≥ 1) and 19,519 unique genes were not expressed ([FPKM] < 1) (Supplemental Data Set 2).
We performed the principal component analysis (PCA) of the transcriptome data from 22 samples at five time points (Fig. 2a). Cells at the same developmental stage were clustered together, further confirming the high reliability of the RNA-seq data.
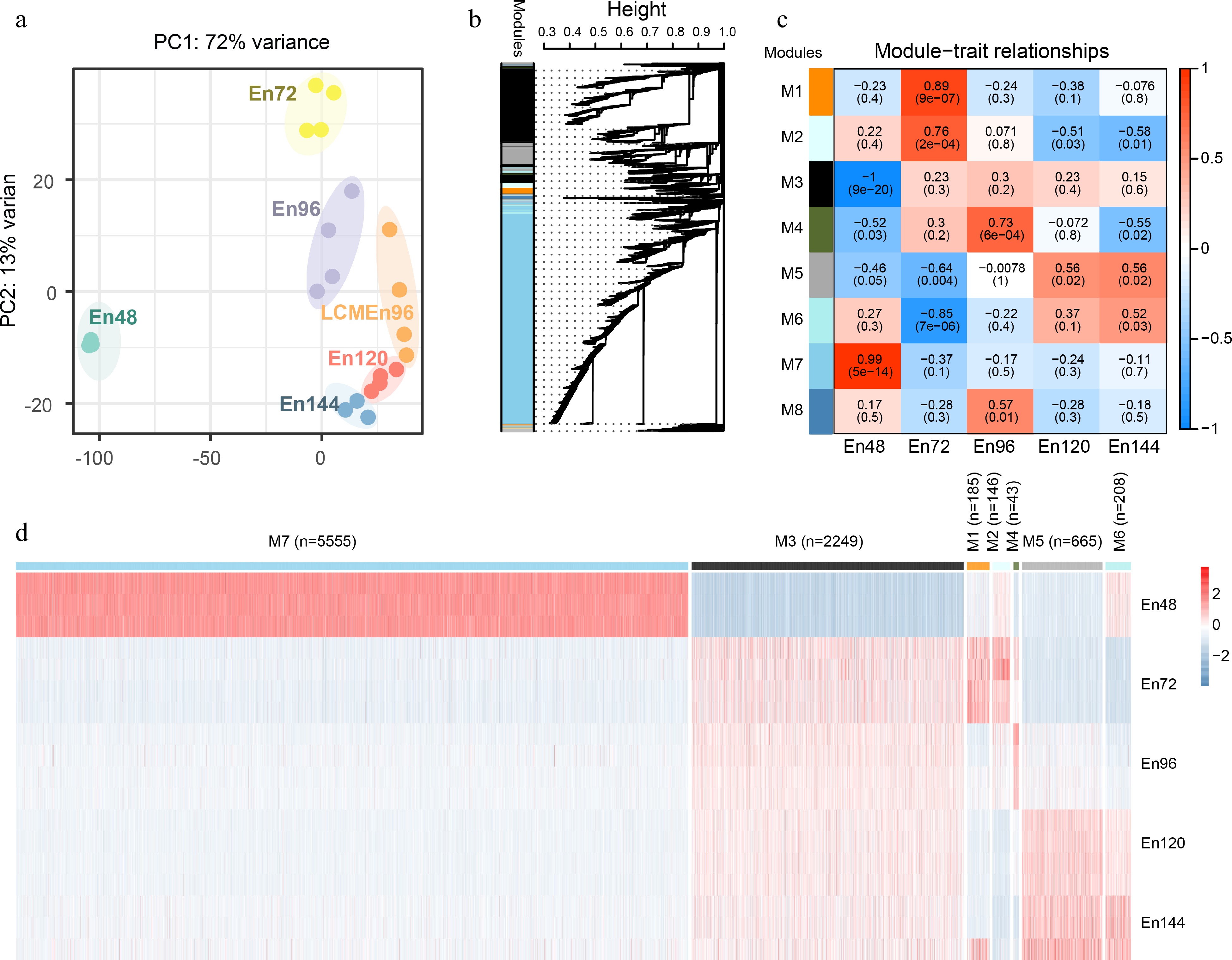
Figure 2.
Global analyses of the transcriptome of early endosperms. (a) Principal component analysis of the transcriptome of early endosperms. (b) WGCNA cluster dendrogram showing co-expression modules identified in 15 samples at five different stages. Modules corresponding to branches are assigned to distinct colors. (c) Correlations between modules and development stages. The correlation is estimated by the Pearson correlation coefficient method. (d) A heatmap showing the spatiotemporal expression pattern of seven stage-specific modules covering all tested stages.
To characterize the transcriptome changes at a system level, weighted gene co-expression network analysis (WGCNA)[24,26] was performed. The set of 22,853 unique expressed genes were filtered with SD ≤ 0.5, and 9,322 genes were obtained as the input gene set for WGCNA. We identified nine distinct modules of co-expressed genes (Fig. 2b, Supplemental Data Set 3). Notably, eight representative co-expression modules were highly correlated with their corresponding developmental stage (Fig. 2c). Genes in these modules displayed a developmental stage-specific expression pattern (Fig. 2d). For example, Module 7 was composed of 5,555 genes that showed the highest expression level in the coenocyte compare with genes in other modules, indicating their important function at this stage. On the contrary, Module 3 was composed of 2,249 genes, which showed the lowest expression level in the coenocyte, but continuously expressed from 72 to 144 HAP. These data indicate that the coenocytic stage is a specific developmental stage.
Biological processes enriched in coenocyte-enriched Module 7
We analyzed the Module 7 gene set in detail, revealing several hub genes that may play an important role in coenocyte formation. To confirm the Module7 gene set, we used Venn analysis to extract the co-downregulated genes set of En72/En48 and En144/En48. We observed significant downregulation of 7,882 genes in En72/En48, 6,834 in En144/En48 (fold change [FC] ≥ 0.5, false discovery rate [FDR] ≤ 0.05) (Supplemental Data Set 4), and 5,826 in both En72/En48 and En144/En48 (hypergeometric test, P = 0). Among the co-downregulated gene set, 4,430 genes (76.0%) belonged to the Module 7 gene set (Fig. 3b). It was much larger than expected by chance alone (hypergeometric test, P = 0), confirming the reliability of the Module 7 gene set.

Figure 3.
The specific expression gene cluster at coenocytic stage. (a) Violin plots showing the dynamic expression of Module 7 gene groups in all tested stages. (b) Comparison of downregulated genes between En144/En48, En72/En48 and Module 7 gene groups. (c) Comparison of genes between Module 7 gene groups and the public RNA-seq data[10]. (d) GO term enrichment analysis of the CE Gene Set. (e) Network of six Hub TFs with regulating genes in CE Gene Set. Color codes indicate the degree of the nodes. plum, 6 Hub TFs; orange, degree ranked 1st; green, degree ranked 2nd; pink, degree ranked 6th.
A comprehensive high temporal-resolution investigation of transcriptomes using data for 31-time points, within the first 6 days of maize seed development, provides an extensive view of transcriptome dynamics for early maize kernel development[10]. However, these samples used for the transcriptomic analysis contained the nucellus and embryo sac, and the nucellus should make the largest contribution to the transcriptome data. So, some specifically expressed genes in the endosperm might be ignored especially between 0 and 48 HAP.
Among the Module 7 gene set, a total of 5,164 genes were also found to be expressed in the nucellus, while 391 genes were not expressed in the nucellus[10]. The 391 genes were collectively named as the Coenocyte Expressed Gene Set (CE Gene Set) (Fig. 3c, Supplemental Data Set 4). There are 268 genes in the CE Gene Set detected in Li et al.'s samples[27], 82.1% were highly expressed at the embryo sac but showed low expression at the ovule (Supplemental Fig. S3, Supplemental Data Set 4), indicating the CE gene set we identified were indeed predominantly expressed in the embryo sac.
Coenocyte is a special stage of early endosperm development, featured by rapid nuclear division and migration without cytokinesis, creating a multinucleate coenocyte[6]. Consistent with this, the CE Gene Set was overrepresented by genes related to DNA-binding transcription factor activity, ethylene-activated signaling pathway, auxin-activated signaling pathway, SCF ubiquitin ligase complex (Fig. 3d, Supplemental Data Set 5). On the other hand, the Module 7 gene set was overrepresented by genes related to auxin-activated signaling pathway, DNA-binding transcription factor activity, and lipid transport (Supplemental Data Set 5). For the 5,555 genes in Module 7 and 391 genes in the CE Gene Set, the enriched GO terms were relatively similar, both focusing on transcriptional regulation and auxin-activated signaling pathways.
We found that 13 auxin pathway genes[28,29] were in the CE Gene Set , including one auxin-responsive factor (ZmARF18 Zm00001d000358), two IAA genes (ZmIAA20 Zm00001d000288, ZmIAA26 Zm00001d021279), one F-box protein-encoding gene (ZmSKP2A Zm00001d000224), one N-acetyltransferase-encoding gene (HLS1 Zm00001d028074), two VAN3-binding protein-encoding (Zm00001d000175, Zm00001d051361 ) and five auxin response genes (ZmSAURs: ZmSAUR32 Zm00001d051127, ZmSAUR39 Zm00001d021454, ZmSAUR41 Zm00001d021062, ZmSAUR71 Zm00001d036623, ZmSAUR72 Zm00001d046986). All these genes were not expressed in the nucellus, indicating an essential role of the auxin pathway in coenocyte formation.
The top 48 hub genes in the CE Gene Set were identified by their high intramodular connectivity value and high degrees (Supplemental Data Sets 4 & 6). These included six TFs, Zm00001d026537 (WUS2), Zm00001d013259 (MADS15), Zm00001d042781 (WIP6), Zm00001d009622 (EREB12), Zm00001d052026 (EREB15) and Zm00001d017677 (BHLH53). The expression of ZmWUS2 was previously detected in the leaf primordia at the P2/P3 stage, indicating ZmWUS2 plays a role in leaf initiation[30]. The mixed overexpression of ZmWUS2 and ZmBbm could significantly increase the transformation efficiency of many previously non-transformable maize inbred lines[31]. Our result suggested that ZmWUS2 might be involved in nuclear division at the coenocytic stage. The functions of other five TFs have not been reported.
WUS2, MADS15, WIP6, EREB12, EREB15 and BHLH53 were predicted to interact with 77, 78, 81, 84, 86, and 90 genes, respectively; some of their potential target genes are overlapped (Fig. 3e). The six TFs co-regulated six genes, including Zm00001d038179, Zm00001d035570, Zm00001d000299, Zm00001d000288, Zm00001d046743 and Zm00001d045582, of which Zm00001d000288 encodes an IAA protein. This again indicates that auxin signaling and transcriptional regulation play a key role in coenocyte development.
Functional transition during early endosperm development
-
From 0 to 144 HAP, endosperm development experiences four stages: coenocyte, cellularization, cell proliferation and differentiation. The single fertilized central cell develops into an endosperm tissue with about 10,000 cells[32,33], which is initially differentiated into four cell types: the embryo surrounding region (ESR), immature starchy endosperm (SE), basal endosperm transfer layer (BETL), and aleurone. Corresponding to this dramatic morphological transition, transcriptomes of different stages had distinct and different gene function enrichment (Fig. 4, Supplemental Data Set 7).

Figure 4.
Expression patterns of genes representing the enrichment GO terms and functional transition over the time course. GO analysis of the specifically expressed genes in each module. The expression patterns of the genes belonging to representative enrichment GO terms were shown.
The stage of endosperm cellularization (Module 1 and 2) is enriched by hormones biosynthetic process and hormonal signaling pathway, including cytokinin-activated signaling pathway (GO:0009736), gibberellin biosynthetic process (GO:0009686), brassinosteroid biosynthetic process (GO:0016132), basipetal auxin transport (GO:0010540). Based on a previous study and our data, auxin signal transduction pathway genes were significantly enriched at both the coenocyte stage and cellularization stage, and the enriched gene ID are different in the two stages[10]. Such as there were two auxin transport genes: Zm00001d005528 and Zm00001d043762 highly expressed in Module 1 cellularization stage. These results reflected the important roles of hormones in endosperm cellularization, especially the auxin. The lipid catabolic process (GO:0016042) was enriched in Module 4, indicating the cell membrane formation during endosperm cellularization. The period of 120 to 144 HAP (Module 5 and 6) was the stage of rapid mitosis and initial cell differentiation of endosperm cells. The differentiation of starchy endosperm cells and the initiation of storage reserve synthesis was consistent with the enrichment of GO classification, including nitrate transport (GO:0015706), amyloplast (GO:0009501), (1->3)-beta-D-glucan binding (GO:0001872), glycogen biosynthetic process (GO:0005978), lipid binding (GO:0008289) and seed oil body biogenesis (GO:0010344). The differentiation of BETL cells is represented by sucrose metabolic process (GO:0005985) and basipetal auxin transport (GO:0010540). These results indicate that the co-expression modules identified by WGCNA have specific function enrichment, corresponding to different endosperm development stages.
Comparison of expressive gene set between En96 and LCMEn96
-
LCM is a powerful method to isolate the pure cell populations. To confirm the transcriptome data of the free-hand samples, we used LCM coupled with RNA-seq to generate the transcriptome of 96-HAP endosperms, designated LCM96En.
The SCC analysis of the En96 and LCMEn96 transcriptome data showed a high correlation within the group (r ≥ 0.92; Fig. 5a) and a relatively low correlation between groups (r ≥ 0.65; Fig. 5a). Transcripts of 19,598 and 15,917 unique genes were detected in En96 and LCMEn96 ([FPKM] < 1). The vast majority expressed genes of En96 (79.3%) and LCMEn96 (97.6%) overlapped (hypergeometric test, P = 0, Fig. 5b). These results indicate that the 96En dataset obtained by both methods were reliable.
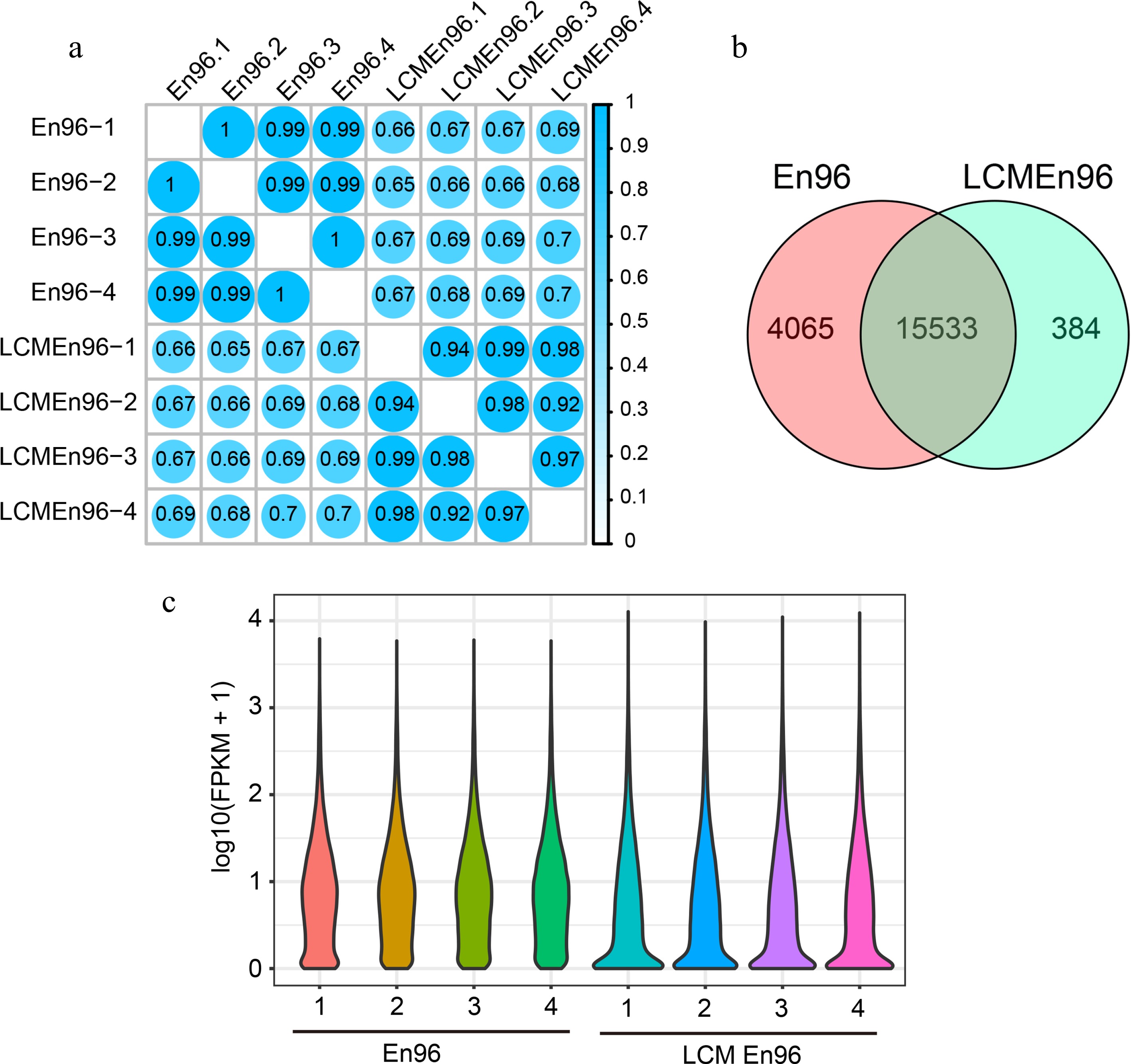
Figure 5.
Comparison of expressive gene sets between En96 and LCMEn96. (a) SCC analysis of the mRNA data for En96 and LCMEn96 using log2-transformed FPKM values. (b) Comparison of expressive genes between En96 and LCMEn96. (c) Violin plots showing the dynamic expression of genes in En96 and LCMEn96. The numbers at the horizontal axis represent the four replicates of each sample.
As shown in the violin plot (Fig. 5c), the expression features were different in En96 or LCMEn96. Compared with En96, LCMEn96 dataset had more elongated distribution, more genes expressed between 0 - 0.2 FPKM, and a higher expression level for highly expressed genes (Fig. 5c). Probably due to a relatively lower RNA quality of LCM samples (RIN: 3.6, Supplemental Fig. S1), and RNA amplified before the RNA library construction, the free-hand samples had higher fidelity than LCM. However, LCM samples could be used as a supplement.
Validation of coenocyte-specific TFs
-
We performed a series of mRNA in situ hybridizations to examine six TFs from Module 7 whether they were specifically expressed at the coenocytic stage (Fig. 3a & c). Among them, three genes were selected from the set of 5,164 genes (Fig. 3c, Supplemental Data Set 3), including Zm00001d043687 (MYB TF), Zm00001d042591 (MADS TF) and Zm00001d029711 (bZIP TF), while the other three genes were selected from the set of 391 CE genes, including Zm00001d000288 (AUX TF), Zm00001d000339 (EREB) and Zm00001d045204 (EREB) (Fig. 3c, Supplemental Data Set 4). All these genes showed a highly specific mRNA localization pattern within the coenocyte at 48 HAP. As a negative control, the corresponding sense probes had no visible signal (Fig. 6). The primers used are list in Supplemental Data Set 8.
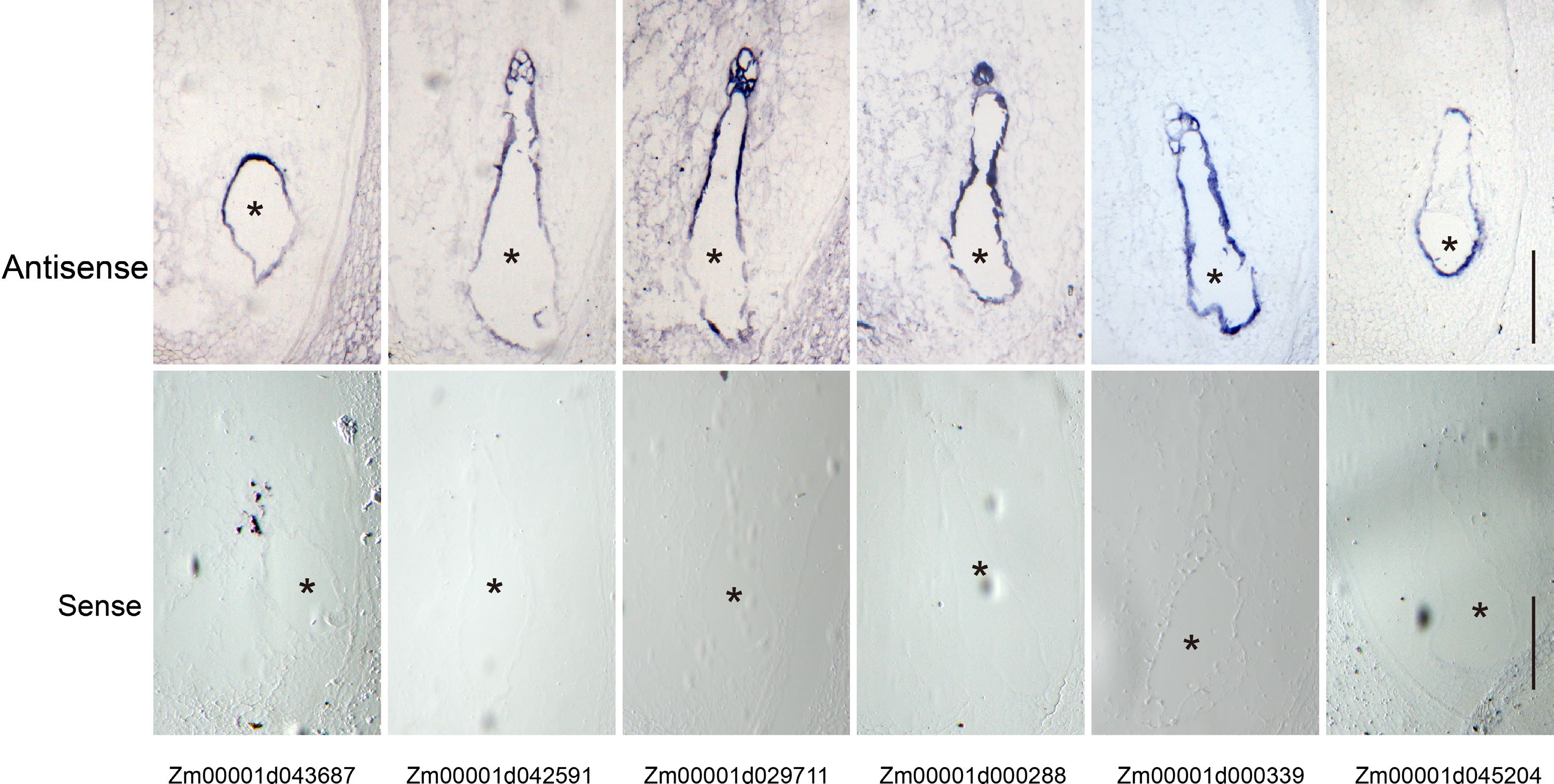
Figure 6.
Validation of the coenocyte-specific genes using in situ hybridization. Upper panel, hybridization with antisense probes; lower panel, hybridization with sense probes. Bar = 200 μm. The endosperms are indicated by black asterisks.
-
The current knowledge on the role of the hormone auxin for endosperm development in monocotyledon and dicotyledon were limited. It was highlighted that the auxin level in the endosperm may determine the timing of endosperm cellularization in dicotyledon[34−36]. The maize mutant de18 (zmyuc1) showed a decreased free IAA level after 8 DAP and a reduced kernel weight at maturity. The reduction in kernel weight is largely due to the reduced cell number and BETL formation, rather than the influence on early development of endosperm[37−39]. It is not clear whether auxin and ZmYUC1 are involved in formation of coenocyte and cellularization. However, the previous study and our data showed that auxin signal transduction pathway genes were significantly enriched at both the coenocyte cellularization stages, and the enriched genes were different at the two stages. Importantly, these genes were not expressed in the nucellus. It was reported that ZmPIN1 transcripts accumulate in the coenocyte around the free nuclei and also later in the cellularization endosperm. About 5-6 DAP, the accumulation of ZmPIN1 transcripts moves toward the chalazal region of the endosperm, where the BETL and ESR begin to differentiate[40]. DR5-RFP has a similar dynamic distribution[13]. These data indicate an important role of the auxin signal transduction pathway in early endosperm development, even though auxin might come from maternal tissues. After 6-8 DAP, endosperm mainly synthesizes auxin by itself. Creation of coenocyte-specific AUX/IAA TF mutants will help to clarify the function of auxin in early endosperm development.
Many approaches were applied to isolate different cell groups in one tissue and then analyze their transcriptomes. Early approaches mainly applied free hand or LCM to isolate individual cell groups for DNA microarray or RNA sequencing[14,41]. Recently, single-cell RNA sequencing (scRNA-seq)[42,43] and spatial transcriptome[44] are powerful techniques to reveal specific and spatial gene expression features for complex cell types at single-cell resolution.
At present, there are still many studies using the free-hand approach to isolate cell groups for RNA seq[45,46]. However, the resolution and reliability of the results from the manual separation depend on the proficiency of an experimental operator. Our data generated from the free hand approach had a better quality than those from LCM in terms of the number and level of the expressed genes. The coenocyte-specifically expressed gene set in our data is overall consistent with the findings of previous studies[10,27]. Furthermore, we mostly, if not completely, eliminated the nucellus and obtained a more precise set of genes that were expressed in early endosperm samples. The newly identified 391 CE genes will be a valuable source for studying coenocyte formation.
Accession numbers
-
The sequencing data have been deposited in the National Genomics Data Center (NGDC;
https://ngdc.cncb.ac.cn/ ) under accession code PRJCA012667. We thank Mrs. Qiong Wang (CAS Center for Excellence in Molecular Plant Sciences, CAS) for technical support. This research was supported by the National Natural Science Foundation of China (32272146 and 31871626 to JC; 31925030 and 31830063 to Y.W; U22A20466 to G.F.) and the Opening Foundation of National Key Laboratory of Crop Science on Wheat and Maize (SKL2022KF08).
-
The authors declare that they have no conflict of interest. Yongrui Wu is the Editorial Board member of Seed Biology who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and his research groups.
-
# These authors contributed equally: Yuxin Fu, Shuai Li
- Supplemental Data Set 1
- Supplemental Fig. S1 Representative quality assessments of RNAs used in sequencing.
- Supplemental Fig. S2 SCC (Spearman's rank Correlation Coefficient) analysis of the mRNA data using log2-transformed FPKM values.
- Supplemental Fig. S3 Expression patterns of CE Gene Set in maize embryo sac and ovule by the public RNA-seq data[27].
- Supplemental Data Set 2
- Supplemental Data Set 1 Data quality and mapping to B73 genome.
- Supplemental Data Set 3
- Supplemental Data Set 2 Expression level of genes in different samples.
- Supplemental Data Set 4
- Supplemental Data Set 3 The expression of WGCNA_modules genes.
- Supplemental Data Set 4 En144 VS En48 downregulated genes set.
- Supplemental Data Set 5
- Supplemental Data Set 5 GO enrichment of the genes in Module 7.
- 6
- Supplemental Data Set 6 Top 50_weight.
- Supplemental Data Set 7 GO terms enriched in the Module 1.
- Supplemental Data Set 7
- Supplemental Data Set 8 The primers for in-situ hybridization of 2D specific genes.
- Supplemental Data Set 8
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press on behalf of Hainan Yazhou Bay Seed Laboratory. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Fu Y, Li S, Xu L, Ji C, Xiao Q, et al. 2023. RNA sequencing of cleanly isolated early endosperms reveals coenocyte-to-cellularization transition features in maize. Seed Biology 2:8 doi: 10.48130/SeedBio-2023-0008 

RNA sequencing of cleanly isolated early endosperms reveals coenocyte-to-cellularization transition features in maize
- Received: 17 February 2023
- Accepted: 11 May 2023
- Published online: 05 July 2023
Abstract: Early endosperm development in maize (Zea mays) is essential for creating a functional endosperm for filling, but its rapid and dynamic process remains largely unknown. The coenocytic stage is a particular stage with rapid nuclear division without cytokinesis. From 48-144 h after pollination (HAP), endosperm mainly undergoes four cellular processes: coenocyte, cellularization, cell proliferation, and differentiation. Although the high temporal-resolution transcriptome data within 144 HAP of maize kernel development have been investigated, due to technical limitations, the samples contained the maternal nucellus and the embryo sac; as a consequence, many endosperm-specifically-expressed genes might be over-looked. In this study, we isolated early endosperms by free hand and laser-capture microdissection (LCM) and generated high-resolution transcriptome data from 48 to 144 HAP with an interval of 24 h. Through weighted gene co-expression network analysis (WGCNA), we identified nine distinct modules of co-expressed gene sets, of which Module 7 was composed of 5,555 genes that showed the highest expression levels at the coenocytic stage. In Module 7, there were 391 genes not expressed in nucellus, and thus were named as the Coenocyte-Expressed (CE) Gene Set. These genes were involved in transcriptional regulation and auxin-activated signaling pathway. Consistent with the stage transition of early endosperm development, the co-expressed gene sets and enriched gene function modules were changed accordingly. We verified the reliability of the transcriptome data by in situ hybridization. Our work provides a valuable gene resource for early endosperm development studies in the future.
-
Key words:
- Maize /
- Coenocyte /
- Transcriptome /
- Early endosperm development