









-
Roses, often referred to as the 'Queen of Flowers', hold significant economic and medicinal value. They are among the world's four major cut flowers. As one of the original centers of rose diversity, China boasts a cultivation history spanning approximately 2,000 years. Roses were widely planted in gardens during the Han Dynasty and reached their zenith in popularity during the Tang Dynasty[1]. In the 18th century, breeders crossed ancient Chinese rose varieties with native European roses, resulting in the creation of Hybrid Tea Roses capable of blooming year-round. This marked a significant transition from Old Garden Roses to Modern Garden Roses[2]. Currently, over 25,000 modern rose varieties have been developed through extensive interspecific and distant hybridization, which has blurred the originally distinct boundaries between varieties. The most widely recognized classification method for roses is the World Federation of Rose Societies (WFRS) classification system, which is based on growth habits and external characteristics. However, the WFRS classification method faces challenges in categorizing novel cultivars developed through extensive hybridization, which often exhibit phenotypic mosaicism. Furthermore, key diagnostic traits such as plant height and flowering period are susceptible to environmental influences - identical genotypes may be misclassified into distinct groups when grown under temperate vs subtropical climatic conditions. Alternatively, molecular marker-based classification methods, such as those using DNA polymorphism, offer advantages as they are not constrained by seasonality, growth stage, or time requirements. Among molecular markers, Simple Sequence Repeats (SSRs) are particularly advantageous due to their low cost, reproducibility, and lack of reliance on radioactive substances. SSR markers have been extensively used in plant phylogenetic studies, genetic map construction, and variety identification[3−5].
To identify conserved SSR markers for rose genetic studies, we performed a comparative analysis of SSR loci across reference genomes of three rose varieties using MISA software[6−8]. Following systematic exclusion of mono-base, di-base, hexa-base, and compound SSR motifs, we identified 3,516 polymorphic SSR loci that were consistently present across all three genomes. These loci included 1,991 tri-nucleotide repeat motifs, 516 tetra-nucleotide repeat motifs, and 1,012 penta-nucleotide repeat motifs (Supplementary Tables S1−S3). Through systematic selection of 384 SSR loci evenly distributed along chromosomes, we designed and synthesized fluorescently labeled primers. Initial screening was conducted through PCR amplification using a bulked DNA pool comprising ten rose cultivars with diverse genetic backgrounds. Following the exclusion of non-polymorphic primers, the remaining 223 candidate primers underwent individual validation via PCR amplification and capillary fluorescence electrophoresis across all ten genotypes. Using GeneMarker software for peak profile analysis and signal quality assessment, we successfully identified 35 highly polymorphic primers that demonstrated amplification of at least three alleles per locus (Supplementary Table S4). The 5' ends of the 35 pairs of polymorphic PCR primers that were screened were labeled with distinct fluorescent markers. PCR amplification and fluorescent capillary electrophoresis detection were then conducted on 364 self-cultivated or introduced rose samples provided by Yunnan Academy of Agricultural Sciences (Supplementary Table S5). The results were analyzed using GeneMarker software to obtain the number of alleles (Na), peak maps, and genotypes for each sample. The results generated by GeneMarker software showed that all primers were able to identify at least three alleles, which is consistent with the results of our pre-screening. A total of 219 alleles were identified among 364 rose germplasms using the identified 35 SSR markers, among which, RcJ120, RcJ248, and RcJ371 identified nine alleles each, while RcJ051 identified the most alleles at ten. The average number of alleles per SSR makers was 6.3, indicating certain polymorphism (Supplementary Table S4). Using these primers, all rose germplasm was successfully distinguished by exhibiting different genotypic compositions at different SSR loci. To demonstrate the discriminatory power of this set of markers more clearly and intuitively, we paired each primer with its corresponding allelic marker, using '1' to indicate the presence of a band and '0' to indicate its absence (Supplementary Table S6). This was compiled into a binary matrix, which was then utilized to generate an SSR fingerprint map for all the rose germplasm. The SSR fingerprint map is highly specific, providing a clear visual representation of genetic differences across samples and serving as a reliable species identification tool (Fig. 1).

Figure 1.
Fingerprints of 364 rose germplasm based on 35 SSR markers.
The 364 rose germplasm were converted into fingerprint profiles in the form of 01 matrices, with one color representing one locus, each row representing one sample, and each column representing one allele.Based on the Genocore software platform, a rose core germplasm collection was established through an allelic maximization strategy[9]. Remarkably, complete allelic coverage (100%) was achieved with a minimal sample size of 28 accessions, representing 7.7% of the original 364 germplasm resources (Fig. 2a). Provenance analysis demonstrated that these core accessions originated from seven internationally renowned breeding institutions, including Delbard, validating the efficacy of global germplasm introduction strategies (Supplementary Table S7). This approach provides significant advantages for optimizing the utilization of rose genetic resources. Principal coordinate analysis (PCoA) conducted in GenAlex indicated partial spatial clustering of the 28-core collection, which did not fully overlap with the dispersion range of the original germplasm (Fig. 2b). This structural deviation likely arose from Genocore algorithmic prioritization of rare allele retention, intentionally selecting accessions carrying low-frequency alleles at the expense of structural mirroring. In contrast, CoreHunter screening at a 20% sampling ratio (72 accessions) produced a core collection with a spatial distribution closely aligned to the original population[10] (Supplementary Table S8). However, despite doubling the sample size, CoreHunter achieved only 96.3% allelic retention. We propose that prioritizing maximal allelic retention with minimal sample size aligns more closely with the core germplasm objective than strict adherence to population structure replication.
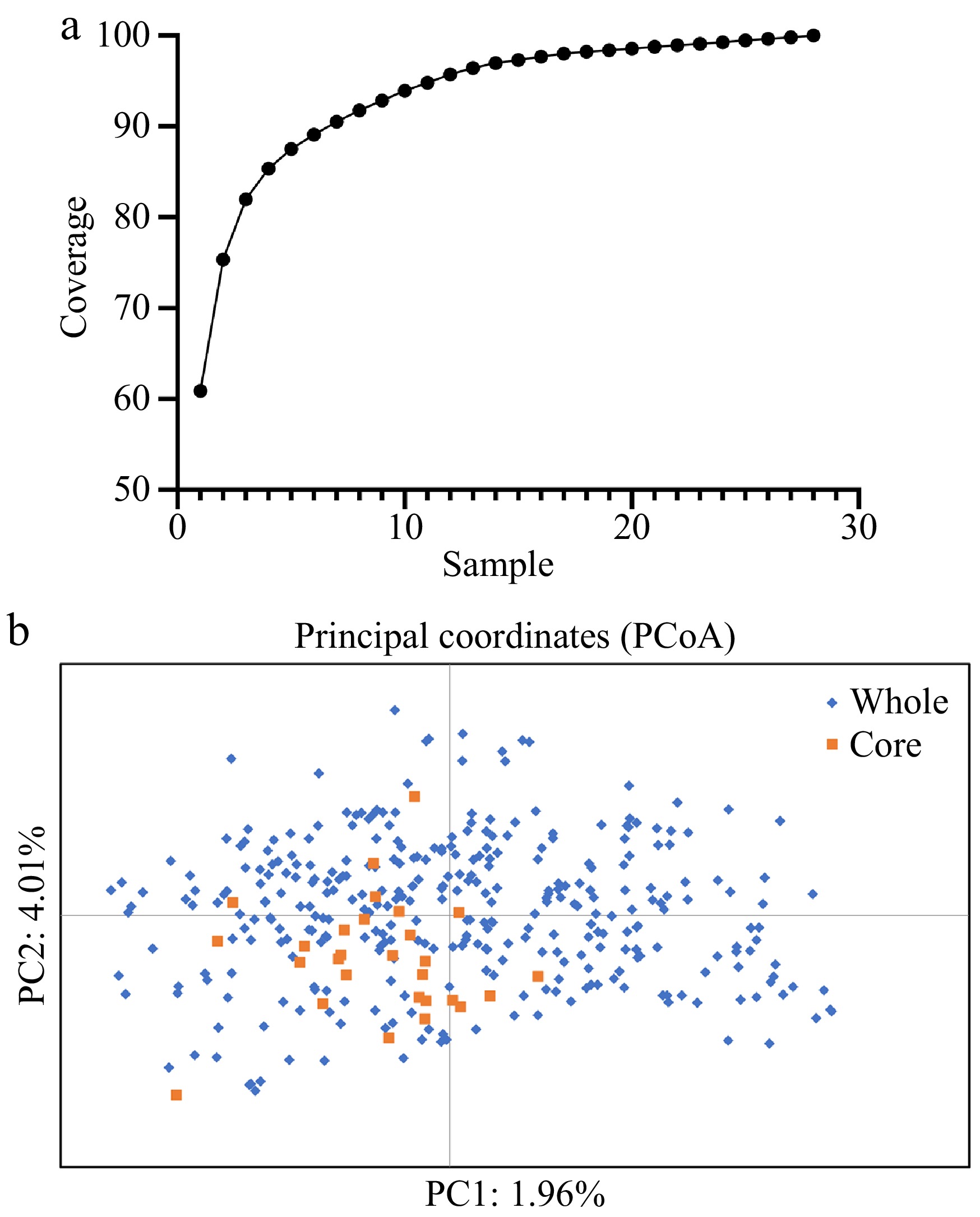
Figure 2.
Core germplasm screening of rose based on genotypic data from SSR markers.
(a) Trends in allele coverage with increasing number of samples. (b) PCoA of the core collection (n = 28) and whole collection (n = 364) were based on genotypic data from SSR markers. The blue represents the variety of whole collection, and the orange represents the core collection.This study analyzed 364 rose accessions, comprising 321 cultivars from eight global breeding companies and 43 Yunnan Academy of Agricultural Sciences varieties, to balance genetic diversity and geographic representation. Despite these systematic efforts, the analysis revealed limited allelic richness, with an average of 6.3 alleles per marker detected across the 364-accession panel. This contrasts sharply with the findings of Guan et al.[4], who reported 13.1 alleles per marker across 192 rose germplasms using ten SSR markers. The discrepancy likely stems from fundamental differences in sample composition - their germplasm pool incorporated both ancient roses and modern hybrids, thereby capturing broader historical genetic strata, whereas our collection focused exclusively on modern rose cultivars. This temporal stratification suggests that intensive modern breeding practices have substantially narrowed the genetic base of contemporary rose varieties through selective sweeps and domestication bottlenecks. These results affirm the imperative of allelic-maximization strategies in core collection development. Breeders must intensify efforts to conserve rare alleles through systematic germplasm curation while advancing their practical utilization. Strengthening core repositories with genetically underrepresented variants will ensure sustainable access to adaptive diversity for future rose improvement.
In summary, 35 SSR markers were selected from three published rose genomes and validated for polymorphism across 364 rose varieties. These markers generated a total of 219 alleles, with each marker amplifying three to ten alleles. Using these polymorphic markers, a comprehensive fingerprint map was constructed, encompassing 364 rose germplasm resources. Additionally, a core collection of 28 rose individuals was established, retaining 100% of the genetic diversity present in the original germplasm. This study provides a reliable and efficient set of SSR molecular markers for rose classification and germplasm management. It also offers a foundational reference for the conservation and sustainable utilization of rose genetic resources.
HTML
This research was funded by the Yunnan Xingdian Talents — Youth Special Project (XDYC-QNRC-2022-0731), Yunnan Xingdian Talents — Special Selection Project for High-level Scientific and Technological Talents and Innovation Teams-Team Specific Project and High-level Talent Introduction Program of Yunnan Province — Industrial Talent Special Project (YNQR-CYRC-2020-004).
-
The authors confirm contribution to the paper as follows: Liu B was responsible for data analysis and visualization, and writing the first draft. Yuan H was responsible for the preparation of experimental materials and data organization. Wani MA was responsible for reviewing and revising the first draft. Jin C was responsible for supervising and leading the experiments. Li F provided the experimental ideas and financial support. All authors reviewed the results and approved the final version of the manuscript.
-
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
-
The authors declare that they have no conflict of interest.
- Supplementary Tables S1 SSRome-Search-Result(Tri).
- Supplementary Tables S2 SSRome-Search-Result(Tetra).
- Supplementary Tables S3 SSRome-Search-Result(Penta).
- Supplementary Tables S4 35 SSR primer information.
- Supplementary Tables S5 364 modern rose sources.
- Supplementary Tables S6 01 matrix based on SSR results.
- Supplementary Tables S7 Core germplasm generated based on Genocore software.
- Supplementary Tables S8 PCoa analysis of core germplasm generated based on CoreHunter software.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press on behalf of Yunnan Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
Liu B, Yuan H, Wani MA, Jin C, Li F. 2025. Fingerprint mapping and core collection construction of modern roses based on SSR markers. Agrobiodiversity 2(1): 9−11 doi: 10.48130/abd-0025-0002


|