










-
The domestication of pigs from wild boars was a consequence of human selection. About one million years ago, the European and Asian boar populations began to diverge[1]. Roughly 9,000 years ago, in the initial phase of pig domestication, distinct populations of domestic pigs in East Asia and Europe started to follow separate breeding and evolutionary paths[2,3]. This divergence resulted in the emergence of unique genetic groups. During this period, extensive artificial selection caused significant phenotypic changes in domestic pigs. Phenotypic changes suggest that specific mutations occurred in genes associated with particular traits. These mutations lead to modifications in the genetic information, known as selection signatures, within the genome.
Identifying genes under selective pressures is a primary objective in the study of pig domestication. Numerous studies have used genomic methodologies to identify genetic changes underlying phenotypic alterations in pigs during domestication. Wang et al. pinpointed a series of genes that are crucial for the performance of several traits[4]. For example, the PTPRM gene is essential in controlling various cellular functions, while the HACD3 gene is pivotal for aiding the accumulation of body fat. Moon et al.[5] identified several genes, such as ABLIM1, CXADR, INSR, RIMS1, and SYNE1, that are connected to the control of growth during development and the formation of anatomical structures. Wang et al.[6] have shown that the color-associated genes MITF and EDNRB have undergone strong selective breeding in the Chinese White-spotted pig breed.
In the domain of pig farming, teat count is a key morphological and reproductive trait. Healthy teats yield milk rich in essential nutrients, providing a vital energy source for the growth and development of young piglets. Zhuang et al.[7] have determined that the VRTN gene's influence on the number of teats may be through linkage with the mutation responsible for the trait. Martins et al.[8] constructed a network that incorporates various biological processes and transcription factors, successfully identifying candidate genes linked to teat number. Within this set, GHR, IFT80, FSTL3, SKOR1, SMURF1, and AKT3 have emerged as the most promising candidates.
The Western pig, characterized by its large White pig breed, has undergone substantial artificial selection during its evolutionary history, leading to marked divergence in teat number breeds. This variation has arisen due to extensive selection processes carried out by companies with unique breeding goals. Several prominent Large White pig strains, such as the French CG, the French Cooperl, and the Dutch Topigs strains, have a higher average teat count of approximately 16 compared to 14 teats of the Danish Large White breed, enabling them to effectively meet the daily lactation requirements of piglets. Numerous studies have explored selection signatures; however, the molecular mechanisms responsible for the higher number of nipples in Large White pig strains under selection pressure need further study.
The present research focuses on identifying selection regions and candidate genes that contribute to the higher number of teats in three distinct Large White pig strains, through an analysis of their selection signatures. Four methods (CLR, nSL, iHS, and XP-EHH) were employed for detecting selection signals to analyze the complete genomes of three prominent domesticated pig breeds: French CG Large White, French Cooperl Large White, and Dutch Topigs Large White. Danish Large White pigs served as the outgroup for comparative analysis. The present findings will shed light on the genomic traits of Large White pigs, offering valuable insights into their selection and breeding in the context of teat number.
-
Phenotypic data was collected on teat number for various Large White pigs, including CG, Topigs, Cooperl, and DanBred from local Chinese companies, originating from different companies and countries. Shandong Huanshan Group had already introduced Topigs Large White pigs from the Dutch Topigs Company in 2011. In 2020, Henan Minwang Company introduced CG Large White pigs from France, and Henan Fengyuan Hepu Company introduced Cooperl Large White pigs from the French Cooperl Company. Liyuan Group introduced Danish Large White pigs from DanBred. Each strain is a purebred core breeding population without crossbreeding. We have partitioned the samples from each breed into families and utilized HIBLUP[9] software to calculate the estimated breeding values for the total teat number trait within each breed. By integrating family data and estimated breeding values, 50 individuals were meticulously selected with an exceptionally high number of teats from each breed.
Genotyping and quality control
-
The Megi Universal Nucleic Acid Extraction Kit was employed to isolate genomic DNA from ear tissue samples. The quantity and quality of the extracted DNA were assessed using a NanoDrop 2000 spectrophotometer, verifying that the OD260/280 ratio was within the range of 1.7 to 2.1 and that the DNA concentration exceeded 50 ng/μL. Genotyping was conducted with the Compass Porcine 50K Plus Breeding Beadchip from Tianjin, China, encompassing 57,466 SNPs across the entire porcine genome. The physical locations of these SNPs were aligned to the Sus scrofa 11.1 assembly (Sscrofa11.1) of the pig reference genome. Quality control measures were implemented using PLINK v1.9 software[10]. Samples with SNP call rates below 90% were excluded. SNPs with genotype-missing rates exceeding 0.1 and a minor allele frequency (MAF) below 0.05 were also discarded. The analysis was restricted to SNPs located on autosomal chromosomes.
In the present research, a dataset comprising whole-genome resequencing from 1,662 pigs formed the basis of the reference panel. This dataset included resequencing information from 1,602 pigs of diverse breeds sourced through the PigGTEX initiative[11], and 60 Suhuai pigs[12]. Beagle software (version 5.2) with its standard settings was utilized to perform haplotype phasing on the reference panel[13] and to impute SNP-chip data to achieve whole-genome resolution[14]. SNPs were filtered out if they had a dosage R-squared (DR2) value below 0.9 and a minor allele frequency (MAF) less than 0.05. Following stringent quality control assessments, a total of 9,705,084, 9,214,835, 8,642,894, and 6,328,251 SNPs corresponding to CG, Cooperl, Topigs, and Danbred were chosen for analysis.
Genetic diversity, linkage disequilibrium analysis, and ROH detection
-
PLINK v1.9[10] was utilized to assess genetic diversity in four distinct Large White pig populations by calculating both observed (Ho), and expected (He) heterozygosity. VCFtools[15] was utilized to calculate nucleotide diversity for each breed using a window of 500 kb with a step of 50 kb. PopLDdecay v3.40 software was employed to evaluate the genome-wide linkage disequilibrium patterns by calculating the r2 values for SNPs pairs across Large White pig populations within a 500 kb window[16].
Analysis of principal components and phylogenetic tree
-
To delve into the genetic connections among cohorts of Large White pigs, we began by refining the marker dataset, eliminating SNPs with substantial relatedness within a 50 SNP window. The window was shifted in intervals of 5 SNPs, with an r2 threshold set at 0.2. After conducting linkage disequilibrium (LD) filtering, PLINK v1.9 software[10] was used to perform principal component analysis (PCA). The PCA plot was then generated using the ggplot2 package in R. The PLINK format file was converted into MEGA format, and a neighbor-joining phylogenetic tree (NJ-tree) was assembled utilizing the p-distance algorithm in MEGA's neighbor-joining module, version 6[17]. The results were visualized using FigTree v1.4.3.
Identification of selection signatures
-
The site-frequency spectrum (SFS) is defined by the frequency at which various allele frequencies occur within a specific target area of the genome. Methods based on the SFS are highly effective at detecting selection signals that have become fixed, with the Composite Likelihood Ratio (CLR) method showing greater detection power than other methods based on the SFS[18]. The CLR test compares the maximum likelihood under neutral conditions (absence of selection sweep) with the maximum likelihood under scenarios of selective sweep[19]. Allele frequency data were utilized to compute CLR test values for scanning regions with complete sweeps. Employing the SweeD software, the CLR was calculated with a 50 kb grid dimension. The highest 1% of the CLR value spectrum from empirical data was used to detect potential regions, and genes located within these regions were designated as candidate genes[20].
Statistical tests based on linkage disequilibrium are highly effective in detecting recent positive selection signatures. These tests are fundamentally rooted in the theory of long-range extended haplotype homozygosity. The construction principles of nSL and iHS (integrated haplotype score) statistics are similar; iHS uses a single marker site to replace the core haplotype in the statistical measure, defining this site as the core site. iHS integrates the genetic distance of the extended haplotypes, where the ancestral alleles are located at the core site and where the newly mutated alleles are located, and calculates the ratio between the two to detect selection signals. The iHS evaluates the relative decay of EHH between ancestral and derived core alleles[21]. Compared to iHS, nSL measures the length of the haplotype homozygosity segment between pairs of haplotypes in the same region of the dataset in terms of the number of mutations, thereby enhancing robustness to recombination and/or variations in mutation rates. The nSL statistic assesses haplotype length based on the number of segregating sites and has shown improved efficacy in detecting soft sweeps[22]. The XP-EHH statistic assesses the extended haplotype homozygosity within the same genomic region across two populations, detecting signals of ongoing or near-fixed selection[23]. Selscan v.1.0.4 software was used to perform nSL, iHS, and XP-EHH scans[24]. The nSL and iHS statistics were calculated with a scaling factor of 20,000, a maximum interval of 200,000, and an EHH threshold of 0.05. The nSL, iHS, and XP-EHH results were normalized by frequency into 100 bins using the 'norm' package included in selscan[24]. The test statistic was computed by calculating the mean normalized value for each 50 kb region. The top 1% of regions exhibiting the highest mean |iHS|, |nSL|, and XP-EHH scores were considered potential targets of positive selection.
Gene annotation
-
To investigate the biological roles of the candidate genes, the Ensembl BioMart tool (
https://asia.ensembl.org/index.html ) was utilized for gene annotation in the selected regions. The Phenome-Wide Association Study (PheWAS) has been widely used to associate genetic variants with phenotypes[25]. The PheWAS module available on the website (http://pigbiobank.farmgtex.org ) was used to explore the connections between the genes of interest identified here and the various complex traits present within the PigBiobank[26]. To enhance the comprehension of the functions of the candidate genes and their related signaling pathways, GO and KEGG pathway enrichment analyses were performed via the KOBAS platform. -
Tthe maximum, minimum, mean, standard error, and coefficient of variation values for teat number across different groups of Large White pigs are presented. Each company collected 50 ear tissue samples, including samples from various family lines (Table 1).
Table 1. Descriptive statistics of the number of teats in 50 Large White pigs of each strain.
Groups N1 Min2 Max3 Mean4 ± SE5 French CG Large White pigs 50 16 20 18.27 ± 0.10 French Cooperl Large White pigs 50 18 20 18.32 ± 0.07 Dutch Topigs Large White pigs 50 18 19 18.26 ± 0.06 Danish Large White pig 50 10 13 12.40 ± 0.09 1 Number of individuals with phenotypic records. 2 Minimum of phenotype. 3 Maximum of phenotype. 4 Mean of phenotype. 5 Standard error. Genetic diversity and linkage disequilibrium
-
After performing quality control procedures, the average Ho, He, and nucleotide diversity were calcuated to evaluate the polymorphism within four Large White pig populations. As shown in Supplementary Table S1, He was significantly lower than the Ho in each population among all the groups analyzed in this study. In the analysis of nucleotide diversity, a substantial difference was identified between the CG and Topigs strains, with the CG strain having the highest nucleotide diversity. The influence of artificial selection on the levels of genome LD within each population was observable. Research indicates that artificial selection increases the degree of linkage disequilibrium in a population. In this study, the LD attenuation rate was highest for the CG strain, followed by the Cooperl and Topigs strains, with the Danbred strain exhibiting the slowest rate (Supplementary Fig. S1).
Analysis of population structure and phylogenetic patterns
-
PCA was conducted to explore clustering patterns among the animals included in the study (Fig. 1). The PCA results demonstrated distinct separation among the Danbred and Topigs strains, and the CG and Cooperl strains. Subsequently, an NJ-tree was built from the matrix of pairwise genetic distance among individuals. The phylogenetic revealed that members of the CG and Cooperl groups clustered closely, forming a distinct branch with a high level of genetic coherence, which is consistent with the observations from PCA.
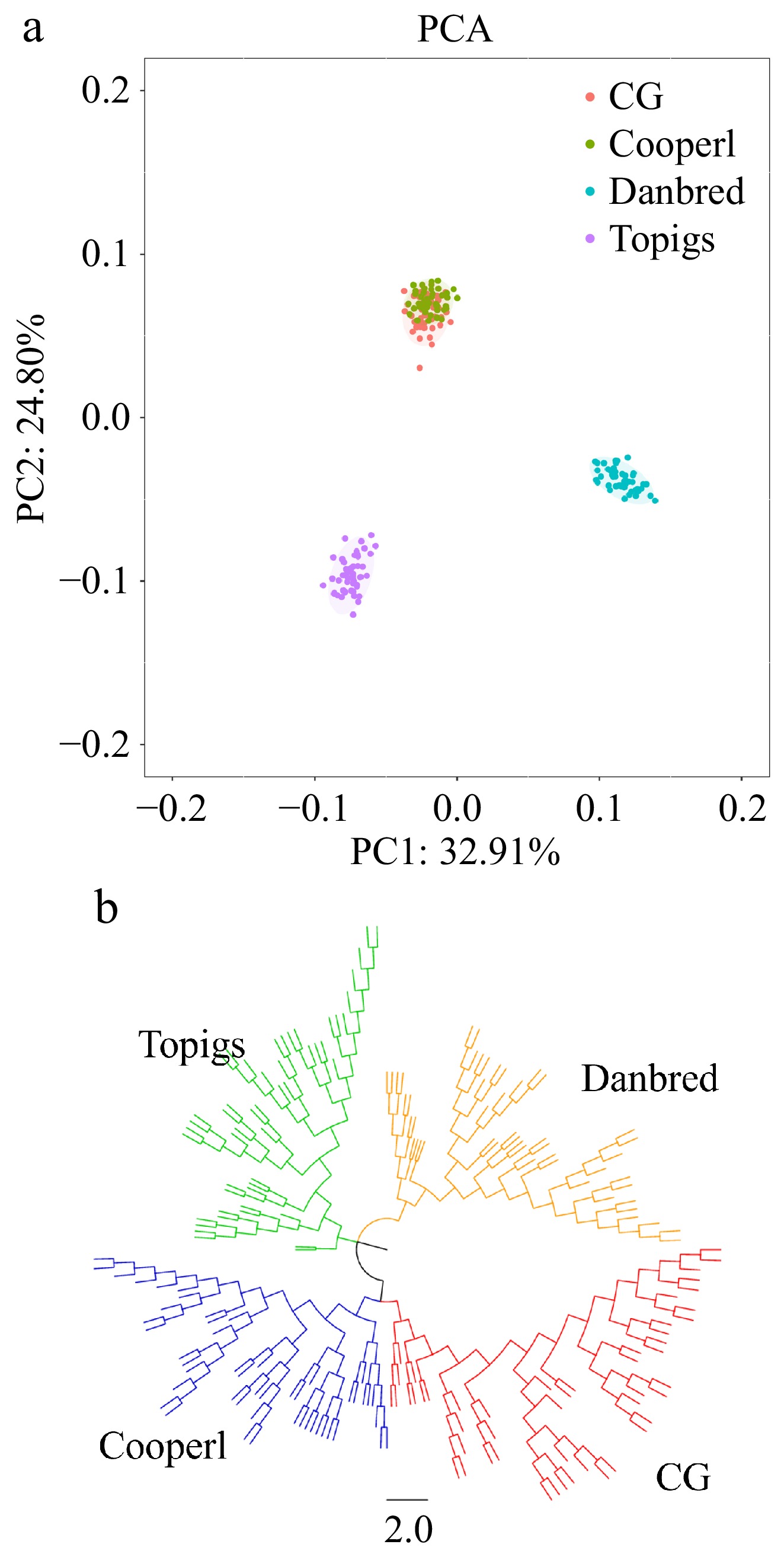
Figure 1.
Analysis of the population structure in four Large White pigs. (a) Principal component analysis. (b) Neighbor-joining tree.
Identification of selection signatures
-
Within this research, four methods: iHS, nSL, CLR, and XP-EHH were employed to identify genomic areas that have undergone recent selection in Large White pigs. Figure 2 presents the SNPs that were determined to be under selection pressure in the CG strain through the conducted tests. In the CG strain, 179,186,252, and 86 candidate genes were identified (Supplementary Table S2) that may be subject to selection according to the CLR, iHS, nSL, and XP-EHH tests, respectively. The results of the four tests conducted on the Cooperl group are displayed in Fig. 3. Within the Cooperl strain, 196,188,275, and 94 candidate genes were found (Supplementary Table S2) as potentially under selection through the CLR, iHS, nSL, and XP-EHH tests, respectively. The Manhattan plot shown in Fig. 4 highlights the discovery of SNPs that may have been influenced by selective pressures within the Topigs strain. These SNPs were evaluated using the CLR, iHS, nSL, and XP-EHH tests. The analysis identified 204, 198, 252, and 113 candidate genes (Supplementary Table S2), respectively, suggesting their potential for being under selection. In this study, genes related to teat number were found by utilizing the Pig QTLdb database (Table 2).
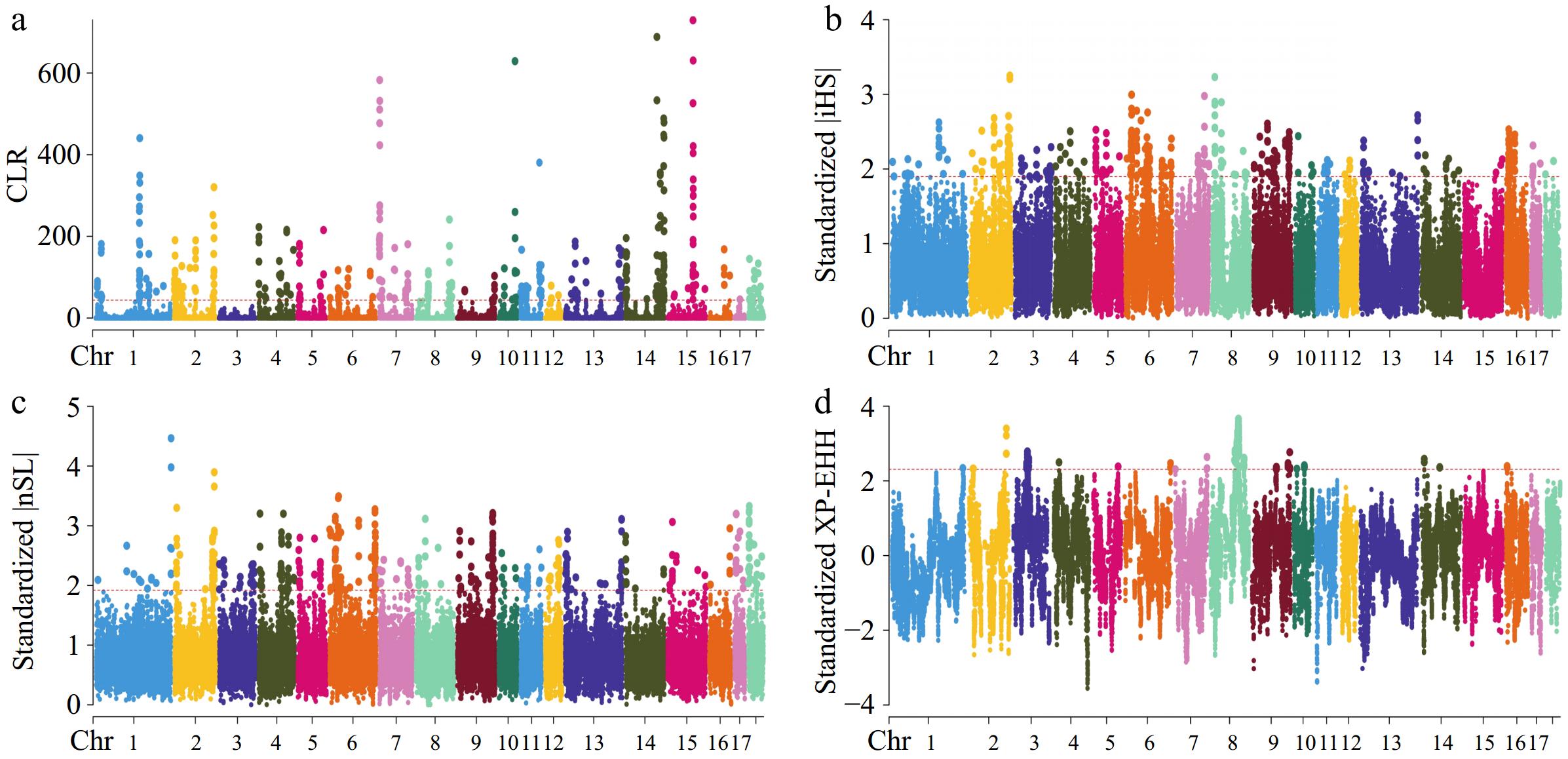
Figure 2.
Manhattan plots for selective sweep analyses of CG strain with the (a) CLR, (b) iHS, (c) nSL, and (d) XP-EHH analyses.
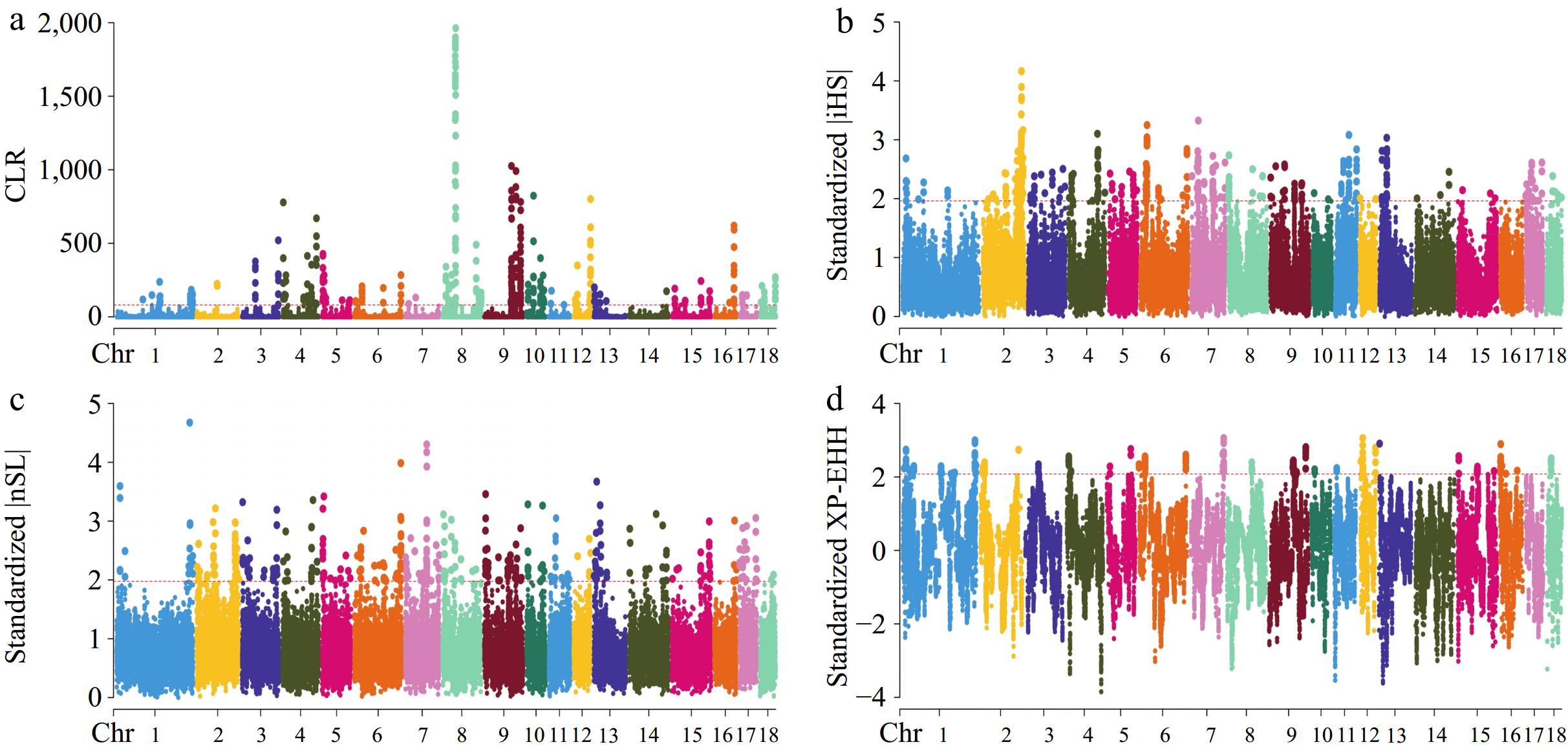
Figure 3.
Manhattan plots for selective sweep analyses of Cooperl strain with the (a) CLR, (b) iHS, (c) nSL, and (d) XP-EHH analyses.
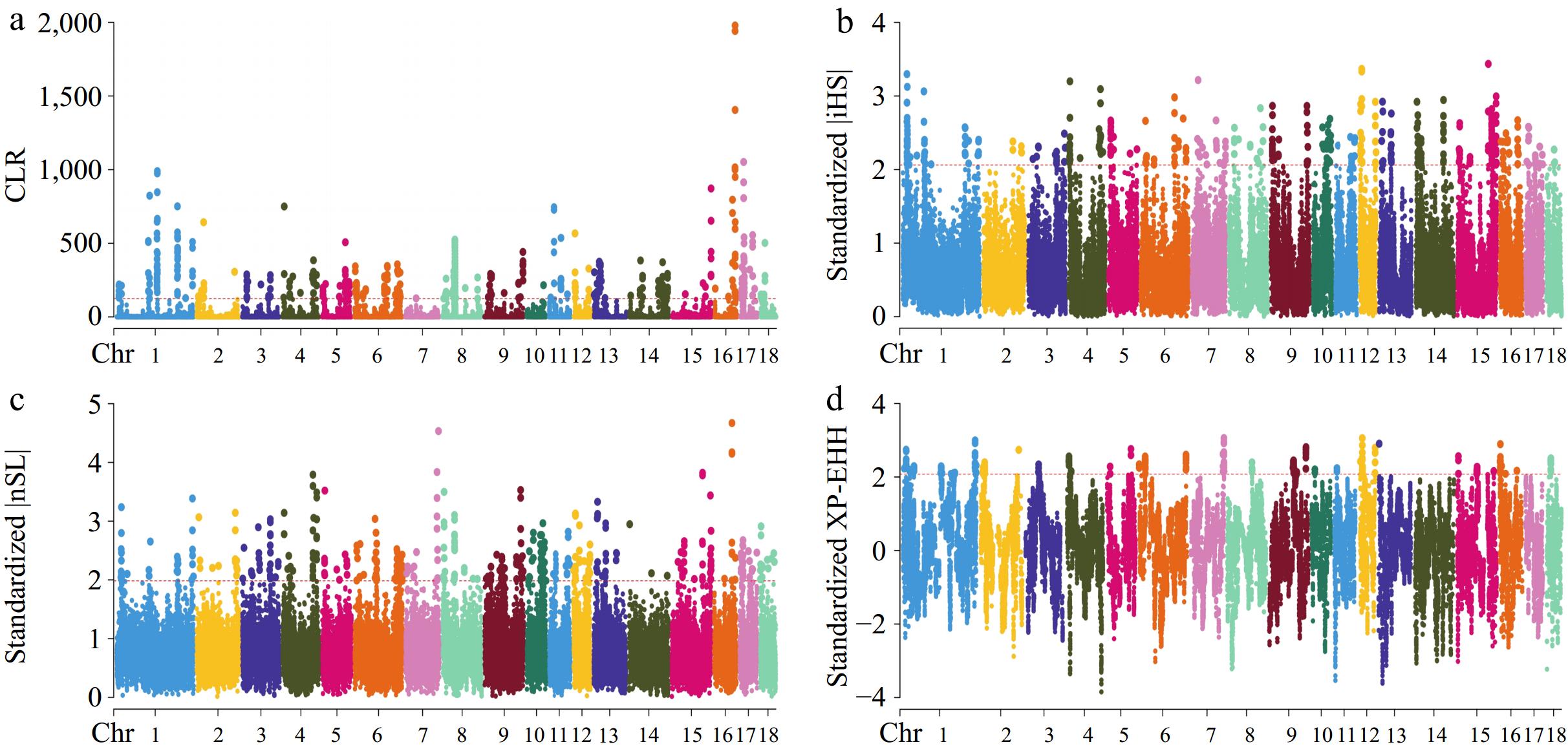
Figure 4.
Manhattan plots for selective sweep analyses of Topigs strain with the (a) CLR, (b) iHS, (c) nSL, and (d) XP-EHH analyses.
Table 2. Potential candidate genes detected using all methods*.
Group Methods Genes Trait CG nSL PAFAH1B1 Teat number UBE2G1 Teat number DNAH12 Teat number PCM1 Teat number PACC1 Teat number iHS CDH13 Teat number CLR JAZF1 Teat number Cooperl CLR ARL4C Teat number RAPGEF2 Teat number Topig nSL DOCK1 Teat number PCM1 Teat number CLR DNAH12 Teat number DOCK1 Teat number PCM1 Teat number * This table is based on the information retrieved in the pig QTLdb database (accessed on 2 April 2024). The CG strain identified four genes (UMODL1, DPP6, ARHGAP26, and COL22A1) (Fig. 5a, Supplementary Table S3) using CLR, nSL, and iHS methods. Additionally, the CG strain detected the presence of four genes (TRPC7, SMIM32, ARMH1, and PROX1) (Fig. 5a, Supplementary Table S3) by simultaneously employing the nSL, iHS, and XP-EHH methods. The Cooperl strain identified three genes (MACROD2, IGSF3, and ZSWIM5) using CLR, nSL, and iHS (Fig. 5b, Supplementary Table S3). Meanwhile, the Cooper strain also identified two genes, TRPC7 and CDH8, using the nSL, iHS, and XP-EHH analysis methods (Fig. 5b, Supplementary Table S3). In the Topigs strain, the analysis using CLR, nSL, and iHS methods revealed seven overlapping genes (TAB2, USP20, FNBP1, NCS1, DYTN, TTC1, and ADRA1B) (Fig. 5c, Supplementary Table S3). Concurrently, by applying the nSL, iHS, and XP-EHH analytical techniques, the Topigs strain identified three genes, ZC3H12D, TAB2, and GRAP2 (Fig. 5c, Supplementary Table S3).
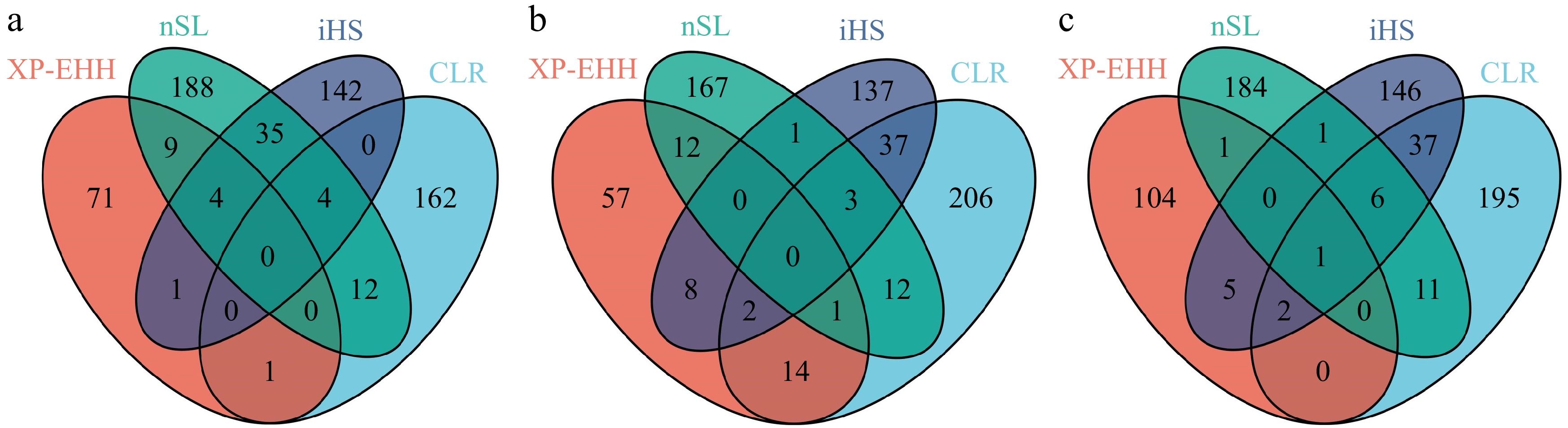
Figure 5.
The Venn diagram illustrates the overlap of genes between four different approaches. (a) CG, (b) Cooperl, (c) Topigs.
To further explore the functions of the candidate genes in pigs, PheWAS analysis was conducted for these genes. Based on GWAS summary statistics, the PheWAS analysis revealed significant correlations between the candidate genes TRPC7 (P = 8.62 × 10−9), DOCK1 (P = 1.81 × 10−8), MRTFA (P = 1.34 × 10−6), and the phenotypic trait of teat number in pigs (Fig. 6). The point plot shows gene-trait associations from gene-based analysis. This result suggests that these three genes are likely candidate genes affecting the trait of teat number in pigs.
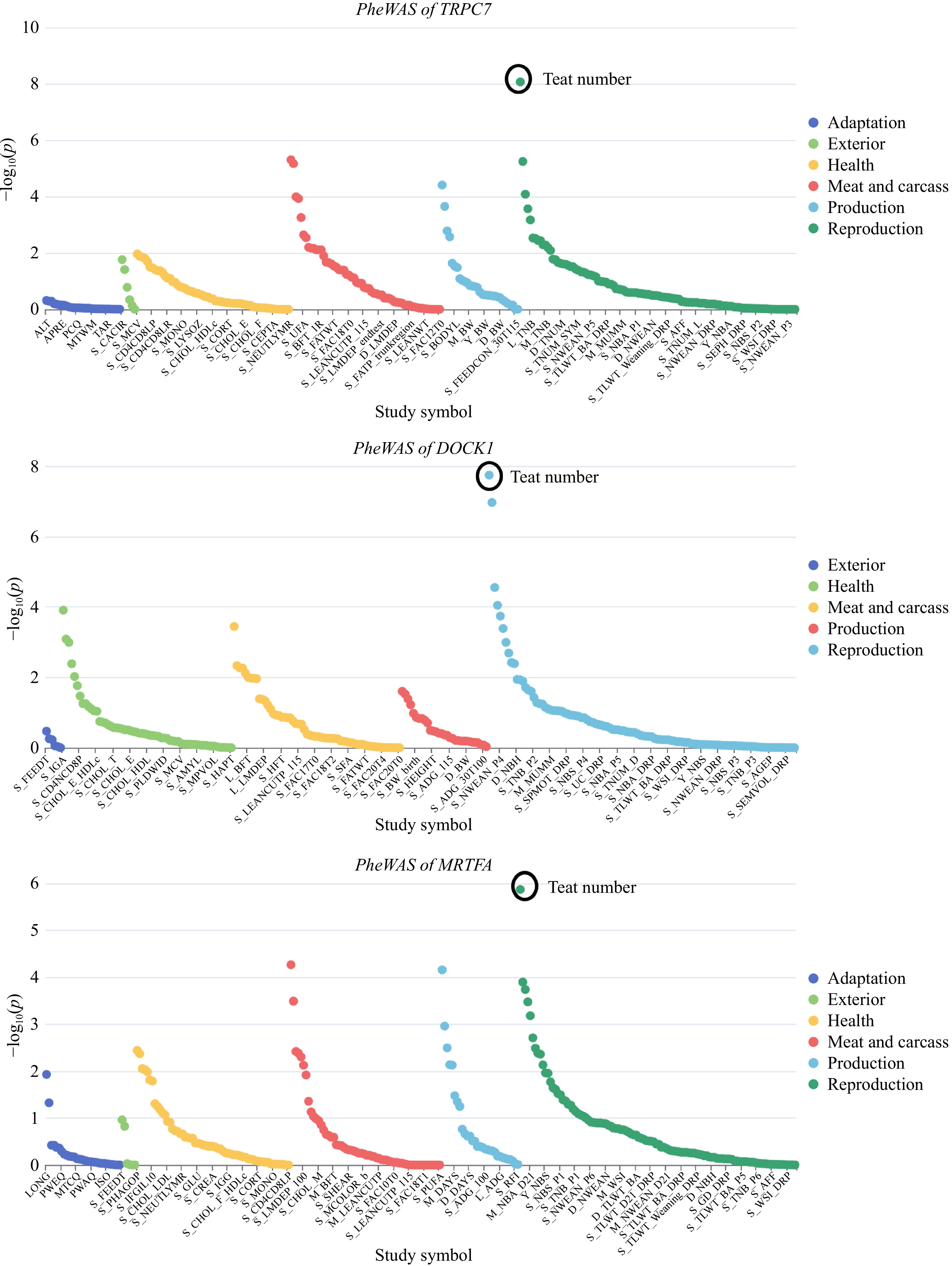
Figure 6.
Results of the PheWAS analysis based on the identified candidate genes.
For each population, GO functional annotation and KEGG pathway analysis were performed on the genes identified through all methods. In the CG strain of Large White pigs, GO and KEGG analyses found 35 Gene Ontology (GO) terms and 11 pathways, including those for animal organ morphogenesis (GO:0009887, p = 0.018), PI3K-Akt signaling pathway[27] (KEGG: ssc04151, p = 0.034), and Jak-STAT signaling pathway[28] (KEGG: ssc04630, p = 0.038) (Supplementary Table S4). The Cooperl strain showed enrichment for the Wnt signaling pathway (KEGG: ssc04310, p = 0.01) (Supplementary Table S4). This pathway plays a crucial role in the developmental processes of mammary gland differentiation[29]. Unfortunately, the pathways related to mammary gland development did not show significant enrichment within the Topigs Large White pig population.
-
The Large White pig, also known as the Yorkshire pig, is a prominent breed of domestic pig originating from England. Selective breeding by different companies has led to the diversification of this breed into a range of unique phenotypes and genetic strains. This deliberate breeding has significantly influenced the genetic makeup of the Large White pig, particularly affecting loci regulated by major genes that determine key traits of the breed[30−32]. By conducting a comparative analysis of different strains of Large White pigs, we have assessed their genetic diversity, population structure, and phylogenetic relationships. Artificial selection might have etched a distinctive imprint into the genomic sequences of individuals in the population[23,33,34]. In the present investigation, different highly sensitive detection techniques were implemented to detect recent selection signatures[35]. The main objective of this research was to pinpoint the potential genes linked to the higher number of teats in Large White pigs as a result of domestication and selective breeding.
The nucleotide diversity in the Cooperl strain of Large White pigs (mean θπ = 0.00145) was found to be similar to that of the CG strain (mean θπ = 0.00146) but greater than that of the Topigs strain of Large White pigs (mean θπ = 0.00138). The present analysis discovered characteristics of the Cooperl strain within Large White pigs exhibited levels of expected and observed heterozygosity comparable to those of the CG strain. This similarity can be attributed to their similar genetic background. Artificial selection can cause a rise in linkage disequilibrium within the population[36,37]. It was observed that the CG strain of Large White pigs exhibited a rapid decline in LD compared to the Cooperl and Topigs strains.
The results of PCA and NJ-tree analysis demonstrate the genetic relationships and differentiation between strains of Large White pigs. In both the PCA and NJ-tree, the CG and Cooperl strains cluster together, which might be associated with their similar genetic backgrounds.
In this study, several genes have been identified that have been reported to influence the count of nipples in pigs. Specifically, PAFAH1B1, RAPGEF2, JAZF1, and ARL4C are identified as influencing teat number in Large White pigs[38−40]. In Landrace pigs, UBE2G1, ARL4C, and CDH13 are associated with teat count[39,41], while DOCK1 affects teat number in Duroc pigs[7]. Additionally, DNAH12, PCM1, and PACC1 are found to influence teat number in Qingping pigs[42]. Notably, this study has identified PCM1 and DOCK1 as potential candidate genes that may affect teat number in Large White pigs.
Through literature review, it was determined that specific genes within potentially selected regions are associated with essential functional traits. In the CG strain, the DPP6 gene is a significant contributor to lactation performance in dairy cows and is a potential candidate for improving milk production traits[43,44]. COL22A1 was identified as a significant candidate gene for milk yield, fat percentage, and protein yield through GWAS, conducted in Chinese Holstein cattle[45]. The significant up-regulation of TRPC7 channels during lactation indicates that these channels are likely essential for facilitating the transport of calcium ions (Ca2+) across mammary epithelial cells[46]. The PROX1 gene is critical for regulating the activity of mammary basal progenitor cells, acting as a negative regulator of mammary stem/progenitor function[47]. Disruption of ROR2 impacts the branching and differentiation of mammary epithelium[48,49]. ABCG1 is essential for modulating lipid transport within the mammary gland, exhibiting distinct physiological functions that change with the gland's functional stage[50].
In the Cooperl strain, the TRPC7 gene was also identified. Moreover, NR3C1 was found to play a pivotal role in regulating cell proliferation during mammary lobuloalveolar development[51]. PPARA may boost the production of monounsaturated fatty acids in goat mammary epithelial cells by modulating the mRNA levels of genes involved in fatty acid synthesis, oxidation, transportation, and triacylglycerol synthesis[52].
In the Topigs strain, several genes have been identified as playing important roles. Notably, DOCK1 plays a significant role in mammary gland involution, and DOCK1-null mammary glands fail to activate Stat3 properly[53]. MRTFA is vital for the proliferation and formation of mammary acini from luminal epithelial cells[54,55]. The TAB2[56] gene is a potential candidate for influencing ectodermal development and is likely to be involved in the early stages of mammary gland development.
CG and Cooperl are well-known French Large White pig breeds. Using iHS and nSL methods, we identified overlapping genes in CG and Cooperl Large White pigs, including TRPC7, SPOCK1, NR3C1, and ARHGAP26. The TRPC7 gene may serve as a critical component in facilitating the transport of calcium ions (Ca2+) across mammary epithelial cells. The NR3C1 gene is essential for regulating cell proliferation during the development of mammary lobules and alveoli. Phenome-wide association studies (PheWAS) is an analytical method that explores the associations between all phenotypes and a certain exposure within the entire phenome. PigBiobank stands as the most extensive and comprehensive database to date, amassing genotype and phenotype data from 71,885 pigs across more than 100 breeds. We leveraged the PheWAS module within this platform for the present analyses and discovered significant associations between the genes TRPC7, MRTFA, and DOCK1 and the trait of teat number. We hypothesize that the TRPC7 gene is a significant candidate influencing teat number in French Large White pigs. Similarly, we propose that the MRTFA and DOCK1 genes are important candidates influencing teat number in Topigs Large White pigs.
Significant enrichment of 18, 11, and 14 genes was identified in the PI3K-Akt, Jak-STAT, and Wnt signaling pathways, respectively. The PI3K/Akt signaling pathway is indispensable for managing key cellular processes such as proliferation, differentiation, and apoptosis in mammary epithelial cells[57]. The JAK-STAT pathway is pivotal in signaling cascades within mammary epithelial tissue, significantly contributing to lactation and mammary gland development[28,58]. The Wnt signaling pathway plays a critical role at every phase of embryonic mammary gland development, exerting canonical influence on both mammary epithelial cells and their associated mesenchyme[59].
-
In this study, it was confirmed that the different strains of Large White pigs have developed their distinct genetic structure features, and several candidate genes were identified, including DPP6, COL22A1, TRPC7, PROX1, ROR2, ABCG1, PPARA, NR3C1, DOCK1, PCM1, and MRTFA, that may exert potential influence on the development of the mammary gland. Among these, DPP6, COL22A1, TRPC7, PROX1, ROR2, ABCG1, PPARA, NR3C1, and MRTFA have been newly identified as genes of interest. The present research sheds new light on the distinct genetic characteristics and evolutionary relationships among various strains of Large White pigs. This study offers novel insights into the genetic mechanisms that govern teat number phenotypes in selected populations.
This work was supported by the Jiangsu Seed Industry Revitalization Project (JBGS[2021]024; JBGS[2021]098), STI 2030 - Major Projects (2023ZD04045), Postdoctoral Fellowship Program of CPSF under Grant Number 2024M751448, and the Jiangsu Funding Program for Excellent Postdoctoral Talent.
-
All animal experiments were performed according to the Guidelines for the Care and Use of Laboratory Animals prepared by the Institutional Animal Welfare and Ethics Committee of Nanjing Agricultural University, Nanjing, China [Certification No.NJAULLSC2022069], approval date: 2022/7/16. The research followed the 'Replacement, Reduction, and Refinement' principles to minimize harm to animals. This article provides details on the housing conditions, care, and pain management for the animals, ensuring that the impact on the animals is minimized during the experiment.
-
The authors confirm contribution to the paper as follows: study conception and design: Huang R, Li P, Zhao Q, Su G; data collection: Cai W, Yu J, Zhou J; analysis and interpretation of results: Ma J, Liu C, Liu Q; draft manuscript preparation: Ma J, Ai X. All authors reviewed the results and approved the final version of the manuscript.
-
The data that support the findings of this study are available in the figshare repository (https://doi.org/10.6084/m9.figshare.26049181.v1).
-
The authors declare that they have no conflict of interest. Ruihuang Huang is the Editorial Board member of Animal Advances who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research groups.
- Supplementary Table S1 Summary of data and statistical analysis in this study.
- Supplementary Table S2 Candidate regions and genes detected using all methods.
- Supplementary Table S3 Overlapping gene.
- Supplementary Table S4 Enrichment analysis of the candidate genes within the CG, Cooperl, and Topig groups was performed using the iHS, CLR, nSL, and XP-EHH methods, along with the GO and KEGG databases.
- Supplementary Fig. S1 The linkage disequilibrium (LD) decay analysis.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press on behalf of Nanjing Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Ma J, Liu C, Cai W, Liu Q, Yu J, et al. 2025. Genome-wide selection signature analysis identified new candidate genes for the teat number in three distinct strains of Large White pigs. Animal Advances 2: e004 doi: 10.48130/animadv-0025-0002 

Genome-wide selection signature analysis identified new candidate genes for the teat number in three distinct strains of Large White pigs
- Received: 20 November 2024
- Revised: 03 January 2025
- Accepted: 08 January 2025
- Published online: 24 February 2025
Abstract: The number of teats is a critical reproductive trait in sows, exerting a direct impact on the survival rate of weaned piglets. Existing studies on selection signals associated with teat numbers in different strains of Large White pigs are scarce. We performed selection signal detection on three distinct strains of Large White pigs with 16 or more nipples: the French CG (Choice Genetics), the French Cooperl, and the Dutch Topigs. Our goal was to identify pivotal candidate genes contributing to the higher nipple count in Large White pigs. A comprehensive analysis, including CLR (Composite Likelihood Ratio), iHS (integrated haplotype score), and nSL (number of segregating sites by length) methods, alongside XP-EHH (Cross-population Extended Haplotype Homozygosity), was undertaken to identify genetic positive selection signatures in Large White pigs. Through our analysis, we identified six key candidate genes in the CG Large White pigs: DPP6, COL22A1, TRPC7, PROX1, ROR2, and ABCG1. In the Cooperl Large White pigs, the key candidate genes were PPARA, TRPC7, and NR3C1. In the Topigs Large White pigs, the key candidate genes were DOCK1, PCM1, and MRTFA. Among the aforementioned genes, PCM1 and DOCK1 have been identified as correlated with the number of teats in pigs, while TRPC7, DOCK1, and MRTFA have shown a significant link with teat number based on PheWAS (Phenome-wide association study) analysis. Our findings show that multiple genes associated with teat development in Large White pigs have experienced significant selection pressure throughout the breeding program. These results deepen our understanding of the progress in breeding Large White pigs and provide a valuable framework for their continued selection.
-
Key words:
- Genetic diversity /
- Selective signature /
- Teat number /
- Pig