










-
Feed efficiency, which refers to the relative ability of animals to convert dietary nutrients into growth, is one of the most critical economic traits in aquaculture[1,2]. The cost of feed is the most significant expenditure in aquaculture, comprising 30% to 70% of the total production costs, and enhancing feed efficiency is pivotal for cost reduction[3]. For instance, in the case of Atlantic salmon, feed expenses constitute 47% of the total production costs, and it is estimated that a mere 2% to 5% improvement in feed efficiency could translate into annual savings ranging from
${\$} $ ${\$} $ The feed conversion ratio (FCR) and feed efficiency ratio (FER) are standard indices for assessing feed efficiency in livestock[8]. However, the FCR and FER, being ratio traits (FCR = FI × BWG−1; FER = BWG × FI−1), do not simply reflect genetic variation in the livestock's weight change and feed consumption, or the potential genetic variation of both, leading to suboptimal selection responses when used directly for breeding purposes[3]. Residual feed intake (RFI) has emerged as a preferred indicator for evaluating feed efficiency in animal breeding programs[9]. RFI is defined as the difference between the actual feed intake and the amount of feed predicted to be required for maintaining normal growth and development based on individual body weight and average daily weight gain; the lower an individual's RFI, the less feed is consumed for maintaining normal growth and development, indicating a higher feed utilization efficiency[10]. Since RFI is independent of the production traits used to estimate it and represents the inherent differences in feed efficiency caused by individual basal metabolic processes, selection based on RFI typically yields better results than selection based on FCR and FER[11]. RFI has been applied in the breeding of fish species such as Oncorhynchus mykiss, Oreochromis niloticus[12], and Dicentrarchus labrax[13].
Alternative splicing (AS) is a vital post-transcriptional modification process in eukaryotes, enabling the generation of diverse transcripts from a single gene through the selective inclusion or exclusion of exons and introns[14]. Initially identified in mammals in 1977, this phenomenon has been confirmed to be prevalent across eukaryotic species, with an estimated 90%–95% of human genes undergoing AS[15,16]. The transcripts produced through AS can either encode distinct functional proteins, thereby enriching the diversity of the eukaryotic proteome, or lack coding potential and are subject to degradation via nonsense-mediated decay (NMD), thereby indirectly modulating gene transcription levels[17]. The interplay between AS and differential expression of genes, both of which are pivotal regulatory mechanisms in eukaryotic biology, remains a contentious issue: some views posit a close relationship, with AS being inherently co-transcriptional and expanding the repertoire of gene expression patterns; others argue for their relative independence, each modulating distinct genes and pathways to exert complementary yet contrasting functions[14,18]. Illumina RNA sequencing has enabled the large-scale identification of AS events and differential gene expression. However, due to sequencing read length limitations, accurate identification and quantification of gene transcripts have been challenging. In addition, the inclusion of transcripts degraded through the NMD pathway in quantification processes has obscured many genes that genuinely exhibit differential expression, diminishing the accuracy of analyses pertaining to the relationship between AS and gene expression[19]. With the development of long-read transcriptome sequencing technologies, various full-length transcripts formed by genes through AS can be completely identified. Functional prediction can distinguish between functional transcripts that encode proteins and nonfunctional transcripts without encoding ability. By integrating short-read transcriptome data, accurate quantification of each transcript is achievable. This technological advancement is expected to facilitate more nuanced and in-depth investigations into the intricate relationship between AS and differential gene expression in the future[18].
Single nucleotide polymorphisms (SNPs) are the most abundant type of sequence variation in eukaryotic genomes, which are characterized by high frequency, co-dominant inheritance, adaptability to high-throughput genotyping, and relatively lower mutation rates compared with microsatellites, making SNPs the preferred markers in population genetics and genomic mapping studies[20]. SNPs can influence AS, with the potential to modify splice sites or splicing efficiency within splicing regions[21]. In the selfing morph of Capsella rubella, two SNPs in the CYP724A1 gene increase intron removal efficiency, leading to increased CYP724A1 expression and consequently reduced petal size, which is more adapted to selfing[22]. There have been numerous reports on SNPs in fish, mainly focusing on germplasm surveys and genetic breeding, but the link between fish SNPs and AS has rarely been reported[23].
The common carp (Cyprinus carpio) is an extremely important economic fish species in aquaculture and is currently farmed in over 100 countries worldwide, accounting for 10% of the annual global freshwater aquaculture production[24]. Jian carp (Cyprinus carpio var. jian), as the first artificially bred new variety of carp in China, is characterized by its high-quality meat, high nutritional value, gentle temperament, robust disease resistance, and broad adaptability[25]. In this study, we integrated PacBio full-length transcriptome sequencing with Illumina transcriptome data to identify AS transcripts and quantify transcripts in Jian carp. Additionally, we employed whole-genome resequencing (WGR) technology to explore the link between SNPs and AS in Jian carp. The objective of this research was to refine the genetic mechanisms affecting feed efficiency in Jian carp, providing a theoretical foundation for further development of its breeding potential and enhancement of its feed efficiency.
-
The experimental Jian carp were obtained from the Freshwater Fisheries Research Center at the Chinese Academy of Fisheries Sciences in Wuxi, China. Selected for their robust reproductive condition, these carp were allowed to spawn naturally during the breeding season. The resulting offspring were reared in ponds to a target weight of about 10 g before being transferred to an indoor recirculating aquaculture system. When the fish reached a uniform weight of approximately 45.5 ± 15.4 g, a total of 168 individuals were chosen for the study and allocated to individual sections within 42 tanks of 300 L, with each tank containing four fish to maintain isolated living environments (separated by partitions). Throughout the study, the water within the tanks was perpetually recirculated, and the water temperature was precisely regulated at 25 ± 2 °C. After a 2-week acclimatization period, the fish were engaged in an 8-week trial. They were fed on large, uniformly sized commercial pellets composed of 31% crude protein and 3.5% crude fat, sourced from Ningbo Tech-Bank Co. Ltd, Ningbo, China. Feed dispensed and remaining amounts were meticulously tracked to accurately calculate individual feed intake (FI). The growth performance of each fish was assessed by recording their weights at the beginning and end of the experiment.
RFI was calculated with the following linear regression model constructed in our previous research[26]:
${\rm{ RFI}}={\rm{DFI}}-\beta _{ \mathrm{0}} -{\beta }_{ \mathrm{1}} \times {\rm{MW}}^{ \mathrm{0.8}}-{\beta }_{ \mathrm{2}} \times {\rm{ADG}} $ In this model, DFI is the daily feed intake (g), β0 is the regression intercept, β1 is the partial regression coefficient of metabolic weight, MW0.8 is the metabolic weight (g), ADG is the average daily weight gain (g/day), and β2 is the partial regression coefficient of ADG. RFI, which signifies the difference between an individual's actual FI and the FI predicted by the model, was calculated using SPSS 27.0 software.
The experimental fish were grouped according to their RFI, with the 30 individuals exhibiting the highest and lowest RFI values being assigned to the low residual feed intake (LRFI) and high residual feed intake (HRFI) groups. After inducing anesthesia with MS-222 at 100 mg/L, liver and blood samples were collected and instantly frozen in liquid nitrogen for subsequent RNA and DNA extraction to prepare sequencing libraries.
Library preparation and sequencing
-
From both the LRFI and HRFI groups, six liver samples were collected to extract total RNA. Subsequently, cDNA libraries were constructed and subjected to Illumina sequencing. The specific experimental procedures followed the research previously conducted in our laboratory[26].
Six liver samples were pooled to form a single sample in the LRFI and HRFI groups for PacBio isoform sequencing (Iso-Seq). The Iso-Seq library preparation adhered to the standard protocol, employing the Clontech SMARTer PCR cDNA Synthesis Kit in conjunction with the BluePippin Size Selection System, following Pacific Biosciences' guidelines (PN 100-092-800-03).
For WGR, 60 blood samples in total from the LRFI and HRFI groups were processed. Genomic DNA of superior quality was isolated from these samples via the phenol–chloroform extraction method. Agarose gel electrophoresis and Agilent 5400 assays were used to assess the concentration, integrity, and purity of the DNA samples. The Covaris sonicator was applied to fragment the purified DNA, and T4 DNA polymerase was used to create blunt ends. An 'A' overhang was added to the 3' ends of the DNA fragments, followed by the ligation of adapters to the phosphorylated ends. The targeted fragments were amplified through polymerase chain reaction polymerase chain reaction (PCR) to finalize construction of the DNA library. These libraries underwent sequencing on the Illumina NovaSeq 6000 platform using 150 paired-end (PE) cycles to achieve high-fidelity sequencing results.
Iso-Seq data filtering
-
SMRTlink version 11.0 was employed to process the sequence data, generating circular consensus sequences (CCS) from BAM files of subreads with the following parameters: a minimum length of 10 bases, 3 minimum passes, a minimum predicted accuracy of 0.99, and a maximum length of 50,000 bases. The resulting CCS.BAM files were then utilized to distinguish between full-length and non-full-length reads using the pbclassify.py script with settings to disregard polyA and a minimum sequence length of 10 bases. The resulting fasta files, both full-length and non-full-length, were subjected to isoform-level clustering (ICE), followed by final polishing with Arrow, applying the following parameters: hq_quiver_min_accuracy, 0.99; bin_by_primer, false; bin_size_kb, 1; qv_trim_5p, 100; qv_trim_3p, 30. To correct any remaining nucleotide errors in the consensus reads, Illumina RNAseq data were integrated using LoRDEC software[27].
Transcript identification
-
Consensus reads were aligned to the genome utilizing GMAP[28] with the following specific parameters: no chimeras; cross-species; expand-offsets, 1; B, 5; K, 50,000; f, samse; n, 1. The TAPIS pipeline[29] was employed for the analysis of gene structure. For gene and transcript identification, the BAM files and gff/gtf format genome annotation files generated by GMAP were utilized. Visualization of the gene structures was accomplished using the gggenes R package. Subsequently, AS and alternative polyadenylation events were analyzed. Fusion transcripts, defined as those mapping across two or more distant genes, were confirmed by at least two Illumina reads.
The functions of unmapped transcripts and those from novel genes were annotated with reference to several databases: NR (NCBI non-redundant protein sequences), NT (NCBI non-redundant nucleotide sequences), Pfam (protein family database), KOG/COG (Clusters of Orthologous Groups of proteins), Swiss-Prot (a manually curated and peer-reviewed protein sequence database), KO [Kyoto Encyclopedia of Genes and Genomes (KEGG) Ortholog database], and GO (Gene Ontology).
BLAST[30] was utilized for searching in the NT database with an e-value threshold of 1e-10. Diamond BLASTX[31] was employed for sequence similarity searching in the NR, KOG, Swiss-Prot, and KEGG databases, also with an e-value threshold of 1e-10. Pfam database analysis was conducted using Hmmscan[32] with the same stringent e-value criterion.
Transcript expression analysis
-
Illumina reads, following the processing pipeline established by our laboratory's prior study[26], underwent quality control, filtering, and alignment to the reference genome. The resulting alignment files were then used to calculate the transcripts' expression abundances. Cuffdiff version 2.1.1 was applied to determine the fragments per kilobase of exon per million fragments mapped (FPKM) for all transcripts across samples. Cuffdiff employs statistical methods based on the negative binomial distribution to assess differential expression in digital transcripts or gene expression data. Transcripts with an adjusted p-value of less than 0.05 were identified as differentially expressed transcripts (DETs). The expression abundance heatmap for all transcripts was created with the pheatmap R package. Volcano plots for DETs and bar charts for the expression abundance of AS transcripts were generated using the ggplot2 R package.
Quantitative real-time PCR
-
To validate the reliability of the transcriptomic data, the expression levels of 12 DETs were examined using quantitative real-time PCR (qRT-PCR), with the corresponding primers listed in Table 1. Additionally, to confirm the differential expression of transcripts of the acsl4a gene between the HRFI and LRFI groups, primers targeting acsl4a_all (encompassing both acsl4a_a and acsl4a_b) and acsl4a_b were specifically designed for qRT-PCR (Table 1). Liver samples from the HRFI and LRFI groups with three biological replicates per group were utilized for qRT-PCR validation. The experimental workflow for qRT-PCR was performed in accordance with the protocol established in our laboratory's prior studies[26].
Table 1. Primer sequences of qRT-PCR.
Transcript ID Primer Sequence (5′–3′) Amplification size (bp) actn4_novel01 F TCAGTCTGGGTTACGACG 227 R CTCACGCCTCAACTCCTC LOC109095800_novel01 F GGCGATAATGTGGACTTG 223 R CCATCTAACTGCCAAAGC LOC109100422_novel05 F ATACGAGGATCGCAAGAG 203 R GTTTCCGTGGAGGAGTAG LOC109105659_novel03 F CGGCATCTACGGCGTGTT 257 R CCTTGCGTTTGCGTGTCT LOC109107915_novel04 F TGGCTTCCAAATCCTTAG 262 R AGGTGCGGAGTCTGTGAT XM_042730762.1 F ACTGGTTGGGACTAAAGG 245 R GCTGCGGTATTTCTTACA XM_042742207.1 F GCTTCCGATTGGTCAGTT 219 R CGTGTCCATTAGGAGGTTT XM_042745279.1 F CTGATGAAATGATGACCCTA 255 R CAGTAATGCTGGAGACCTT XM_042745964.1 F GCTCTGTATCGCCCACTC 275 R CGTCACGGCTCTTCTTGT XM_042765333.1 F ACCCACTAAAGACTCCACA 190 R ACCCGATAGACACTGACCT XM_042771159.1 F AATGGGCGATTCAGTAGCA 243 R GGATGGTGGTGATGGTGG XM_042777294.1 F CTCGTGGCAAGCTGTCTA 228 R GGAACTTAGGGTTACTCTGG acsl4a_all F CGTGGACAGGAAGAGCAT 142 R AACAGCGAGGTCTGATGG acsl4a_b F TTGTTTCTTCGTTGCACATT 196 R TGTACTGGGAACAAAAGAAC β-actin F CGTGATGGACTCTGGTGATG 162 R TCGGCTGTGGTGGTGAAG GO and KEGG enrichment analysis
-
Genes exhibiting AS and possessing DETs were filtered for GO and KEGG enrichment analysis. GO enrichment analysis was conducted using the GOseq R package, which accounts for gene length bias. GO terms with a corrected p-value of less than 0.05 were identified as significantly enriched. KEGG serves as a comprehensive database resource that elucidates the high-level functions and utilities of biological systems, including cells, organisms, and ecosystems, on the basis of molecular-level information, particularly from large-scale molecular datasets derived from genome sequencing and other high-throughput experimental technologies. For testing the statistical enrichment of genes within KEGG pathways, KOBAS software[33] was employed. Visualization of the GO and KEGG enrichment results was achieved using the ggplot2 R package.
Sequence analysis of genes
-
The nucleotide and deduced amino acid sequences of the genes were examined utilizing Tbtools[34]. Protein structures and functional domains were predicted with the SMART (
http://smart.embl-heidelberg.de/ ). Three-dimensional protein structure models were constructed using SWISSMODEL (https://swissmodel.expasy.org/ ) and visualized with Swiss-PdbViewer.Data processing of WGR
-
Quality control and alignment of WGR data, as well as the subsequent identification and annotation of SNPs, were performed according to the protocols described in previous studies conducted in our laboratory[35]. The threshold parameters for calling SNPs were configured as follows: minor allele frequency (MAF) = 0.05; max-missing rate = 0.7; minimum read depth (minDP) = 4; maximum read depth (maxDP) = 1,000; minimum quality score (minQ) = 30; minimum genotype quality (minGQ) = 80.
-
In this study, a total of 23,997 and 23,938 AS events were detected in the LRFI and HRFI groups of Jian carp, respectively. Upon identification and classification of these events, the types of AS in both groups were ranked by their proportion from highest to lowest as follows: alternative 3' splice site (A3) > skipped exon (SE) > alternative first exon (AF) > alternative 5' splice site (A5) > retained intron (RI) > alternative last exon (AL) > mutually exclusive exons (MX). No significant differences were observed between the groups (Supplementary Fig. S1).
Transcript expression patterns
-
The expression abundance of all transcripts in the liver samples from 12 carp in total from the LRFI and HRFI groups was calculated, resulting in the identification of 70,422 transcripts with detectable expression abundance. Cluster analysis based on the expression abundance of all transcripts across the 12 carp revealed that, except for one individual from the LRFI group, the remaining 11 individuals tended to cluster with their corresponding group members (Fig. 1a), indicating significant differences in the overall transcript expression patterns between individuals of the LRFI and HRFI groups. In total, 200 differentially expressed transcripts (DETs) were identified with Cuffdiff software, with 92 DETs showing increased expression and 108 DETs showing decreased expression in the LRFI group relative to the HRFI group (Fig. 1b).
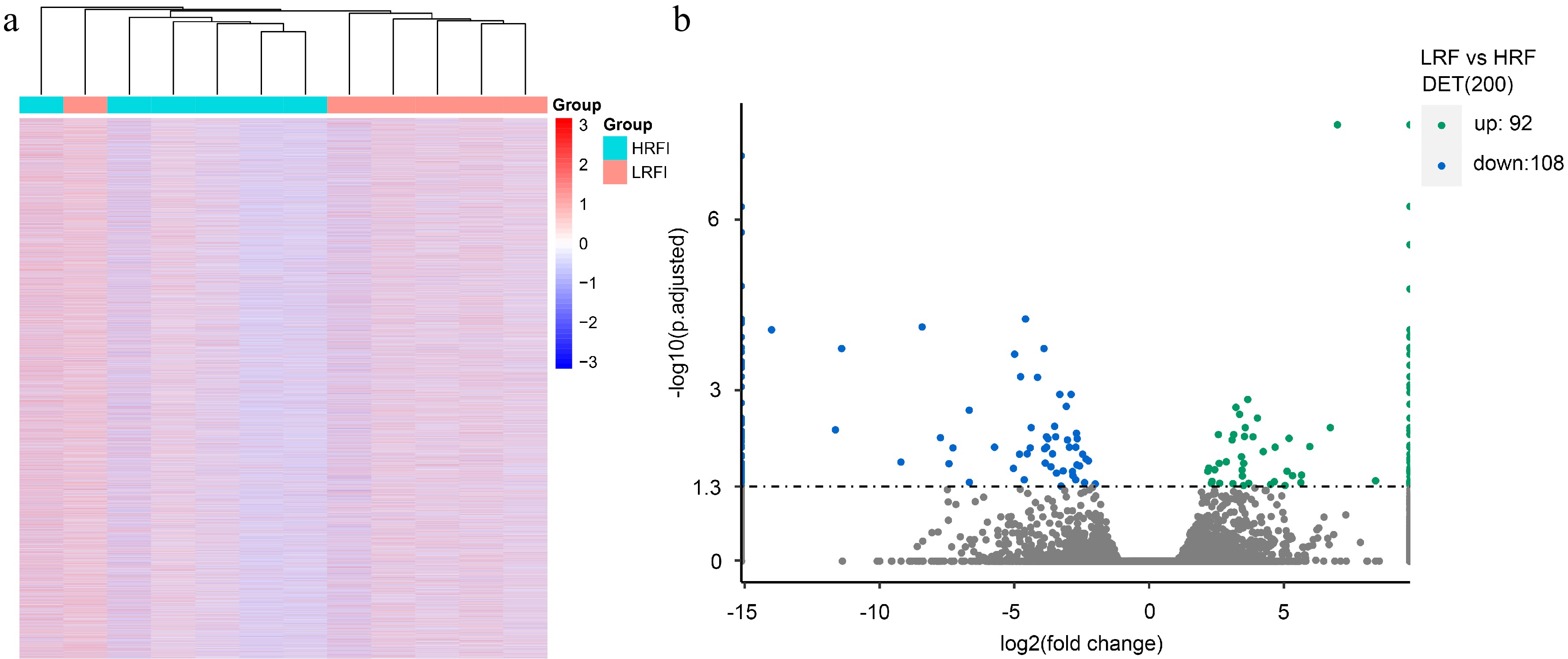
Figure 1.
The expression profiles of all transcripts from the low residual feed intake (LRFI) and high residual feed intake (HRFI) groups of Jian carp. (a) Heatmap of the transcripts' expression. (b) Volcano plot of the transcripts' expression.
GO and KEGG enrichment analysis
-
Among the 200 DETs, 71 transcripts were considered to be AS transcripts, originating from 64 genes. These genes, referred to as differential alternative splicing genes (DASGs), were subjected to GO and KEGG enrichment analysis. In the GO enrichment analysis, DASGs were primarily involved in processes such as polysaccharide binding, translational elongation, negative regulation of endopeptidase activity, and endopeptidase inhibitor activity. Additionally, a considerable number of genes were enriched in terms such as the extracellular region (10), extracellular space (10), and protein binding (9), although the scores of fold enrichment for these terms were relatively low (Fig. 2). In the KEGG enrichment analysis, DASGs were enriched in pathways such as ferroptosis, the HIF-1 signaling pathway, and systemic lupus erythematosus with a lower adjusted p-value (Fig. 3). Metabolic pathways closely related to feed efficiency, such as lysine degradation and fatty acid degradation, were also enriched, suggesting a potential association between these pathways and the differences in feed efficiency among individuals of Jian carp.
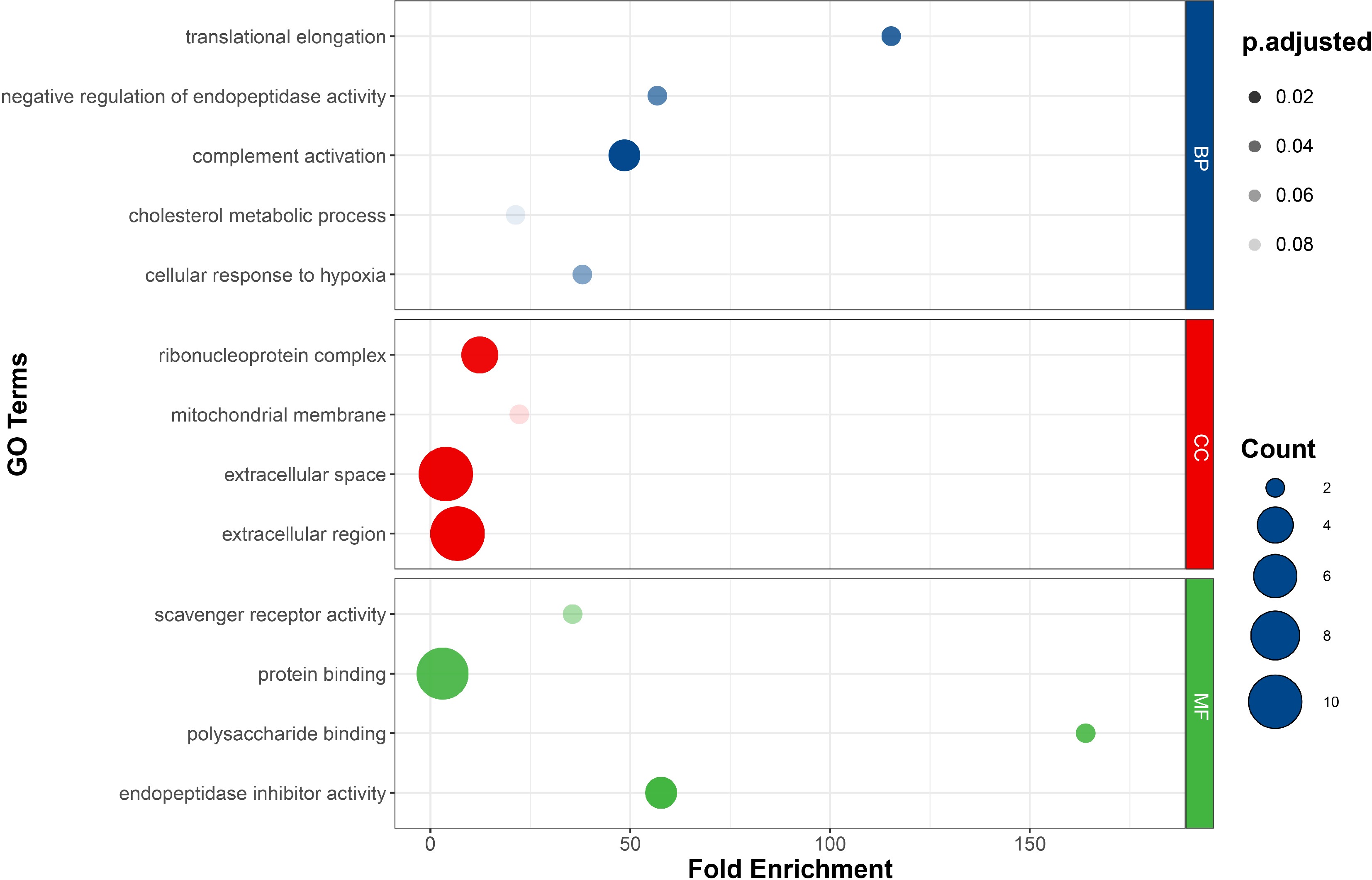
Figure 2.
The GO signaling pathway annotation of differential alternative splicing genes (DASGs).
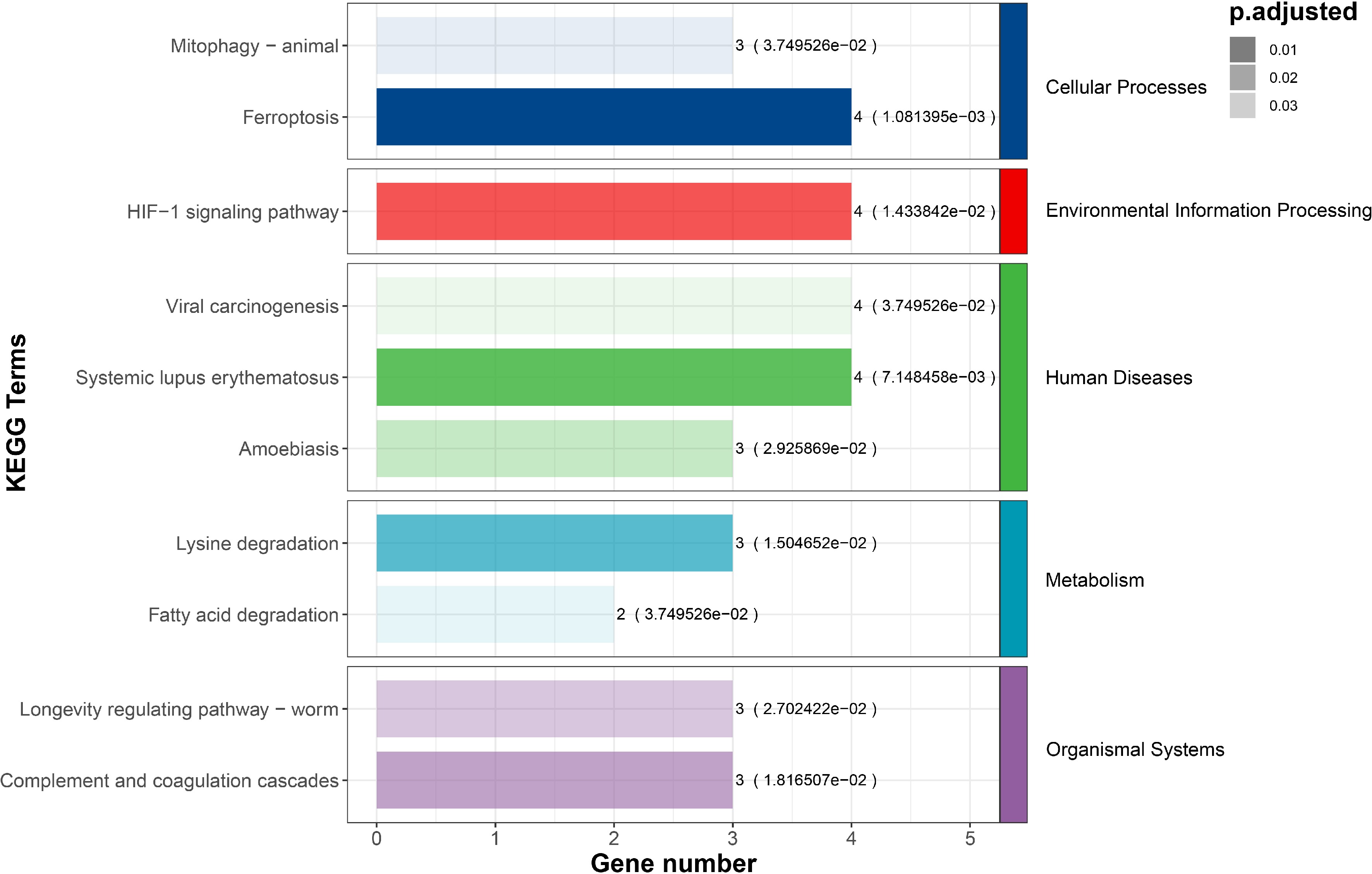
Figure 3.
The KEGG signaling pathway annotation of DASGs.
Gene analysis
-
Among the 64 DASGs, the genes acsl4a and Hadha, which are implicated in the metabolic processes of organisms and are hypothesized to be key genes affecting the differences in feed efficiency in Jian carp, were further analyzed on the basis of annotation information and existing literature reports. The acsl4a gene had two transcripts, acsl4a_a and acsl4a_b, both spliced from 15 exons, but the first exon of acsl4a_a was longer than that of acsl4a_b (Fig. 4a). The predicted open reading frames (ORFs) and protein domains indicated that acsl4a_a encoded short peptides (< 100 aa) without identifiable protein domains, while acsl4a_b encoded a 738-aa polypeptide with a transmembrane region and adenosine monophosphate (AMP)-binding domains, and showed significantly higher expression in the LRFI group (Fig. 5a). The Hadha gene had four transcripts, Hadha_a, Hadha_b, Hadha_c, and Hadha_d, spliced from 18, 19, 20 and 9 exons, respectively (Fig. 4a). Hadha_a and Hadha_b encoded short peptides (< 100 aa) without identifiable protein domains; Hadha_c encoded a 761-aa polypeptide with ECH_2, 3HCDH_N, 3HCDH, and low complexity domains; Hadha_d encoded a 394 aa polypeptide with 3HCDH_N, and two 3HCDH domains (Fig. 4b). According to three-dimensional structure predictions, the overall structures of the Hadha_c and Hadha_d proteins were similar, both containing a V-shaped groove that could be the binding site for interacting molecules. However, the Hadha_d protein lacked a loose long peptide chain at the N-terminus, and the groove of which was wider than that in the Hadha_c protein. In the LRFI and HRFI groups, Hadha_c did not show significant differences in expression levels, while Hadha_d showed significantly higher expression in the HRFI group (Fig. 5b).
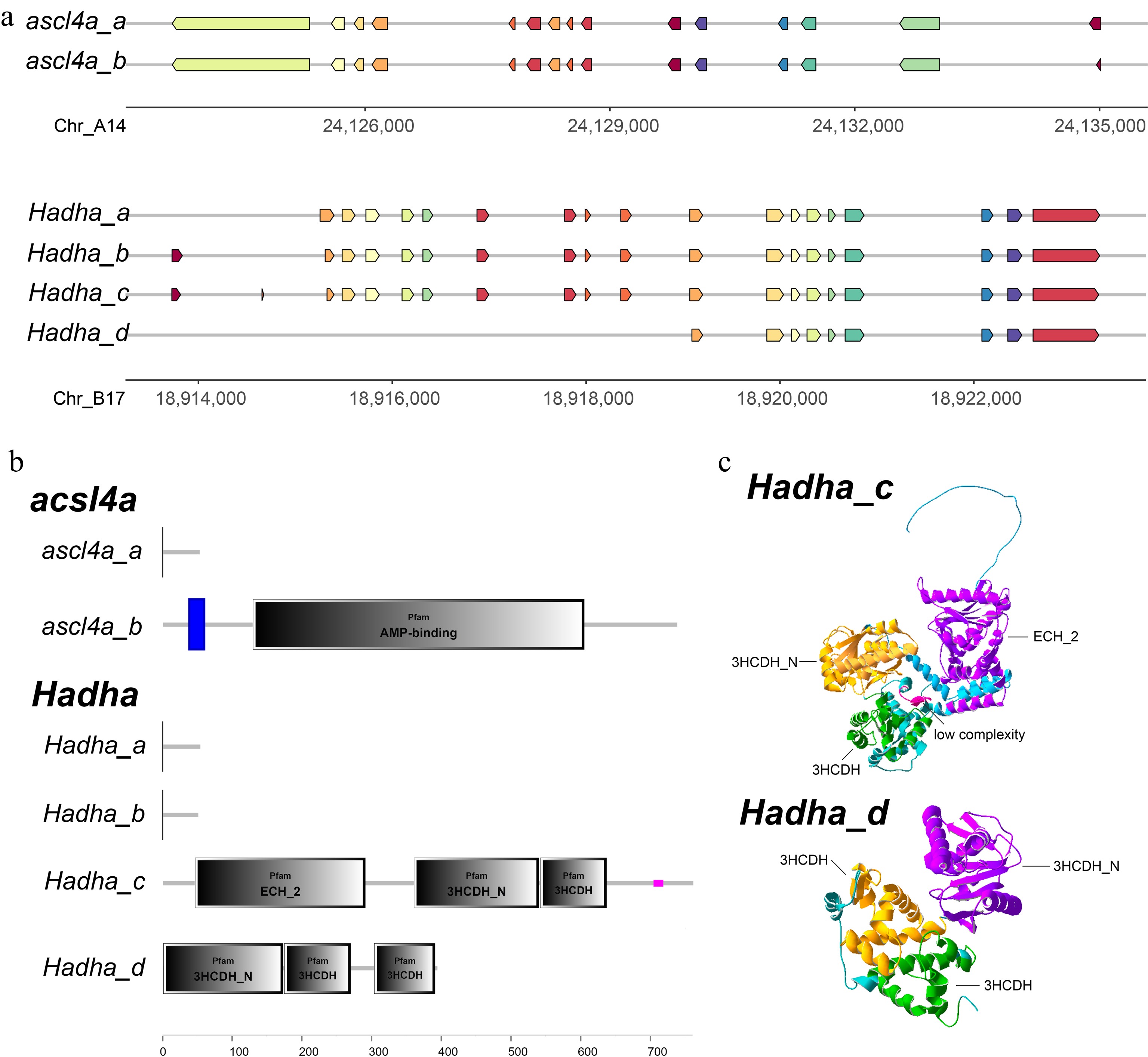
Figure 4.
The characteristics of the acsl4a and Hadha genes. (a) The exonic disparity of transcripts derived from acsl4a and Hadha. (b) The protein structural domains of different splice variants of acsl4a and Hadha. (c) The three-dimensional structure of Hadha_c and Hadha_d proteins.
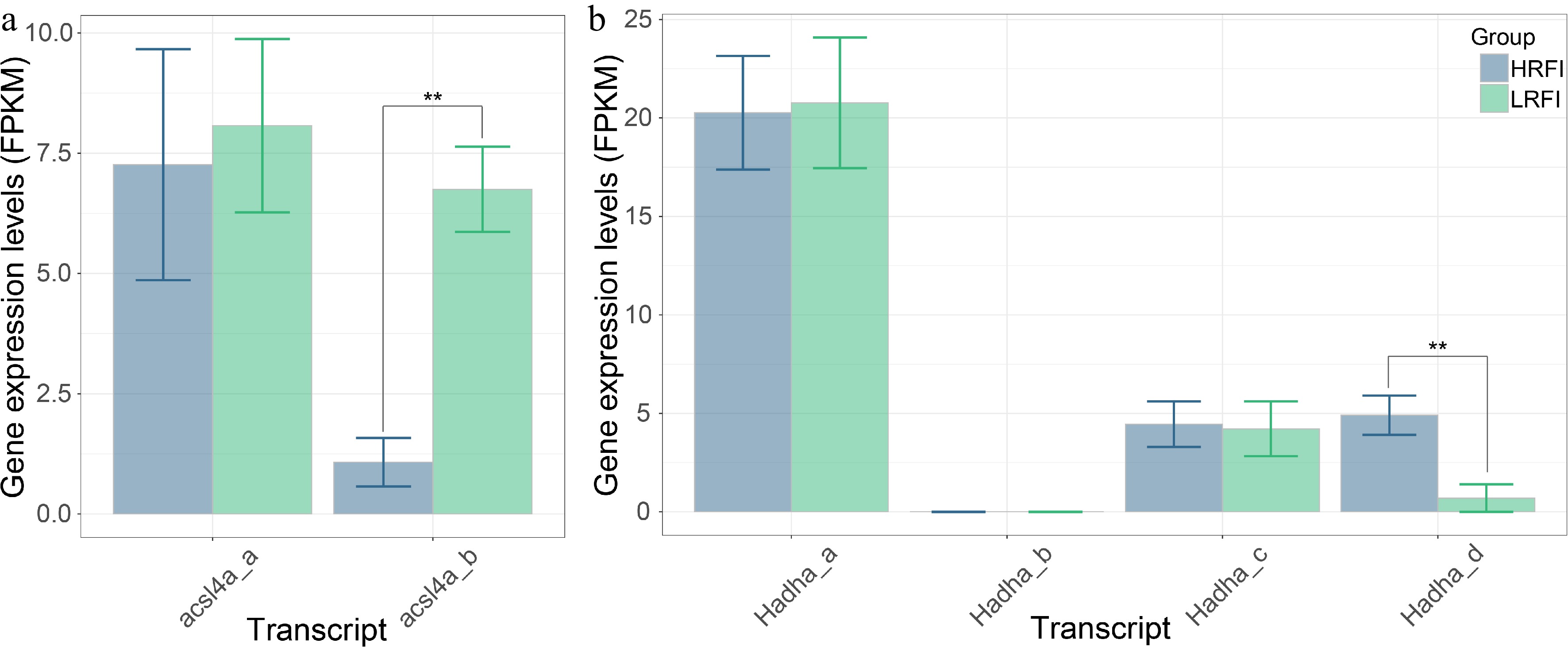
Figure 5.
The expression profiles of transcripts derived from acsl4a (a) and Hadha (b) in the LRFI and HRFI groups.
The reliability of the transcriptomic analyses was further corroborated by qRT-PCR validation. Among the 12 DETs examined, 11 DETs exhibited consistent expression patterns between the HRFI and LRFI groups across both the transcriptomic and qRT-PCR datasets, with the exception of XM_042745279.1 (Fig. 6a). Furthermore, qRT-PCR analysis revealed that the expression of acsl4a_b was significantly lower in the HRFI group compared with the LRFI group, consistent with the transcriptome-derived results (Figs 5b and 6b). For Hadha, transcript-specific qRT-PCR validation could not be performed due to technical limitations in primer design. This challenge arose from the high isoform multiplicity of Hadha, which precluded the identification of unique sequences for individual transcripts.
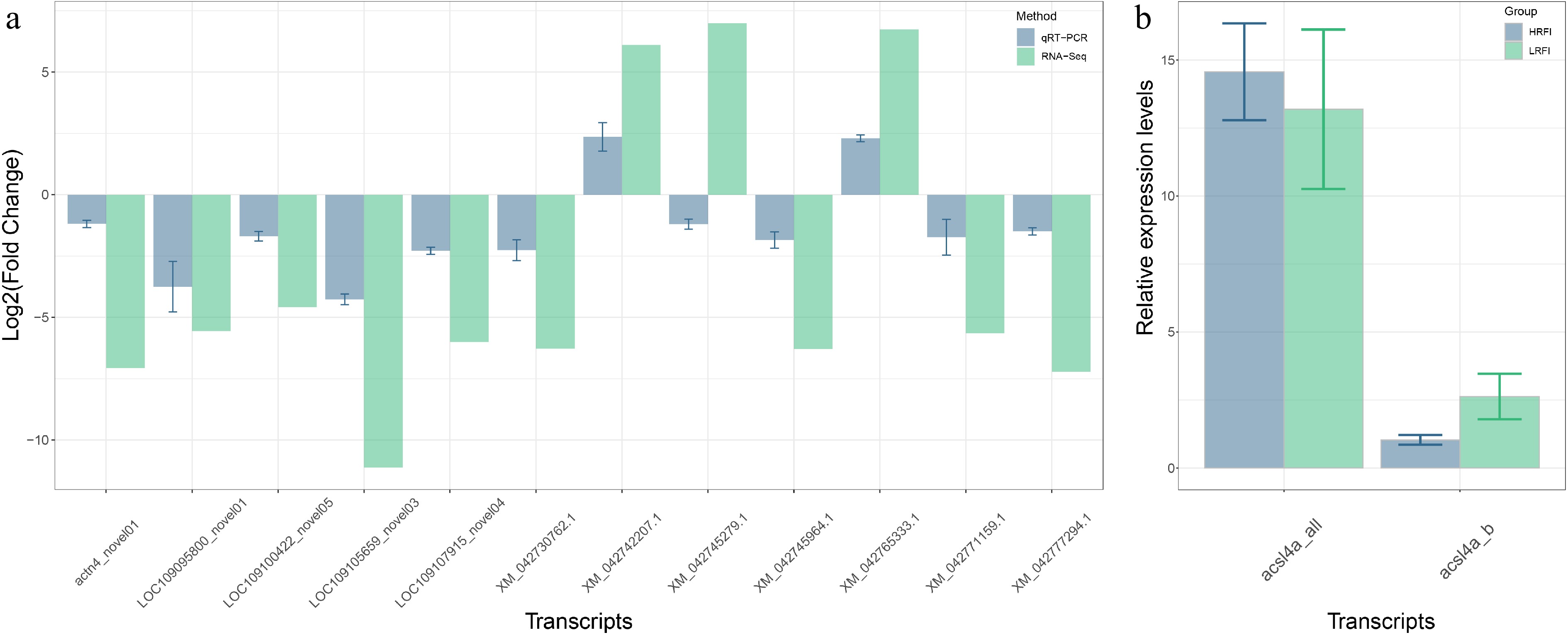
Figure 6.
Quantitative real-time PCR (qRT-PCR) validation. (a) The expression profiles of 12 differentially expressed transcripts (DETs). (b) The relative expression levels of transcripts derived from acsl4a detected through qRT-PCR.
SNP type analysis
-
Through WGR, a total of 32,593,671 SNPs were identified across the 50 chromosomes and mitochondria of the C. carpio genome (Supplementary Fig. S2), and we classified and counted the types of these SNPs. SNPs located in the 64 DASGs and the intergenic regions flanking DASGs were extracted, and 70,218 SNPs were obtained. Comparing these SNPs with all SNPs in the type analysis revealed that the proportion of SNPs causing changes in the splicing region of transcripts and mis-sense mutations in the SNPs associated with DASGs (1.32% and 3.76%) were significantly higher than over all SNPs (0.67% and 1.90%) (Fig. 7).
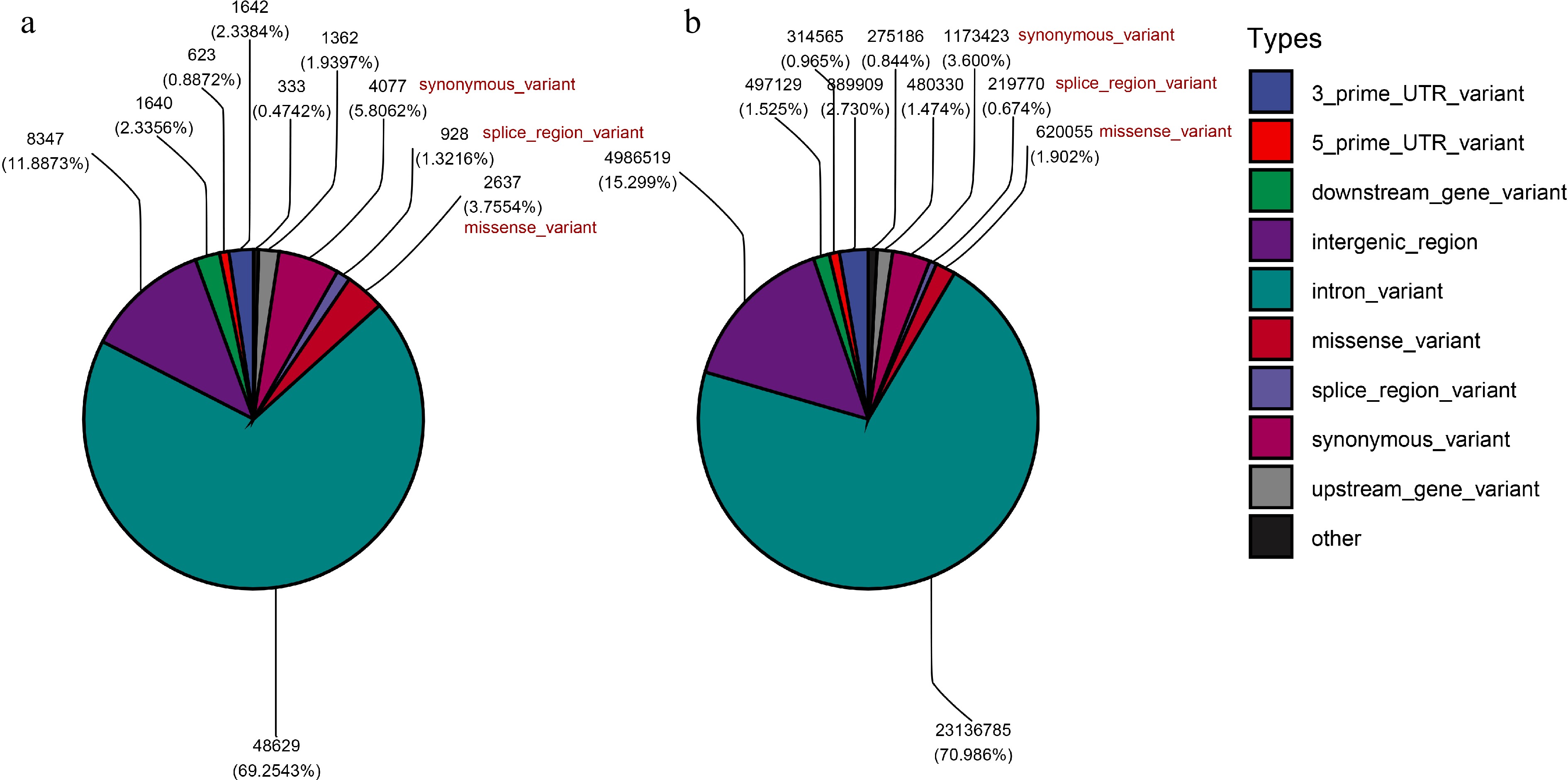
Figure 7.
Classification of SNP types. (a) The proportion of various SNP types among SNPs associated with DASGs. (b) The proportion of various SNP types among all SNPs.
-
Improving feed efficiency is key to reducing the economic costs associated with aquaculture and transitioning the industry towards environmentally sustainable practices[3]. Although the measurement of RFI is costly and time-consuming, it is an ideal indicator for evaluating feed efficiency due to its accuracy. In this study, we conducted an 8-week feeding trial to calculate the RFI of Jian carp and performed group division. By leveraging a multi-omics approach that integrated PacBio full-length transcriptome, Illumina transcriptome, and WGR data, we explored the genetic mechanisms through which AS modulated feed efficiency in Jian carp. Additionally, we attempted to elucidate the interplay among AS, gene expression levels, and SNPs in fish.
AS is a vital biological mechanism by which organisms process immature mRNA to produce different transcript isoforms, thereby regulating protein function[36]. It has been reported that AS is involved in regulating growth and development, sex determination, environmental adaptation, biological rhythms, aging, carcinogenesis, and other life activities in organisms[37−41]. Studies of AS based on Illumina RNA sequencing are limited by short read lengths and permit only the statistical analysis of AS events such as A3, SE, AF, A5, RI, AL, and MX within the genome. These constraints prevent the acquisition of full-length sequences of AS transcripts, thereby hindering the analysis of their expression levels and functions[42]. This study overcame the sequencing read length limitations by integrating PacBio full-length transcriptome and Illumina transcriptome data, yielding 70,422 full-length transcripts and performing quantification. In the differential expression analysis of the transcripts, 200 transcripts were found to be significantly differentially expressed between the LRFI and HRFI groups, with 71 of these transcripts being AS transcripts derived from 64 DASGs. The differential transcriptomic analysis results described above were validated by qRT-PCR of 12 DETs. GO and KEGG annotation of the DASGs indicated that the lysine degradation and fatty acid degradation signaling pathways may be involved in regulating feed efficiency in Jian carp. Similar results have been reported in Oncorhynchus tshawytscha, where differentially abundant proteins in the liver and muscle tissues of salmon with varying levels of feed efficiency were mainly enriched in the protein metabolism and lipid metabolism pathways according to the GO annotations[2]. Amino acid metabolism and fatty acid metabolism in organisms play crucial roles in energy production and tissue protein synthesis, and are closely associated with feed efficiency[43]. Most higher organisms are incapable of synthesizing lysine, making it an essential amino acid (IAA), and adequate levels of lysine must be maintained for protein synthesis[44]. Studies have shown that low-RFI cattle have a significantly higher liver lysine content and differential expression of lysine metabolism-related genes compared with high-RFI cattle, and similar results have been observed in sheep, with low-RFI sheep exhibiting higher serum lysine levels[43,45]. In rainbow trout farming experiments, trout fed lysine-sufficient diets displayed significantly higher feed efficiency than those fed lysine-deficient diets[46]. In weaned calves, increasing the lysine content in the feed from 6 to 8 g/kg or from 7 to 9 g/kg led to a linear increase in feed efficiency[47]. The total body fat content in livestock is generally inversely correlated with feed efficiency, with individuals having a higher body fat content often showing lower feed efficiency, and body fat content has become an indirect indicator for assessing feed efficiency[1,3]. A study on Nellore bulls regarding feed efficiency revealed that bulls with high feed efficiency consumed less and had less subcutaneous and visceral fat deposition compared with bulls with low feed efficiency, and transcriptome data from their livers indicated that the differentially expressed genes were mainly enriched in fat metabolism pathways[5]. In summary, amino acid metabolism and fatty acid metabolism are closely related to feed efficiency. This study identified genes with AS and differential transcript expression between the LRFI and HRFI groups of Jian carp, which were enriched in the lysine and fatty acid degradation pathways, suggesting that AS might regulate feed efficiency in Jian carp by modulating the lysine and fatty acid metabolic processes.
In this study, two key candidate genes affecting feed efficiency in Jian carp, acsl4a and Hadha, were further analyzed to explore their gene structure, AS events, and transcript expression profiles. The acsl4a gene belongs to the achaete–scute complex-like (ACSL) family, which encodes proteins that catalyze the conversion of free long-chain fatty acids into fatty acyl-CoA, representing the first step in most fatty acid processing and metabolic pathways[48]. In Danio rerio, acsl4a plays a pivotal role in regulating the proper development of the dorsal–ventral axis. Disruption of acsl4a results in diminished bone morphogenetic protein (BMP) signaling, leading to dorsalization of the embryo[49]. In this study, acsl4a formed two transcripts through AS, of which the functional acsl4a_b exhibited significantly higher expression in the LRFI group compared with the HRFI group. This suggested that the LRFI group exhibited more vigorous conversion of long-chain fatty acids, which was less prone to deposition, aligning with the negative correlation between fat deposition and feed efficiency. The Hadha gene encodes the mitochondrial trifunctional enzyme subunit-alpha, which, together with the mitochondrial trifunctional enzyme subunit-beta encoded by the Hadhb gene, forms a heteroctameric protein: the mitochondrial trifunctional protein (MTP)[50]. MTP catalyzes the final three steps of mitochondrial long-chain fatty acid oxidation, including enoyl-CoA hydratase, 3-hydroxyacyl-CoA dehydrogenase, and 3-ketoacyl-CoA thiolase activities, and individuals lacking MTP have impaired long-chain fatty acid metabolism, leading to fat deposition[51]. In this study, two transcripts of Hadha, Hadha_c and Hadha_d, were found to encode proteins. Hadha_c did not show significant differences in expression between the LRFI and HRFI groups, while Hadha_d exhibited significantly higher expression in the HRFI group. Although the overall structure of the Hadha_c and Hadha_d proteins was similar, the amino acid sequence of Hadha_c was nearly twice as long as that of Hadha_d, with more complex structural features. We hypothesize that the Hadha_c protein is the primary catalyst, while the Hadha_d protein acts as a competitive inhibitor for substrate binding, serving as a negative regulatory factor for Hadha_c. The elevated expression of Hadha_d in HRFI carp likely suppresses the function of Hadha_c, facilitating the deposition of long-chain fatty acids within the bodies of HRFI carp.
SNPs are the most prevalent form of genetic variation in organisms and have emerged as key molecular markers for mapping traits and conducting genome-wide association studies in economically significant species[52]. Traditionally, the investigation of genetic variations, encompassing SNPs, has been predominantly centered on their influence on epigenetic patterns or the regulation of transcription. Nevertheless, there is a burgeoning appreciation for the pivotal role that SNPs play in the modulation of post-transcriptional mechanisms, such as AS. SNPs have the potential to modify diverse facets of splicing regulation, including cis-regulatory elements, the expression or functionality of trans-acting factors, and the interplay among these elements[53]. In humans, a SNP located in Exon 8 of the CD46 gene, designated rs2724374, influences the AS of CD46, and splice variants are implicated in the pathogenesis of hemolytic uremic syndrome[54]. In Duroc pigs, a SNP within Intron 9 of the PHKG1 gene, denoted g.8283C>A, resulted in the generation of aberrant PHKG1 transcripts, leading to premature termination of protein coding, which diminishesd the glycogenolytic capacity in Duroc pigs, significantly increasing the protein content in their pork[55]. In the present study, SNPs associated with DASGs were more inclined to alter splice junctions within transcripts and to induce mis-sense mutations compared with the SNPs overall, highlighting the significant role of SNPs in influencing AS events in organisms.
-
In summary, the present study focused on the cultivated breed of common carp, Jian carp, as the subject of investigation, to investigate the correlation between feed efficiency and AS events through a multi-omics approach. We measured the RFI of Jian carp as the indicator of feed efficiency and divided the carp into two groups: the LRFI and HRFI groups. By integrating PacBio full-length transcriptome sequencing with Illumina transcriptome sequencing, we obtained 70,422 full-length transcripts in the liver of Jian carp, with 200 DETs showing significant differences in expression between the LRFI and HRFI groups. In the analysis of key candidate genes affecting feed efficiency in the Jian carp, acsl4a and Hadha, we discovered that the proteins encoded by the DETs of these two genes exhibited substantial structural differences compared with other transcript proteins. We propose that AS events are instrumental in modulating the functionality of the acsl4a and Hadha genes. This research sheds light on the influence of AS events on feed efficiency in fish, offering novel insights into the feed efficiency regulatory network in fish. Moreover, the DASGs identified in this study, including acsl4a and Hadha, can be developed as potential molecular markers for breeding.
This study was supported by the Central Public Interest Scientific Institution Basal Research Fund CAFS (2024JBFR09), the earmarked fund for CARS (CARS-45), the Central Public Interest Scientific Institution Basal Research Fund CAFS (2023TD40), Hebei Province Modern Agriculture Industry Technology System Freshwater Aquaculture Innovation Team (HBCT2023230203), and the National Key Research and Development program (2022YFF0608203).
-
The use of animals in this study was performed in accordance with the guidelines for the care and use of animals for scientific purposes set by the Animal Ethics Committee of the Freshwater Fisheries Research Center, Chinese Academy of Fishery Sciences (Wuxi, China) identification number: LAECFFRC-2023-06-12, approval date: 2023/6/12.
-
The authors confirm their contributions to the paper as follows: writing – original draft: Xu Y; visualization: Xu Y, Liu Y, Wang W, Feng W; validation: Xu Y, Liu Y, Wang W; software: Xu Y, Wang W, Feng W; methodology: Xu Y, Liu Y, Wang W, Zhang Z, Jiang K, Li J, Sewo DY; investigation: Xu Y, Liu Y, Zhang Z, Jiang K, Li J, Sewo DY, Tang Y; formal analysis: Xu Y, Wang W, Jiang K; resources: Zhang Z, Li J; writing – review & editing, supervision, project administration, funding acquisition, and conceptualization: Tang Y. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
-
The authors declare that they have no conflict of interest.
- Supplementary Fig. S1 Location of SNPs among 50 chromosomes and mitochondria of Jian carp.
- Supplementary Fig. S2 The Classification of alternative splicing AS types.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of Nanjing Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Xu Y, Liu Y, Wang W, Zhang Z, Jiang K, et al. 2026. The full-length transcriptome reveals alternative splicing events involved in regulating feed efficiency in Jian carp (Cyprinus carpio var. jian). Animal Advances 3: e001 doi: 10.48130/animadv-0025-0030 

The full-length transcriptome reveals alternative splicing events involved in regulating feed efficiency in Jian carp (Cyprinus carpio var. jian)
- Received: 06 June 2025
- Revised: 29 June 2025
- Accepted: 08 July 2025
- Published online: 09 January 2026
Abstract: Feed efficiency is a pivotal trait impacting aquaculture's profitability. This study focused on Jian carp to explore the genetic mechanisms underlying the feed efficiency of fish from the perspective of alternative splicing (AS). Through the combined analysis of PacBio full-length transcriptome sequencing and Illumina transcriptome sequencing data, a total of 70,422 full-length transcripts were identified from the liver of Jian carp, among which 200 transcripts showed significant expression differences between the low residual feed intake (LRFI) and high residual feed intake (HRFI) groups. A subset of 71 differentially expressed transcripts (DETs) were identified as alternatively spliced transcripts derived from 64 genes. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotations suggested that these AS events might influence feed efficiency in Jian carp by influencing the lysine degradation and fatty acid degradation pathways. Prediction of amino acid sequences, identification of structural domains, and three-dimensional protein structure predictions revealed significant structural differences between proteins encoded by differentially expressed alternatively spliced transcripts and other transcripts for key candidate genes: acsl4a and Hadha. Whole-genome resequencing was employed to identify single nucleotide polymorphisms (SNPs) within the genome of Jian carp. SNP annotation using SnpEff indicated that a higher proportion of SNPs associated with the 64 differentially spliced genes altered the transcript splicing regions and introduced nonsense mutations compared with the general SNP population. This study unveiled the involvement of AS events in regulating the function of specific genes, thereby affecting the feed efficiency of Jian carp. The findings contribute to the comprehension of the regulatory network governing fishes' feed efficiency and provide potential directions for exploring methods to improve feed efficiency in these fish.
-
Key words:
- Alternative splicing /
- Feed efficiency /
- Jian carp