









-
The intestinal tract, a central organ for nutrient digestion and absorption and a crucial immunological barrier in monogastric animals, harbors diverse microbial communities including bacteria, archaea, eukaryotes, viruses, and fungi. These microbes engage in intricate cross-kingdom signaling with the host. Through these interactions, they form complex networks that play essential roles in maintaining nutritional metabolic homeostasis and host health[1]. The development of the archaea domain's phylogenetic classification system by Carl Woese revolutionized microbiological research[2]. Among archaea, anaerobic methanogens have drawn significant attention because of their unique molecular features. These organisms possess conserved 16S rRNA sequences that distinguish them from eubacteria, lack peptidoglycan cell walls, and have membranes composed of isoprenoid-derived lipids linked to glycerol-1-phosphate via ether bonds[3,4]. In terms of energy metabolism, methanogens perform methanogenesis, an anaerobic respiratory process that reduces carbon dioxide, acetate, and methyl compounds to produce methane. This process relies on archaea-specific cofactors, such as Coenzymes M and F420[5]. Although methanogens have a relatively low abundance in the gut microbiota, constituting approximately 1% of the porcine intestinal microbiome and approximately 10% of human gut anaerobes, they significantly influence the host's metabolic homeostasis[6,7] as hidden architects of intestinal ecology. They achieve this through mechanisms such as modulating the partial pressure of microenvironmental hydrogen and remodeling the microbial interaction network[8,9].
Methanogens exhibit a dual nature in their ecological functions. On one hand, their hydrogenotrophic metabolism is beneficial, as it removes excess molecular hydrogen generated during fermentation. This process helps maintain transmembrane proton gradients, thereby promoting the growth of fibrolytic bacteria and enhancing the efficiency of dietary fiber degradation[10]. As a result, methanogens can improve nutrient absorption, particularly in malnourished hosts. On the other hand, methanogens have been associated with various metabolic disorders including irritable bowel syndrome (IBS), chronic constipation, and obesity[11]. Clinical studies show that individuals with more abundant methanogens often have elevated hepatic triglyceride levels. Moreover, obese individuals typically harbor more methanogens than those with a normal body weight, and bariatric surgery can reduce methanogen populations[10,12]. Given this metabolic complexity, the concept of "archaebiotics" has emerged. This strategy aims to dynamically regulate methanogen populations according to the host's metabolic phenotype, ultimately restoring the metabolic network's balance.
Despite significant progress in gut microbiome research, several principal limitations persist. The overwhelming dominance of bacteria in terms of abundance within the animal intestine has skewed research efforts predominantly towards functional analyses of the bacterial domain. Consequently, the contributions of methanogens to host physiological regulation have remained largely under the radar, leaving a critical knowledge gap in understanding their multifaceted roles. From a clinical perspective, the use of germ-free animal models has yet to establish a clear pathogenic threshold of methanogens[6,13]. This lack of definition complicates the diagnosis and management of potential methanogen-associated disorders. Moreover, the existing body of research is heavily concentrated on human and porcine intestinal ecosystems[7,14]. The scarcity of comparative metagenomic studies across other monogastric species hinders the development of a comprehensive understanding of methanogens' ecology and function across diverse hosts. This review endeavors to bridge these gaps by comprehensively examining methanogens' taxonomic characteristics, factors influencing their abundance, metabolic pathway intricacies, interactions with the host's metabolic and immune systems, and synergistic relationships with bacterial communities.
-
The methodologies employed in the taxonomy of methanogens have undergone substantial evolution, paralleling the rapid advancements in microbiome research technologies. In the early stages of investigation, 16S rRNA gene-based sequencing served as the cornerstone for delineating phylogenetic lineages among methanogens[15]. However, the subsequent advent of sophisticated techniques such as metagenomics, flow cytometry, quantitative real-time polymerase chain reaction (qPCR), and matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) has revolutionized the field, markedly enhancing both the detection efficiency and taxonomic resolution[16,17]. For instance, flow cytometry capitalizes on the autofluorescence property of Coenzyme F420, a key cofactor in the methanogenesis pathway with a maximal absorption peak at 420 nm[18]. This characteristic allows for the rapid and accurate quantification of methanogens within complex microbial communities[16]. Similarly, qPCR offers highly sensitive detection capabilities by targeting specific functional genes associated with methanogenesis, such as mcrA, which encodes methyl-Coenzyme M reductase[19]. The Genome Taxonomy Database (GTDB) acts as a comprehensive resource, currently containing 2,339 archaeal genomes distributed across 19 phyla, over 70% of which represent uncultured organisms[20]. Notably, the establishment of a curated repository containing 1,167 archaeal genomes of the human gut has provided invaluable material for in-depth investigations into the structural organization and functional dynamics of methanogen communities[6].
From a phylogenetic perspective, methanogens are currently classified into nine distinct orders, namely Methanobacteriales, Methanococcales, Methanomicrobiales, Methanosarcinales, Methanocellales, Methanopyrales, Methanomassiliicoccales (formally recognized as the seventh methanogen order by the International Committee on Systematics of Prokaryotes in 2013[21])[22−24], and the more recently described Methanonatronarchaeales[25] and Methanoliparales[23]. Among these, the Methanomassiliicoccales order exhibits unique evolutionary divergence compared with the other methanogen groups. It is further divided into two families, the free-living clade Methanomethylophilaceae, which includes Methanomassiliicoccus luminyensis and Candidatus Methanomassiliicoccus intestinalis[8], and the host-associated clade Methanomassiliicoccaceae, encompassing organisms such as Candidatus Methanomethylophilus alvus[6,8]. Within the Methanobacteriales order, Methanobrevibacter smithii is classified into two species-level clades, 'smithii' and 'smithii_A', according to the GTDB classification system[20], highlighting the intricate taxonomic diversity within this group.
Morphologically, methanogens can be classified into six distinct cellular architectures. These include rod-shaped forms, exemplified by Methanobacterium and Methanothermobacter; coccoid shapes, such as Methanococcus and Methanosphaera; spiral structures, as seen in Methanospirillum; tetrad-forming cells, typified by Methanosarcina; plate-shaped organisms like Methanoplanus; and filamentous types, represented by Methanosaeta[18,26]. This morphological diversity provides a fundamental basis for initial taxonomic identification and offers insights into the physical adaptations of methanogens to various ecological niches.
In terms of metabolism, methanogens are commonly categorized according to their substrate utilization patterns into three major groups: hydrogenotrophic, aceticlastic, and methylotrophic. Among these, hydrogenotrophic lineages are the most prevalent (Table 1)[4,27]. Some methanogen taxa exhibit strict substrate specificity. For example, Methanosphaera spp., with Methanosphaera stadtmanae as a representative[28], exclusively reduce methanol, utilizing H2 as an electron donor[29]. In sharp contrast, members of the Methanosarcinales order show remarkable metabolic versatility, being able to utilize multiple substrates, including acetate and methyl compounds[23]. This ability to employ diverse substrates defies simple classification into a single metabolic category. The functional diversity among methanogens not only underscores the evolutionary adaptation of these organisms to specific ecological niches but also uncovers potential molecular targets for modulating their ecological roles. Understanding these metabolic distinctions is crucial for predicting how methanogens interact with other members of the microbiota and for devising strategies to manipulate their activities in both natural and engineered ecosystems.
Table 1. The substrates available to methanogens from different methanogenesis pathways
Order Family Genus Species Substrates Ref. Methanobacteriales Methanobacteriaceae Methanobrevibacter M. smithii H2, methanol [19] M. oralis H2 [59] M. arboriphilus H2, formate [60] Methanosphaera M. stadtmanae H2, methanol [29] Methanomicrobiales Methanomicrobiaceae Methanogenium H2, formate [61] Methanosarcinales Methanosarcinaceae Methanosarcina M. barkeri Acetate [62] Methanimicrococcus Methylamine [58] Methanomassiliicoccales Methanomethylophilaceae Candidatus Methanomethylophilus M. alvus Methanol [63] Methanomassiliicoccaceae Methanomassiliicoccus M. intestinalis Methylamines [36] M. luminyensis Methylamines [64] -
Methanogens are widely distributed throughout the gut of monogastric animals, and their presence and abundance are influenced by a multitude of factors, including the growth stage of the animal, its dietary composition, the surrounding environment, and its physiological state.
Distribution of methanogens in the intestines of monogastric animals
-
Multi-species studies aimed at exploring the distribution of methanogens within the intestinal ecosystems of animals have identified five key archaeal lineages. These lineages, namely Methanobrevibacter and Methanosphaera (both belonging to the Methanobacteriales order), Methanomethylophilaceae (from the Methanomassiliicoccales order), Methanocorpusculum (of the Methanomicrobiales order), and Methanimicrococcus (from the Methanosarcinales order), are recognized as the dominant archaeal components in the gut. Collectively, they account for more than 90% of the archaeal communities[15,30]. Among these, Methanobrevibacter[31] stands out as the most prevalent genus across the intestinal tracts of various animals, succeeded by the candidate taxon Ca. Methanomethylophilaceae[15]. Meanwhile, although Methanosphaera has a wide distribution across different animal species, its abundance is relatively lower[31].
In the context of the human gut microbiome, methanogens typically constitute approximately 10% of the anaerobic communities in healthy individuals[32]. Metagenomic analyses have shown that the Methanobacteriales (87.15%) and Methanomassiliicoccales (12.43%) are the predominant orders, with Methanobrevibacter being the dominat genus, accounting for 85% of the archaeal population[6]. At the species level, profiling has identified Methanobrevibacter smithii, Methanobrevibacter oralis, Methanosphaera stadtmanae, Methanomassiliicoccus luminyensis, Candidatus Methanomassiliicoccus enteris, and Candidatus Methanomethylophilus alvus as the core methanogens in the human gut[33]. Methanobrevibacter smithii exhibits an almost universal colonization rate (97.5% prevalence) and constitutes 84% of the adult archaeomes, with 16% belonging to the clade Methanobrevibacter smithii_A and 68% to the clade Methanobrevibacter smithii. On the other hand, Methanosphaera stadtmanae has a mean abundance of 13% and a prevalence of 29%[6,8,34]. Additionally, other taxa such as multiple species from the Methanomassiliicoccales order[35], Ca. Methanomassiliicoccus intestinalis[36], and Methanosphaera cuniculi (originally isolated from rabbit intestines)[37] often inhabit the human gut. When considering the strains Mx02, Mx03, and Mx06, the cumulative prevalence of these additional taxa reaches 80%[38].
The archaeal communities in pigs closely resemble those in humans in terms of composition. Methanobacteriales (57%–80%) and Methanomassiliicoccales (15.07%) are the dominant orders, with Methanobrevibacter (57%) and Methanosphaera (3%–14%) being the principal genera[7,39,40]. Methanobrevibacter smithii is detected in nearly all samples from the porcine colon (99.7%) and feces (99.9%)[7,41]. However, the dominance patterns of Methanobrevibacter smithii can vary, depending on the analytical methodology employed. qPCR often identifies it as the most prevalent archaeal species[30], whereas amplicon sequencing reveals that Methanobrevibacter smithii has a relatively minor prevalence, with Methanobrevibacter millerae, Methanosphaera cuniculi, and Methanobrevibacter boviskoreanii collectively accounting for 80%–90% of the total archaeal abundance[9]. Spatial analysis of the porcine gut shows a progressive increase in the relative abundance of Methanobrevibacter (comprising 44.2%–59% of archaeal communities) from the ileum to the colon. In contrast, methylotrophic Methanomethylophilaceae archaea remain scarce, accounting for less than 0.1% of the archaeal population throughout the porcine gastrointestinal tract[9].
In lagomorphs, archaeal communities are predominantly composed of Methanobrevibacter species[42,43]. In companion animals, such as dogs (where archaeal communities constitute 25% of the gut microbiota), cats (16.66%), and horses (4.16%), Methanobrevibacter smithii co-occurs with other archaeal species, including Methanocorpusculum aggregans, Methanocorpusculum labreanum, Methanobrevibacter millerae, Methanobrevibacter thaueri, and Methanobrevibacter olleyae[30]. These findings highlight the species-specific variations in the composition and abundance of methanogens across different monogastric animals, which may be associated with their unique dietary habits, digestive physiologies, and ecological niches.
Factors influencing the abundance of methanogens
-
The diversity and functionality of mammalian gut archaea are intricately regulated by a combination of the host's phylogeny, dietary habits, fiber content, and intestinal physiological characteristics[15]. These factors interact in complex ways to shape the composition and dynamics of methanogen communities within the gut ecosystem.
In addition to the previously mentioned differences in the prevalence of methanogens among different animal species, significant variations in the gut methanogen community structure are also observed among different breeds within the same species. For example, in pigs, the diversity of methanogens in the gut of the fat-type Erhualian breed is notably lower than that in the lean-type Landrace pigs[39].
The developmental stage of the host significantly impacts the structure of archaeal communities. In mammals, neonatal colonization by Methanobrevibacter smithii is likely facilitated by maternal transmission, primarily through breast milk[44]. As the host matures into adulthood, the abundance of Methanobrevibacter smithii increases substantially, while geriatric populations tend to exhibit an enrichment of Methanomassiliicoccales[45]. Similar trends are observed in pigs, where the dominant position of Methanobacteriales is gradually weakened by Methanomassiliicoccales[46]. Adult pigs generally display higher archaeal α-diversity compared with piglets. However, the weaning and growth phases in piglets are associated with a notable increase in archaeal richness. This change is largely attribute to dietary adjustments, such as an increased intake of fiber[7]. Weaning presents a pivotal transition point in the gut ecosystem, triggering a succession of archaeal communities. Suckling and nursery-stage piglets are predominantly colonized by Methanobrevibacter smithii, which is gradually replaced by Methanobrevibacter boviskoreanii and members of the Methanomassiliicoccales after weaning[7,47]. These developmental changes underscore the dynamic and adaptive nature of host–microbe interactions, which aim to optimize nutritional metabolism while navigating niche competition.
Dietary fiber content plays a central role in determining the distribution and function of methanogens. The development of methanogen communities relies on the presence of anaerobic environments and diverse carbohydrate sources[45,48]. In pigs, high-fiber diets indirectly boost the abundance of certain methanogens, such as Methanobrevibacter sp900769095, by promoting the growth of hydrogen-producing bacteria. Methane production in this context is positively correlated with the fiber-degrading activities of the gut microbiota but negatively associated with starch metabolism[7]. A decrease in the fiber-to-starch ratio in the diet can lead to the accumulation of lactic acid and a subsequent drop in gut pH. This shift favors the conversion of lactate to propionate, a process that competes with methanogenesis for nicotinamide adenine dinucleotide (NADH)-reducing equivalents, ultimately suppressing methane production[49,50]. Moreover, high-fiber diets stimulate the production of bacterial methylamine, providing a substrate for methylotrophic methanogens, such as those belonging to the Methanomassiliicoccales[51]. Similarly, diets rich in protein have been shown to increase the overall abundance of methanogens[52], highlighting the multifaceted impact of dietary components on methanogens' ecology.
Disease states have a profound impact on the homeostasis of methanogen communities within the gut. While conditions such as colorectal cancer, polypectomy, and IBS have shown minimal effects on the abundance of human methanogens, inflammatory bowel diseases (IBD), including ulcerative colitis and Crohn's disease, are associated with reduced methanogen colonization[53,54]. This disruption suggests that chronic inflammation in the gut can alter the ecological niche, making it less hospitable for methanogens. In swine infected with influenza A, significant shifts occur in the archaeal community. The abundance of Methanobrevibacter boviskoreanii and Methanosphaera cuniculi decreases, while Methanobrevibacter millerae and Methanomethylophilaceae increase. Additionally, Methanosphaera stadtmanae is detected specifically in diseased pigs[8]. Notably, Methanobrevibacter species exhibit remarkable tolerance to antibiotics that target bacterial RNA and protein synthesis, and cell wall formation. This characteristic may influence the outcome of clinical interventions[55], as the persistence of methanogens during antibiotic treatment could potentially affect the recovery of the gut microbiota and host health.
Cross-regional studies of swine microbiota have revealed significant variations in archaeal diversity and community composition. Chinese swine populations exhibit lower archaeal diversity compared with their Danish and French counterparts, with distinct dominant taxa in each region. Methanobrevibacter is the predominant genus in Chinese (44.94%) and French (15.41%) swine, while Candidatus Methanomethylophilus alvus dominates in Danish herds (14.32%)[46]. Similarly, the marked enrichment of Methanomassiliicoccales Mx06 in non-Westernized human populations highlights the role of lifestyle factors, including diet and environmental exposures, in shaping the biogeography of methanogens[6]. These findings underscore the complex interplay among environmental factors, host characteristics, and methanogen communities, which has important implications for understanding the ecological dynamics of the gut microbiota and its impact on host health.
-
Methanogenesis stands as one of the most ancient energy-conserving metabolic processes, exerting direct physiological effects on gastrointestinal systems[56]. Similar to the final workers in an industrial assembly line, methanogens occupy the terminal position in microbial trophic chains. They utilize the end-products of dietary substrate fermentation to produce methane[31,57] (Fig. 1). Acting as the ultimate electron acceptors, methanogens metabolize byproducts from bacteria and eukaryotes, such as H2, CO2, acetate, and methylamines, which are generated during the breakdown of dietary polymers, including short-chain fatty acids (SCFAs) and alcohols (Table 1). There are three primary methanogenic pathways: hydrogenotrophic (dependent on H2 and CO2), acetoclastic (involving acetate cleavage), and methylotrophic (utilizing methanol or methylamines)[58]. The metabolic flexibility of Methanobrevibacter is a key factor contributing to its dominance within the archaeal communities in the human gut[19].
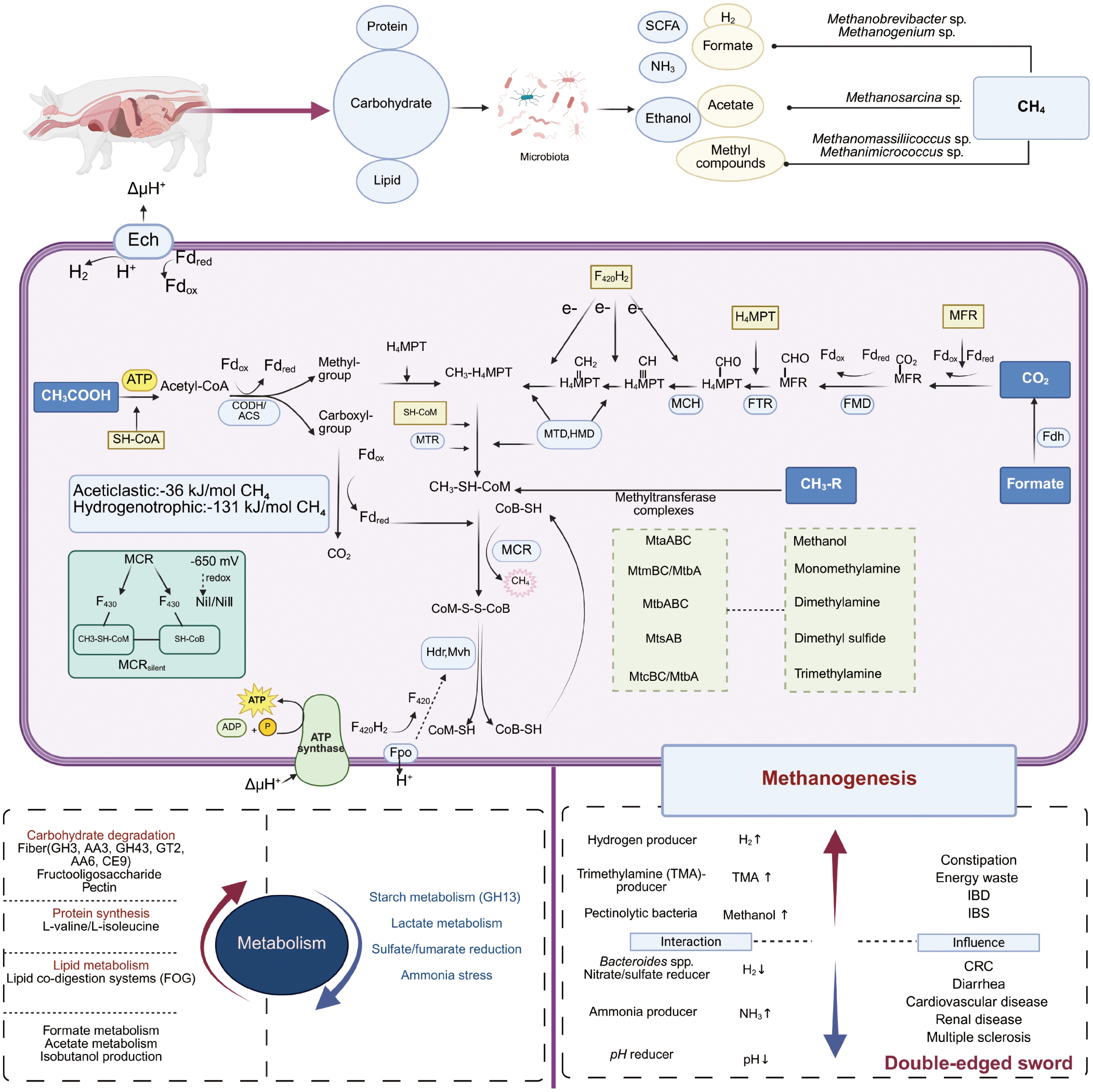
Figure 1.
Effects of methanogens on the host's metabolism and health and the mechanisms of methanogenesis.
Central to all methanogenic pathways is the terminal reaction catalyzed by methyl-Coenzyme M reductase (MCR). This reaction involves the reduction of CH3–S-Methyl-Coenzyme M (CoM) using 7-mercaptoheptanoylthreonine phosphate (CoB-SH), resulting in the formation of methane and the heterodisulfide CoM-S-S-CoB (HDS)[65]. Encoded by the mcrA gene[66], MCR exists as an (αβγ)2 heterohexamer[67], featuring two Ni-centered (Ni I/Ni II) F430 active sites. These active sites are derived from 5-aminolevulinate[18] and are formed at the subunit interfaces (α/α′/β/γ and α′/α/β′/γ′)[67].
The MCR active sites bind SH-CoM and SH-CoB in a sequential manner, triggering a conformational change that locks the enzyme into an inactive state (MCRsilent) with the formation of the CoM–CoB heterodisulfide[67]. The enzymatic activity of MCR is critically dependent on the redox state of Ni. The NiI-MCR form, with a midpoint potential of –650 mV, catalyzes the methanogenesis reaction. In contrast, the NiII-MCR form requires reductive activation, which is facilitated by dithiothreitol, adenosine triphosphate (ATP)-binding proteins (A2), and Fe-S complexes (A3a)[68,69]. This sensitivity of Ni to redox changes is fundamental to the strict anaerobic nature of methanogens and their vulnerability to inhibitors such as 3-nitrooxypropanol (3-NOP) and bromoethanesulfonate (BES). These inhibitors function by oxidizing the Ni center, thereby disrupting the methanogenic process[70,71].
The heterodisulfide reductase (HdrABC) and the methyl-viologen-reducing hydrogenase complex (MvhAGD) universally mediate the reduction of HDS, regenerating the coenzymes CoM-SH and CoB-SH[72,73]. HdrABC, a membrane-associated Fe-S protein complex, collaborates with F420H2 dehydrogenase (Fpo) under hydrogenotrophic conditions[58]. HdrA contains an electron-bifurcating flavin adenine dinucleotide (FAD) moiety, while HdrB forms the catalytic core responsible for the reduction of HDS[74,75]. Meanwhile, MvhA and MvhG constitute the conserved [NiFe] hydrogenase module, with MvhD facilitating the transfer of reducing equivalents to Hdr[73].
Methanogens possess two evolutionarily conserved Hdr systems, flavin-based electron bifurcation (FBeB), which is predominant in hydrogenotrophic methanogens, and cytochrome-dependent electron transfer (CDeT), which is characteristic of methylotrophic and acetotrophic methanogens[27]. FBeB enzyme complexes, such as HdrABC-MvhAGD[76], utilize flavin cofactors (FAD/FMN) for electron bifurcation[77]. These flavoproteins accept electron pairs from NAD(P)H, F420H2, H2, or formate, generating low-potential electrons that are transferred via ferredoxin (Fd). The reaction can be represented as 2H2 + Fdox + CoM-S-S-CoB → reduced ferredoxin (Fdred2) + CoM-SH + CoB-SH + 2H+. This electron-bifurcating mechanism enables methane production with a minimal ATP requirement of ≤ 1 ATP per molecule by coupling Fdred to CO2 reduction, effectively closing the metabolic loop of the Wolfe cycle[78]. In CDeT systems, the reduction of the lipophilic carrier methanophenazine (Mp) is coupled with H+ electrochemical potential (ΔμH+), driving both the generation of Fdred and ATP synthesis[27].
In addition to these core modules, substrate-specific adaptations diversify the auxiliary pathways of methanogenesis. The methanogenic metabolic network integrates crucial methyl carriers, including methanofuran (MFR), tetrahydromethanopterin (H4MPT) (derived via the Wood–Ljungdahl pathway), N5-methyltetrahydromethanopterin, and enzyme complexes such as Coenzyme M methyltransferase (MTR), MCR, formate dehydrogenase (FMD), and methanophenazine-dependent enzymes (tetrahydromethanopterin formyltransferase [FTR], methenyltetrahydromethanopterin cyclohydrolase, methylenetetrahydromethanopterin dehydrogenase/methenyltetrahydromethanopterin hydrogenase)[23,27,79], highlighting the evolutionary plasticity and functional versatility of these metabolic components within the archaeal domain.
Hydrogenotrophic methanogenesis
-
In methanogenic archaea, the vast majority of hydrogenotrophic lineages, including Methanococcales, Methanopyrales, Methanobacteriales, Methanomicrobiales, Methanocellales, and Methanosarcinales (excluding Methanomassiliicoccales), use the Wood–Ljungdahl pathway for CO2 fixation[80]. These organisms utilize H2 as an electron donor to sequentially reduce CO2 to CH4[81]. For example, the metabolic activity of dominant methanogens in the porcine gut, such as Methanobrevibacter sp900769095), is intricately linked to hydrogenotrophic methanogenesis[7].
Notably, certain methanogens can utilize formate as an alternative electron carrier[4,82]. Formate dehydrogenase (FDH) oxidizes four formate molecules to CO2[58], and subsequent hydrogenlyase (such as Hdr)-mediated reactions generate H2[7]. Many members of the Methanomicrobiales lack genes encoding hydrogenase, rendering formate metabolism essential for providing reducing equivalents. In Methanothermobacter speices, for instance, Hdr and FDH exhibit activity only when formate serves as the electron donor, and the activity of formate hydrogenlyase is crucial for supporting Hdr reactions[82]. Additionally, some methanogens can utilize secondary alcohols, such as 2-propanol, 2-butanol, and cyclopentanol, or ethanol as electron donors[27].
Hydrogenotrophic methanogenesis commences with the reduction of CO2 to formylmethanofuran (CHO-MFR), a reaction catalyzed by formylmethanofuran dehydrogenase (FMD)[83]. This initial step occurs under conditions where low-potential Fdred is available, generated through either the FBeB or CDeT systems[58]. The endergonic nature of this reaction is energetically coupled with the generation of an ion gradient via the membrane-bound energy-conserving hydrogenase (Ech)[58]. Subsequently, formylmethanofuran: FTR transfers the formyl group from CHO-MFR to H4MPT, forming CHO-H4MPT. This cofactor-dependent reaction necessitates the formation of a ternary complex involving formyl-MFR, H4MPT, and the apoenzyme[84]. The resulting CHO-H4MPT undergoes a series of sequential transformations. First, methenyltetrahydromethanopterin cyclohydrolase catalyzes a dehydration reaction, yielding N5,N10-methenyl-H4MPT (CH≡H4MPT). This is followed by F420H2-dependent reduction steps mediated by methylenetetrahydromethanopterin dehydrogenase[85] and methenyltetrahydromethanopterin hydrogenase[86], which convert CH2=H4MPT into N5-methyl-H4MPT (CH3-H4MPT) and then N5-methyl-H4MPT (CH3-HMPT). Throughout these dehydration and reduction processes, F420H2 provides the necessary electrons.
The methyl group from CH3-H4MPT is transferred to CoM-SH by MTR, resulting in the formation of CH3-S-CoM[87]. MCR then catalyzes the terminal reductive demethylation of CH3-S-CoM to produce CH4, using CoB-SH as the electron donor. This reaction simultaneously generates the heterodisulfide CoM-S-S-CoB[67]. The metabolic cycle is completed through reduction of CoM-S-S-CoB back to SH-CoM and SH-CoB, a process mediated by Hdr complexes[72,73]. Electrons for this final reduction step are sourced from F420H2 or H2[67], ensuring the continuous operation of the hydrogenotrophic methanogenesis pathway.
Aceticlastic methanogenesis
-
Acetotrophic methanogens employ a core mechanism to cleave acetate into methane and CO2, a process involving intramolecular electron transfer from the methyl group to the carboxyl carbon[79]. Presently, only two genera, Methanosarcina (within the Methanosarcinales order)[88] and Methanothrix (previously known as Methanosaeta)[89], are recognized for their ability to perform this metabolic function. Methanothrix represents a strictly acetoclastic lineage that reduces acetate to CH4 through direct interspecies electron transfer (DIET) with syntrophic bacteria, such as Geobacter metallireducens. This unique pathway is the only known means by which Methanothrix can carry out autotrophic respiration, as it allows for the direct uptake of extracellular electrons from organic donors[89,90]. In contrast, Methanosarcina acetivorans displays metabolic versatility. It can oxidize carbon monoxide to CO2 while simultaneously reducing CO2 to CH4 via the Wood–Ljungdahl pathway[91]. The metabolism of CO in M. acetivorans generates auxiliary substrates, such as acetate and formate, which are integrated into the methanogenic metabolic network to conserve energy. Despite the occasional colonization of animal intestines by low-abundance Methanosarcina strains, the physiological roles of these organisms in acetoclastic methanogenesis within the gut remain largely uncharacterized[15].
In the process of acetoclastic methanogenesis, acetate is first activated through an ATP-dependent reaction to form acetyl-CoA[92]. Subsequently, acetyl-CoA is enzymatically cleaved into enzyme-bound methyl and carbonyl moieties[93]. The methyl group is transferred to H4MPT, entering the final two steps of the pathway that are shared with hydrogenotrophic methanogens. Meanwhile, the carbonyl moiety is oxidized to CO2, with ferredoxin (Fdox) serving as an the electron acceptor and generating reduced ferredoxin (Fdred)[93]. The Fdred then participates in the reduction of the heterodisulfide (CoM-S-S-CoB) during the catalytic cycle of MCR[94]. The change in free energy associated with acetoclastic methanogenesis is –36 kJ/mol of CH4[95], which is markedly lower than that of hydrogenotrophic pathways (–131 kJ/mol of CH4[75]). This lower energy yield results in reduced energy capture efficiency, necessitating compensatory adaptations in acetotrophic methanogens. Such adaptation may include high substrate affinity or syntrophic metabolic coupling, enabling these organisms to persist within their ecological niches.
Methylotrophic methanogenesis
-
Methylotrophic methanogens catalyze the reductive demethylation of various methyl compounds, including methanol, monomethylamine, dimethylamine, trimethylamine, dimethyl sulfide, and methanethiol, through the methylotrophic methanogenesis pathway[94]. This metabolic process comprises two consecutive methyl transfer reactions. First, substrate-specific methyltransferase complexes, such as MtsA/MtsB for dimethyl sulfide, MtaABC for methanol, and MtmBC/MtbA for monomethylamine, transfer the methyl group from CH3-R donors to the corresponding corrinoid proteins, forming CH3-corrinoid intermediates. Second, the subsequent transfer of the methyl group to Coenzyme M (CoM-SH) results in the formation of methyl-Coenzyme M (CH3-S-CoM)[94]. When H2 is abundant, MCR reduces CH3-S-CoM to CH4. However, under conditions of electron donor limitation, the reverse Wood–Ljungdahl pathway oxidizes CH3-S-CoM to CO2, releasing electrons that support the subsequent reduction of methyl groups. This establishes an autocatalytic electron cycling mechanism, ensuring the metabolic pathway's continuous operation[96−98].
Methanogens that utilize methyl compounds in the presence of H2 represent a substantial proportion of archaeal populations within animal microbiomes[8,64]. For example, members of the Methanomassiliicoccales order employ methylated amines, such as trimethylamine, as methanogenic substrates[99,100]. The model species Methanomassiliicoccus luminyensis couples H2 oxidation with heterodisulfide reduction via the membrane-bound Fpo-HdrD electron transport chain. This coupling generates a proton motive force (ΔμH+) that is essential for ATP synthesis, while simultaneously eliminating the organism's dependence on sodium ions, thereby enabling efficient energy conservation[64].
Methanosphaera stadtmanae, which lacks carbon monoxide dehydrogenase/acetyl-CoA synthase, relies exclusively on methanol and H2 for methanogenesis. This process is mediated by the methanol: coenzyme M methyltransferase encoded by the mtaABC genes[28,101]. In porcine gut, M. smithii may engage in methanogenesis through methyl metabolic bypass pathways[7]. These methanogenic strains employ specialized enzymatic systems tailored to specific substrates, energy-coupling strategies, and reverse reaction electron cycling mechanisms. These adaptations allow them to maintain metabolic activity under variable conditions of methyl compound availability and energy constraints, highlighting their resilience and versatility within the gut ecosystem.
-
In porcine models, developmental stages and dietary regimes exert profound regulatory effects on methanogenic activity. Suckling piglets display low gene abundance related to the acetoclastic pathway, which rapidly increases to parental levels during the nursery phase. In contrast, genes associated with the methylotrophic pathway decline with age[7]. Metatranscriptomic analyses have revealed that Methanobrevibacter dominates in hydrogenotrophic metabolism, whereas Methanosphaera relies on methyl reduction pathways[9]. Notably, the transcriptional activity of these taxa significantly surpasses their genomic abundance. For instance, the transcript levels of Methanosphaera cuniculi and Methanosphaera stadtmanae exceed their genomic abundance by 27- and 30-fold, respectively[7,102], underscoring their central role in H2/methyl metabolism.
The metabolic networks of methanogens intricately interact with the host's nutritional metabolism. A high-fiber diet can enhance the activities of fibrolytic enzymes (GH3) and the metabolic pathway of formate in the porcine gut[7]. Furthermore, in vitro studies shows that Methanobrevibacter can maintain a high abundance under fecal microbiota co-culture conditions with high concentrations of oligofructose and pectin[103]. Conversely, hydrogenotrophic methanogenesis is negatively correlated with starchase (GH13) and lactate metabolism[7]. Methanogenic activity is positively associated with the intestinal concentrations of formate and acetate[57]. In the microbiomes of piglets, the activation of the sulfate/fumarate reduction pathway reduces the acetate/propionate ratios and suppresses methanogenesis, indicating ecological competition for hydrogen sinks[7]. Additionally, ammonia inhibition disrupts acetoclastic methanogenesis and syntrophic chains by binding to coenzymes (such as Coenzyme M) or blocking the active sites of MCR[104].
Specific methanogen strains have been shown to correlate with host phenotypes. The abundances of Methanobrevibacter smithii and Methanobrevibacter sp900769095 are positively associated with porcine body weight[105]. The symbiotic strain Candidatus Methanomethylophilus alvus Mx1201 potentially modulates the host's protein synthesis and lipid metabolism through the regulation of the shikimate pathway and bile resistance genes[100]. The metabolic repertoires of methanogens include L-valine/L-isoleucine biosynthesis, isobutanol production, and carbohydrate-active enzyme (CAZyme) families (AA3, GH43, GT2, AA6, CE9), indicating their potential in amino acid and carbohydrate metabolism[106]. The dominance of acetotrophic Methanosarcina in FOG (Fats, Oil, and Grease) co-digestion systems highlights its role in lipid metabolism[107]. In rat models, the depletion of methanogens induced by bromochloromethane increases daily weight gain and adiposity, suggesting that methanogen-targeted interventions could be useful for weight management. The diversity of methanogens is positively correlated with the intensity of fiber fermentation in the porcine hindgut[108], and their redox-balancing metabolism affects the host's energy allocation and adipogenesis[39]. These findings elucidate the profound metabolic plasticity of methanogens in the host's energy partitioning and lay the molecular foundations for the targeted modulation of intestinal methane emissions and nutrient utilization efficiency.
Methanogens and gut health
-
Methanogens have been found to be disproportionately abundant in patients suffering from IBD, periodontal disease, obesity, cancer and diverticulosis[109,110]. The ecological functions of gut methanogens and their associations with various diseases have become crucial areas of focus in clinical microbiological research.
Recent scientific progress has established intestinal methanogen overgrowth (IMO) as a distinct pathological condition that is independent of small intestinal bacterial overgrowth (SIBO). IMO is characterized by the excessive proliferation of methanogens and elevated levels of methane in the breath (≥ 10 parts per million [ppm])[111]. This condition has a strong correlation with constipation and an extended colonic transit time[109,112]. This phenomenon highlights the dual metabolic impacts of methanogenesis. On one hand, the scavenging of hydrogen by methanogens alleviates metabolic inhibition. On the other hand, the associated energy expenditure might exacerbate the metabolic burden on the host. Studies in horses have demonstrated a positive association between long-term colonization by Methanobrevibacter and mortality[113], while syntrophic interactions between Christensenella and methanogens have been linked to weight loss in humans[114].
In the context of specific disease, patients with constipation-predominant irritable bowel syndrome (IBS-C) show increased fecal microbial α-diversity and a higher abundance of Methanobrevibacter, especially Methanobrevibacter smithii[115,116]. Breath testing has revealed that in individuals who are high methane emitters (with CH4 levels ranging from 5 to 75 ppm), there is a 1,000-fold enrichment of Methanobrevibacter smithii[57]. Mechanistically, mevalonate pathway inhibitors, such as lovastatin, can alleviate constipation by suppressing the methanogenic activity[117]. Paradoxically, in patients with IBD, there is a dysbiosis in the methanogen community. The total abundance of methanogens in IBD patients exceeds that in healthy controls[110], yet the core species Methanobrevibacter smithii is depleted[54], while Methanosphaera stadtmanae experiences proliferation[118]. This pathogen activates the TLR8-dependent NLRP3 inflammasome pathways in monocyte-derived dendritic cells (moDCs), which, in turn, triggers the release of pro-inflammatory cytokines and leads to hyperactivation of the innate immune system[119,120]. In colorectal cancer (CRC) patients, the abundance of Methanobacterium and Methanosarcina is reduced, and Methanocaldococcus and Methanotorris are depleted in the advanced stages of CRC. This suggests that the exhaustion of methanogens may accelerate the process of tumorigenesis[121]. These findings emphasize the functional heterogeneity among methanogens within inflammatory microenvironments.
The influence of methanogens on the host's metabolic health exhibits bidirectional regulation (Fig. 1). Zhou et al. reported that an increase in fumarate reductase activity leads to the accumulation of succinate in the intestines of piglets, which can contribute to post-weaning diarrhea[122]. In contrast, Chen et al. observed a sharp decline in fumarate reductase expression in healthy piglets after weaning[7]. These findings suggest that methanogens may reshape their intestinal H2 consumption patterns through hydrogenotrophic methanogenesis, thereby competing with fumarate reductase. Consequently, targeting this interaction may represent a potential therapeutic strategy for alleviating post-weaning diarrhea in piglets. The enrichment of Methanobrevibacter associated with anorexia may adapt to hypocaloric states through H2 oxidation-induced thermogenesis, contributing to the maintenance of metabolic homeostasis[114,123]. Members of the Methanomassiliicoccales order, such as Methanomassiliicoccus luminyensis, metabolize trimethylamine (TMA) through pyrrolysine-dependent methyltransferase systems[38,124] by methylotrophic methanogenesis. In this way, they inhibit the conversion of TMA into the pro-atherogenic trimethylamine-N-oxide (TMAO)[125], thereby presenting potential therapeutic applications for cardiovascular and renal diseases[100,126]. In patients with multiple sclerosis (MS), there is a negative correlation between the abundances of Methanobrevibacter smithii and Methanobrevibacter sp900766745 and disease severity. Additionally, treatment with dimethyl fumarate increases the colonization levels of these methanogens, accompanied by weight reduction[127]. Conversely, long-term colonization by methanogens has been inversely correlated with host longevity, potentially accelerating the aging process via the depletion of redox potential[113].
From an immunological perspective, ether lipid vesicles (archaeosomes) derived from Methanobrevibacter smithii can induce influenza hemagglutinin-specific CD8+ T cell responses and facilitate the vertical transfer of maternal antibodies[128,129]. In MS patients, Methanobrevibacter smithii activates the TLR8-NLRP3 inflammasome pathway, leading to the upregulation of genes such as CASP1, TRAF5, and STAT5B, which are associated with interferon (IFN) signaling, IL-2 pathways, and PPAR/RXR regulation[130]. It also significantly alters the expression of antimicrobial peptide genes in moDCs[120]. Similarly, Methanobrevibacter stadtmanae can induce the robust release of pro-inflammatory cytokines in moDCs[120]. Antimicrobial peptides (AMPs) are a crucial component of intestinal immunity. They exert immune functions not only against bacteria and fungi but also against methanogens. Bang et al.[131,132] compared the sensitivity of three methanogenic archaea—Methanobrevibacter smithii, Methanomassiliicoccus luminyensis, and Methanosphaera stadtmanae—with human cathelicidin-derived peptides LL32 and LL20, as well as the antimicrobial peptide NK-lysin. The tested methanogens exhibited different levels of sensitivity, with M. smithii being the most susceptible. These findings clearly demonstrate that the antimicrobial peptides released by human innate immune cells target not only bacteria and fungi but also archaea.
Developmental studies have shown that intestinal methanogenesis in piglets, as indicated by the abundance of the mcrA gene, is lower than that in adult pigs. Elevated activities of sulfate reductase (encoded by asrA and aprA) and fumarate reductase (encoded by frdA) suggest that H2 is preferentially diverted towards sulfate and fumarate reduction processes[7]. Notably, the accumulation of succinate mediated by fumarate reductase can trigger weaning-associated diarrhea in piglets, indicating the potential of modulating H2 sinks as a therapeutic strategy[7,122]. These findings systematically elucidate the involvement of methanogens in the host's pathophysiology through mechanisms such as metabolic network remodeling, immunophenotypic regulation, and energy homeostasis modulation. As a result, they provide the molecular basis for targeted microbiome engineering and the development of novel therapeutic approaches.
-
Metabolic interactions between methanogens and bacteria significantly influence methanogenic efficiency through intricate hydrogen metabolism and electron transfer mechanisms. Methanogens play a pivotal role in sustaining syntrophic bacterial activity by maintaining extremely low hydrogen partial pressures (H2 < 0.1 Pa), thereby establishing cross-domain metabolic coupling[133]. The hydrogen generated by carbohydrate-fermenting bacteria, including Mogibacterium, Pyramidobacter, Christensenella, Anaerostipes, Ruminococcus, and Aminipila, serves as a substrate for methanogens (such as Methanobrevibacter species) to reduce CO2 to CH4[57,113,134]. Conversely, Bacteroides species can alter hydrogen's availability by recycling mucin glycans, thus fueling nitrate/sulfate-reducing bacteria and subsequently suppressing methanogenesis[135]. Sulfate-reducing bacteria (e.g., Desulfovibrio) and Fibrobacter succinogenes (through phosphotransacetylase-driven succinate/propionate synthesis) further limit the accessibility of H2 through substrate competition[45,113,135]. Methylotrophic methanogens, like Methanosphaera stadtmanae, can inhibit hydrogenotrophic methanogens by reducing the H2 concentration below 0.1 Pa, inducing interspecific metabolic suppression[136]. Additionally, the ammonium produced from bacterial protein degradation is assimilated by methanogens (e.g., Methanobrevibacter smithii) via ammonium transporters (AmtB, encoded by MSM0234)[10].
Associations between health or disease states and the gut microbiota reveal diverse correlations between methanogen abundance and specific bacterial taxa. In healthy individuals, Akkermansia, Phascolarctobacterium, and Eubacterium exhibit positive associations with methanogens, whereas Bacteroidetes and Veillonellaceae show negative correlations[137]. In patients with IBS, the positive associations between methanogens' abundance and bacterial diversity/richness are more pronounced. Co-occurring taxa, such as Christensenella and members of the Ruminococcaceae family, synergistically contribute to metabolic dysregulation[57,138]. Notably, the abundance of Methanomassiliicoccales is correlated with TMA-producing bacteria[100], and Bacteroides fragilis may modulate methanogens' distribution by regulating the colonic tumor microenvironment[121]. Mathematical modeling has demonstrated that sulfate-reducing bacteria compete more strongly with methanogens for H2 than reductive acetogens in the human intestine[139]. Methylotrophic archaea (e.g., Methanosphaera stadtmanae) engage in metabolic coupling with pectinolytic bacteria (e.g., Bacteroides) by utilizing the methanol released by the latter[28]. Overgrowth of Lachnospiraceae, Lactobacillaceae, and Streptococcus can suppress methanogens' activity by reducing the pH, thereby decreasing CH4 production[140]. These complex interaction networks shed light on the dynamic equilibria of carbon, hydrogen, and electron fluxes within gut microbiomes, offering valuable ecological insights for the targeted modulation of methanogenic modulation.
-
Methanogens, as integral archaeal constituents of the gut microbiota in monogastric animals, display distinct host-specific distribution patterns. Methanobrevibacter smithii is predominant in the intestines of both humans and pigs, whereas Methanomassiliicoccales and Methanosphaera assume specialized ecological roles in rabbits and companion animals. The abundance of methanogens is intricately influenced by multiple factors. The host's developmental stages play a crucial role, as evidenced by maternal transmission in neonates and significant shifts post-weaning. Dietary components, such as high-fiber diets that promote the growth of hydrogenotrophic methanogens, also profoundly impact their population dynamics. Disease states have differential effects. For instance, IBD leads to reduced methanogen colonization, while IBS and obesity are associated with methanogen overgrowth.
The three primary methanogenic pathways (hydrogenotrophic, acetoclastic, and methylotrophic) exemplify the metabolic duality of methanogens in modulating host health. The hydrogenotrophic pathway, which enhances fiber degradation, has been linked to constipation in certain contexts. The acetoclastic pathway is mainly involved in syntrophic lipid digestion and is restricted to specific methanogen lineages. The methylotrophic pathway can reduce the toxicity of trimethylamine but may also trigger inflammatory responses. Methanogens engage in syntrophic interactions with fibrolytic bacteria, such as Christensenella, by efficiently scavenging H2. However, they also compete with sulfate-reducing bacteria and acetogens for substrates, influencing the overall metabolic balance within the gut microbiota.
Notwithstanding the significant progress in the field, several research gaps remain. Current studies often exhibit a bacterial-centric bias, overlooking the unique contributions and functions of methanogens. The pathogenic thresholds of methanogens in various host conditions are yet to be precisely defined, and cross-species comparisons are relatively limited. To fully elucidate the roles of methanogens in the microbiota–host axis, future research endeavors should focus on integrating multi-omics approaches to comprehensively map methanogens' metabolic networks. Developing 'archaebiotics' for targeted modulation of methanogen communities and engineering ecological strategies, such as enhancing hydrogen sinks, hold promise for mitigating methane-related disorders. Unraveling these dynamics will not only advance the development of novel therapies for metabolic diseases but also optimize their utilization in animal production and contribute to reducing environmental methane emissions, thereby addressing both health and environmental challenges.
This study was funded by National Natural Science Foundation of China (31872369). Figures are created in
https://BioRender.com .-
Not applicable.
-
The authors confirm contributions to the paper as follows: study conception, manuscript revison and funding acquisition: Luo Y; writing − original draft: He J, Dai Z; writing − review and editing: Deng X, Li H, Huang S, Wu A, Mao X. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Xiaofeng Deng, Hua Li, Shuangmei Huang
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of Nanjing Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Deng X, Li H, Huang S, He J, Dai Z, et al. 2026. Methanogens as hidden architects of intestinal ecology in monogastric animals: distribution and function. Animal Advances 3: e003 doi: 10.48130/animadv-0025-0039 

Methanogens as hidden architects of intestinal ecology in monogastric animals: distribution and function
- Received: 18 June 2025
- Revised: 23 July 2025
- Accepted: 28 July 2025
- Published online: 23 January 2026
Abstract: Methanogens, strictly anaerobic archaea within the gut microbiota of monogastric animals, play dualistic roles in host health through their unique molecular and metabolic characteristics. Distinguished by conserved 16S rRNA sequences, ether-linked membrane lipids, and archaea-specific cofactors (e.g., Coenzymes M and F420), these microorganisms drive methanogenesis via hydrogenotrophic, aceticlastic, and methylotrophic pathways. Despite their low abundance (~1%–10% of gut anaerobes), methanogens critically regulate the host's metabolic homeostasis by scavenging hydrogen to enhance fibrolytic bacterial activity, improving dietary fiber degradation and nutrient absorption. However, their overgrowth correlates with metabolic disorders such as irritable bowel syndrome (IBS), inflammatory bowel disease (IBD), obesity, and chronic constipation, underscoring a functional duality. Host-specific distribution patterns reveal the dominance of Methanobrevibacter smithii in humans and pigs, while Methanomassiliicoccales and Methanosphaera occupy niche roles in rabbits and companion animals. Their abundance is shaped by developmental stages (e.g., maternal transmission, post-weaning shifts), dietary fiber intake, physiological states (e.g., IBD-linked reduction, IBS/obesity-associated proliferation), and environmental stressors (e.g., ammonia tolerance). Current research limitations include bacterial-centric biases, undefined pathogenic thresholds, and scarce cross-species comparisons. Future directions emphasize multi-omics integration to elucidate methanogen–host interactions, develop 'archaebiotics' for targeted population modulation, and engineer ecological strategies (e.g., enhancing hydrogen sinks) to mitigate methane-related disorders. Advancing this knowledge will optimize therapeutic interventions for metabolic diseases, improve nutrient utilization, and reduce environmental methane emissions.
-
Key words:
- Monogastric animals /
- Methanogens /
- Metabolism /
- Health /
- Interaction