








-
C.H. Waddington first defined epigenetics as developmental processes between the genotype and phenotype. Advancements in molecular biology and high-throughput sequencing propelled epigenetic studies into the molecular and big-data eras. Today, epigenetics is a cornerstone of genetics, investigating heritable gene expression changes without DNA sequence alterations, including DNA methylation, histone modifications, non-coding RNAs (ncRNAs), chromatin remodeling factors, and others. Epigenetic modifications in germ cells can be transmitted to offspring during gametogenesis and fertilization, enabling somatic cells of the offspring to carry epigenetic modifications similar to those of the parent, thereby achieving heritability[1]. In plants, certain somatic cell epigenetic modifications can be passed to offspring through vegetative propagation. For example, in plant varieties propagated through methods such as cuttings and tillering, the epigenetic modifications in somatic cells can be stably transmitted to offspring plants[2].
With a short life cycle, mutant libraries, and high transformation efficiency, Arabidopsis thaliana has contributed to unraveling the mechanisms of epigenetic regulation, leading to the identification of over 100 key regulators to date. This review focuses on epigenetic regulatory mechanisms and their applications in plant biology (Fig. 1). These findings will provide evolutionary and functional clues for identifying analogous mechanisms across organisms or uncovering universal epigenetic principles, ultimately advancing application of epigenetics in agriculture and biotechnology.
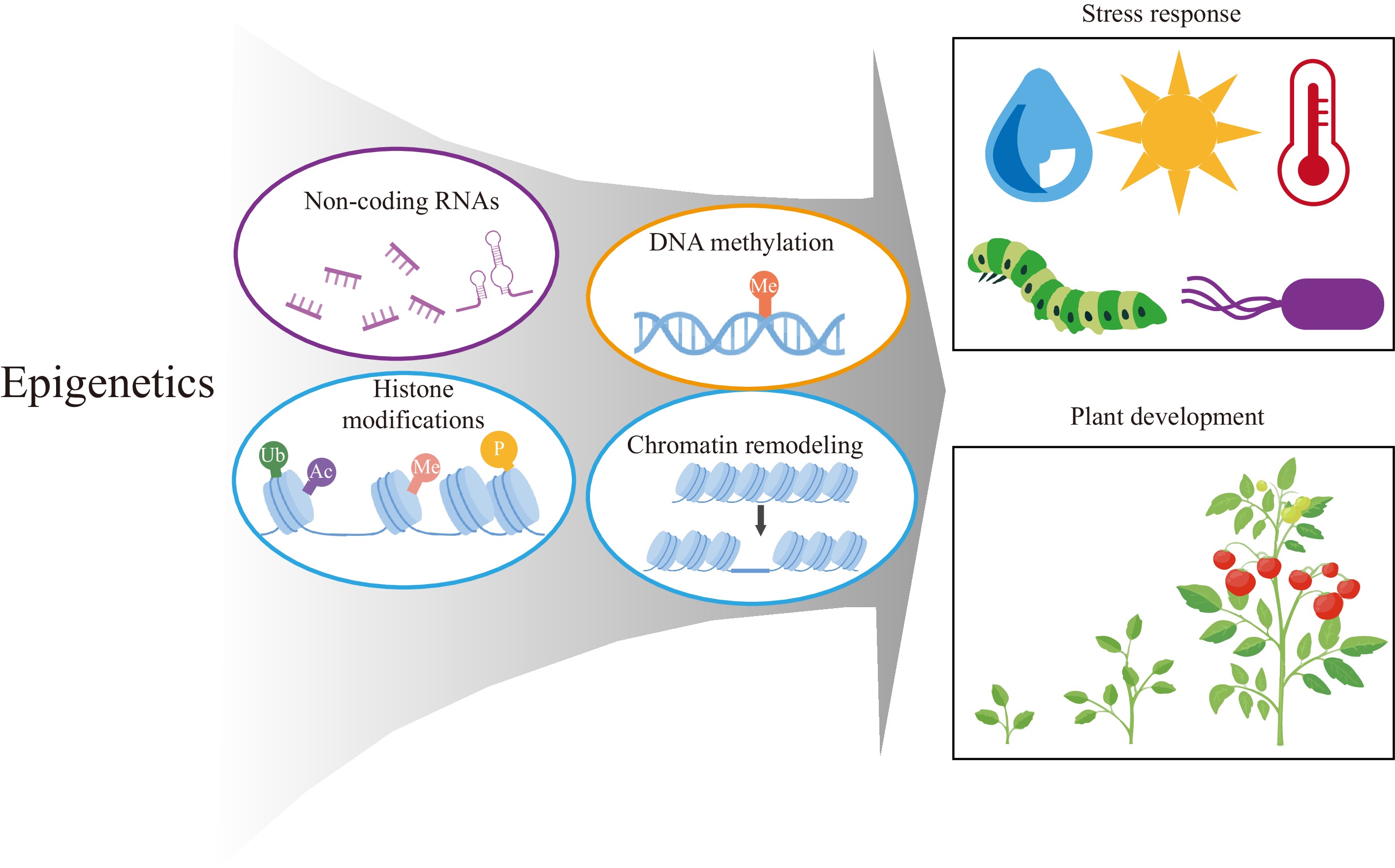
Figure 1.
Framework of epigenetic research in plants. The dynamic regulatory roles of DNA methylation, histone modifications, non-coding RNAs, and chromatin remodeling factors in plant development and stress responses.
-
DNA methylation, which occurs primarily at cytosine bases (5-methylcytosine, 5mC), is a common DNA modification that plays a critical regulatory role within eukaryotic genomes. Whereas DNA methylation mainly occurs at CG dinucleotides in mammals, plants exhibit DNA methylation in three sequence contexts: CG, CHG, and CHH (H = A/T/C), each mediated by distinct enzymatic machinery. The methylation landscape at specific loci is dynamically modulated through the combined actions of DNA methyltransferases and demethylases, which are responsible for establishing, maintenance and demethylation[3].
RNA-directed DNA methylation (RdDM) is a plant-specific mechanism where small interfering RNAs (siRNAs) recruit the DNA methyltransferase DRM2 to specific locations to establish de novo DNA methylation. The maintenance of DNA methylation is also a crucial epigenetic process in plants, ensuring the heritability of gene expression patterns and genome stability. In Arabidopsis, such maintenance relies on different mechanisms for each sequence context: CG methylation is predominantly maintained by MET1, a methyltransferase that identifies hemimethylated CG sites during replication; for CHG methylation, its maintenance relies on chromomethylase 3 (CMT3); the maintenance of CHH methylation is mediated by DRM2 or CMT2 in different genomic regions[4]. In plants, active demethylation depends primarily on bifunctional DNA glycosylases (ROS1, DME, DML2, and DML3) that replace the excised 5mC with an unmodified cytosine through the base excision repair pathway to accomplish the active demethylation[5].
DNA methylation plays a crucial role in regulating gene expression and silencing transposable elements, which are essential for maintaining normal growth, development, and genome stability. Compared to Arabidopsis thaliana, crop species with larger genomes often have a much higher TE content and a larger number of TEs near to genic regions. Consequently, crop DNA methylation mutants usually display severe developmental defects or lethality, highlighting the fundamental role of epigenetic regulation in maintaining the genomic integrity of agriculturally important species[6,7]. In Arabidopsis thaliana, DNA methylation in the promoter of the FWA gene silences its expression and promotes timely flowering. In fwa epi mutants, the loss of DNA methylation in the promoter region triggers the FWA expression, resulting in a late-flowering phenotype. This characteristic establishes FWA as a pivotal reporter for studying DNA methylation dynamics. Its methylation-sensitive expression patterns and associated phenotypic changes enable direct evaluation of epigenetic editing efficacy on DNA methylation states for researchers, making it a benchmark for validating the efficacy and precision of novel epigenetic editing tools. Furthermore, FWA serves as both a mechanistic model for dissecting DNA methylation-mediated regulation and a screening platform to identify new epigenetic regulators[8].
DNA demethylases are pivotal regulators in multiple plant developmental pathways. In Arabidopsis, the demethylase ROS1 regulates stomatal development and seed dormancy, functions in plant–microbe interactions, and plays a core regulatory role in various stress responses. In rice (Oryza sativa), OsROS1a and its homologs DNG701/DNG704 regulate DNA methylation in gametes and zygotes, affecting the development of the embryo and the endosperm aleurone layer, thereby enhancing nutritional content[9]. Like ROS1, DME also plays a role in plant development, particularly in reproductive processes. DME-mediated pericentromeric demethylation enables transposon reactivation and male gametophyte development in pollen vegetative cells of Arabidopsis[10]. DME establishes maternal hypomethylation in the endosperm, which influences gene imprinting and seed development. DME acts synergistically with ROS1 to ensure male fertility in Arabidopsis. Both enzymes are essential for the normal growth of pollen tubes, functioning semi-redundantly in pollen vegetative cells to mediate DNA demethylation[11].
DNA methylation also has roles in fruit ripening. For example, the ripening of tomato fruit is driven by DNA demethylation via SlDML2, which activates the promoters of key genes such as RIN, NOR, and PSY1[12]. In addition, low temperatures delay fruit ripening by inhibiting the demethylation activity of SlDML2. The m6A demethylase SlALKBH2 also influences tomato fruit ripening by controlling the stability of SlDML2 mRNA. By contrast, the ripening of orange is associated with global hypermethylation, which is caused by the downregulation of demethylases[13].
-
Histone modifications, mainly occurring on the N-terminal tails of core histones (H2A, H2B, H3, and H4), are covalent changes that regulate chromatin structure and gene expression. These modifications include various types such as methylation, acetylation, phosphorylation, ubiquitination, SUMOylation, and ADP-ribosylation[14], with methylation and acetylation being the most well-characterized. Histone methylation occurs at two residues: arginine and lysine. Histone acetylation typically occurs on lysine residues in the N-terminal regions of histones, weakening the histone–DNA interaction and facilitating transcription factor binding to DNA to activate gene expression.
Histone modifications are dynamically regulated by site-specific methylation levels and acetylation states of specific amino acids, which collectively orchestrate transcriptional regulation. Methylation and acetylation are established by methyltransferases and acetyltransferases, removed by demethylases and deacetylases, respectively. Thus, the equilibrium of these modifications is maintained by a 'writer-eraser-reader' system, in which writers establish modifications and erasers remove them, enabling precise epigenetic control. In terms of writers, histone methylation is primarily performed by SET-domain histone methyltransferases and members of the protein arginine N-methyltransferase (PRMT) family, such as enhancer of zeste homolog 2 (EZH2) and ARABIDOPSIS HOMOLOG OF TRITHORAX (ATX) proteins in Arabidopsis. Acetylation is performed by histone acetyltransferases (HATs), such as the HAC family in Arabidopsis. In terms of erasers, methylation is removed by lysine-specific demethylase 1 (LSD1) and jumonji domain-containing proteins (JMJs) with the JmjC domain, such as REF6/JMJ12 in Arabidopsis and JMJ706 in rice[15], and acetylation is removed by histone deacetylases (HDACs) such as the HDAC family proteins in rice. Reader proteins recognize a variety of specific histone modifications through reader domains, including plant homeodomain (PHD), bromo adjacent homology (BAH), tudor, chromodomain, bromodomain, and proline-tryptophan-tryptophan-proline (PWWP) domains. These readers recruit chromatin remodelers or transcriptional machinery to translate epigenetic signals into biological outputs such as transcriptional activation or repression[16].
Histone modifications are dynamically modulated during developmental processes and in response to environmental changes. In Arabidopsis, the flowering repressor FLOWERING LOCUS C (FLC) is regulated by complex histone modifications. Before vernalization, the chromatin at the FLC locus exists in active histone modifications such as H3K4me3. Under non-vernalizing conditions, these activation-associated histone marks (H3K4me3, H3K36me3, and H3 acetylation) are maintained through the recruitment of histone-modifying enzymes by the FRI (FRIGIDA) complex, which stabilizes an open chromatin conformation to ensure sustained high expression of FLC, thereby inhibiting flowering; after vernalization, multiple pathways establish and maintain H3K27me3 to repress FLC expression and thus promote flowering. The Polycomb Repressive Complex 2 (PRC2) is a central mediator of FLC silencing during vernalization. Upon cold exposure, VAL1 or VAL2 proteins bind to the cis-acting cold memory element (CME) at the FLC locus, facilitating the recruitment of PRC2. This complex catalyzes the trimethylation of histone H3 at lysine 27 (H3K27me3), establishing a transcriptionally repressive heterochromatic structure that epigenetically silences FLC expression[17]. During vernalization, the histone acetylation landscape at the FLC chromatin undergoes dynamic remodeling. For example, the deacetylation of H3K9 and H3K14 can inhibit the expression of FLC. In the early stage of vernalization, the histone deacetylation mediated by VAL1 promotes the silencing of FLC, and subsequently, the PRC2-mediated H3K27me3 modification further maintains the silenced state, providing a robust epigenetic mechanism for FLC expression repression. During wheat embryo development, the dynamic regulation of H3K27 trimethylation (H3K27me3) and H3K27 acetylation (H3K27ac) ensures stage-specific transcriptional control of gene expression[18].
Changes in histone modifications also modulate gene expression to regulate plant resistance to abiotic and biotic stresses. Recent studies have characterized a novel histone acylation modification (H4K8hib) in rice, demonstrating that the histone deacetylase HDA705 serves as the key enzyme responsible for removing H4K8hib marks. Disruption of HDA705 significantly boosts H4K8hib deposition at defense-related genes, upregulates their expression and enhances blast resistance. This discovery highlights an epigenetic link between histone acylation dynamics and plant immunity, offering new epigenetic strategies for crop disease resistance engineering[19].
-
Non-coding RNAs (NcRNAs) represent functional RNA molecules that do not undergo translation into proteins. The most extensively studied are the regulatory ncRNAs, which include long ncRNAs (lncRNAs), microRNAs (miRNAs), and siRNAs. LncRNAs are generally defined as transcripts longer than 200 nucleotides in length, whereas miRNAs and siRNAs are distinct classes of short ncRNAs, typically 20–30 nucleotides in length.
NcRNAs exhibit diverse genomic origins, arising from intergenic, promoter, exonic, or intronic regions of genes. They can be associated with transposable elements, enhancers, or transcription start sites and may derive from either the sense or antisense strands[20]. Numerous lncRNAs have crucial regulatory roles in plant growth and reproductive development, participating in fundamental biological processes such as photomorphogenesis, vascular tissue development, stress responses, reproductive development, and fruit ripening. At the DNA and chromosome levels, lncRNAs can regulate gene expression through histone modification[21]. Some lncRNAs serve as central platforms for recruiting epigenetic modifier complexes to specific genomic regions, thereby altering DNA or chromatin modifications and modulating the expression of target genes.
At the transcriptional level, lncRNAs can recruit protein complexes to chromatin and guide their localization to regulate gene expression. For instance, APOLO in Arabidopsis regulates polar auxin transport by influencing the formation of chromatin loops[22]. Some lncRNAs regulate gene expression by affecting transcriptional initiation, elongation, termination, or post-transcriptional processes. A notable example is the Arabidopsis lncRNA IPS1, which functions as a target mimic for the microRNA miR399. By binding to miR399, IPS1 inhibits its RNA silencing effect on the target gene PHO2, thereby increasing PHO2 expression levels[23]. Other specific lncRNAs participate in alternative splicing of mRNAs, regulating plant development by modifying mRNA splicing patterns[24].
miRNAs are phylogenetically conserved small RNAs (20–24 nucleotides) that predominantly regulate critical transcription factors, playing essential roles in plant development and stress response. In plants, miRNAs are encoded by MIR genes and transcribed by RNA polymerase II (Pol II) into long primary miRNA transcripts (pri-miRNAs) containing stem-loop structures. miRNA precursors undergo endonucleolytic cleavage by Dicer-like 1 (DCL1), generating precursor miRNAs (pre-miRNAs) with stem-loop architecture. Subsequent processing by DCL1 produces miRNA/miRNA* duplexes, which are stabilized by 3′ terminal methylation mediated by HUA ENHANCER 1 (HEN1) to prevent uridylation-dependent degradation. The mature miRNA strand is then incorporated into the RNA-induced silencing complex (RISC), which contains ARGONAUTE (AGO) proteins. Through sequence complementarity, the RISC complex post-transcriptionally regulates target gene expression through mRNA cleavage or translational repression[25].
miRNAs typically target and negatively regulate important transcription factors. For example, Arabidopsis thaliana miR156 targets the transcription factor SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) and participates in the regulation of processes such as flowering, shade avoidance, insect resistance, drought resistance, heat-shock memory, and plant maturation. In rice, the miR156–SPLs module controls key agricultural traits such as ideal plant architecture, disease resistance, grain quality, and seed dormancy. In maize, miR156 regulates leaf development and the differentiation of reproductive organs by targeting different members of the SPL family[26,27]. These findings suggest that microRNA regulatory networks exhibit both conservation and functional specificity across different species.
siRNAs are categorized into two main classes: RNA-dependent RNA polymerase 6 (RDR6)-dependent secondary siRNAs and RNA polymerase IV-dependent siRNAs (P4-siRNAs)[28]. Secondary siRNAs are derived from non-coding genes and protein-coding genes in large gene families. Their dsRNA precursors are processed by DCLs to produce phased siRNAs that silence both the original locus in cis and homologous loci in trans[29]. Such phasiRNAs (phased secondary small interfering RNAs) have important roles in the regulation of plant development and stress responses[30]. P4-siRNAs are typically 24 nt in length and are generated from heterochromatic regions and transposable elements. They participate in RdDM, inducing transcriptional gene silencing. In rice, a MITE transposon upstream of the PigmS (Pigm Susceptible) gene produces siRNAs that suppress the expression of PigmS in leaves through DNA methylation, thereby releasing the disease-resistance gene PigmR (Pigm Resistant) to promote broad-spectrum resistance to rice blast[31]. Transposon-derived siRNAs also play important regulatory roles in the bacterial blight resistance of rice and drought resistance of maize[32]. With the advancement of research, it has been recognized that small RNAs are closely associated with key agronomic traits. Techniques such as Short Tandem Target Mimic (STTM) for silencing miRNAs by mimicking short tandem target fragments, or CRISPR/CAS9-based gene editing to generate miRNA mutants, enable precise regulation of miRNA expression levels, thereby modulating agricultural traits. These approaches have already been successfully implemented in certain crop species, such as in rice MIR396ef mutants, in which both grain size and panicle branching were significantly increased; Simultaneous silencing of MIR482b and MIR482c in tomato enhanced resistance to pathogenic bacteria; In cotton, multiple mutant lines generated through random mutagenesis of MIR482 showed improved resistance to Verticillium dahliae[33]. These advances have emerged as transformative approaches for crop breeding and trait improvement.
-
Chromatin is a complex structure composed of genomic DNA, histones, and non-histone proteins in eukaryotes; its structure and state play a crucial role in regulating gene expression. Chromatin remodeling factors are indispensable for multiple plant biological processes. Chromatin remodelers harness the energy from ATP hydrolysis to remodel DNA–histone interactions, dynamically regulating chromatin architecture. Chromatin remodeling factors can be divided into four major families (SWI/SNF, ISWI, CHD, and INO80/SWR1) according to the structure and function of their ATPase subunits[34]. Members of these families are highly conserved in yeast, fruit flies, and humans, and corresponding homologous genes are also present in plants. Arabidopsis contains three classes of SWI/SNF chromatin remodeling factors: BRAHMA(BRM), SPLAYED(SYD), and MINUSCULE1/2(MINU1/2). Knockout of any class of SWI/SNF remodeling factors in Arabidopsis is either lethal or causes severe pleiotropic growth and developmental defects, demonstrating their essential roles in growth and development[35]. Other remodelers have also been reported to participate in the regulation of plant hormone signaling pathways and biotic and abiotic stress responses by influencing transcription, DNA replication, and genome stability[34].
Chromosome Conformation Capture (3C) technology and its derivative methods, including Circularized Chromosome Conformation Capture (4C), Chromosome Conformation Capture Carbon Copy (5C), and High-throughput Chromosome Conformation Capture (Hi-C) technology, can capture the three-dimensional structural information of chromatin with high resolution throughout the entire genome. The three-dimensional genomic structure of plants includes nuclear compartments, topologically associating domains (TADs), and chromatin loops. These 3D genomic structures regulate gene expression via compartmentalization (A/B compartments), TAD-mediated enhancer-promoter interactions, and chromatin loop formation connecting regulatory elements to target gene promoters[36]. Research on chromatin remodeling factors and 3D genomes has identified key regulators of crop yield. For example, the OsINO80 remodeler maintains transposon silencing through H3K27me3 and H3K9me2 deposition, enhancing genomic stability under stress and offering targets for stress-resistant breeding[37]. In tomato, the HSFA1a transcription factor remodels 3D chromatin architecture to activate heat-responsive genes under heat stress[38]. Although certain progress has been made in understanding the mechanisms and applications of chromatin remodeling factors and the 3D genome in plants, there are still many unknown areas that need to be explored.
-
Spontaneous epigenetic variations are common in crops, and the importance of epigenetic modifications for crop domestication and improvement is increasingly recognized[39]. Epigenetic modifications can rapidly adapt to environmental stimuli; therefore, epigenetic variations are highly prevalent in biological evolutionary processes and exhibit a significantly higher mutation frequency compared to genetic variations. Numerous instances of natural epigenetic variation exist across plant species. In Linaria vulgaris, hypermethylation of the Lcyc gene leads to transcriptional silencing, altering the floral symmetry. In rice, the methylation level of the RAV6 promoter region affects leaf angle by modulating BR homeostasis. In the tomato Cnr mutant, DNA methylation in the CNR promoter represses its expression, influencing fruit ripening[4]. During cotton evolution, demethylation of COL2 upregulated its expression, promoting photoperiodic flowering and global adaptability[40]. These examples suggest that epialleles control important agronomic traits in various crops. In addition, the development of epigenetic recombinant inbred lines (epiRILs) has demonstrated the potential use of epigenetic variations in breeding programs[41], which highlights the heritability of methylation patterns and phenotypic diversity.
The above examples illustrate spontaneous mutations in plants. Currently, advanced gene-editing tools enable precise editing of epigenetic modifications. The CRISPR system, initially identified in bacteria and archaea, is a revolutionary gene-editing tool that uses a guide RNA (gRNA) to target specific DNA sequences. The CRISPR/dCas9 system has been reengineered for epigenome editing, which retains its DNA-binding capacity but cannot perform cleavage. Epigenetic effectors can be linked to dCas9 to enable locus-specific epigenetic changes, offering broad engineering advantages. By fusing catalytic domains, such as Dnmt3a, Dnmt3L, and TET1, with dCas9, targeted DNA methylation or demethylation at specific genomic loci can be achieved[8]. These dCas9-guided DNA methylation and demethylation systems, termed 'CRISPRoff' and 'CRISPRon' systems, enable gene silencing and activation, respectively[42]. Effector domains, including acetyltransferases (such as p300) and deacetylases (such as HDAC3), can be used to regulate gene expression. Fusion proteins such as dCas9-KRAB and dCas9-p300 have been used to analyze the functions of promoters and enhancers[43]. Such epigenetic effectors have also been successfully used for DNA methylation editing and gene expression regulation in Arabidopsis[8]. Notably, epigenetic regulation of target genes via DNA methylation or demethylation in plants exhibits transgenerational heritability, thereby transmitting dynamically modulated gene expression patterns to subsequent generations[44,45].
The following examples illustrate the practical applications of epigenetic modifications in plants, highlighting the potential of epigenome editing technologies to alter plant traits. In Arabidopsis, H3K4me3 recruits DNA demethylases to enhance locus-specific DNA demethylation, a mechanism that has been leveraged to develop efficient epigenome-editing tools[46]. In chrysanthemums, epigenetic alleles of CmMYB6 regulate flower color variation, and targeted epigenome editing enables modulation of its expression to control pigmentation intensity[47].
Spontaneous DNA methylation mutations are non-random across genomes, indicating that some genomic regions have more unstable methylation. This poses challenges for targeted editing. Epigenetic changes, affected by environmental factors, can make plant traits unstable across generations, impacting breeding sustainability. For instance, soybean MSH1-induced epi alleles are stable for three generations under controlled conditions; however, environmental changes may compromise their stability[48]. The complexity of genetic mechanisms presents challenges in predicting and controlling epigenetic modifications in breeding. Further work is required to improve the efficiency and specificity of epigenomic editing. To make this technology suitable for crop breeding, it will be necessary to reduce off-target effects, optimize the expression levels of gRNAs, effector domains, and dCas protein variants, and enhance the transgenerational stability of the edits.
-
Research on plant epigenetic regulation is burgeoning and promises to have broad applications in plant biology, breeding, agricultural production, and related fields. Continued advances in sequencing, proteomics, and associated technologies will enable the more widespread performance of multi-omics studies in which epigenomic, transcriptomic, proteomic, metabolomic, and single-cell multi-omics data are deeply integrated. By integrating these approaches, researchers will ultimately gain a deeper understanding of how epigenetic regulation interacts with gene expression, protein function, and metabolic networks at different stages of plant development and under various environmental conditions. With advances in technology and research, additional epigenetic modifications such as 4mC have been identified in eukaryotes. During sperm maturation, genome-wide 4mC modifications emerge, critically regulating spermatogenesis and functional maturation[49]. In addition to characterizing genome-wide regulatory patterns, detailed analysis of locus-specific mechanisms could provide robust scientific support for precision breeding in plants.
Precise modulation of the epigenetic states of genes that govern key agronomic traits, such as yield, quality, and stress tolerance, may facilitate the development of crop varieties with superior productivity, enhanced nutritional profiles, and improved environmental adaptability. However, precise regulation of individual target genes also requires a more detailed understanding of their cis- and trans-regulatory elements. While current epigenetic editing tools still have some limitations, ongoing research will undoubtedly improve their efficiency and precision. It may lead to the development of new tools that can precisely edit epigenetic marks in specific genomic regions and cell types, as well as technologies for real-time monitoring and regulation of epigenetic states. Such innovations could significantly advance both the fundamental research and practical applications of plant epigenetics. Unlike transgenic technology, epigenome editing does not alter the genomic DNA sequence but regulates gene expression through modulation of epigenetic modifications. In crop breeding, hybridization and selfing strategies allow the segregation of progeny that are free of exogenous genetic elements. As for off-target effects, unlike the high precision required in gene therapy, crop breeding can utilize molecular and phenotypic screening to select target plants[50]. Strengthening scientific communication to enhance public understanding can facilitate their agricultural application.
Epigenetic modifications exhibit both conserved and species-specific regulatory patterns. For example, the 3D architecture of chromatin varies significantly among plants with different genome sizes, and the content of transposable elements also differs significantly among species[36]. Comparative studies investigating epigenetic changes across diverse taxa are valuable. In contrast to diploid model plants like Arabidopsis, maize, and rice, many crops – such as wheat, rapeseed, and cotton – are polyploids. Complex subgenome interactions governed by epigenetic regulation in polyploid species present unique challenges and opportunities. Research on such species not only supports agricultural production but also holds promise for deciphering mechanisms of epigenetic regulation in polyploid genomes.
Epigenomics research is currently undergoing rapid expansion. Future efforts must prioritize staying abreast of cutting-edge innovations, particularly the integration of artificial intelligence (AI). Integrating AI, especially machine learning for multi-omics data analysis and predictive modeling of epigenetic networks, shows great potential. AI can be used to discover enhanced gene-targeting systems, design precise sgRNAs with minimal off-target effects, and engineer novel effector or nuclease proteins. Effectively leveraging AI could accelerate the application of epigenomic strategies in breeding high-quality crop varieties.
This research was supported by the National Key R&D Program of China (grant numbers: 2022YFD2100100 and 2021YFA1300404 to ZL), Guangdong S&T Program (grant numbers: 2024B1111130001 to ZL), Shenzhen Science and Technology Program (grant numbers: JCYJ20241202125311016 and KQTD20240729102038044 to ZL). Some templates used in the figure were downloaded from
BioRender.com .-
The authors confirm contribution to the paper as follows: writing mauscript: Lang Z, Wu S, Hou Y, Ma Y; reviewing manuscript and providing advice: Wang P. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Siqun Wu, Yawen Hou
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wu S, Hou Y, Ma Y, Wang P, Lang Z. 2025. Epigenetic regulation and its applications in plants. Epigenetics Insights 18: e010 doi: 10.48130/epi-0025-0009 

Epigenetic regulation and its applications in plants
- Received: 22 March 2025
- Revised: 30 May 2025
- Accepted: 16 June 2025
- Published online: 08 July 2025
Abstract: Plant research has made numerous significant contributions to the field of epigenetics, including the discoveries of paramutation, parental imprinting, nucleolar dominance, and the RNA-directed DNA methylation (RdDM) pathway. In this review, we summarize the regulatory mechanisms of DNA methylation, histone modification, non-coding RNAs, chromatin remodeling factors, and 3D genome organization, highlighting their functions in plant growth, development, and responses to stress. Meanwhile, we describe the effects of epigenetic variations on key agronomic traits in crops and discuss potential applications of precise epigenomic editing in crop breeding. Finally, we summarize current limitations in epigenetic research, outline future challenges, and propose prospective research directions. With continued scientific advancement and societal progress, epigenetics will contribute more effectively to various aspects of human society - not only in plant breeding but also in therapeutic applications for human diseases.
-
Key words:
- Plant epigenetics /
- Epigenetic regulation /
- Epigenetic application /
- Epigenome editing /
- Crop breeding