








-
The epigenome—encompassing DNA modifications (e.g., 5-methylcytosine), histone variants, non-coding RNAs, and emerging RNA base marks—dynamically regulates gene expression, cellular identity, and environmental responses[1]. Aberrant epigenetic patterns drive diseases ranging from cancer to neurodevelopmental disorders[2], while natural variation underpins critical traits in agriculture[3]. Consequently, high-resolution epigenetic profiling is essential for deciphering biological mechanisms, and advancing therapeutic and agricultural innovation[4,5].
Critical limitations of current methodologies, however, impede accurate epigenome interrogation. Bisulfite sequencing (WGBS/RRBS), the gold standard for DNA methylation, relies on harsh bisulfite treatment to convert unmethylated cytosines to uracil. This process fragments DNA, destroys sequence context, and prevents recovery of intact molecules—resulting in low genomic coverage, regional biases, and challenges with limited clinical samples[6,7]. Crucially, it cannot distinguish 5 mC from 5 hmC, a key demethylation intermediate[8,9], nor detect non-cytosine modifications like N6-methyladenosine (m6A). Although enzymatic approaches (e.g., EM-Seq) mitigate DNA degradation and reduce input requirements[10], their conversion efficiency remains suboptimal for quantitative accuracy. Chromatin Immunoprecipitation sequencing (ChIP-seq) faces distinct hurdles: its dependence on antibody specificity and empirical optimization of cross-linking/sonication introduces lab-to-lab variability, masks cell-type-specific heterogeneity, and struggles with transient histone marks[11−13]. Microarray-based methods (e.g., MeDIP-chip) avoid bisulfite treatment but suffer from probe-dependent resolution limits, preventing discovery of novel modified regions[14], and cannot pinpoint exact modified bases[15]. Collectively, these constraints obscure native epigenetic states and dynamic processes.
Emerging as a paradigm-shifting alternative, Oxford Nanopore Technologies (ONT) directly sequences native DNA and RNA by monitoring ionic current changes during molecule translocation through protein nanopores[16]. This approach uniquely preserves endogenous epigenetic modifications without chemical perturbation or cross-linking, generates ultra-long reads for resolving complex genomic architectures, and enables real-time analysis. Here, ONT’s capabilities are systematically evaluated against conventional methods to demonstrate its superior fidelity in capturing the full spectrum of epigenetic information[14,17].
-
Unlike bisulfite sequencing—which irreversibly alters DNA through chemical conversion—Oxford Nanopore Technologies (ONT) sequences native nucleic acids without modification, enabling direct detection of base modifications via characteristic disruptions in ionic current[16−18]. This approach resolves DNA methylation at single-base resolution within individual molecules, eliminating context loss, and preserving intact genomic architecture[16,17]. Critically, ONT distinguishes 5-methylcytosine (5 mC) from 5-hydroxymethylcytosine (5 hmC) based on distinct current signatures: 5 mC reduces ionic current, while 5 hmC produces an intermediate signal, allowing unambiguous discrimination of these marks in active demethylation pathways (e.g., mammalian stem cells)[19,20].
Furthermore, ONT’s long-read capability facilitates allele-specific methylation analysis by spanning full gene loci, promoters, and enhancers in a single read[21,22]. When combined with the BIND&MODIFY protocol, ONT enables simultaneous profiling of histone modifications and DNA methylation at single-molecule resolution. Here, antibodies targeting specific histone marks recruit protein A fused to EcoGII/HIA5 6mA methyltransferases, converting local adenosines to 6mA as a surrogate mark. Subsequent ONT sequencing maps both the original histone modification (via 6mA deposition sites), and native DNA methylation patterns genome-wide[23,24], providing integrated chromatin state insights (Fig. 1).
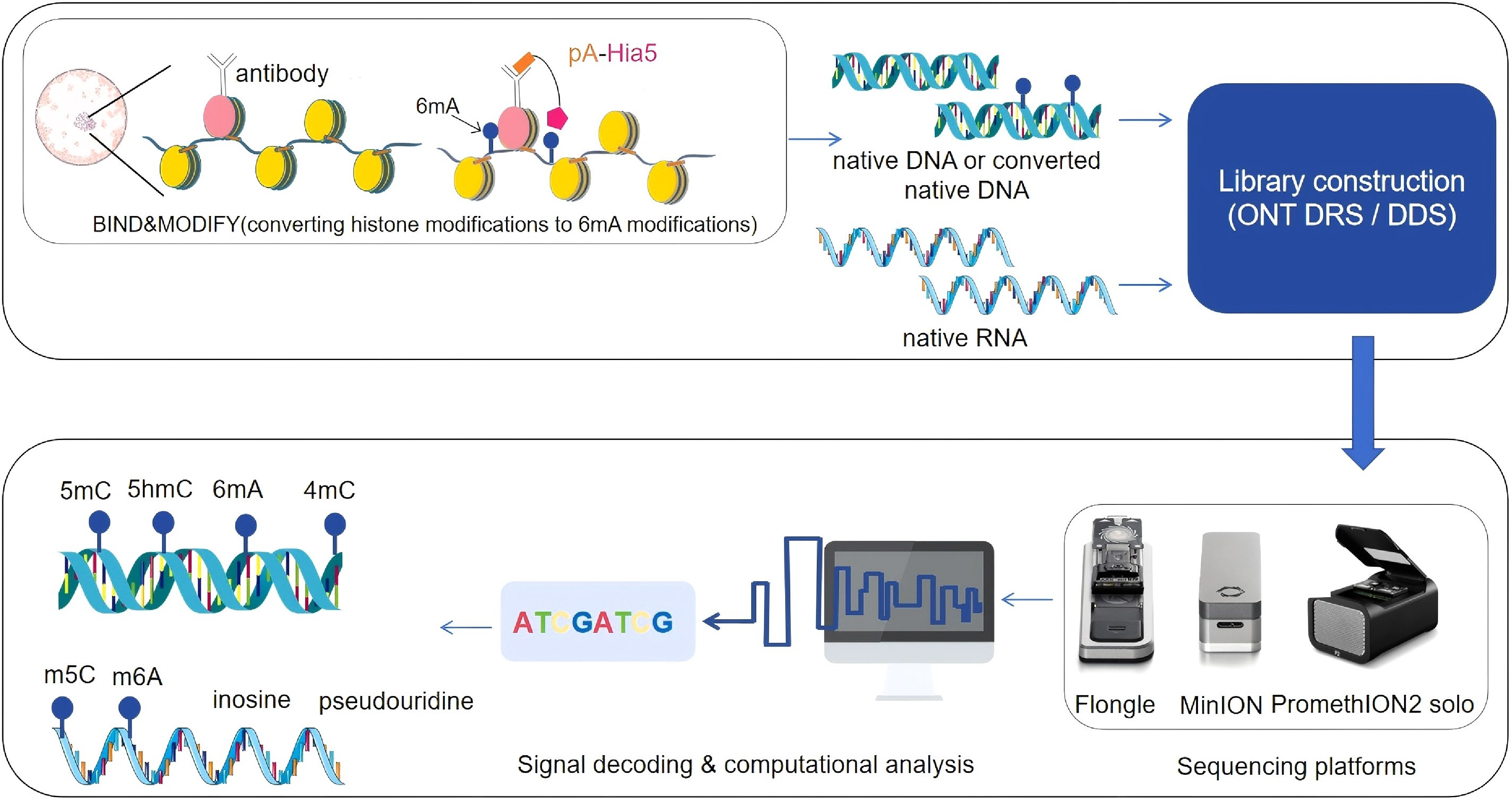
Figure 1.
A New paradigm shift for epigenetic profiling with ONT DDS/DRS, for simultanoeusly profiling DNA methylation and histone modifications at single molecular level, antibodies against histone modifications and proteinA fused with 6 mA methyltransferase to convert histone modifications to 6 mA marked genomic DNA, then the extracted DNA with BIND&MODIFY treated or without treated native DNA or native RNA can be subjected to library preparation, following sequencing platform (Flongle, MinION or PromethION) sequencing, the acquired ionic current signals can be decoded to the sequences of DNA or RNA with native modifications. The drawing for the figure got the support from BioIcons techniques and the images of sequencers are from Oxford Nanopore Technologies.
Ultra-long reads: resolving complex genomic architectures
-
ONT’s ability to generate ultra-long reads (> 2 Mb) overcomes fundamental limitations of short-read technologies (e.g., Illumina) in repetitive regions like pericentromeres, telomeres, and nucleolar organizer regions[22,25]. This capability enabled the first high-resolution mapping of centromeric methylation in Arabidopsis[22], and provides three transformative advantages: (1) Allele-Specific Phasing: Long reads assign methylation states to parental alleles, revealing monoallelic phenomena (e.g., genomic imprinting at IG-DMR)[21]. (2) Structural Variant-Epigenetic Linkage: Simultaneous detection of structural variants (SVs) and co-localized methylation changes exposes how SVs disrupt regulatory landscapes[26,27]. (3) Single-Molecule Heterogeneity: Direct observation of mixed methylation states within a locus captures tumor mosaicism missed by bulk methods[7,28,29].
Clinically, this was validated in cancer, where ONT identified allelic methylation imbalances at BRCA1 in 80% of tumors—undetectable by Illumina WGBS due to short-read mapping ambiguities[28].
Multi-omics integration: holistic epigenome interrogation
-
ONT uniquely enables integrated 'hologenome' analysis from a single sample, correlating DNA methylation, RNA expression, and protein activity via barcoding[30]. By sequencing genomic DNA and cDNA in parallel from identical cell populations, experimental batch effects are eliminated, clarifying causal relationships between epigenetic states and transcriptional outcomes[30]. Adding Direct RNA Sequencing (DRS) further illuminates: (1) mRNA/lncRNA dynamics (splicing, polyadenylation, tail length) influenced by DNA methylation. (2) Native base modifications (e.g., m6A, pseudouridine) on regulatory RNAs that modulate epigenetic machinery (e.g., Polycomb complexes)[31−34]. This multi-layered view was demonstrated in glioblastoma, where integrated ONT DDS/DRS profiling linked hypermethylated tumor suppressors to oncogene amplifications, enabling patient stratification[4]. ONT also serves as the ideal validation platform for epigenetic editing (e.g., dCas9-DNMT3A/TET1), providing precise methylation readouts at edited loci[1,4,5].
Real-time data and field applicability: democratizing epigenetics
-
ONT’s portable MinION/Flongle platforms deliver real-time data acquisition, accelerating experimental optimization and clinical decision-making[35]. Applications include: (1) Point-of-care detection of epigenetic biomarkers (e.g., SEPT9 methylation for colorectal cancer screening)[36−38]. (2) On-site environmental epigenomics (e.g., plant stress responses under drought/heat)[3]. (3) Space biology studies monitoring astronaut epigenetic drift during missions[39]. In contrast, short-read systems require off-site lab infrastructure, delaying results by days—a critical limitation for time-sensitive diagnostics or dynamic biological processes.
-
To systematically evaluate the strengths and limitations of Oxford Nanopore direct DNA/RNA sequencing (ONT DDS/DRS) for epigenetic profiling, a head-to-head comparison was conducted against established methodologies (Table 1). This analysis highlights critical trade-offs in sensitivity, resolution, scalability, and biological fidelity across technologies.
Table 1. Comparative performance of ONT direct sequencing vs traditional epigenetic technologies.
Epigenetic parameter ONT direct sequencing (DDS/DRS) Traditional technologies
(WGBS/ChIP-seq/meRNA-IP, etc.)DNA methylation Advantages • Simultaneous base-resolution detection of 5 mC, 5 hmC, 6 mA[40] • Gold standard for 5mC (bisulfite sequencing) • Resolves repetitive regions (e.g., centromeres)[22] • Lower cost per sample • Single-molecule resolution[7,40,41] • Mature bioinformatics pipelines Disadvantages • Requires high-input DNA quality • Limited to 5 mC (requires additional assays for
5 hmC/6 mA)• Basecalling accuracy for modifications needs improvement[16,17] • Poor performance in repetitive regions • Computationally intensive (GPU-dependent)[16] • Bulk averaging masks single-molecule heterogeneity RNA base modifications Advantages • Direct detection without enrichment/conversion[18] • Lower cost • Multi-modification profiling in single reads[16,19] • Tolerates moderate RNA degradation • Single-molecule resolution[18,31] • Established protocols Disadvantages • Technical expertise required • Antibody-dependent (bias risk) • Higher cost per run • Single-modification per experiment • Underdeveloped modification-detection algorithms[16] • Labor-intensive workflow Histone modifications Advantages • BIND&MODIFY: Simultaneous DNA methylation + histone mark profiling[24] • Field standard for histone marks • Single-cell epigenetic state resolution[23] • Lower cost • Direct correlation of histone-DNA crosstalk[23,24] • Mature analysis tools[13] Disadvantages • Requires nuclear isolation & protein engineering • GC bias in sequencing depth • Needs dual-modification algorithm development • Bulk averaging obscures cellular heterogeneity • Histone-to-6 mA conversion step adds complexity • No DNA methylation co-profiling Alternative splicing Advantages • Full-length transcript isoforms[18,31] • Lower cost • Single-molecule splicing patterns • Abundant third-party software • Multi-process integration (splicing + modifications)[31] Disadvantages • Higher cost than short-read RNA-seq • Assembly errors in isoform reconstruction • Data quality-dependent • No direct modification/splicing linkage • Limited statistical tools • Population-averaged data Poly(A) tail length Advantages • Multi-parameter profiling (tail length + splicing/modifications)[18] • Lower cost • Isoform-specific tail length detection • Detects tail heterogeneity • No additional experimental steps • Simple analysis Disadvantages • Higher cost than PAL-seq[42] • Isolated measurement (no multi-process context) • Insensitive to tail heterogeneity • Population-averaged data • Algorithmic limitations in estimation • Isoform-agnostic quantification Non-coding
RNA (ncRNA)Advantages • Full-length lncRNA characterization • Universal ncRNA compatibility • ncRNA processing/modification detection[31] • Standardized protocols Disadvantages • Poly(A)-tail dependency limits non-polyadenylated ncRNAs • Fragmented long ncRNA assembly • Poor small ncRNA performance without adaptations • No direct modification detection • Higher cost • No single-molecule resolution -
Despite its transformative potential, ONT direct sequencing faces critical hurdles that must be addressed to fully realize its utility in epigenetics:
Key Challenges: (1) Basecalling Accuracy & Bias: While Dorado’s sup model achieves high accuracy for DNA modifications (e.g., 99.5% for CpG methylation, 98.7% for 5 hmC), and RNA modifications (98.7%–99.7% across contexts), it remains inferior to Illumina/PacBio for standard base calling (~99.9% to 99.99%)[40,41]. Crucially, residual biases persist—early models systematically undercalled modified bases[16], and even current algorithms require extensive training on modification-specific datasets to minimize context-dependent errors (e.g., sequence motif effects). This limits reliable detection of low-prevalence marks in heterogeneous samples like tumors. (2) Library Preparation Artifacts: Optimized protocols for native nucleic acid library preparation are less mature than those for bisulfite-converted or immunoprecipitated samples[16,38]. High variability in sequencing yields from identical total RNA inputs indicates significant technical noise, likely stemming from inconsistent end-repair, adapter ligation, or motor protein activity during translocation. Standardized, low-bias workflows are urgently needed to ensure reproducibility, particularly for single-cell applications. (3) Fragmented Bioinformatics Ecosystem: The absence of universal analysis tools impedes reproducibility. Most modification callers (e.g., deepsignal-plants, Xpore, m6Anet) are instrument- or flowcell-specific (e.g., trained exclusively on R9.4.1 or RNA002 data), failing with newer chemistries like R10.4 or RNA004[16]. This siloed landscape forces researchers into laborious tool-switching and manual retraining, hindering scalable epigenetic analysis.
Future Priorities: To overcome these barriers, efforts should prioritize: (1) Context-Aware Basecallers: Development of machine learning models trained on diverse epigenetic landscapes (e.g., mixed 5 mC/5 hmC regions, repetitive DNA) to improve detection of rare or complex modifications. (2) Hybrid Enrichment Strategies: Integration of targeted capture (e.g., CpG island probes) with ONT sequencing to deepen coverage of clinically relevant loci without sacrificing long-read advantages[43]. (3) CRISPR epigenetics Synergy: Coupling ONT with dCas9-effector systems (e.g., TET1 demethylases) for real-time validation of epigenetic edits at endogenous loci[1]. (4) Cost Reduction and Scalability: Engineering streamlined workflows to enable large-scale studies of epigenetic drift in aging, cancer evolution, and population genomics.
-
Oxford Nanopore direct sequencing represents a paradigm shift in epigenetic profiling, directly overcoming the fragmented, context-loss limitations of short-read technologies. By analyzing native nucleic acids at single-molecule resolution, generating ultra-long reads to resolve complex loci, integrating multi-omics layers (DNA methylation, histone marks, RNA dynamics), and enabling low-input/single-cell analyses, ONT provides an unprecedented lens into dynamic epigenetic processes central to health and disease. Although challenges in basecalling fidelity, library prep robustness, and bioinformatic interoperability remain, rapid advancements in nanopore chemistry, AI-driven signal processing, and community-driven tool development are accelerating ONT toward becoming the gold standard for comprehensive, biologically faithful epigenomic characterization. For researchers decoding the functional impact of epigenetic variation, ONT delivers not merely data—but the clarity, length, and depth required to resolve the epigenome’s true complexity.
This work was supported by the following grants: Yunnan Provincial Major Basic Research Projects (202401BC070003) to SZ; National Natural Science Foundation of China (NSFC) Regional Science Foundation Projects (32460487) to SZ; Yunnan Provincial Major Science and Technology Special Projects (202502AE090006) to SZ. National Natural Science Foundation of China (32170362) to ML; Guangdong Natural Science Funds for Distinguished Young Scholars (2022B1515020026) to ML; Youth Innovation Promotion Association, Chinese Academy of Sciences (Y2021094) to ML.
-
Not applicable.
-
The authors confirm their contributions to the paper as follows: conceived and designed the study: Zhang S, Luo M; drafted the manuscript: Zhang S, Lu Z, Zhang H; contributed literature curation: Bai G, Luo M. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study; all data generated or analyzed during this study are included in this published article.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Zhihui Lu, Hanxue Zhang
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Lu Z, Zhang H, Bai G, Luo M, Zhang S. 2026. Advantages of Oxford Nanopore Technologies direct DNA/RNA sequencing for epigenetic profiling over traditional technologies. Epigenetics Insights 19: e003 doi: 10.48130/epi-0025-0017 

Advantages of Oxford Nanopore Technologies direct DNA/RNA sequencing for epigenetic profiling over traditional technologies
- Received: 27 August 2025
- Revised: 11 November 2025
- Accepted: 22 December 2025
- Published online: 02 February 2026
Abstract: Epigenetic regulation—orchestrated by DNA methylation, histone modifications, non-coding RNAs, and emerging RNA base modifications—dictates gene expression, cell identity, and environmental responses. Yet, conventional methods like bisulfite sequencing and ChIP-seq impose critical limitations: they require harsh chemical treatments (e.g., bisulfite conversion) or artificial cross-linking that distort in vivo epigenetic states, obscure dynamic modifications, and sacrifice strand specificity. Here, Oxford Nanopore Technologies (ONT) direct sequencing is advocated as a transformative solution. By enabling native, amplification-free interrogation of DNA and RNA with ultra-long reads and real-time analysis, ONT uniquely captures the epigenome in near-physiological condition. It directly detects diverse base modifications—including 5 mC, 5 hmC, m6A, and pseudouridine, etc.—without engineered antibodies or destructive chemistry, while preserving critical strand orientation and phasing adjacent marks. Crucially, the present comparative analysis confirms ONT’s superior accuracy for modified base calling, enhanced sensitivity to low-abundance epigenetic variants, and unparalleled scalability for multi-omic integration. This fidelity to native molecular states positions ONT to redefine epigenetic research across basic, translational, and applied biology, establishing a new paradigm for resolving dynamic epigenetic landscapes in health and disease, as well as crop adaptation to internal/external cues.