









-
Ascorbic acid (AsA), known as vitamin C, is a vital water-soluble antioxidant that plays essential roles in plant growth, development, and stress response, which functioned as a powerful scavenger of free radicals, activating the plant's antioxidant system to reduce the concentration of oxygen free radicals and mitigate the damage caused by reactive oxygen species (ROS)[1]. In addition to its antioxidant function, AsA is involved in various critical physiological processes[2]. For instance, it serves as an enzymatic cofactor in photosynthesis and acts as a photoprotectant to dissipate excess light energy[3]. AsA also contributes to cell division and expansion, osmotic adjustment, and hormone biosynthesis[4]. Moreover, it significantly enhances plant stress tolerance, such as improving salt tolerance by alleviating salt stress effects[5] and protecting against cold stress by neutralizing excess ROS[6]. Additionally, ascorbic acid regulates fruit ripening by efficiently eliminating ROS and modulating the cellular redox state, making it a key indicator of fruit nutritional value[7]. Understanding the mechanisms underlying vitamin C accumulation not only improves fruit quality but also facilitates the breeding of fruits with enhanced nutrition and agronomic traits[8]. Notably, ascorbate oxidase (AAO) and ascorbate peroxidase (APX) are essential antioxidant enzymes in plants, involved in the regeneration and degradation of ascorbic acid, thereby influencing its accumulation and, consequently, fruit ripening[9]. The major biosynthesis pathways of AsA currently recognized include the L-galactose pathway, L-gulose pathway, D-galacturonic acid pathway, and the inositol pathway[10]. APX exhibits the highest affinity among hydrogen peroxide-metabolizing enzymes[11]. It utilizes AsA as a specific electron donor to catalyze the reduction of hydrogen peroxide (H2O2) to water and oxygen, while AsA itself is oxidized to monodehydroascorbate (MDHA), thereby mitigating oxidative damage. APX plays a critical role during both fruit ripening and post-harvest, as it regulates H2O2 and AsA levels, which directly affect fruit quality[9]. In contrast, AAO utilizes AsA as a substrate to generate ROS and also converts AsA to MDHA[4]. A decline in AsA content is temporally aligned with the onset of fruit ripening, indicated by color alterations, and is strongly associated with the upregulation of AAO activity[12]. This implies that AAO contributes critically to the ripening process by modulating AsA accumulation. Understanding the interaction between AAO and APX during fruit ripening offers valuable insights into the accumulation and stability of ascorbic acid.
In recent years, the APX gene family has been identified in various plants, with 11 in Populus trichocarpa[13] and eight in Cannabis sativa[14]. Based on subcellular localization, APX proteins are distributed in the cytoplasm, chloroplasts, peroxisomes, and mitochondria[15]. Increasing evidence suggests that APX proteins are pivotal for plant growth, development, and responses to abiotic stress[16]. Numerous studies have demonstrated that overexpression of APX genes enhances plant tolerance to abiotic stress, including heat, cold, drought, and salinity. For example, Wang et al. reported that overexpression of an APX gene family member from Camellia azalea increased heat and cold stress tolerance in tobacco plants[17]. Similarly, Liu et al. found that overexpression of Apium graveolens APX1 enhanced drought tolerance and increased ascorbic acid content in Arabidopsis[18]. Additionally, Zhang et al. demonstrated that loss of function in OsAPX2 resulted in semi-dwarf seedlings, yellow-green leaves, leaf lesion mimics, and sterility in rice[19]. In contrast, studies on the AAO gene family remain limited, with existing research primarily focusing on Gossypium hirsutum[20] and Beta vulgaris[21]. However, AAO genes play multiple roles in plants, particularly in rapidly growing tissues and young fruits. Zhang et al. found that suppression of AAO expression resulted in increased ascorbic acid accumulation and improved fruit yield[22], whereas upregulation of AAO expression led to reduced stomatal aperture in tobacco. Despite substantial progress in understanding the roles of APX and AAO in plants, their molecular mechanisms and specific functions in fruit development, ripening, and ascorbic acid accumulation remain unclear.
The kiwi berry (Actinidia arguta), a perennial vine of the Actinidiaceae family, is native to regions spanning from southwest China to the Russian Far East[23]. Often referred to as hardy kiwi, it differs from other kiwifruits by its typically sweeter flavor and the ability to be consumed whole without peeling. Additionally, A. arguta is rich in vitamin C and contains polyphenols, dietary fiber, and various other nutrients, which contribute to its potential in preventing chronic diseases and promoting human health[24]. Due to its favorable agricultural traits, including small fruit size and exceptional cold tolerance, along with its high nutritive value, A. arguta has gained increasing popularity among consumers[25]. Although significant progress has been made in various aspects of A. arguta research, such as its genetic characterization—exemplified by the recent genome assembly of A. arguta, which has paved the way for future functional genomics studies—there is still much to explore in terms of its bioactive ingredients[23]. For example, Leontowicz et al. compared the bioactivity and nutritional properties of A. arguta with those of A. deliciosa and A. eriantha and found that A. arguta exhibited superior bioactivity and nutritional value[26]. Antioxidants and nutrients, particularly ascorbic acid, have been among the most extensively studied compounds in A. arguta. However, most research has focused on comparisons with other Actinidia species and the dynamic changes in these compounds over time. It remains unclear how ascorbic acid accumulation is associated with the enzymes AAO and APX in A. arguta, particularly during its growth and development. While Lin et al. found significant correlations between AAO, APX, and AsA pool, the mechanisms underlying their interaction in regulating ascorbic acid accumulation and metabolism are still poorly understood[27].
This study aims to identify all members of the APX and AAO gene families in A. arguta potentially involved in ascorbic acid regulation and to investigate their expression patterns and relationships to ascorbic acid accumulation during fruit development, with the goal of elucidating molecular mechanisms regulating ascorbic acid accumulation. To further validate the patterns of gene expression, two A. arguta cultivars with distinct fruit coloration were selected. 'Kukuwa' (KKW), a Japanese cultivar, is characterized by its green flesh, elevated ascorbate content, and high antioxidant capacity, whereas 'Xiangziguang No. 1' (XZG), a Chinese cultivar, is distinguished by its red fruit pigmentation. These findings provide valuable insights into ascorbic acid regulation and may offer potential genetic resources for enhancing nutritional content and antioxidant capacity in A. arguta.
-
All APX and AAO protein sequences of A. thaliana were obtained from the TAIR database and used as query sequences. Candidate APX and AAO genes in A. arguta (A. arguta 'M', Supplementary Table S1) were identified by local BLASTP v2.15.0+ searches (E-value ≤ 1e-5) against the A. arguta genome. The resulting sequences were further examined with HMMER v3.3.2 and the PFAM database to confirm the presence of APX or AAO conserved domain[28,29]. Finally, sequences lacking the corresponding conserved structural domains were manually excluded from further analysis. Additionally, APX and AAO homologs from four Actinidia species (A. chinensis cv. Hongyang, A. eriantha, A. hemsleyana, and A. rufa) were consistently identified using the same approach as in A. arguta and retrieved from their respective genome databases (Supplementary Table S1). The isoelectric point (pI), molecular weight (MW), and instability indices of the identified AaAPX and AaAAO proteins were calculated by the ExPASy-ProtParam tool[30]. Subcellular localization of AaAPXs and AaAAOs was predicted using CELLO v.2.5[31].
Phylogenetic analysis
-
Protein sequences of APX and AAO from A. thaliana and five Actinidia species were aligned using MUSCLE[32]. Phylogenetic trees were constructed with the neighbor-joining method in MEGA v7.0[33], and classification of A. arguta family members was guided by previously reported clades in Arabidopsis. The resulting trees were visualized using the ggtree package in R v4.4.3[34].
Gene structure, conserved motifs, and cis-acting element analysis
-
Structural information of AaAPXs and AaAAOs was extracted from the A. arguta 'M' genome annotation (GFF files). Gene exon–intron structure was visualized utilizing the Gene Structure Display Server (
http://gsds.cbi.pku.edu.cn ). Protein sequences were aligned with ClustalW, and the alignments were displayed using TBtools v2.084[35,36]. Conserved motifs were identified through the MEME Suite[37]. For cis-regulatory analysis, 2.0 kb upstream promoter sequences of each AaAPX and AaAAO gene were retrieved from the genome and scanned for cis-acting elements using PlantCARE[38]. The identified elements were curated, formatted as BED files, and subsequently visualized with GSDS 2.0.Collinearity and selective pressure analysis
-
Syntenic relationships among gene family members were examined using MCScanX implemented in TBtools v2.084 with default settings[35,39]. Protein sequences were aligned in ParaAT v2.0 with MUSCLE[40], and nonsynonymous (Ka) and synonymous (Ks) substitution rates were estimated using KaKs_Calculator v2.0[41]. Selection pressure was inferred based on the Ka/Ks ratio, with values < 1 indicating purifying selection and > 1 indicating positive selection.
Differential expression analysis of A. arguta APX and AAO gene family
-
To investigate the molecular mechanisms underlying antioxidant activity during kiwifruit development, previously generated transcriptomic and metabolomic datasets from the flesh of two A. arguta cultivars, Kukuwa (KKW, full-green flesh) and Xianziguang No. 1 (XZG, red) (unpublished data from our laboratory), were analyzed. Fruit samples were collected from three replicate lianas at early fruit (T1, May 15th, approximately 20 d after full bloom), fruit ripening (T2, June 19th), and fully ripe (T3, September 15th) stages to represent the progression from early to full maturity, in the kiwifruit germplasm resource nursery at the Institute of Botany of Shaanxi province, Xi'an, China. A total of 18 samples (three biological replicates per developmental stage) were analyzed, with transcriptomic data generated using the Illumina sequencing platform and metabolomic profiles obtained through comprehensive metabolite analysis by Metware Biotechnology Co, Ltd. (Wuhan, China). In the present study, these datasets were primarily examined to assess the expression of selected genes and the accumulation patterns of corresponding metabolites.
To explore the expression dynamics of AaAPX and AaAAO genes across the developmental stages of two A. arguta cultivars, clean reads were mapped to the A. arguta 'M' reference genome with HISAT2 v2.2.1[42]. Subsequently, transcript abundance was quantified with HTSeq and normalized as FPKM[43]. Differential expression between stages was assessed in DESeq2, applying |log2FC| > 1 & q value < 0.05 as cutoffs[44]. Visualization of expression profiles was carried out by generating heatmaps in TBtools v2.084[35]. Finally, functional enrichment of the differentially expressed genes was examined through Gene Ontology (GO) analysis in the R package clusterProfiler v4.0[45], with results plotted in ggplot2 under R v4.4.3[46].
Validation of key expressed genes by qRT-PCR
-
To examine the expression patterns of key genes (AaAPX2, AaAPX3, AaAPX4, AaAPX14, AaAAO15, and AaAAO16) in relation to key metabolites across two A. arguta varieties by qRT-PCR, total RNA was extracted utilizing TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and then reverse-transcribed into complementary DNA (cDNA) using the FastQuant First Strand cDNA Synthesis Kit (Tiangen, Beijing, China). Primers for the target genes were designed utilizing Primer3Plus (
www.primer3plus.com ) (Supplementary Table S2). Actinidia β-actin was chosen as the reference gene for normalization[47]. The relative gene expression levels were determined by the 2−ΔΔCᴛ method. -
In the present study, bioinformatics tools were employed to perform a genome-wide analysis of AAO and APX genes present in five Actinidia species and A. thaliana, as shown in Supplementary Table S3. A total of 14 to 44 AAOs and six to 25 APXs were identified in Actinidia plants, respectively (Supplementary Table S3). In the A. arguta 'M' genome, 25 AaAAOs (AaAAO1-AaAAO25) and 44 AaAPXs (AaAPX1-AaAPX44) genes were identified based on their order in chromosomes of the A and D sub-genomes, and dispersed across five and nine distinct chromosomes, respectively. Notably, the majority of these chromosomes contained only a single AaAAO or AaAPX gene. In contrast, two to three AaAPX genes were concurrently localized on chromosomes 21A-D and 11A-D, whereas two AaAAO genes were found on chromosomes 28A-D and 15A-B. Moreover, specific subsets of AaAAO (e.g., AaAAO11–14) and AaAPX (e.g., AaAPX25–28) genes exhibited co-localization on chromosomes 13, 23, and 28 within the A and D sub-genomes (Fig. 1).

Figure 1.
Distribution of AaAAO and AaAPX genes on chromosome of A. arguta.
The physicochemical properties analysis showed that the AaAAO proteins are composed of 475–799 amino acids and their molecular weights ranged from 53,245.32–88,873.57 Da, with isoelectric points of 5.7–6.96 for eight acidic proteins and 7.35–9.07 for 17 alkaline proteins. Similarly, the AaAPX proteins comprised 158–453 amino acids with molecular weights ranging from 18,078.65–48,956.51 Da, and isoelectric points ranging from 5.49–6.95 for 21 acidic proteins and from 7.57–9.6 for 23 alkaline proteins (Supplementary Tables S4, S5).
Phylogenetic analysis
-
To investigate the evolutionary relationships of AAO and APX proteins among Actinidia plants, two separate phylogenetic trees were constructed: one for 69 AAOs and the other for 116 APXs from A. thaliana, A. arguta, A. chinensis 'Hongyang', A. eriantha, A. hemsleyana, and A. rufa. The model plant A. thaliana contains three putative AO genes, namely At4g39830, At5g21105, and At5g21100[48]. Further, classification results of the AAOs fell into three subfamily (Class I–III), encompassing 28, 12, and 29 proteins, respectively, and each labeled with a different colour (Fig. 2a). The APXs were divided into five subfamily (Class I–V), containing 34, 26, 14, 10, and 30 proteins, respectively (Fig. 2b). According to A. thaliana APXs[49], clade I and II comprise cytosolic APXs, and clade III, V, IV mainly include chloroplastic APXs, as predicted by subcellular localization. The phylogenetic results obtained in this study are consistent with those reported previously[50].
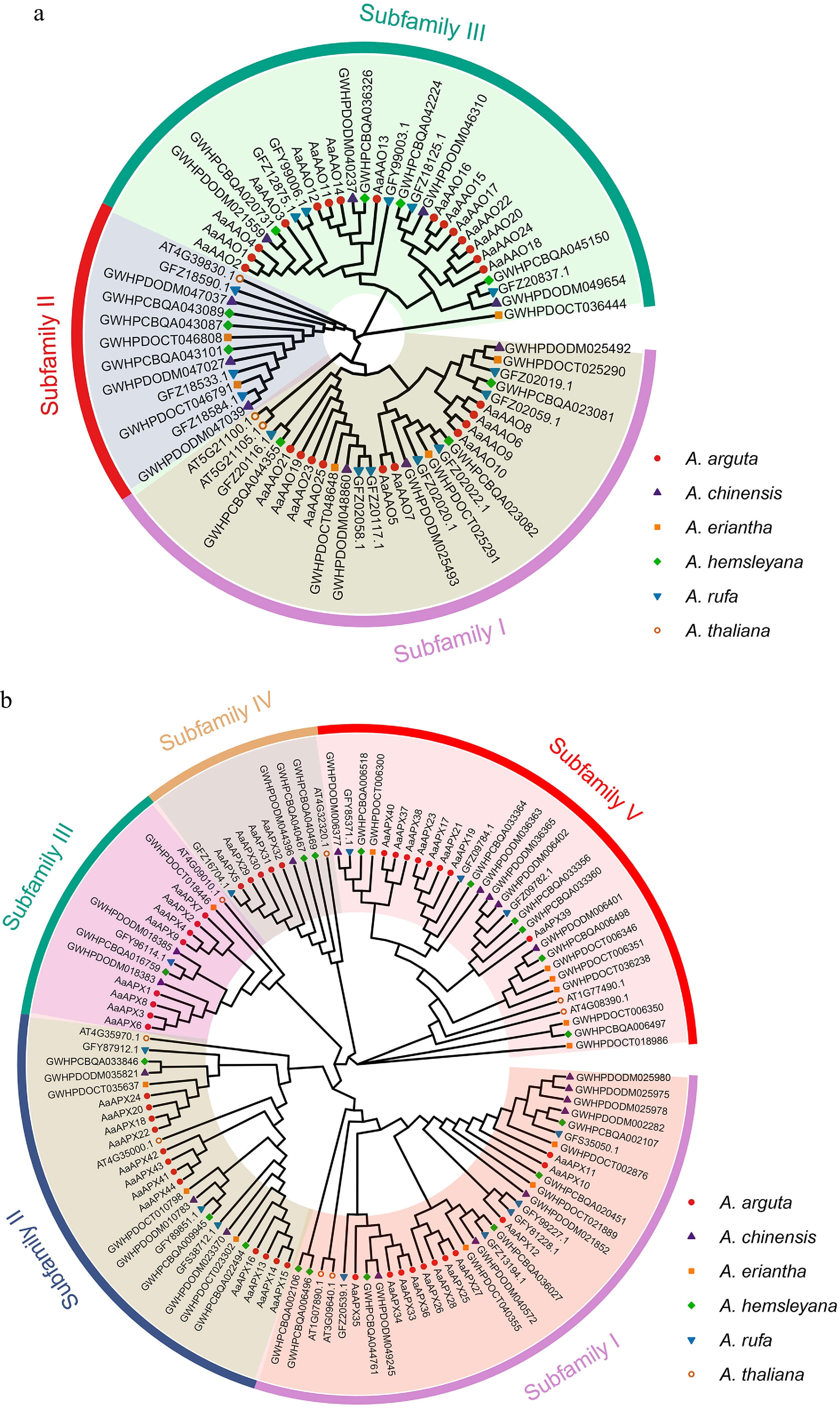
Figure 2.
Phylogenetic analyses of the (a) AAO, and (b) APX families from five Actinidia species and Arabidopsis. Colored outer rings represent diverse subfamilies.
Gene structure and conserved motifs analysis
-
The phylogenetic trees of all 25 AaAAOs (protein sequences) and 44 AaAPXs (protein sequences) in A. arguta were conducted separately, and their topological structures were consistent with those shown in Fig. 2. MEME was utilized to conduct further analysis of conserved motifs type and arrangement in AaAAO and AaAPX genes, and the results revealed that motifs ranged from six to ten in all AAO genes, with over half of the genes possessing ten conserved motifs, small half contained eight conserved motifs, among which motifs (1, 2, 3, 5, 7, 10) were the most conserved. In terms of the motifs in APX, nine conserved motifs were identified within these genes, and most of them demonstrated high similarity; however, several genes exhibited varying degrees of deletion. For instance, AaAPX5 only contained motif 1 and motif 10, and AaAPX22 encompassed motif 2, motif 3, motif 7, and motif 9. Besides, three conserved domains, PF07732, PF00394, and PF07731, were identified in all AAO gene family, PF00141 was identified in all APX gene family, indicating the accuracy of these genes (Fig. 3). A comparative analysis of exon–intron architectures was performed to characterize the evolutionary differences between the AAO and APX gene families in A. arguta. The structural characterization revealed that AAO genes generally exhibited simpler organizations, with most members containing only a single exon. In contrast, APX genes displayed significantly greater structural complexity, with exon numbers ranging from three to eight across different members. The sole exception was AaAPX15, which contained only two exons (Fig. 3).
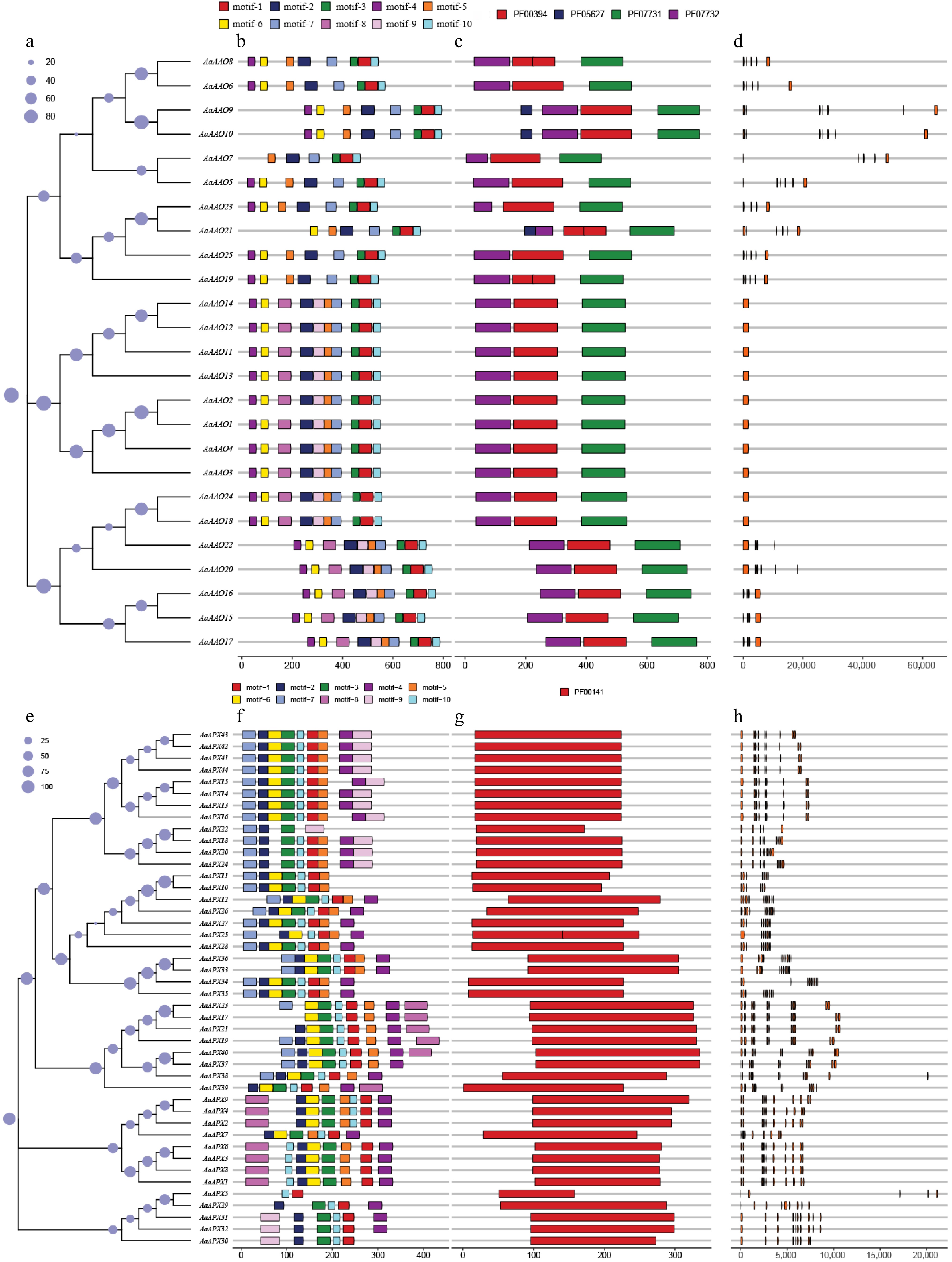
Figure 3.
(a)–(d) shows the analysis of AaAAOs genes, and (e)–(h) shows the analysis of AaAPXs genes. (a), (e) Neighbor-joining trees constructed for AaAAOs and AaAPXs genes, respectively. (b), (f) Protein motifs, with motifs 1–10 displayed in different colored boxes. (c), (g) Conserved domains, represented by different colored boxes. (d), (h) Gene structures, where orange boxes represent CDS.
Analysis of cis-acting elements
-
A comprehensive cis-acting element analysis was performed on the promoter regions of AAOs and APXs in A. arguta to explore their potential regulatory roles. The identified cis-elements were primarily classified into four functional categories: hormone-responsive, light-responsive, stress-responsive, and growth and development-related elements. In addition, several elements were associated with flavonoid biosynthesis, metabolic regulation, and MYB binding, indicating potential regulatory roles of these genes in stress adaptation and secondary metabolism (Fig. 4). Interestingly, the number of cis-acting elements identified in APXs was higher than that in AAOs, implying that APXs may have a broader regulatory role in plant development and abiotic stress adaptation. Furthermore, ABRE elements—related to abscisic acid (ABA) responsiveness—were found in all AaAAO genes, except AaAAO10 and AaAAO20, highlighting their potential involvement in hormone signaling pathways. Meanwhile, Box 4 elements, indicative of light responsiveness, were present in all AaAPX genes, except AaAPX7 and AaAPX9. These findings emphasize the likely participation of AAO and APX genes in hormonal regulation, light signaling, and stress-responsive networks during A. arguta fruit development (Fig. 4).

Figure 4.
Predicted cis-elements within the 2,000 bp upstream region of AaAAO and AaAPX genes. Different colors indicate distinct cis-element categories, with numbers displayed in the corresponding boxes. (a) AAO gene family. (b) APX gene family.
Collinearity and selective pressure analysis
-
To clarify the expansion history of the AAO and APX gene families in A. arguta, duplication mode analysis was conducted. Most members were traced back to whole-genome (WGD) or segmental duplication events, suggesting that large-scale genome duplications were the primary driving force in their expansion. Additionally, two AAO genes (AaAAO6 and AaAAO8) exhibited tandem duplication (TD), while four APX genes (AaAPX2, AaAPX4, AaAPX7, and AaAPX9) experienced proximal duplication (PD) (Supplementary Table S6), further supporting the contribution of both large-scale and local duplication events to gene family expansion in A. arguta (Supplementary Table S6). Ka (non-synonymous substitution rate) and Ks (synonymous substitution rate) values were also calculated for homologous gene pairs within A. arguta, and the results showed that most of the Ka/Ks ratios were < 1, indicating that these gene pairs have undergone purifying selection during evolution in A. arguta (Supplementary Table S7).
To further explore the origins and evolutionary history of the AAO and APX gene families in A. arguta and the other three species, including A. chinensis, A. eriantha, and A. thaliana, a comparative synteny analysis was conducted. The analysis identified 18, 22, and three orthologous genomic APX gene pairs between A. thaliana and A. arguta, A. chinensis and A. arguta, A. eriantha and A. arguta, respectively. In contrast, there were no AAOs synteny results found between A. arguta and A. eriantha. A much higher degree of synteny was observed between A. chinensis and A. arguta, particularly on chromosome 28, which contained more orthologous gene pairs. Furthermore, the comparative analysis revealed that the number of APX genes was consistently lower than that of AAO genes across all four species, suggesting a differential expansion pattern between the two gene families (Fig. 5).
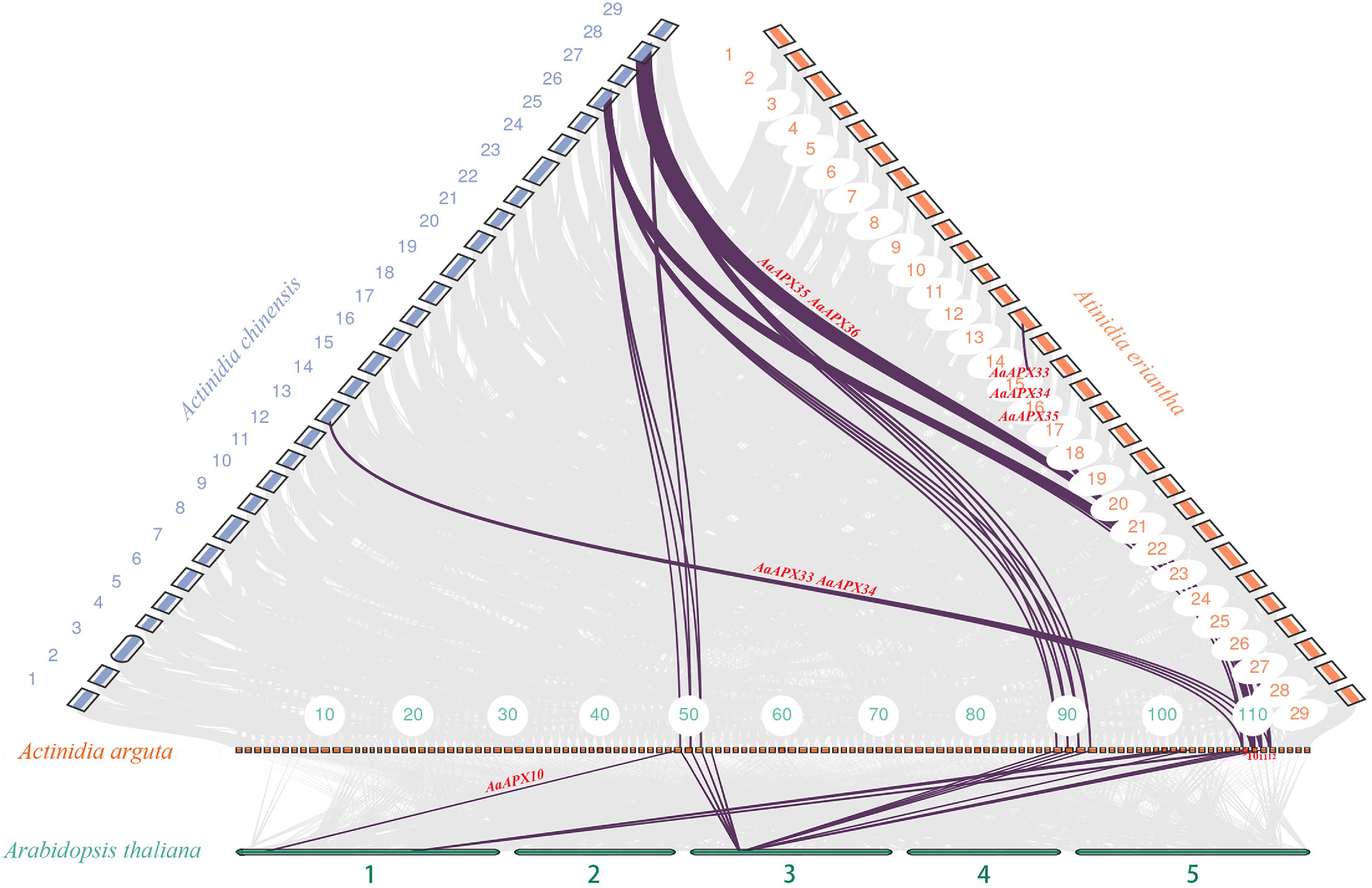
Figure 5.
Collinearity analysis of the APX gene family between A. arguta and three related species (A. thaliana, A. chinensis, and A. eriantha). Gray lines indicate overall collinear regions, while colored lines denote orthologous gene pairs.
Prediction of signal peptides
-
SignalP 6.0 was used to predict signal peptides in the AaAAO and AaAPX to evaluate their potential roles in secretion pathways. The AaAAO proteins exhibited a high likelihood of containing N-terminal signal peptides, with most genes showing signal peptide probabilities (SP(Sec/SPI)scores) exceeding 0.999, and predicted cleavage sites between amino acid positions 21 and 27. These findings suggest that AaAAO proteins are likely secreted, potentially playing a role in extracellular antioxidant activity. In contrast, the majority of AaAPX proteins lacked predicted signal peptides, exhibiting signal peptide probabilities near zero, indicating their retention within the cytosol. However, AaAPX26 was an exception, with an SP probability of 0.999589 and a predicted cleavage site at positions 21–22, suggesting a potential extracellular function for this protein (Supplementary Table S8). These results imply that AaAAO proteins may be involved in extracellular antioxidant defense, whereas AaAPX proteins primarily function within the cell, indicating their distinct subcellular roles in oxidative stress responses.
Differential expression pattern of AAO and APX gene family
-
To investigate the potential role of the AAO and APX gene families in regulating L-ascorbate metabolism, expression patterns of these genes were analyzed in two A. arguta varieties (KKW and XZG) across three developmental stages. For example, AaAPX14, AaAPX21, and AaAPX23 were predominantly downregulated in KKW at T1 but exhibited variable expression in other stages. In contrast, AaAPX1, AaAPX6, AaAPX8, and AaAPX9 showed high expression levels in XZG at T1 in the Vitamin C biosynthesis (Fig. 6a, b). Notably, some antioxidant genes displayed consistent expression trends across different stages. For instance, AaAPX38, AaAPX10, and AaAPX44 were significantly upregulated, whereas AaAAO9, AaAPX39, and AaAPX34 were specifically downregulated at the T1, T2, and T3 stages, respectively. Interestingly, several genes exhibited significantly different expression patterns between the two varieties. For instance, AaAAO1 and AaAAO13 showed high expression levels at the first developmental stage in KKW, whereas AaAAO19, AaAAO25, and AaAAO23 were highly expressed at the same stage in XZG (Fig. 6a, c). Pearson correlation coefficient analysis indicated that their expression levels were highly correlated (Pearson's r > 0.85, p < 0.05), such as AaAAO10/AaAA23, AaAPX1/AaAPX6, and AaAAO8/AaAPX25 (Supplementary Fig. S1). However, the expression patterns of AaAAO and AaAPX genes varied between the two A. arguta varieties during the kiwifruit developmental stage (Supplementary Figs S2, S3). Specifically, in comparisons between XZG and KKW, 16, 11, and nine genes were significantly upregulated, while six, 15, and ten genes were significantly downregulated at the T1, T2, and T3 stages, respectively (Supplementary Fig. S3). Furthermore, the distribution of transcription factor (TF) families between different developmental stages of KKW and XZG was explored, and the results revealed that the bHLH and AP2/ERF families show a high frequency of gene involvement. In contrast, other TF families like WRKY, MYB, and NAC display more moderate and consistent involvement across the stages, suggesting their stable regulatory roles throughout development (Supplementary Fig. S4).
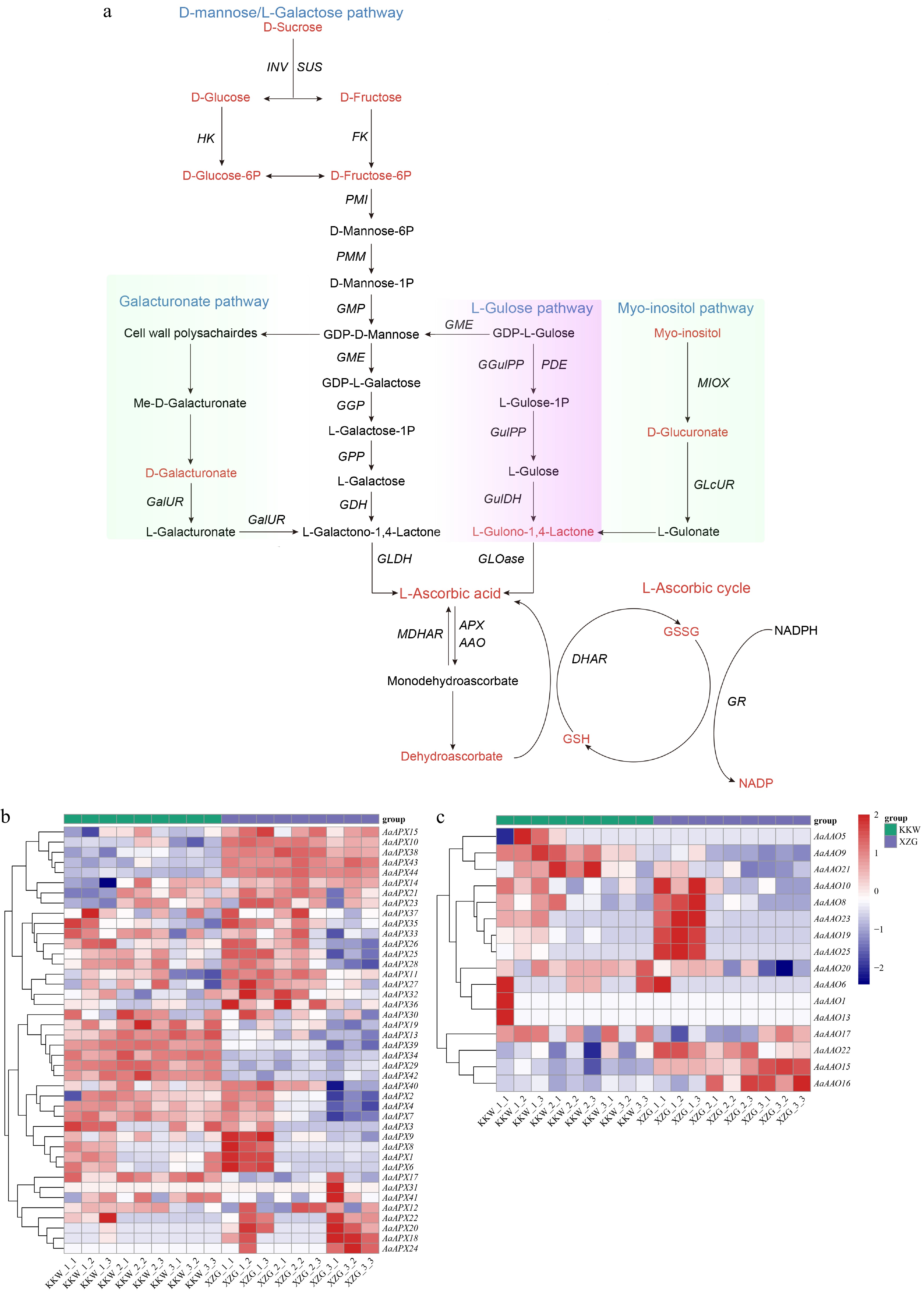
Figure 6.
(a) Overview of the AsA metabolome profiles in two kiwifruit varieties. (b) Differential expression of AaAPX gene family members. (c) Differential expression of AaAAO gene family members.
To verify the accuracy of screened genes, six DEGs (AaAPX2, AaAPX3, AaAPX4, AaAPX14, AaAAO15, and AaAAO16) were selected for qRT-PCR analysis (Figs. 7, 8). The relative expression levels of AaAPX3, AaAPX4, AaAPX14, AaAAO15, and AaAAO16 were higher in KKW than in XZG, indicating that these genes may be involved in cultivar-specific regulation. These results suggest that ascorbate-related oxidoreductases genes play complex regulatory and diverse roles in kiwifruit. Furthermore, the potential roles of APX and AAO gene family members were investigated; all identified gene members were subject to GO enrichment analyses. Except for partial gene members, such as AaAAO16_25, that can't be annotated with corresponding functions, most of them were predominantly classified into four categories: L−ascorbate peroxidase activity, response to ROS, cellular response to oxidative stress, and hydrogen peroxide catabolic process (Supplementary Fig. S5).
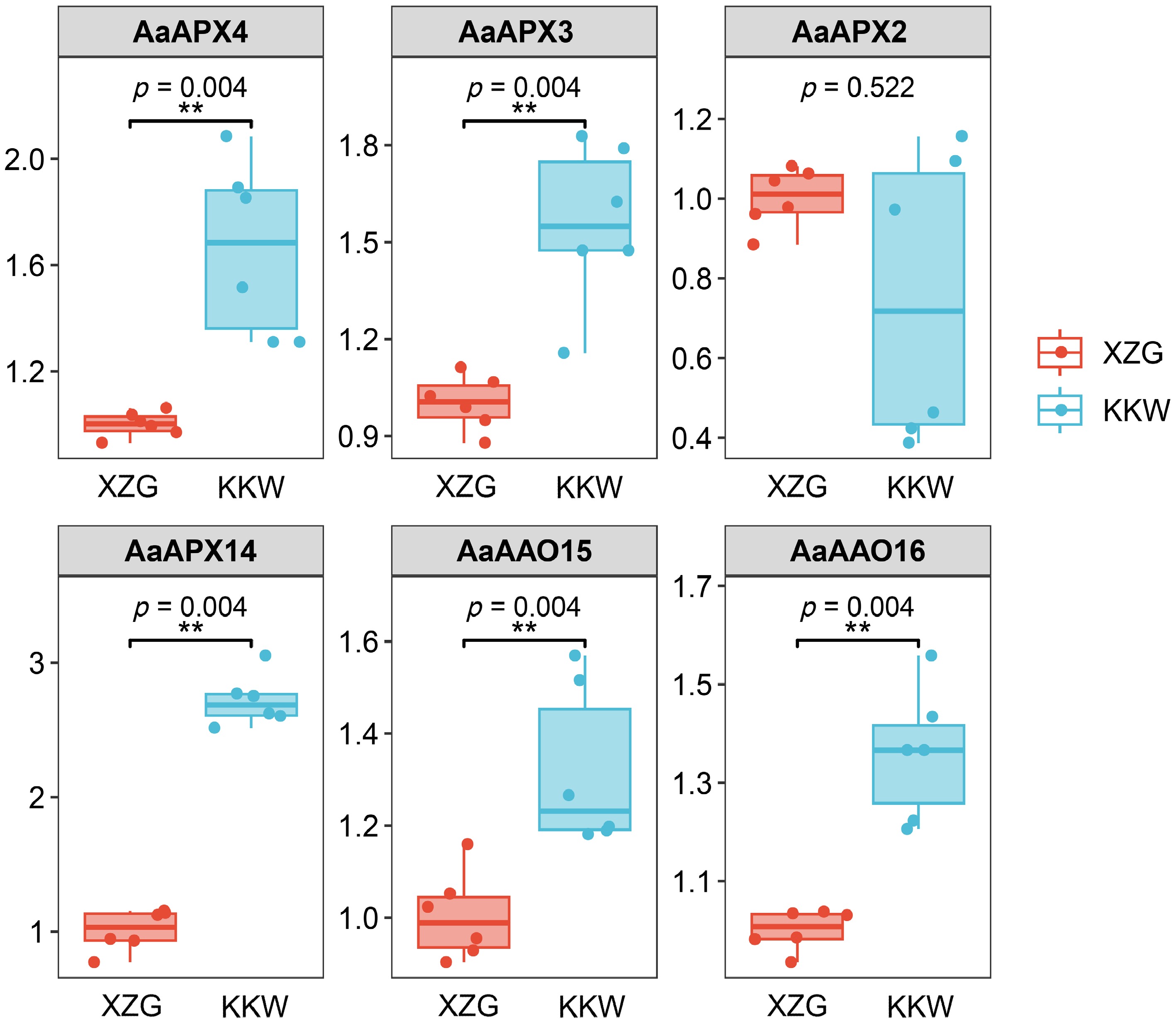
Figure 7.
Results of qPCR analysis of the six selected AaAPXs in KKW and XZG.
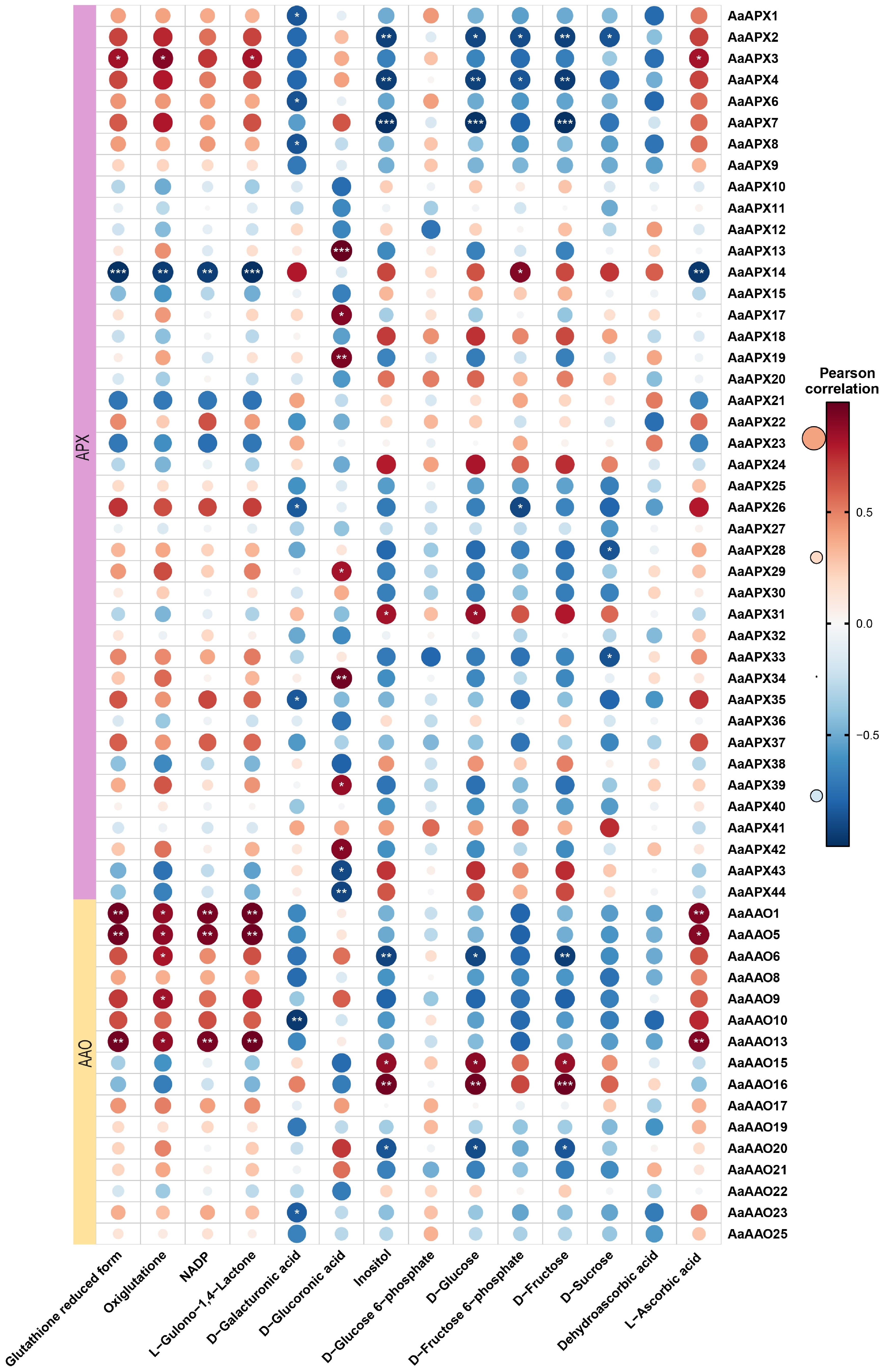
Figure 8.
Correlation analysis between the expression of AaAAO and AaAPX genes and ascorbic acid-related metabolites in A. arguta. Red and blue color notes positive and negative correlations with gene expression, respectively.
Correlation analysis of ascorbate-related genes during kiwifruit ripening
-
To investigate whether the expression levels of AaAAO and AaAPX genes are associated with L-ascorbate metabolism in kiwifruit, Pearson correlation coefficients (PCCs) were calculated between 69 genes and 13 related metabolites, including L-ascorbic acid, dehydroascorbic acid, and D-galacturonic acid, based on the four major AsA biosynthesis and recycling pathways (Fig. 8, Supplementary Table S9). The analysis revealed that AaAAO1, AaAAO5, AaAAO13, and AaAPX3 were positively correlated with L-ascorbic acid levels, whereas AaAPX14 showed a negative correlation with L-ascorbic acid.
Notably, it was observed that L-ascorbate displayed the highest accumulation at the developmental stage in KKW and then gradually declined during subsequent kiwifruit ripening, with a higher content than the other varieties at the T1 stage (Fig. 9). In contrast, XZG samples exhibited higher levels of fructose and glucose compared to KKW. Correlation analysis showed that AaAAO15 and AaAAO16 were positively correlated with fructose content, whereas AaAAO6, AaAAO20, AaAPX2, AaAPX4, and AaAPX7 were negatively correlated with fructose (Fig. 8). Our analysis revealed strong connectivity between the AaAAO and AaAPX genes and related metabolites, highlighting their potential role in regulating L-ascorbate metabolism during kiwifruit ripening.
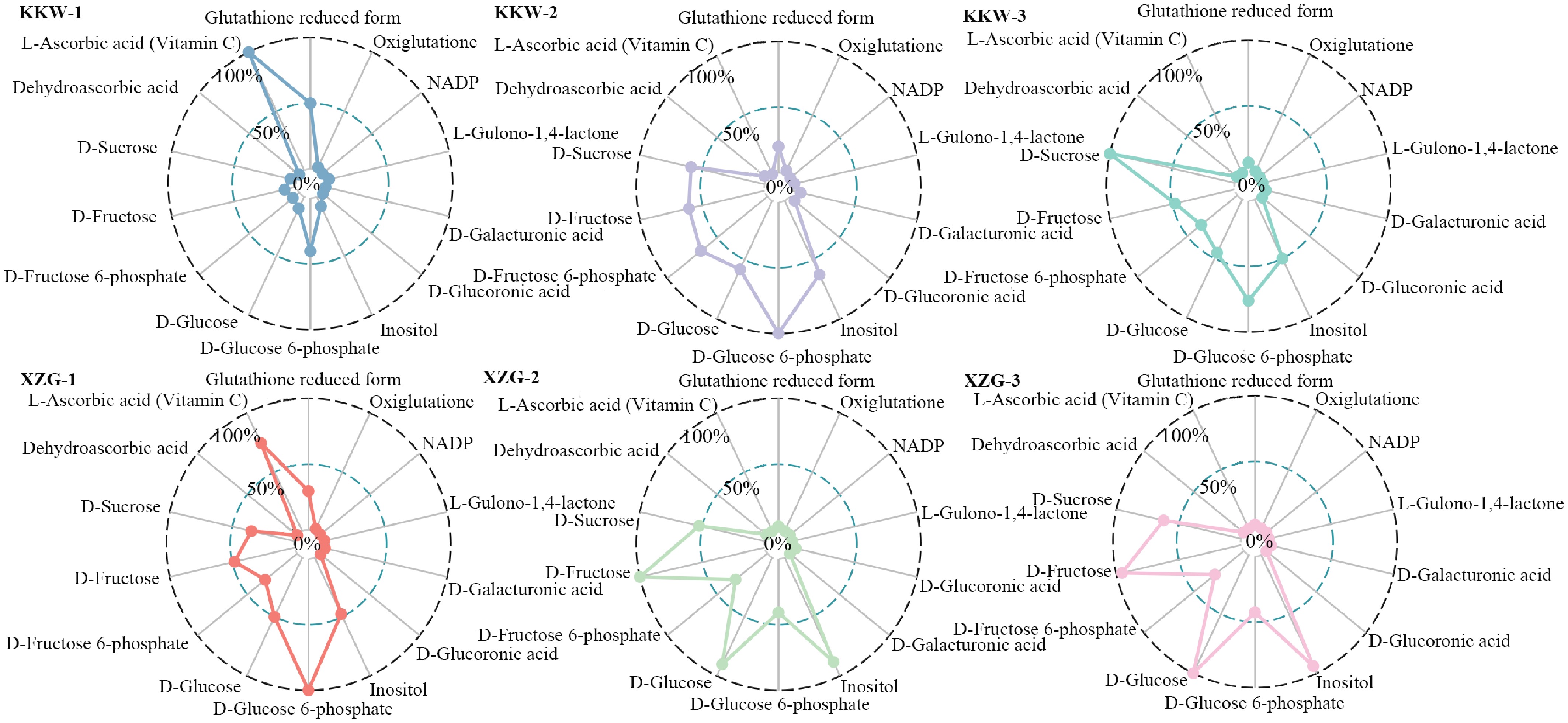
Figure 9.
Comparison of the 13 representative metabolites between two kiwifruit varieties.
-
Ascorbic acid is essential for multiple physiological functions, especially in plant development, and serves as a powerful antioxidant that alleviates ROS accumulation[1]. Nevertheless, the functional interaction between AAO and APX in ascorbic acid homeostasis, and their influence on its fluctuation and fruit quality, remains poorly understood. While the genome-wide identification of APX genes has been extensively documented in various plant species, research on AAO has been relatively limited, with only a few studies reported in G. hirsutum and B. vulgaris[20,21]. Genome-wide identification in A. arguta uncovered 44 APX genes and 25 AAO genes. The isoelectric points (pI) of APX and AAO ranged from 5.49–9.71 and 5.7–9.07, respectively, consistent with previous findings in other species[20,21]. For instance, the pI range of CsAPX was reported to be 5.54–8.87[14], while that of GhAO was 5.65–8.63[20]. Subcellular localization analysis revealed that APX proteins were predominantly localized in the chloroplast (19/44) and cytoplasm (24/44), with only AaAPX7 detected in the nucleus. In contrast, AAO proteins were mainly distributed in the vacuole (10/25), nucleus (7/25), and chloroplast (6/25), suggesting their potential functional roles in distinct organelles[51]. Furthermore, an uneven genomic distribution of AAO and APX gene family members was observed. For example, nine AaAPX genes were identified on chromosome 11, while no AaAPX genes were detected on chromosome 15. Similarly, AaAAO genes exhibited a clustered distribution, with eight genes located on chromosome 28 and none on chromosome 25. This uneven distribution may mirror the genetic variation and evolutionary trajectory of A. arguta, possibly driven by specific gene duplication events[52].
Phylogenetic analysis provides key insights into gene family evolution, enabling the identification of orthologs, functional inference, and the detection of lineage-specific variations[53]. For instance, Liao et al. demonstrated that APX genes in A. chinensis could be classified into four phylogenetic groups, with genes sharing similar intron numbers clustering together[1]. Similarly, Pang et al. reported that nine CaAPX genes from pepper were grouped into five clades alongside APX genes from A. thaliana, rice, tomato, and potato[16]. In this study, 44 APX genes in the A. arguta genome were identified, which were classified into five distinct subfamilies similar to the previous study. Notably, genes such as AaAPX1_4, AaAPX6, and AaAPX8_9, localized to the chloroplast, clustered within the same subfamily, while others, including AaAPX12, AaAPX17, and AaAPX19, were grouped into different subfamilies. Additionally, 25 AAO genes were identified, which were also classified into three groups. Intriguingly, all vacuole-localized AAO proteins (e.g., AaAAO1_4 and AaAAO11_14) clustered within subfamily III, alongside other members with distinct subcellular localizations. This phylogenetic pattern suggests a possible link between protein subfamily classification and localization, though experimental validation is required to confirm this hypothesis[53].
Gene duplication events, including tandem repeats and segmental duplications, are critical mechanisms for expanding gene families by generating homologous genes and increasing gene copy numbers[54]. In this study, 44 AaAPX and 25 AaAAO genes were identified, most of which originated from whole-genome duplication (WGD) events, highlighting the significant role of WGD in the expansion of AaAPX and AaAAO gene families in A. arguta. Furthermore, the Ka/Ks analysis revealed that the majority of homologous gene pairs exhibited Ka/Ks ratios < 1 (Supplementary Table S7), indicating that AaAPX and AaAAO genes have undergone purifying selection during their evolutionary history. Cis-acting elements are critical regulators of plant gene expression and play an indispensable role in mediating plant responses to environmental changes and evolutionary adaptations[55]. It's widely reported that members of the APX gene family are involved in abiotic stress responses. For instance, Li et al. reported that the loss of any APX genes in A. thaliana led to reduced stress tolerance during both germination and maturation stages, underscoring the functional importance of these genes in stress adaptation[56]. In this study, Box 4, ARE, and ABRE were identified as the predominant cis-regulatory elements in the promoters of APX genes, with 142, 75, and 68 occurrences, respectively. These elements are primarily associated with light response, stress response, and hormone signaling (Fig. 4), highlighting the pivotal role of APX genes in regulating key physiological processes in plants[57]. As a species characteristically thriving in and well-adapted to sun-exposed environments, the high abundance of light-responsive cis-regulatory elements in its APX genes may reflect an evolutionary adaptation to optimize light utilization and enhance stress tolerance. Interestingly, it was also observed that ABRE (50 occurrences), Box 4 (47 occurrences), and ARE (43 occurrences) were the dominant cis-regulatory elements in the promoters of AAO genes. The predominance of ABRE elements, which are associated with hormone signaling, suggests that APX and AAO genes may share overlapping functional roles in mediating plant responses to hormonal and environmental stimuli. Notably, the light-responsive ATCT motif and Gap box were uniquely predicted in AaAAO5 and AaAPX3, respectively. The unique presence of these regulatory elements may be associated with their putative functions as key regulators. These findings provide valuable insights into the regulatory mechanisms underlying the stress-responsive and hormone-mediated functions of APX and AAO gene families in A. arguta. Further investigation into the specific roles of these cis-regulatory elements could enhance our understanding of their contributions to plant development, adaptation, and evolution[58].
Extensive studies have documented the variation in APX gene expression among different tissues in multiple plant species. For instance, Pang et al. investigated the expression profiles of CaAPX genes in different tissues of pepper and found that CaAPX8 exhibited consistently high expression levels across all tissues except at the F-Dev5 stage[16]. Similarly, Aleem et al. utilized RNA-seq data to analyze the expression patterns of APX genes in fourteen soybean tissues, revealing that the majority of APX genes displayed tissue-specific expression patterns, highlighting their functional diversity[59]. Here, expression patterns of APX and AAO genes were examined across three developmental stages in A. arguta, and AsA content was detected in the three stages to explore the relationship between the accumulating level and the degradation/regeneration of AsA. In fact, the AsA content across the three developmental stages represents a net outcome of the balance between its de novo biosynthesis, degradation, and regeneration. In a previous study, AcAO1 and AcAPX2 were identified as key genes in the AsA recycling pathway. At 10 DPA, despite high expression levels of biosynthetic genes, the AsA content was low—a phenomenon that coincided with the peak expression of both AcAO1 and AcAPX2 and the highest accumulation of L-dehydroascorbate. Functional genomic validation subsequently confirmed that the AsA pool at this stage is primarily regulated by the recycling pathway[60]. In Stage I, the observed peak in AsA accumulation is driven by active biosynthesis to support rapid cell division and morphogenesis. Critically, this stage was characterized by the most robust transcriptional activity, as the majority of genes within both the APX and AAO gene families exhibited their highest expression levels, with the greatest number of highly expressed genes occurring in Stage I compared to the subsequent stages (Fig. 6b, c). The concurrent high expression of APX and AAO genes in Stage I reflects a dynamic balance in AsA metabolism. APX-driven regeneration maintains the reduced AsA pool, whereas AAO-mediated degradation likely serves to actively fine-tune the pool size and redox signaling for proper development. Specifically, AaAAO1, AaAAO13, AaAAO19, and AaAAO25, exhibited significantly higher expression levels at the first developmental stage in both KKW and XZG cultivars compared to the other two stages (Fig. 6c). In contrast, during Stages II and III, the significant decrease in AsA content correlates with a predicted decline in biosynthetic activity. This period was marked by a widespread downregulation in both AAO and APX gene expression, indicating a coordinated reduction in the entire metabolic flux of AsA encompassing both its regenerative turnover and its degradative consumption. This collective downregulation reflects a metabolic shift away from the high demand of active growth towards a lower maintenance requirement in maturing tissues. However, Specific APX genes, such as AaAPX18, AaAPX24, and AaAPX31, displayed the highest expression levels during the third developmental stage. As supported by Liu et al., the initial low accumulation at 10 DPA can be attributed to intense degradation and regeneration, evidenced by the high expression of APXs and AAOs, particularly AAOs[60]. The subsequent peak around 20 DPA was primarily driven by active biosynthesis, coupled with a relative decline in APX and AAO expression (though their levels remained substantial). Finally, the decrease in AsA during ripening was mainly caused by reduced biosynthesis, with degradation/regeneration processes also contributing.
GO enrichment analysis indicated that these genes are mainly associated with responses to ROS (Supplementary Fig. S5), implying a potential role in ROS scavenging and oxidative stress regulation during fruit development. Correlation analysis revealed strong co-expression between AaAPX18 and AaAPX24 (Supplementary Fig. S1), suggesting a coordinated role in antioxidant metabolism. Phylogenetic analysis further indicates that both genes cluster with the A. thaliana APX gene At4g35000 (Fig. 2b), which is known to participate in ROS detoxification and redox homeostasis[16]. This homology implies that AaAPX18 and AaAPX24 may serve conserved roles in regulating oxidative balance during fruit development, meriting further functional validation. Moreover, the positive correlations of AaAAO1, AaAAO5, and AaAPX3 with L-ascorbic acid suggest their involvement in ascorbate accumulation, whereas the negative correlation of AaAPX14 may indicate a role in ascorbate turnover. While this study documented dynamic changes in AsA content and identified key candidate genes in the two varieties, the molecular mechanisms responsible for the varietal differences are not yet fully understood. Elucidating the precise roles of these genes, including their specific contributions to the regeneration and degradation processes that govern AsA dynamics, will require further molecular validation through techniques such as functional genomics. Collectively, these findings shed light on the temporal regulation of APX and AAO gene expression and their roles in stress responses and developmental processes in A. arguta. Further functional studies are needed to clarify their contributions to redox homeostasis, stress tolerance, and ascorbic acid biosynthesis in kiwifruit.
-
In this study, a comprehensive genome-wide analysis of APX and AAO genes in A. arguta was performed, identifying 44 APX and 25 AAO members. Phylogenetic analysis revealed that both APX and AAO genes could be classified into five and three distinct groups. A potential correlation between protein subfamily classification and subcellular localization was observed, although further experimental validation is required to confirm this hypothesis. Additionally, APX and AAO gene family members were unevenly distributed across different chromosomes, a pattern likely attributable to gene duplication events during evolution. Expression analysis of APX and AAO genes across three developmental stages revealed that AAO and APX genes exhibited relatively higher expression levels in the first stage in both KKW and XZG cultivars. Notably, AaAPX18 and AaAPX24 displayed the highest expression levels during the third developmental stage, suggesting a potential synergistic effect in mediating ROS scavenging and mitigating oxidative damage. The AsA level peaked at 20 DPA, driven predominantly by active biosynthesis, and subsequently declined during ripening mainly due to attenuated biosynthesis. Consistently, the expression of AAO and APX genes, which was relatively strong in the initial phase, also weakened alongside the biosynthetic process. This finding highlights their critical role in maintaining cellular homeostasis during late developmental stages. These results provide comprehensive insights into the genomic organization, evolutionary dynamics, and functional roles of APX and AAO gene families in A. arguta. Further investigation into the regulatory mechanisms and synergistic interactions of these genes could enhance our understanding of their contributions to antioxidant defense, ascorbate accumulation, and biosynthesis.
This research was supported by the National Natural Science Foundation of China (Grant No. 32300314), the Shaanxi Academy of Science Research Funding Project (Grant Nos 2022K-09, 2024p-12), and the Xi'an Botanical Garden of Shaanxi Province Young Talent Funding Project (Grant No. 2024TJ-02).
-
The authors confirm contribution to the paper as follows: study conception and design: Jia Y; data collection: Qiang X; analysis and interpretation of results: Jia Y, Qiang X, Jiang X, Yan C; technical assistance: Qiang X, Ren T, Yang Y, Chang X, Zhang Y; draft manuscript preparation: Jia Y, Jiang X, Yan C. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and supplementary information files.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Xin Jiang, Chunli Yan
- Supplementary Table S1 Genome information for genome-wide identification of AaAPX and AaAAO genes in this study.
- Supplementary Table S2 Primers for the qRT-PCR experiment.
- Supplementary Table S3 The number of AaAPXs and AaAAOs genes in Arabidopsis and Actinidia used in this study.
- Supplementary Table S4 Characteristics of 44 APX proteins in A. arguta.
- Supplementary Table S5 Characteristics of 25 AAO proteins in A. arguta.
- Supplementary Table S6 The three duplicated types of AaAPX and AaAAO genes in kiwifruit genome.
- Supplementary Table S7 Homologous APX and AAO gene pairs and Ka/Ks values in A. arguta.
- Supplementary Table S8 Results of SignalP.
- Supplementary Table S9 Representative metabolites involved in Vc metabolism in the two kiwifruit varieties.
- Supplementary Fig. S1 Correlation analysis of AaAPX genes and AaAAO genes in kiwifruit.
- Supplementary Fig. S2 Comparison of AAO expression pattern between two varieties and different stages.
- Supplementary Fig. S3 Differential expression analysis of APX and AAO gene families across KKW and XZG individuals.
- Supplementary Fig. S4 Heatmaps of differentially expressed TFs among the two kiwifruit varieties.
- Supplementary Fig. S5 GO Enrichment of AaAAOs and AaAPXs gene families.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Jiang X, Yan C, Qiang X, Ren T, Yang Y, et al. 2026. Genome-wide analysis of ascorbate-related oxidoreductases genes in Actinidia arguta provides new insights into ascorbic acid metabolic regulation during kiwifruit development. Fruit Research 6: e003 doi: 10.48130/frures-0025-0042 

Genome-wide analysis of ascorbate-related oxidoreductases genes in Actinidia arguta provides new insights into ascorbic acid metabolic regulation during kiwifruit development
- Received: 18 July 2025
- Revised: 03 November 2025
- Accepted: 25 November 2025
- Published online: 21 January 2026
Abstract: The ascorbate oxidase (AAO) and ascorbate peroxidase (APX) gene families are essential for the metabolic regulation of ascorbic acid. In this study, 25 AAO and 44 APX genes were identified in A. arguta, which were classified into three and five subfamilies, respectively. Syntenic analysis revealed that the expansion of these gene families was primarily driven by whole-genome duplication. Transcriptome analysis showed differential expression of AAO and APX genes across developmental stages in two kiwifruit cultivars. In addition, several key metabolites involved in the L-ascorbic acid pathway were detected, including L-ascorbic acid (AsA), dehydroascorbic acid, and oxidized glutathione. Notably, AaAAO1, AaAAO5, and AaAPX3 showed correlations with AsA content. Furthermore, the expression patterns of six selected genes (AaAPX2, AaAPX3, AaAPX4, AaAPX14, AaAAO15, and AaAAO16) were further validated in two A. arguta varieties utilizing qRT-PCR. Overall, this study provides new insights into the identification and functional roles of AAO and APX gene families in A. arguta and highlights their role in regulating ascorbic acid metabolism.
-
Key words:
- Actinidia arguta /
- Ascorbic acid /
- Gene family /
- Differentially expressed genes /
- Metabolites