








-
With the advances in long-read sequencing technology, we can easily generate a large amount of high-quality sequencing data using efficient sequencing platforms such as PacBio Revio (
www.pacb.com/revio ) and Nanopore duplex[1]. With these high-quality data and state-of-the-art assembly algorithms (such as hifiasm[2] and verkko[3]), we can conveniently generate a high-quality, error-free, and more continuous genome assembly than before. In general, a high-quality genome assembly is generated by following a combined strategy of scaffolding, polishing, and gap-filling processes, then the genome annotation could be considered the most important process for further in-depth analysis. Nowadays, genome annotation is facing challenges of the partial conservation of sequence patterns, variable intron length, the different distances between the genes, alternative splicing, TE insertions, and pseudogenes[4]. Accurate genome annotation is crucial and provides basic information to focus on species evolution, population genetic analysis, functional genomics, and so on.There are a large number of well-established and widely used genome annotation pipelines, such as MAKER[5], MAKER-P[6], BRAKER[7], GETA (
https://github.com/chenlianfu/geta ), EvidenceModeler[8], EGAP (Eukaryotic Genome Annotation Pipeline) (https://github.com/ncbi/egapx ), and so on. Although these pipelines are different in detail (for example, the different pipelines combine different software/tools), the core idea is almost the same: combining the different types of evidence and synthesizing them into non-redundant genome annotations. These different types of evidence include ab initio (de novo) evidence, expressed sequence tags (ESTs), or RNA-seq evidence, and homology-based evidence. Each type of evidence can be generated by corresponding input data and approaches. For ab initio evidence, we could generate the results by using the RNA-seq data and high-quality homologous protein sequences to train the gene models for the researchers' specific case, through AUGUSTUS[9], SNAP (https://github.com/KorfLab/SNAP ), Helixer[10], and so on. For ESTs or RNA-seq evidence, we could obtain the gene models by using the RNA-seq data through the Program to Assemble Spliced Alignments (PASA)[8], StringTie[11], TransDecoder (https://github.com/TransDecoder/TransDecoder ), exonerate (https://github.com/nathanweeks/exonerate ), and so on, to generate the transcriptome information to support genome annotation. For the homology-based evidence, we use the homologous protein sequences to generate the results through miniprot[12], exonerate, GenomeThreader[13], and so on. Finally, these different types of evidence are combined to obtain final non-redundant high-quality genome annotation, usually, this combining process could be varied if researchers use different pipelines. Here we have summarized the commonly used genome annotation pipelines and the widely used tools in each procedure (Fig. 1).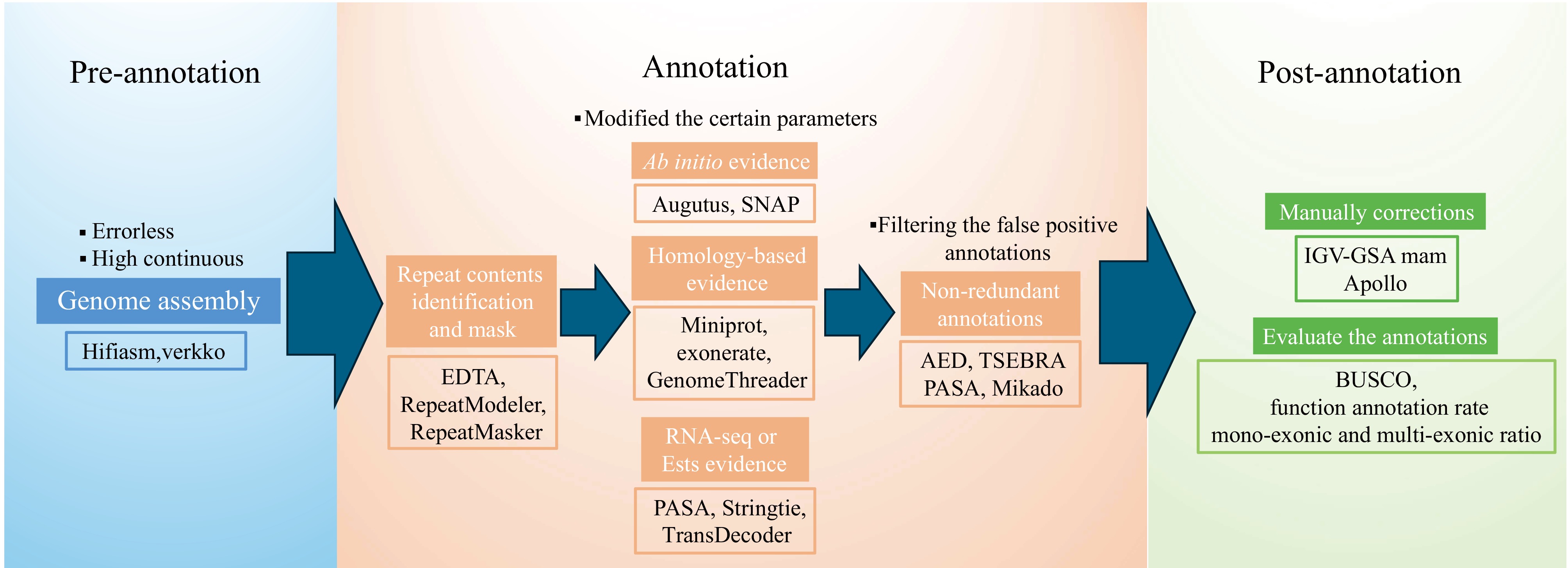
Figure 1.
Recommended flowchart for genome annotation. The tools/pipelines listed in the figure only represent part of those commonly used in genome annotations. Researchers need to carefully consider and use the appropriate tools/pipelines for their specific scenario.
It is widely known that different evidence and pipelines could cause significantly varied annotation results[14], and the different parameters used in certain tools/pipelines may also generate slightly different annotation results. For example, for different cut-off e-values used in read/sequence alignment (e.g. the default e-value for blastx in MAKER is 1e-06, researchers can manually change it as 1e-5 or any other value), the different alignment results may slightly change the evidence, and finally change the annotation results. This means that even if we have chosen good benchmarked tools/pipelines and high confidence and sufficient evidence, the annotation results will still have prediction errors, including loss of genes[15], incorrect exon and gene boundaries[16], retention of non-coding sequences in coding exons[17], and fragmentation or fusion of gene models[18]. These errors can seriously affect downstream analysis, leading to bias in large-scale genomic research[19−21]. Here, we briefly discuss reasonable procedures to reduce annotation errors (Fig. 2).
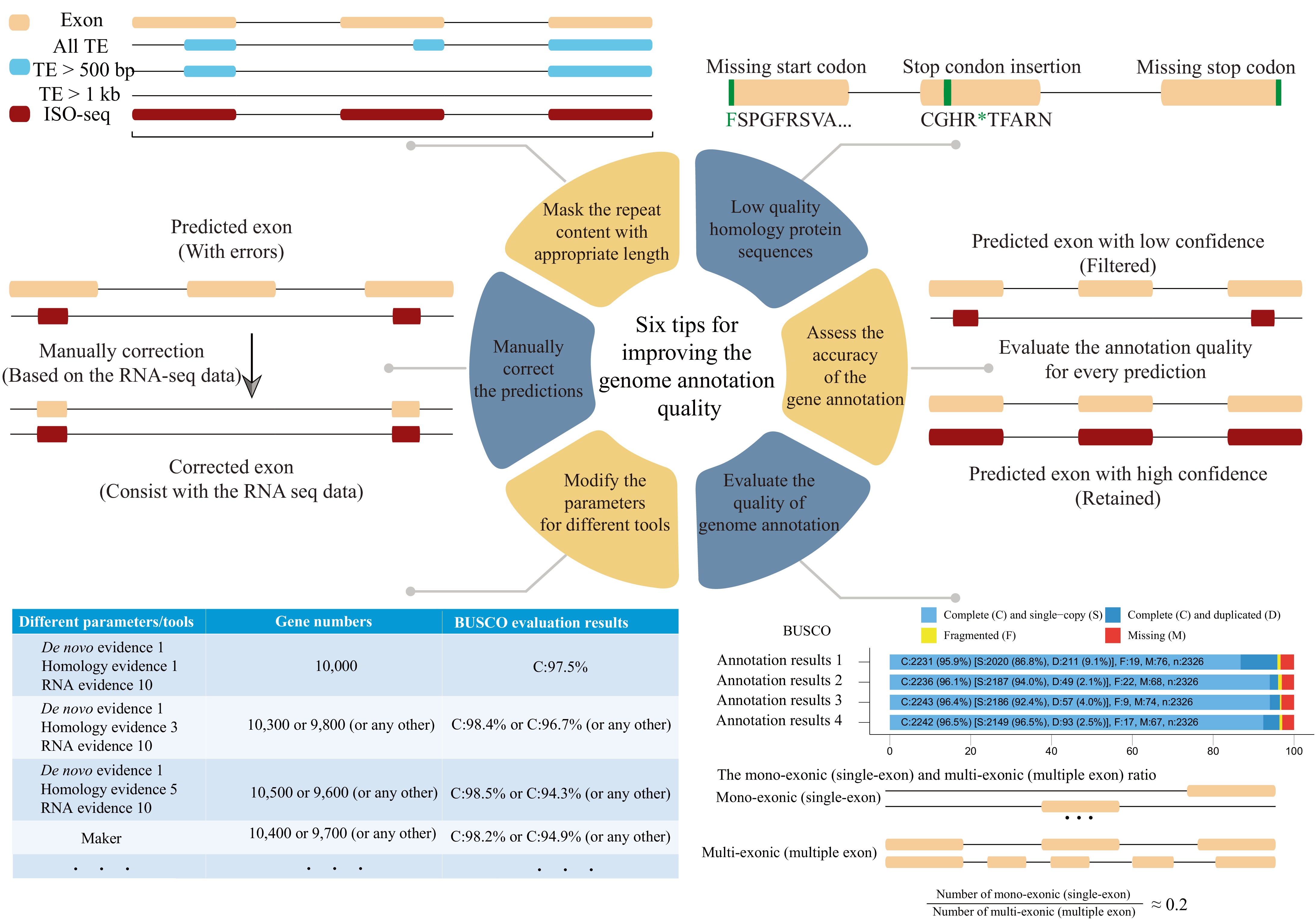
Figure 2.
Six tips for improving genome annotation quality.
First, the identification and masking of repeat regions is critical, for the repeat regions could lead to false evidence for gene annotation[22]. For example, the transposable elements (TEs) inserted into the genic region could be erroneously identified as extra exons. In maize, researchers have found that the TEs near or within genes would fragment gene space or even miss gene annotations[23]. By masking the TEs with different lengths (≥ 1 kb, ≥ 500 bp, and ≥ 80 bp), researchers found that unmasking short TEs (< 1 kb) would effectively prevent the fragmentation of genic regions and improve the annotation quality according to the evaluation results of Benchmarking Universal Single-Copy Ortholog (BUSCO)[24] in maize[23]. Therefore, we recommend that the researchers mask the TEs of limited length rather than all TEs when performing genome annotation. Moreover, one of the most commonly used repeat content annotation pipelines, Extensive de novo TE Annotator (EDTA), its default output of repeat-masked genome sequences named '[genome_prefix].fa.mod.MAKER.masked', masked the TE > 1 kb which means you could mask the genome by filtering different TE lengths. By masking the TE with different lengths, it is possible to explore reasonable and suitable annotation results for your specific genomes.
Second, erroneous gene predictions could be propagated while we generate homology-based evidence. The low-quality proteins from the public databases containing the sequence or prediction errors could lead to inaccurate annotation results[25,26], for example, some genes may contain multiple stop codons. Other annotation processes may further use these errors to generate the new annotation results and eventually obtain a wide dissemination. To address the challenges caused by low-quality homology evidence, we recommend utilizing the high-confidence protein from uniprot (
www.uniprot.org/help/downloads ) and the already known well-annotated results from related species or perform quality control by yourself before the annotation process.Third, genome annotation results using different pipelines may vary and cause bias. There are already benchmarking works showing that MAKER has a lower performance than the combination pipeline of TSEBRA[27] and BRAKER[4,7]. Even using the same pipeline, e.g. EvidenceModeler, users must manually select the weight value for different evidence to generate the appropriate results, while modifying the weight value will undoubtedly slightly change the annotation results. In practice, we recommend that researchers set the weight value from the EST or RNA-seq evidence to 10 and the other set to 1. Also, the researchers could modify the weight value for their specific case, which would lead to slightly different annotation results, and then choose the best one as the final annotation result. Moreover, we need to be cautious of default parameters used in certain tools. Some species may have a specific genome architecture, such as the Chinese pine containing many genes with an extremely long intron (> 10 kb)[28], when we use Trinity[29] to assemble the RNA-seq data on the genome-guide mode, the '--genome_guided_max_intron' should use the larger value instead of the default value of 10,000. In practice, we may need to try several iterations of different parameters, or even different tools/pipelines, and compare the different annotation results and choosing the outperformed one as the final result.
Fourth, we need to use appropriate evaluation methods to assess the accuracy of the gene annotation. If the researchers use MAKER, they could calculate the annotation edit distance (AED), which can be used to measure the congruence between the annotation and its supporting evidence[30]. The AED value ranges from 0 to 1, the annotations with an AED of 0 referring to the perfect agreement with the evidence provided, while 1 refers to the complete lack of evidence to support the annotation. In practice, the annotations with AED values lower than 0.5 (≤ 0.5) could be considered good annotations, and lower than 0.3 could be considered high-quality annotations[5]; so we can filter out the annotations with large AED values to reduce the false positive annotation results. For those who used the BRAKER pipeline, we could use TSEBRA (
https://github.com/Gaius-Augustus/TSEBRA ) to filter out the low-confidence annotations according to the provided evidence and increase the accuracy. Furthermore, PASA can be used to update annotation results, and incorporate PASA alignment evidence to correct exon boundaries for any annotation pipeline, as well as Mikado[31] dose.Fifth, the genome annotation results could be manually corrected by Apollo[32], IGV‐GSAman (Genome Sequence Annotation Manipulator, an extended version of Integrative Genomics Viewer) (
https://gitee.com/CJchen/IGV-sRNA ), and so on. Based on the datasets from RNA-seq and homologous proteins, we could jointly use them to manually correct the annotations. By visually checking each annotation, a significant improvement could be observed compared to the original annotations[33,34].Sixth, evaluate the quality of genome annotation results globally. The BUSCO is the most commonly used tool to assess the quality. Empirically, the BUSCO complete value should be at least 90% to be considered as a good annotation result. The BUSCO evaluation process works by evaluating the presence of predefined highly conserved orthologous genes, and there are already 462 available datasets that could be used for evaluation. The most important thing is that we need to choose the appropriate datasets to evaluate the results, for example, the same annotation results could get different BUSCO completeness scores through the different BUSCO datasets[35]. We could also calculate the mono-exonic (single-exon), and multi-exonic (multiple-exon) ratios to evaluate the annotation quality, which can be done by gFACs[36]. By assessing a large set of model plant genomes' annotation results, the ideal ratio was near 0.2[4,37]. This means that, theoretically, the more the ratio deviates from 0.2, the higher the likelihood of erroneous annotations. Moreover, we could also compare the annotated genes with the public databases and calculate the rates. Higher rates of over 80% by the different databases indicate better prediction results[4].
From the above six tips, you would notice that the most important dataset is the RNA-seq data. Whatever the tools/pipelines we use for the genome annotation or correction, high-confidence evidence is always needed, and in most cases the sufficient depth, multiple tissues, and different periods of RNA-seq data is always treated as the most high-confidence evidence, for some genes may be expressed only in specific tissues or developmental period.
It is also worth mentioning that how the long-read transcriptome data was used in the genome annotation. In general, short-read data could produce predicting errors because of error mapping, as the single read cannot cover the full length of the gene. The use of long-read sequencing can increase the accuracy of automated genome annotation by improving genome mapping of sequencing data, correctly identifying intron-exon boundaries, directly identifying alternatively spliced transcripts, identifying transcription start and end sites, and providing accurate strand orientation for single exon genes[38,39]; and several studies have already shown that long reads paired with short reads can improve annotation quality[40,41], also benchmarking work has proven that using high-quality long reads could significantly improve the annotation results compared to the short-read-based annotations[42]. In addition, deep learning and machine learning approaches have recently been adopted for genomic annotation (reviewed by Chen et al.[43]). We hope that this discussion could inspire researchers in the annotation process and help them to choose the optimal pipeline/parameters to obtain annotation results with fewer errors for further downstream analysis.
HTML
This work was funded by the Chinese Academy of Agricultural Sciences Elite Youth Program (110243160001007), The Agricultural Science and Technology Innovation Program, and the Funding of Major Scientific Research Tasks, Kunpeng Institute of Modern Agriculture at Foshan (KIMA-ZXFR2024004).
-
The authors confirm contribution to the paper as follows: study conception and design: Lan L, Wu Z, Li C; draft manuscript preparation: Lan L, Hu H, Jia Y, Zhang X, Jia M. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
-
The authors declare that they have no conflict of interest. Dr. Zhiqiang Wu is the Editorial Board member of Genomics Communications who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research groups.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
Lan L, Hu H, Jia Y, Zhang X, Jia M, et al. 2025. Tips for improving genome annotation quality. Genomics Communications 2: e005 doi: 10.48130/gcomm-0025-0006


|