








-
An R-loop is a three-stranded nucleic acid structure composed of a DNA-RNA hybrid strand and an unpaired single-stranded DNA (ssDNA) (Fig. 1a)[1]. First observed in vitro via electron microscopy in 1976[2], R-loops are now recognized as widespread genomic features across diverse species, and play multifaceted roles in key biological processes, including transcription, replication, antibody class switching, DNA repair, and genome stability[1,3]. As an epigenetic factor, R-loops can influence other epigenomic marks, such as DNA methylation patterns, histone modifications, RNA modifications, and higher-order chromatin architectures[4]. Moreover, recent studies highlight R-loop's crucial roles in driving cytosolic DNA-RNA hybrid and activating immunity[5,6].
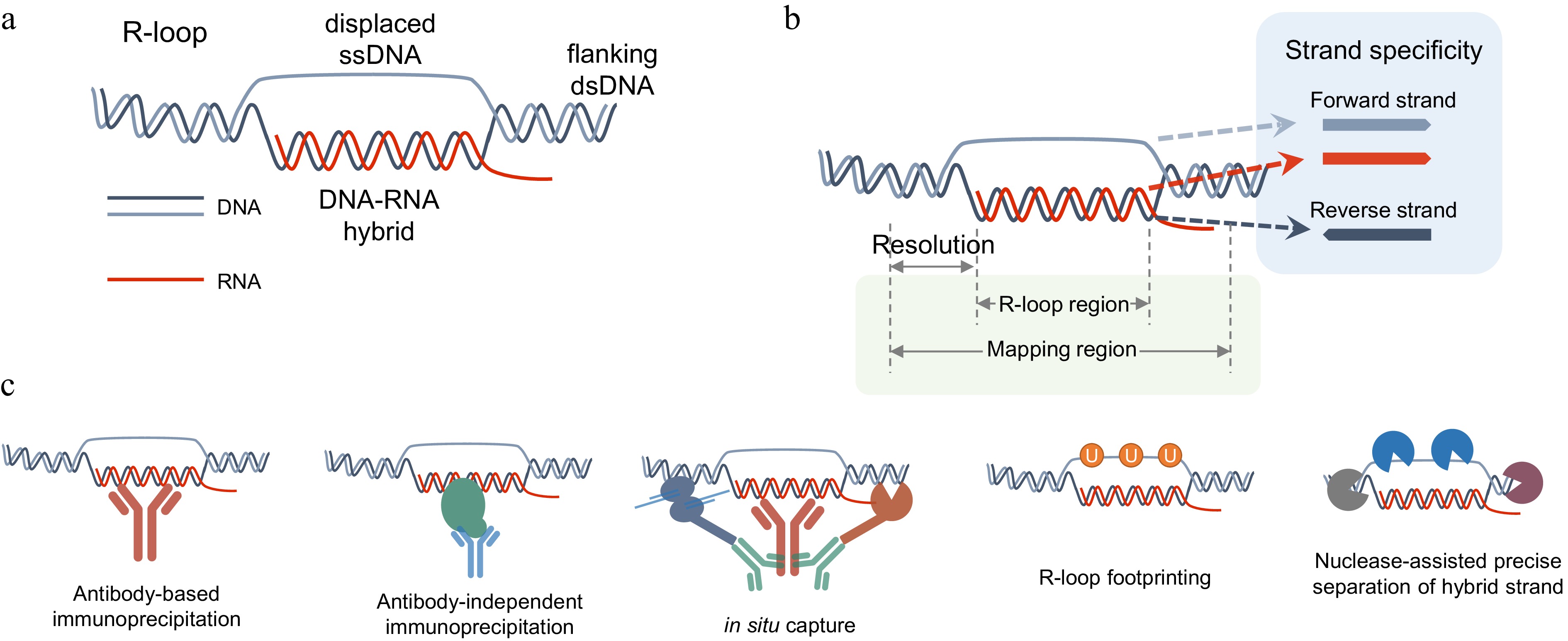
Figure 1.
R-loop structure and profiling methods. (a) Sketch map of R-loop. (b) Schematic diagram illustrating the resolution and strand-specificity provided by different R-loop mapping methods. Resolution is defined as the distance between the experimentally identified edges of R-loop fragments via isolation or labeling and their actual genomic boundaries. Strand-specificity refers to the orientation of an R-loop, which can be defined based on the sequence of the ssDNA strand or the hybrid DNA/RNA strand. (c) Diagrammatic representation of methodological strategies adopted by R-loop mapping methods.
R-loops in the genome can be categorized into distinct types according to their origin and structural characteristics: co-transcriptional R-loops, which form when nascent RNA hybridizes with its template DNA; trans R-loops, in which the RNA strand is not directly transcribed from the DNA strand it binds to, but originates from other transcripts with sequence similarity; replication-associated R-loops, derived from RNA primers of Okazaki fragment synthesized during DNA replication, and repair-associated R-loops, formed by transcription at DNA break sites during DNA repair.
This diversity in origin and structure complicates the development of robust R-loop detection technologies. To study the genomic distribution and dynamics of R-loops, a series of R-loop profiling methods has been established. The essential information required to define an R-loop comprises: genomic coordinates, typically defined by the region of its DNA-RNA hybrid, or the ssDNA segment (Fig. 1b); strand specificity, represented by the orientation of the hybrid DNA strand, the hybrid RNA strand, or the unpaired ssDNA (Fig. 1b); and abundance, encompassing both relative and absolute quantification. Relative quantification involves normalization to determine the level of a specific R-loop relative to all R-loops, whereas absolute quantification utilizes spike-in standards to calculate the exact molecular count or the percentage of R-loops relative to total genomic DNA. Genomic coordinates constitute the most fundamental information among these technologies; however, strand specificity and absolute quantification are often optional and not universally supported. Based on their mapping strategies, current R-loop mapping technologies can be broadly grouped into five categories: (1), antibody-based immunoprecipitation methods; (2) antibody-independent immunoprecipitation methods; (3) in situ capturing methods; (4), footprinting-assisted methods; (5) methods based on nuclease-assisted precise separation of hybrid strands.
-
The S9.6 antibody, an anti-DNA-RNA hybrid antibody[7,8], is a cornerstone tool for R-loop research. Using this antibody, techniques such as immunofluorescence[9], dot/slot blot[10], and DRIP-qPCR[11] have been developed to detect R-loops. However, these approaches provide only bulk quantification and cannot resolve the precise genomic locations of individual R-loops.
To enable genome-wide mapping of R-loops, DRIP-seq (DNA:RNA immunoprecipitation coupled to sequencing) was developed[11]. DRIP-seq employs the S9.6 antibody to specifically enrich DNA-RNA hybrid fragments by immunoprecipitation, which are then analyzed by high-throughput sequencing[11]. DRIP-seq provided the first comprehensive landscapes of R-loop distributions and revealed many of their key genomic features. It has since become one of the most widely used R-loop mapping approaches, substantially advancing the field of R-loop biology. Nevertheless, DRIP-seq has several limitations: its resolution is constrained by the spacing of restriction enzyme cleavage sites, resulting in broad genomic intervals rather than precise R-loop boundaries. It also requires substantial input material and lacks R-loop strand-specific information. In addition, since DNA-RNA hybrid strands are typically resistant to restriction enzyme digestion, longer R-loop molecules may remain substantially lengthy after fragmentation, posing a challenge to efficient detection by next-generation sequencing. These technical limitations hinder the application of DRIP-seq across many research domains. To overcome these shortcomings, multiple derivatives of DRIP-seq have been developed, such as S1-DRIP-seq[12], ssDRIP-seq[13], ULI-ssDRIP-seq[14], mDRIP-seq[15], qDRIP-seq[16], RDIP-seq[17], and DRIPc-seq[18].
S1-DRIP-seq (S1 nuclease DNA:RNA immunoprecipitation sequencing) is a refinement of the standard DRIP-seq method that enhances specificity by digesting genomic DNA with S1 nuclease prior to sonication to remove non-template ssDNA from R-loops[12]. S1-DRIP-seq achieves enhanced resolution by utilizing sonication, which generates shorter DNA fragments than the low-frequency restriction enzymes used in standard DRIP-seq. However, since a normal dsDNA library preparation method with dsDNA adaptor ligation was employed[12], S1-DRIP-seq could not profile the strand information of DNA-RNA hybrids. Moreover, hybrid strands without dsDNA ends could be generated from long R-loops after sonication, which are undetectable by standard dsDNA library preparation.
To achieve strand-specific R-loop mapping, ssDRIP-seq was developed[13]. By using a cocktail of frequent-cutting restriction enzymes, ssDRIP-seq achieves resolution below 100 bp. The shorter dsDNA arms flanking the R-loop by frequent-cutting restriction enzymes also increase the elution efficiency of the displaced ssDNA of R-loops, thereby enabling selective enrichment of DNA-RNA hybrid strands alone[19]. The DRIPed hybrids are further fragmented by sonication, reducing detection bias against R-loops of varying lengths. Combining with the use of an ssDNA ligase Adaptase, to map the ssDNA strand from DRIPed DNA-RNA hybrid, it enables ssDRIP-seq as a strand-specific, high-resolution method for genome-wide R-loop mapping. While resolution is substantially improved, ssDRIP-seq still cannot be used for precise mapping of R-loop boundaries. Moreover, its sensitivity remains insufficient for profiling ultra-low-input samples.
To address these issues above, ssDRIP-seq was further optimized into ULI-ssDRIP-seq, which is an ultra-low-input R-loop mapping technique[14]. With mung bean nuclease, entire hybrid strands are precisely separated for further steps of ULI-ssDRIP-seq, promoting the resolution to a near-single-base level[14]. By using ssDNA adaptor pre-ligation strategy, and rearranging the workflow to a one-tube pipeline, ULI-ssDRIP-seq can map R-loops from as few as 1,000 animal cells[14], and it has been used to profile R-loop landscapes and dynamics during vertebrate early embryogenesis[14]. The introduction of exogenous gDNA from a phylogenetically distant species as a spike-in, coupled with the comparison of read counts pre- and post-DRIP, enables ULI-ssDRIP-seq to facilitate inter-sample quantitative comparisons of R-loop level[14], though absolute quantification remains unavailable.
To enable absolute quantification, qDRIP-seq was established by utilizing a synthetic DNA-RNA hybrid spike-in[16]. This method provided the first genome-wide estimates of both the absolute number of R-loop hybrids per cell and their half-lives[16]. Since qDRIP-seq employs a library construction approach similar to ssDRIP-seq, which directly detects the template ssDNA strand of the hybrid with Adaptase, it can also profile the strand specificity of an R-loop[16].
Apart from DNA, RNA also provides a viable molecular target for the characterization of R-loops. RDIP-seq[17] and DRIPc-seq[18] map R-loops by detecting RNA strands from DRIPed R-loop formations, giving them the ability of strand-specific R-loop profiling. However, this approach may introduce potential signal interference from double-stranded RNA (dsRNA)[20]. Another constraint inherent to RNA-based R-loop mapping approaches is their incapacity to precisely localize trans R-loops, as these techniques rely on sequencing RNA strands rather than determining its genomic binding loci.
With the accelerated development of omics big data studies, there is a growing and urgent requirement for high-throughput R-loop profiling methods capable of handling large-scale samples. To further enhance the throughput of R-loop mapping, mDRIP-seq was established based on ssDRIP-seq and ULI-ssDRIP-seq, by adopting a barcoding-and-pooling strategy[15]. The first adaptors with barcode sequences are ligated to gDNA fragments by splint strands and T4 DNA ligase, and different barcoded samples are then pooled into one tube, followed with DRIP, and the rest of the ssDNA library construction steps. Following demultiplexing based on the sequences of barcode, the sequencing data is assigned to their respective samples[15]. mDRIP-seq preserves the high-resolution and strand-specific features of ULI-ssDRIP-seq, yet achieves substantially higher library construction efficiency[15].
DRIP-seq and its derivatives constitute a foundational suite of tools for R-loop profiling, collectively providing a wealth of information that has been indispensable for advancing the understanding of R-loop biology.
-
While the S9.6 antibody has been widely used for R-loop detection, concerns persist regarding its specificity. Studies report that S9.6 binds not only DNA-RNA hybrids but also double-stranded RNA (dsRNA), and its affinity for hybrids exhibits sequence-dependent variation[20,21]. This affinity for dsRNA and bias of binding sequences may compromise the accuracy and reliability of the S9.6-based R-loop mapping methods. Thus, to replace the S9.6 antibody, several R-loop-recognizing proteins are employed to establish antibody-independent R-loop mapping techniques. Ribonuclease H (RNase H) enzymes, including RNase H1 and RNase H2, specifically resolve DNA-RNA hybrids by cleaving the RNA strand[22,23]. RNase H contains a hybrid-binding domain (HBD) and an RNase endonuclease domain[24]. Its selective affinity for binding RNA-DNA hybrids makes RNase H a valuable tool for R-loop identification and detection.
DRIVE-seq (DNA:RNA in vitro enrichment and sequencing), developed by Ginno et al., employs catalytically inactive RNase H1 (dRNH1) for in vitro enrichment of DNA-RNA hybrids as an alternative to the S9.6 antibody[11]. This strategy avoids dsRNA cross-reactivity and offers cost efficiency through bacterial protein expression of dRNH1. However, despite being reported alongside DRIP-seq, DRIVE-seq has seen limited adoption, primarily because of its substantially lower sensitivity compared to DRIP-seq[11], which might be due to the differential affinities of dRNH1 and the S9.6 antibody.
To improve the sensitivity of the dRNH1-based R-loop mapping method, R-ChIP (R-loop Chromatin Immunoprecipitation) was developed by recognizing R-loops in vivo and integrating the workflow with ChIP-seq[25]. R-ChIP also utilizes a catalytically inactive RNase H1 (D210N) for genome-wide R-loop mapping by expressing the D210N mutant in live cells, followed by the strand-specific ChIP-seq library construction[25]. R-ChIP utilizes sonication to fragment the genome prior to immunoprecipitation, resulting in enriched R-loop signals with higher resolution. A similar strategy is employed in RR-ChIP, with a difference that it detects the RNA strand of the hybrid rather than the DNA strand[26]. As in vivo R-loop mapping methods, R-ChIP and RR-ChIP are hypothesized to exhibit enhanced capture efficiency for unstable R-loops relative to DRIP-based methods, which may lose signals from transient R-loop structures during DNA extraction and associated protein removal. Conversely, the ability of dRNH1 to unbiasedly detect all R-loops in living cells presents a substantial challenge, given that the R-loop recognition of dRNH1 may be affected by cellular co-factors like RPA[27], or the accessibility of R-loops.
Besides RNase H1, another RNase H family protein, RNase H3, is also capable of R-loop recognition. An R-loop profiling method using enDR3, which is a fusion of two N-terminal domains of RNase H3 with point mutations is developed for both ex vivo/in vitro and in vivo R-loop capture[28]. Combing DRIPc-seq, enDR3-DRIPc-seq utilized enDR3 for ex vivo R-loop mapping, obtaining R-loop data similar to those from DRIPc-seq with S9.6 capture. By expressing enDR3 in human cells and combining it with the ChIP-seq method, enDR3-ChIP-seq also enables in vivo profiling of R-loop landscapes, similarly to R-ChIP.
-
In addition to immunoprecipitation-based techniques, in situ capture strategies have been widely adopted in various epigenomic methods in recent years, as exemplified by methods such as CUT&RUN[29] and CUT&Tag[30].
MapR is a method that uses RNase H1 in combination with CUT&RUN to detect R-loops in the genome[31]. This technique employs a fusion protein (RH∆-MNase) comprising catalytically inactive RNase H1 (RH∆) and micrococcal nuclease (MNase), which specifically binds R-loops and cleaves adjacent DNA in situ, followed by dsDNA library construction and sequencing[31]. The primary advantage of MapR lies in its lower time-cost compared to most immunoprecipitation-based R-loop mapping methods. As a typical dsDNA library construction is used in MapR, the efficiency and accuracy of MapR in R-loop profiling is constrained. This is because MNase digestion might produce hybrid-only fragments without dsDNA ends, rendering them undetectable in conventional dsDNA library workflows.
CUT&Tag is another widely used in situ method, by using antibody-guided Tn5 transposase to cut target gDNA and ligate a dsDNA adaptor simultaneously[30]. R-loop CUT&Tag is established by merging the CUT&Tag method with R-loop-specific affinity reagents (S9.6 antibody or RNase H1 HBD domain) to achieve in situ capture and sequencing of R-loops[32]. Due to the inherent advantages of the CUT&Tag method, R-loop CUT&Tag demonstrates excellent performance in terms of sensitivity and resolution. However, these in situ capture methods lacks the capability to supply R-loop strand-specific information, which results from the non-stranded dsDNA library construction strategies used. Although it was reported that Tn5 can be inserted into hybrid strands and subsequently enable strand-specific library construction via reverse transcriptase[33], this strategy was not implemented in R-loop CUT&Tag. Instead, R-loop CUT&Tag employs a conventional Tn5-based dsDNA library preparation protocol. Consequently, strand-specific information is lost, while the potential for signal interference from open chromatin regions flanking R-loops is increased. Despite both techniques employing in situ detection strategies, the overlap between R-loop peaks by MapR and R loop CUT&Tag is modest[32,34], which might be attributed to differences in the enzymatic preferences of Tn5 vs MNase.
-
In addition to DNA-RNA hybrids, the unpaired ssDNA portion can be used for R-loop mapping. Native bisulfite sequencing methods based on Sanger sequencing has been used widely for R-loop footprinting, in which the unpaired cytosine is converted to uracil while other C bases in dsDNA or DNA-RNA hybrids is unconverted[35]. Based on the C-to-T conversion information, the ssDNA region is detected with high resolution to profile the R- loop footprints.
bisDRIP-seq (bisulfite-assisted DNA:RNA immunoprecipitation sequencing) integrates DRIP with on-bead bisulfite treatment to profile R-loops genome-wide, at near-nucleotide resolution[36]. In bisDRIP-seq, gDNA fragments are converted by native bisulfite treatment under non-denaturing conditions, followed by S9.6 DRIP-seq. The C-to-T or G-to-A mutations provide strand-specific information for the displaced ssDNA strand. A similar strategy has been applied to address the strand information limitation of MapR. BisMapR was developed by combining native bisulfite treatment and the MapR method, generating higher resolution strand-specific R-loop profiling data, compared to MapR[37]. SMRF-seq (Single-molecule R-loop footprint sequencing) is the first third-generation-sequencing based R-loop mapping method that combines native bisulfite conversion and PacBio single-molecule sequencing, which is used to reveal genome-wide R-loop profiles at near single-nucleotide resolution[38].
Given that ssDNA-labeling methods can identify R-loops, could R-loop mapping be achieved solely through such approaches, without reliance on any R-loop-recognizing proteins? Although native bisulfite-based techniques offer high resolution and adaptability, their application is constrained by DNA damage introduced during bisulfite treatment. N3-kethoxal, an azido derivative of kethoxal that selectively labels guanine in unpaired ssDNA, is used by KAS-seq (kethoxal-assisted ssDNA sequencing) for genome wide ssDNA mapping in situ[39]. Building on KAS-seq, spKAS-seq (strand-specific kethoxal-assisted ssDNA sequencing) was developed as an antibody-free, low-input technique that labels ssDNA by N3-kethoxal, enabling the mapping of the ssDNA of R-loops with strand specificity[34]. Notably, even in the absence of the S9.6 antibody immunoprecipitation, the R-loop profiles generated by spKAS-seq exhibit substantial concordance with DRIP-seq signals[34]. Nevertheless, these footprinting-assisted methods also detect non-canonical single-stranded motifs, such as D-loops, G-quadruplex, or DNA hairpins, potentially inflating R-loop estimates. Moreover, ssDNA-binding proteins might block bisulfite and kethoxal labeling, yielding false negatives and limiting the technology's applicability.
-
In the methods described above, the gDNA fragmentation steps are derived from other reported common epigenomic methods, including sonication, restriction enzyme digestion, or Tn5 fragmentation. None of these fragmentation methods could precisely cleave R-loop boundaries, which constrain the ultimate resolution of R-loop mapping.
Mung bean nuclease is a single-strand-specific endonuclease that cleaves both DNA and RNA, which could be used for removing single-stranded parts of DNA, and leaving blunt dsDNA ends. In ULI-ssDRIP-seq and mDRIP-seq, the mung bean nuclease is used for separating the intact hybrid strand from the R-loop, with its ability to cut the junction part between dsDNA and the hybrid, followed by S9.6 DRIP and ssDNA sequencing[14]. Although the intact hybrid is first isolated, it must be further fragmented by sonication into uniformly sized fragments to accommodate the limited read lengths of next-generation sequencing. Consequently, none of them can directly and precisely map the boundaries of partially overlapping R-loops from different genomics copies.
Further, RIAN-seq (R-loop Identification Assisted by Nucleases-sequencing) employs an antibody-free strategy by utilizing a combination of endonucleases and exonucleases to remove non-hybrid components of gDNA, without requiring enrichment or capture steps[40]. After gDNA fragmentation with four restriction enzymes, Nuclease P1 (for ssRNA/ssDNA digestion), T5 exonuclease (for ssDNA/dsDNA resolution), and lambda exonuclease (for supercoiled dsDNA degradation and dsDNA-to-ssDNA conversion), are used in RIAN-seq to separate intact DNA-RNA hybrids from R-loops[40]. Compared to other methods, much shorter R-loop fragments (from 60 to 130 bp) are mapped with RIAN-seq[40]. This combined digestion strategy obtains intact DNA-RNA hybrids with blunt ends that closely reflect the R-loop regions. However, because the library construction in RIAN-seq relies on Tn5, which inserts adapters internally rather than ligating them to fragmented ends, the R-loop mapping regions are shortened compared to the real R-loop regions. Furthermore, similar to R loop CUT&Tag, RIAN-seq does not provide strand-specific information of R-loops.
-
The diverse techniques developed to date have enabled multi-faceted characterization of R-loops. Nevertheless, persistent debates center on the authenticity of reported R-loop signals. Significant discrepancies exist among data obtained from different techniques when applied to the same biological material. For instance, ex vivo/in vitro methods such as DRIP-seq and DRIPc-seq primarily yield signals concentrated in the gene body, whereas in vivo techniques such as R-ChIP predominantly detect signals around the transcription start site (TSS).
Initially, technical biases were often attributed to nonspecific recognition by the S9.6 antibody, spurring the development of alternative R-loop-binding proteins such as RNase H variants. However, subsequent studies revealed that when employing identical experimental strategies, the sequencing outcomes obtained using either S9.6 or RNase H proteins are highly similar[28,32]. In contrast, when detection strategies differ, particularly between ex vivo/in vitro and in situ/in vivo protocols, even the same recognition protein can produce divergent results[28,32]. Notably, the exception is that the R-loop landscapes profiled by spKAS-seq, an in vivo/in situ method, are closer to those obtained by in vitro DRIP-seq than other in vivo/in situ methods[34].
One plausible explanation is that cellular R-loops exist in multiple states. Based on stability, they may be classified as: (i) stable R-loops, which maintain structure independently of accessory factors; and (ii) unstable R-loops, whose integrity depends on other cellular components. Furthermore, based on accessibility, R-loops may be categorized as highly accessible or poorly accessible. Unstable, factor-dependent R-loops may dissociate during gDNA extraction, leading to signal loss in ex vivo assays. Conversely, poorly accessible R-loops may be difficult for recognition proteins to bind to in living cells, resulting in underestimation by in vivo or in situ methods. It should be emphasized that the above speculations have not yet been fully verified through experiments, but they still offer valuable insights. It can be reasonably inferred that no single technique is likely optimal in an absolute sense, as different methods are suitable for mapping distinct structural subsets of R-loops. Similar debates have emerged in the methodological development of histone modification profiling[41]. Integrating multiple technologies, therefore, offers a more comprehensive strategy for evaluating R-loop distributions.
The key characteristics of each technique are summarized in Fig. 2 to facilitate the informed selection based on experimental needs. Below, we outline practical considerations for method optimization. (1) Fragmentation method: Restriction enzyme–based fragmentation employs relatively mild reaction conditions, minimizing mechanical disruption of R-loop structures. However, restriction enzymes often fail to cleave simple sequence repeats (e.g., [GA]n), and some are sensitive to DNA methylation, leading to incomplete fragmentation in repeat-rich or highly methylated genomic regions. In contrast, sonication provides consistent fragmentation independent of sequence composition or epigenetic modifications. Its disadvantage is the potential for mechanical disruption to R-loops. (2) Library construction strategy: The dsDNA library strategy benefits from being the most commonly used DNA library preparation method, making it suitable for broad technical adoption. However, it requires intact R-loop structures with long flanking dsDNA arms, which limits resolution improvement. The ssDNA library strategy enables strand-specific detection by targeting ssDNA or DNA-RNA hybrid strands, allowing higher-resolution mapping. A disadvantage is that non-R-loop hybrids, such as retrotransposon DNA-RNA hybrid intermediates may also be captured. RNA-focused library construction prioritizes information from the RNA component of R-loops, facilitating identification of RNA origins in trans R-loops. However, it increases susceptibility to contamination by dsRNA or residual RNA. (3) R-loop sensor design: In vivo expression of R-loop sensors reduces artifacts associated with DNA extraction but may introduce binding bias in the presence of endogenous R-loop associated proteins. In vitro recognition strategies remove protein interference and improve accessibility uniformity across genomic regions, though harsh extraction conditions can compromise the stability of fragile R-loops.

Figure 2.
Overview of R-loop profiling methods.
The advancement of epigenomic technologies promises exciting future directions for R-loop omics. Third-generation sequencing platforms are increasingly applied in epigenomics. By combining in situ labeling systems with the ability to directly read base methylation, long-read sequencing has enabled detection of diverse epigenetic marks, including DNA methylation, histone modifications, and chromatin accessibility[42]. The long-read capability is particularly advantageous for mapping complex genomic regions such as repetitive sequences, which are often enriched with R-loops. Integrating R-loop profiling with third-generation sequencing therefore represents a promising frontier.
Single-cell sequencing and spatial omics have also emerged as vibrant research areas, dramatically advancing the understanding of cellular heterogeneity and tissue-scale organization in contexts such as embryogenesis and tumor biology[43]. Single-cell epigenomic methods have successfully profiled various chromatin features, and spatial epigenomics has recently achieved several breakthroughs[43]. Profiling R-loop landscapes at single-cell resolution and within tissue architecture will widen the conceptual framework and refine the mechanistic understanding of R-loop biology.
This work was supported by Basic Research Center, Innovation Program of Chinese Academy of Agricultural Sciences (CAAS-BRC-AFIS-2025-02), the opening Foundation of State Key Laboratory of Crop Gene Resources and Breeding (CGRB-2025-04), Central Public-interest Scientific Institution Basal Research Fund (Grant No. Y2022QC33), and National Natural Science Foundation of China (Grant No. 32071437 and 31900302).
-
The authors confirm their contributions to the paper as follows: study conception and design: Xu W, Sun C; draft manuscript preparation: Xu W, Wang X. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing is not applicable as no datasets were generated or analyzed during this review.
-
The authors declare that they have no conflict of interest.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang X, Sun C, Xu W. 2026. Genome-wide R-loop profiling methods. Genomics Communications 3: e005 doi: 10.48130/gcomm-0026-0004 

Genome-wide R-loop profiling methods
- Received: 14 December 2025
- Revised: 31 January 2026
- Accepted: 04 February 2026
- Published online: 18 March 2026
Abstract: R-loops, three-stranded nucleic acid structures comprising a DNA-RNA hybrid and displaced ssDNA, play various and important roles in genomes. Accurately mapping the genomic distribution of R-loops remains challenging due to their structural diversity and dynamic nature. Current genome-wide R-loop profiling technologies were reviewd with various strategies, including antibody-based immunoprecipitation, antibody-independent immunoprecipitation, in situ capture, footprinting, and nuclease-mediated hybrid separation. The principles, advancements, and inherent limitations of these methods are detailed, highlighting their complementary potential. Furthermore, we discuss the ongoing controversies regarding signal specificity and the complementary nature of different techniques, proposing that integrating multiple methods offers the most comprehensive strategy. Together, these rapidly evolving technologies are deepening our mechanistic insight into R-loop functions and their roles in genome regulation.
-
Key words:
- R-loop /
- Next-generation sequencing /
- Non-coding RNA /
- Omics