









-
The plastid genome (plastome) and mitochondrial genome (mitogenome) of angiosperms exhibit distinctive evolutionary characteristics. The plastid point mutation rate is several times higher relative to the mitochondrial rate[1,2], but in most other aspects—size, gene order, gene content, intron content, and repeat content—the plastome is more stable in comparison with the mitogenome, and often substantially so, at least among photosynthetic plants[3,4].
A typical angiosperm plastome is 120–160 kb in size, with about 80 encoded proteins, 21 introns, and two copies of a large inverted repeat (IR), all of which are stably arranged such that collinearity can be directly compared across the diversity of most land plants, although the plastomes from some lineages are more variable in these characteristics[5,6]. Smaller repeats, which are notably rare to absent from most plastomes[7], have been shown to promote recombination when present[8,9], suggesting evolutionary pressure to limit these potentially deleterious rearrangements.
In contrast, angiosperm mitogenomes are much more variable, ranging from hundreds of kilobases to >10 megabases in size, encoding 30–40 protein-coding genes and harboring 20–25 introns[4,10]. Additionally, unlike plastomes, mitochondrial repeat content tends to include an abundance of larger (>1 kb) and smaller (<1 kb) repeats, many of which promote recombination, yet the specific repeats are highly lineage-specific[11]. Plant mitogenomes also typically incorporate fragments of plastid DNA, and these MIPTs (mitochondrial DNA of plastid origin) can sometimes occupy a sizeable fraction of the mitogenome[12].
To date, plastomes have been sequenced from thousands of angiosperms, including dense sampling of closely related species within many genera and families, substantiating the stability of these features among most photosynthetic species while also identifying a growing list of lineages that diverge in some respects from this general stability[5,13]. In contrast, the hundreds of sequenced angiosperm mitogenomes vary substantially even among closely related species (e.g., studies[14,15−23]), demonstrating that the angiosperm mitogenome is more structurally labile than the plastome despite the slower mitochondrial point mutation rate[3,4,24]. Although these general evolutionary tendencies between the angiosperm mitogenome and plastome are clear, few studies have densely sampled both organellar genomes in parallel to directly compare their rates of functional and structural change.
To make such direct comparisons, we focused our sampling efforts on the Digitalideae, a small tribe within the Plantaginaceae containing two genera, Digitalis and Erinus[25], with predominant distributions around the Mediterranean through Europe and Western Asia[26]. The monotypic Erinus includes E. alpinus, the fairy foxglove, which is a niche ornamental suited for rock gardens. Digitalis, or foxgloves, usually have showy inflorescences with numerous, colorful flowers, and several species (particularly D. purpurea) are sold as ornamentals[27,28]. Some Digitalis species also have a complex history of medical usage with severe side-effects, as the plants produce cardiac glycosides that have a narrow dosage range between therapeutic benefit and toxicity[27,28].
Digitalis, which has subsumed species formerly placed in Isoplexis, is now subdivided into five sections (Digitalis, Frutescentes, Globiflorae, Isoplexis, and Macranthae), although these results are based on few loci whose phylogenetic signals are not fully congruent[26,29]. Moreover, single-marker analyses of the plastid trnL/trnF or the nuclear internal transcribed spacer (ITS) of the ribosomal DNA (rDNA) repeat suggested that at least two species may be polyphyletic: For D. ferruginea, two sampled subspecies (subsp. ferruginea and subsp. schischkinii) did not form a monophyletic group, whereas for D. lutea subsp. australis, some individuals were grouped within section Macranthae while an Italian individual was grouped instead within section Globiflorae[26]. These uncertainties, and the overall limited availability of molecular data for Digitalideae, indicate that deeper molecular sampling is needed to resolve the phylogeny of Digitalideae.
Interestingly, the toxic properties of Digitalis have provided impetus for nucleotide sequencing, as genomic data are sensitive markers for detecting trace amounts of toxic plant material in foods and supplements[30−32]. Additionally, plastomes from D. lanata and D. purpurea were recently made available[32,33], and their phylogenetic analysis indicated the close relationship of Digitalideae and Plantagineae[33], a result also recovered for several mitochondrial genes and nuclear rDNA[34]. By expanding on these emerging molecular resources, we endeavored to generate a set of complete plastomes, mitogenomes, and nuclear rDNA for six Digitalideae species (D. ferruginea, D. grandiflora, D. lanata, D. lutea, D. purpurea, and E. alpinus), aiming to examine the relative rates of structural and functional evolution in the organellar genomes and the phylogenetic utility of these genomes and nuclear rDNA.
-
Sanger reads from D. purpurea (Supplementary Table S1) were assembled with PCAP[35] using the autopcap option. Illumina reads from D. ferruginea, D. grandiflora, D. lutea, D. lanata, and E. alpinus (Supplementary Table S1) were assembled with SPAdes v4.2.0[36] and Velvet v1.2.10[37], essentially as described previously[38,39]. For both assemblers, the specified range of kmers depended on read length and quantity: Longer Illumina reads (>150 bp) with larger datasets (>5 Gb) were assembled with larger kmers (79, 95, 111, 127), whereas shorter reads (< 150 bp) or smaller datasets (< 5 Gb) were assembled with shorter kmers (61, 71, 81, 91). SPAdes assemblies used the careful option and set the coverage cutoff to either 10 (for > 5-Gb datasets) or 5 (for < 5-Gb datasets). Velvet assemblies were run multiple times for each species using pairwise combinations of kmer and expected coverage value (set to 50, 100, 200, 500, or 1,000), with no scaffolding and minimum coverage set to 10% of the expected coverage.
Final sequence assembly
-
Assembled contigs containing plastid DNA, mitochondrial DNA, or nuclear rDNA were identified by blastn v2.15.0 searches with the default settings using the plastome (GenBank accession MW877561), annotated mitochondrial genes (OK514181), and the nuclear rDNA (OK523399) from Aragoa abietina as queries. For each species, the nuclear rDNA cluster containing intact 18S, 5.8S, and 26S rDNA genes was usually present within a single identified contig; otherwise, minimal manual assembly was required by overlapping the ends of two identified contigs. The plastome was also recovered in one or a few overlapping contigs, and the IR was inferred from default blastn searches using these plastid contigs as both query and subject, identifying the position at which a sequence fragment (the IR) had two alternative flanking sequences, i.e., the large single-copy (LSC) and small single-copy (SSC) regions at each end.
Scaffolding of the identified mitochondrial contigs involved default blastn searches to identify overlapping contig ends among the mitochondrial contigs. Mitochondrial contigs with twice the depth of read coverage (as reported by SPAdes and Velvet) were inferred to be repetitive and used twice during scaffolding. The remaining unresolved mitochondrial scaffold ends invariably matched the plastome, indicating the presence of a MIPT. MIPT sequences were iteratively reassembled with SPAdes, using the careful and only-assembler options, from a filtered subset of reads that either matched with higher similarity to the mitochondrial scaffold ends than to the plastome or matched nonidentically to the plastomic region from which the MIPT was derived.
All mitogenomic and plastomic contigs were ultimately assembled into circular arrangements, with no mitochondrial contigs left unresolved, indicating completeness. To verify the assembly and inferred repeats for each species, the depth of read coverage was calculated by mapping the full dataset of reads from that species to the final mitochondrial and plastid assemblies with blastn, requiring a minimum match similarity of 90% and a minimum match length of 90% of the length of the mapped read. Coverage depth was calculated using 100-bp sliding windows with 100-bp steps.
Sequence annotation
-
Nuclear rDNA was annotated automatically during GenBank submission. Plastomes were annotated by propagating the annotations from the Digitalis purpurea plastome (OL977691) during GenBank submission, which required manual correction of a few start and stop codons. Mitochondrial genes for proteins and rRNAs were annotated by blastn searches, using genes from the A. abietina mitogenome (OK514181) as queries, with manual correction of some start and stop codons as needed. Mitochondrial tRNAs were identified using tRNAscan-SE v2.0.7[40] in organelle mode as implemented online by GeSeq[41].
Taxon verification
-
Two studies have extensively sampled a set of taxa from Digitalideae, including the plastid trnL/trnF intergenic region, the nuclear ITS region[26], and the nuclear progesterone 5β-reductase (P5βR) gene[29]. These sequences were extracted from the initial assembled contigs by blastn searches with default settings, and homologous sequences were collected from GenBank (Supplementary Table S2). The regions were aligned with MAFFT v7.525[42], trimmed manually to remove gappy regions, and concatenated with Sequence Matrix v1.10[43] to generate a three-locus dataset containing 46 taxa. The concatenated dataset was analyzed phylogenetically with IQ-TREE v2.2.2.6[44] using 1,000 fast bootstrap replicates for branch support and a maximum likelihood (ML) model of best fit (TN + F + G4) that was auto-selected during the run.
The plastid matK and rbcL genes have also been widely sampled from many Digitalideae species. These two gene sequences were collected from GenBank and extracted from the initial genomic assemblies (Supplementary Table S2). The sequences were then aligned, trimmed, concatenated, and phylogenetically analyzed as described above using the best-fitting auto-selected ML model (K3Pu + F + I).
Collinearity analyses
-
Plastomic collinearity was assessed and visualized with the online implementation of mVISTA (
https://genome.lbl.gov/vista/mvista/submit.shtml ) using the Shuffle-LAGAN option, with the D. ferruginea plastome as the focal genome. The collinearities of the entire mitogenomes, the large mitogenomic repeats, and the large plastid repeats were compared using blastn with default parameters. Homologous blocks for the entire mitogenomes (a minimum of 1,000 bp and 90% identity) and the large mitochondrial and plastid repeats (a minimum of 100 bp and 90% identity) were plotted with Circos v0.69-10[45].Quantification of repeats and MIPTs
-
Repeat boundaries in the mitogenomes and plastomes were identified using blastn searches, with a minimum length of 100 bp and a minimum sequence identity of 90%. MIPT boundaries in the mitogenomes were identified by using blastn searches comparing the plastome and mitogenome from each species, with a minimum 100-bp length and 90% identity, which avoided false positive matches between mitogenomic and plastomic paralogs (e.g., mitochondrial atp1 and plastid atpA, or mitochondrial nad5 and plastid ndhF).
Modeling repeat turnover
-
If repeat content has experienced no turnover between two species, then the amount of shared repetitive DNA between two genomes (R) should be the same; that is, R is expected to be static (RStatic) as defined by the following equation:
$ {R}_{{{Static}}} = {L}_{{{Shared}}}*{F}_{A} = {L}_{{{Shared}}}*{F}_{B} $ (1) where, LShared is the amount of DNA shared between the two genomes, and FA and FB are the fractions of Genome A and Genome B that are repetitive. In contrast, if repeat content has fully turned over, such that ancestral repeats were lost in one or both descendant genomes and new repeats were gained, then R under a model of full turnover (RTurnover) is expected to be defined as the product of FA and FB according to the following equation:
$ {R}_{{{Turnover}}} = {L}_{{{Shared}}}*{F}_{A}*{F}_{B} $ (2) These two alternative models, no turnover (RStatic) and complete turnover (RTurnover), set the upper and lower bounds of R expected on average between any two genomes. To test the fit of a group of genomes to either model, the actual amount of repetitive DNA shared between each pair of species (RActual) was assessed by blastn searches (a minimum of 100 bp in length and 90% identity) and this value was compared with the expected RStatic (Eq. 1) or RTurnorver (Eq. 2) using an online implementation of the one-tailed Wilcoxon signed-rank test (
www.statskingdom.com/175wilcoxon_signed_ranks.html ).Phylogenetic analyses
-
Individual mitochondrial genes and introns (including 500 bp for each end of the trans-spliced introns), complete plastomes (after removal of one copy of the IR to avoid double weighting of the IR), and entire rDNA clusters were collected from the six Digitalideae and additional Plantaginaceae species (Supplementary Table S3). MAFFT was used in 'einsi' mode to align individual mitochondrial genes and introns, in ‘fftnsi’ mode to align plastomes, and in ‘ginsi’ mode to align rDNA clusters. Plastomic and rDNA alignments were trimmed using Gblocks v0.91b[46] with relaxed parameters (−t = d −b1 = half −b2 = half −b3 = 8 −b4 = 5 −b5 = half). A concatenated mitochondrial alignment was generated from the trimmed mitochondrial gene and intron alignments using Gblocks in batch mode (−a = y) with relaxed parameters. Phylogenetic trees were constructed with IQ-TREE using an ML model of best fit that was auto-selected during the run (model GTR + F + I + G4 for rDNA and mitochondria, TVM + F + I + G4 for plastomes) and 1,000 fast bootstrap replicates.
-
We assembled Sanger read pairs from D. purpurea and Illumina read pairs from nine individuals of six Digitalideae species: D. ferruginea (three individuals) and D. lanata from section Globiflorae, D. grandiflora and D. lutea (two individuals) from section Macranthae, D. purpurea from section Digitalis, and E. alpinus as the sole member of Erinus. To verify the species identity for all sampled read datasets, we extracted the plastid trnL/trnF intergenic region, the nuclear rDNA region containing ITS1 + 5.8S rDNA + ITS2, and the nuclear P5βR gene from initial assemblies and compared them with the available sequences (Supplementary Table S2) that were previously sampled from diverse Digitalideae species[26,29]. A second marker set was generated from two common plant DNA barcodes, plastid matK and rbcL, which were extracted from our assemblies and compared with available Digitalideae sequences in GenBank (Supplementary Table S2).
Phylogenetic analyses of both sets of marker data were consistent for the placement of all nine sampled taxa in this analysis, in which all but one taxon clustered essentially in the expected locations (Supplementary Fig. S1). Only D. lutea URI10 grouped unexpectedly in section Globiflorae with D. ferruginea, instead of with the other D. lutea sequences in section Macranthae. Specifically, the trnL/trnF and ITS sequences from D. lutea URI10 are nearly identical (> 99.8% similarity) to those of D. ferruginea subsp. schischkinii, quite similar (98.4%–99.1% similarity) to most other D. ferruginea individuals, but less similar (< 96%) to D. ferruginea subsp. ferruginea and other published D. lutea sequences[26,47]. Thus, phylogenetic and sequence similarity suggest that the URI10 individual may be from D. ferruginea subsp. schischkinii (or a closely related species in section Globiflorae) rather than D. lutea. However, the lack of monophyly for both D. lutea and D. ferruginea, in this and previous work[26], limits any conclusions about the species identity of URI10 and indicates that the circumscription of both species needs to be investigated. Given this uncertainty, we will continue to refer to URI10 as D. lutea URI10 in this study.
Digitalideae plastomes are very similar in size, structure, and content
-
Four Digitalideae plastomes (D. ferruginea, D. grandiflora, D. lutea URI10, and E. alpinus) were newly assembled, and their sequences were compared with published plastomes from two additional species (D. lanata, D. purpurea). All six plastomes were similar in size (152–153 kb) and contained the same set of 79 coding sequences (CDSs), 4 rRNAs, 30 tRNAs, and 21 introns, which were organized into a structure with the LSC and SSC regions separated by two IR copies, as exemplified by D. ferruginea (Fig. 1a). Duplication of the IR in the assembly was verified by its approximately doubled depth of coverage relative to the LSC and SSC (Fig. 1b). The sequences of all six plastomes were entirely collinear (Fig. 1c). The repeat content (> 100 bp) for all plastomes comprised just the IR, which was collinear and nearly identical in size among species (Fig. 1d), with the IR–LSC boundary situated between rpl2 and either rps19 or trnH-GUG and the IR–SSC boundary falling within the ndhF and ycf1 genes (Fig. 1a).
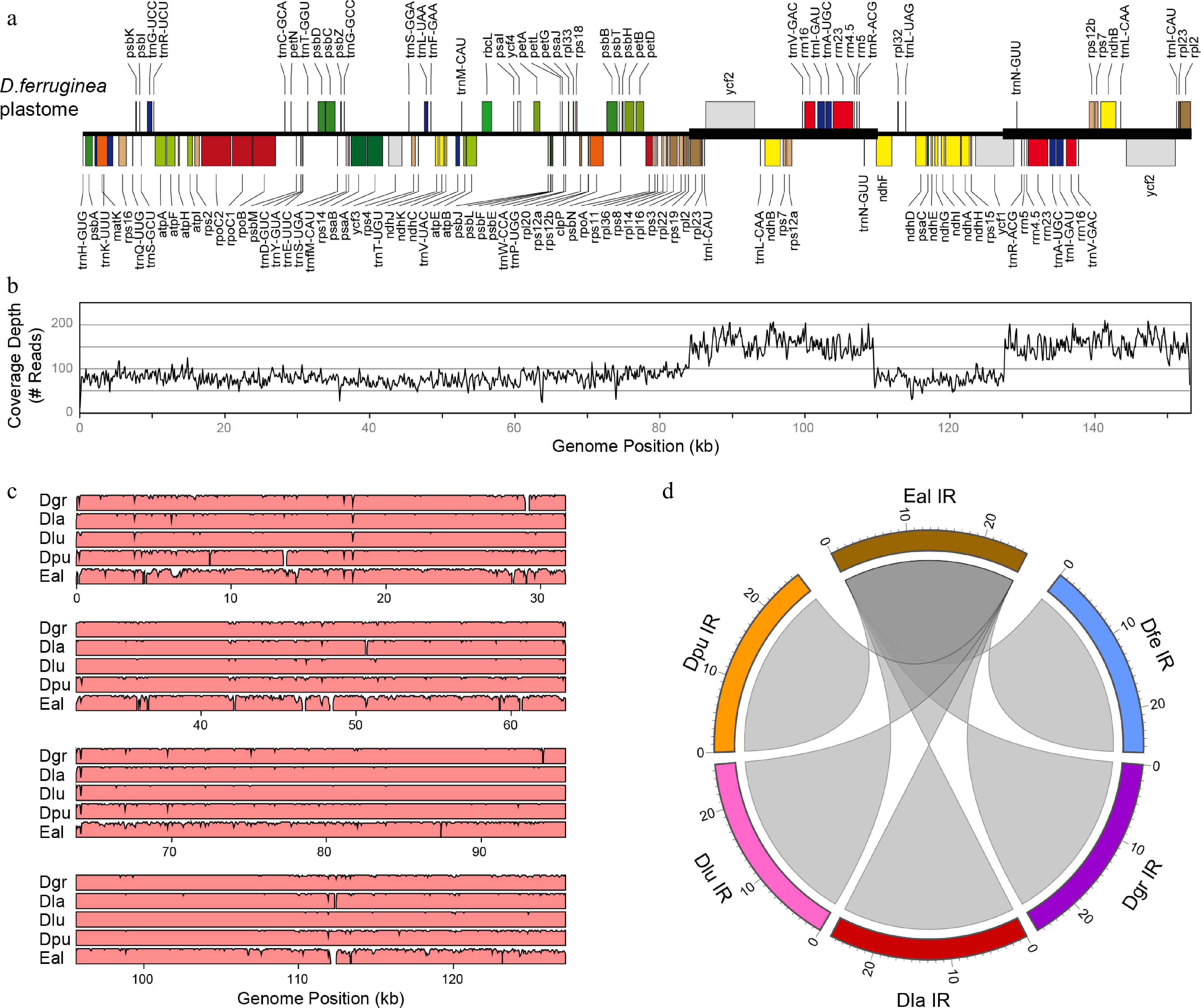
Figure 1.
Plastome analyses in Digitalideae. (a) The annotated linear genome map. The locations of the IR copies are shown with a thick horizontal line. (b) Plot showing the depth of read coverage. (c) mVISTA plot showing the sequence similarity (%) of the D. ferruginea plastome compared with the five other Digitalideae plastomes. (d) Circos plot showing the sequence homology of the E. alpinus IR compared with the five Digitalis IRs. Rulers are in kb.
Size and structural composition of Digitalideae mitogenomes
-
Six Digitalideae mitogenomes were newly assembled, with sizes ranging from 320 kb for E. alpinus to 384–570 kb in the five Digitalis species, which is similar to the mitogenomic size variation found for other Lamiales (Table 1). Digitalideae mitogenomes exhibited minor variation in GC percentage (44.7%–45.1%) and MIPT content (2.0%–3.2%), and more substantial variation in repetitiveness (0.3%–11.1%), compared with other Plantaginaceae and the more distantly related Lamiales mitogenomes (Table 1).
Table 1. Comparison of mitogenomic characteristics for Digitalideae and related species.
Species Chromosome (#) Size (kb) GC Repeat MIPT CDS (#) rRNA (#) tRNA (#) Introns (#) Digitalideae Digitalis ferruginea 1 431 44.9% 3.3% 2.4% 36 3 19 21 Digitalis grandiflora 1 570 44.7% 5.0% 3.2% 36 3 19 21 Digitalis lanata 1 437 45.1% 0.3% 2.5% 36 3 18 21 Digitalis lutea 1 468 44.9% 11.1% 2.3% 36 3 19 21 Digitalis purpurea 1 384 45.1% 6.5% 2.3% 36 3 18 21 Erinus alpinus 1 320 44.9% 2.2% 2.0% 35 3 19 21 Other Plantaginaceae Aragoa abietina 1 365 45.0% 0.5% 1.2% 34 3 16 22 Linaria vulgaris 2 468 44.6% 5.8% 0.5% 34 3 14 22 Misopates orontium 1 570 44.6% 1.0% 3.3% 35 3 19 22 Other Lamiales Boea hygrometrica 1 511 43.3% 0.9% 9.1% 35 3 22 23 Castilleja paramensis 1 495 43.5% 2.3% 16.1% 36 3 21 23 Dolichandrone spathacea 1 670 44.9% 3.6% 1.2% 36 3 17 22 Erythranthe guttata 1 526 45.1% 8.4% 2.8% 34 3 22 22 Rotheca serrata 1 482 45.5% 0.3% 2.7% 33 3 18 23 The most notable differences among the Digitalideae mitogenomes are the presence of distinct large repeats and frequent structural rearrangements (Fig. 2). The positions of the largest (> 1 kb) repeats and MIPTs are annotated for the exemplar D. ferruginea mitogenome (Fig. 2a) and are also shown for the mitogenomes of the other five species (Supplementary Fig. S2). Importantly, all large repeats in all Digitalideae mitogenomes were verified by depth of coverage mapping, showing that the large repeated sequences in each genome were present at twice the depth of coverage relative to nonrepetitive regions, and confirming the absence of any large mitochondrial repeat in D. lanata (Fig. 2b, Supplementary Fig. S2).
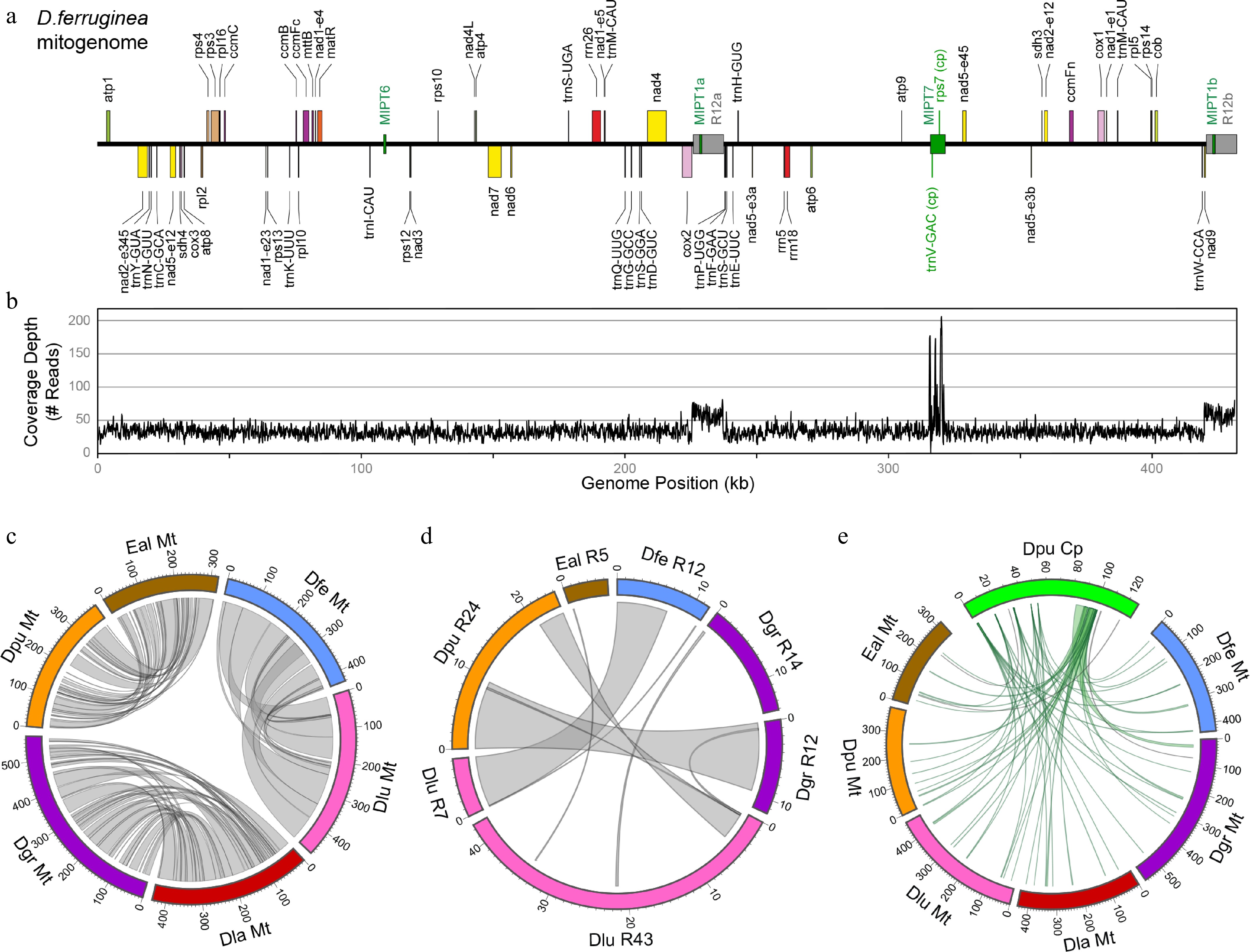
Figure 2.
Analyses of the Digitalideae mitogenome. (a) The annotated linear genome map. The locations of the large (> 1 kb) repeats (gray) and MIPTs (green) are shown as boxes centered on the main horizontal axis. (b) Depth of the read coverage plot. (c) Circos plot showing homologous segments (> 1 kb) between pairs of Digitalideae mitogenomes. (d) Circos plot showing homologous segments (> 100 bp) among the large repeats in Digitalideae mitogenomes. (e) Circos plot showing MIPTs (> 100 bp) in Digitalideae mitogenomes. Rulers are in kb for all Circos plots.
Pairwise comparisons of mitogenomes at different evolutionary depths showed the extensive rearrangement of homologous blocks (a minimum of 1 kb) and a progressive fragmentation of contiguity over time (Fig. 2c). The more closely related pair (D. ferruginea and D. lutea URI10) shared 13 blocks averaging 36.4 kb in length, the intermediately related pair (D. grandiflora and D. lanata) shared 38 blocks averaging 11.3 kb, and the more distantly related pair (D. purpurea and E. alpinus) shared 31 blocks averaging 7.1 kb (Supplementary Table S4).
The size and number of large (> 1 kb) and small repeats (100–1,000 bp) varied substantially among the Digitalideae mitogenomes (Supplementary Table S5). Overall, mitogenomic repetitiveness ranged from 0.3% in D. lanata, which harbors no large repeats, to 11.1% in D. lutea URI10, which contains > 51 kb of repetitive DNA, primarily in two large repeats. Most of the large mitogenomic repeats are species-specific (Fig. 2d), except for a 9.6-kb portion shared between the 24.1-kb repeat in D. purpurea and the 11.7-kb repeat in D. grandiflora, a 7.1-kb portion shared between the 11.8-kb repeat in D. ferruginea and the 7.3-kb repeat in D. lutea URI10, a 3.7-kb portion shared between the 24.1-kb repeat in D. purpurea and the 42.6-kb repeat in D. lutea URI10, and a few small segments (104–336 bp) shared among various larger repeats (Supplementary Table S6).
MIPTs have remained quite stable among the Digitalideae (Fig. 2e). Of the eleven MIPT blocks (> 100 bp) originating from distinct plastomic regions, four are partially or fully shared between Erinus and most Digitalis species, another four are shared among most or all Digitalis species but not Erinus, and only three were uniquely present in a single species (Supplementary Table S7).
Functional content among Digitalideae mitogenomes
-
Plant mitogenomic gene content varies among species, but they typically contain genes for 30–40 proteins, 15–25 tRNAs, and 3 rRNAs[10,48]. All five Digitalis mitogenomes encode the same set of 36 CDSs, but E. alpinus has only 35 because of the pseudogenization of rps10 (Table 1; Fig. 3a). Of the 41 mitochondrial protein-coding genes found among angiosperms[10], all Digitalideae mitogenomes lack five genes: rps1, rps2, rps7 and rps11 are absent from all Plantaginaceae, whereas a possibly functional rps19 gene is still present in Misopates orontium. Most mitochondrial CDSs are present in a single copy in each genome; however, exact duplicates were found for atp6 in D. lutea, Exon 5 of nad1 in D. grandiflora and D. purpurea, and Exon 3 of nad5 in four Digitalis species (all but D. purpurea). Interestingly, all Digitalis mitogenomes carry an intact plastid-derived rps7 gene in one shared MIPT. However, mapping analysis of RNA-seq datasets from D. purpurea (NCBI Sequence Read Archive accessions SRR24974225 and SRR25582083) and D. ferruginea (SRR8895219) failed to find a single read that mapped specifically to this MIPT copy of plastid rps7. Thus, the MIPT copy of rps7 is unlikely to be expressed or functional.
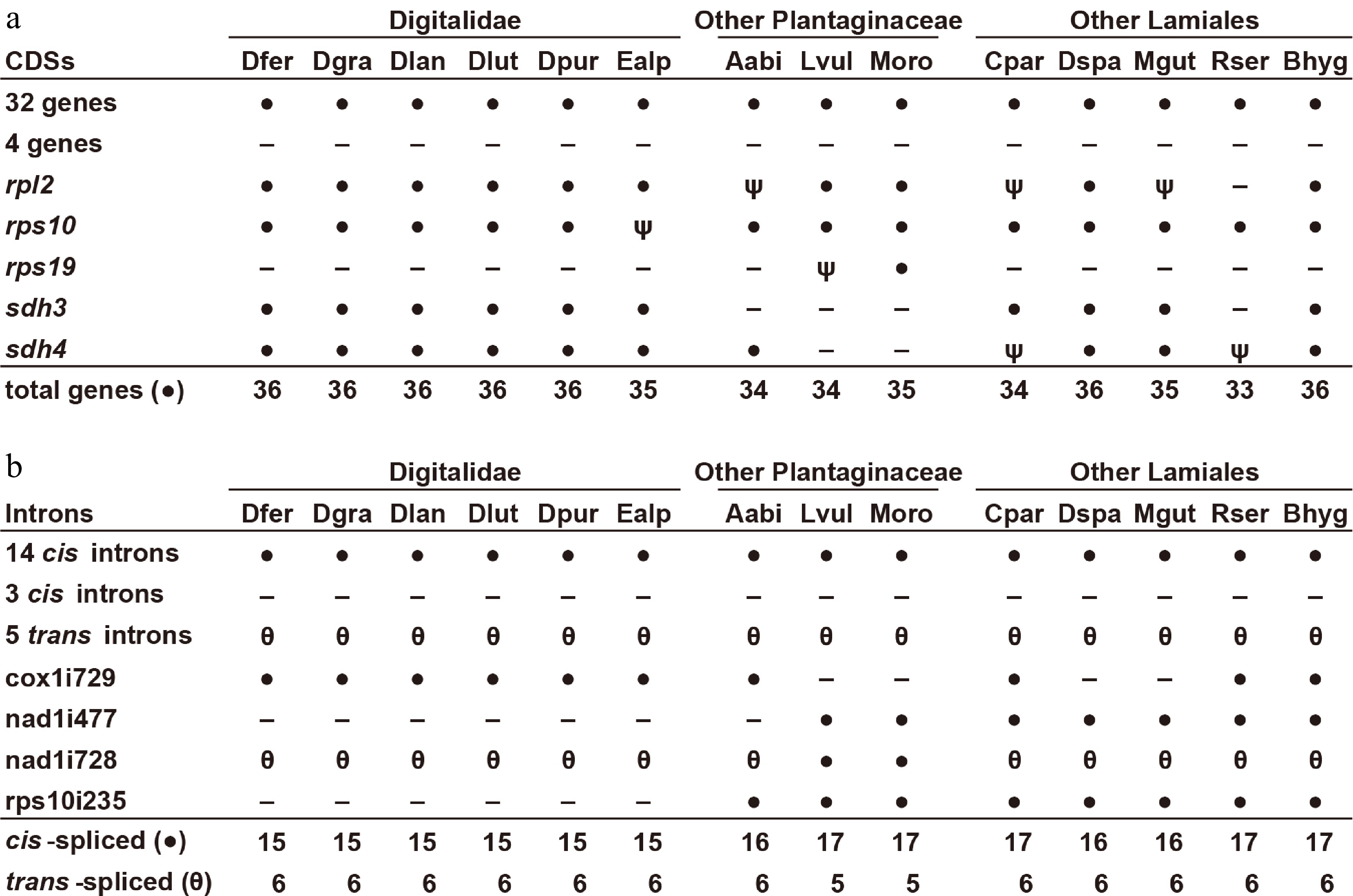
Figure 3.
Gene and intron content among Digitalideae mitogenomes. (a) CDS content. The 32 genes present (●) in all species include atp1, -4, -6, -8, and -9; ccmB, -C, -Fc, and -Fn; cob; cox1, -2, and -3; matR; mttB; nad1, -2, -3, -4, -4L, -5, -6, -7, and -9; rpl5, -10, -16; and rps3, -4, -12, -13, and -14. The four genes absent (−) from all species include rps1, -2, -7, and -11. Inferred pseudogenes (ψ) are marked. (b) Intron content. The 14 cis-spliced introns (●) present in all species include ccmFCi829; cox2i691; nad2i156, -i709, and -i1282; nad4i461, -i976, and -i1399; nad5i230 and -i1872; nad7i140, -i209, and -i917; and rps3i74. The five trans-spliced introns (θ) present in all species include nad1i394, nad1i669, nad2i542, and nad5-i1455 and -i1477. The three cis-spliced introns lost (−) from all species include cox2i373, nad7i676, and rpl2i846.
All six Digitalideae mitogenomes have retained all three rRNA genes and a very similar set of tRNAs covering 18 distinct anticodons for 15 amino acids, of which rrn26 and several tRNAs are present in extra copies in some species (Supplementary Table S8). A few MIPT-derived tRNAs are present in some Digitalideae mitogenomes, but RNA-seq read mapping (SRR8895219) failed to provide evidence of trnV-GAC being expressed in D. ferruginea.
Most angiosperms have 20–25 mitochondrial introns, of which 5 or 6 have a split arrangement that requires trans-splicing for their removal[10]. All six Digitalideae mitogenomes have the same set of 21 introns, including a single Group I intron (cox1i729) and 6 introns that require trans-splicing (Table 1; Fig. 3b). Of the five lost introns, three are absent from all Lamiales (cox2i373, nad7i676, rpl2i846), whereas the loss of nad1i477 is specific to Digitalideae and Aragoa, and the loss of rps10i235 is restricted to Digitalideae. The intron nad1i728 has been repeatedly split into a trans-spliced arrangement during seed plant evolution[49,50], and this split has variously occurred upstream or downstream of matR, a maturase encoded within this intron. In Digitalideae, the intron was split between matR and nad1 Exon 5, whereas nad1i728 has remained in a cis-spliced arrangement in two Antirrhineae species (Linaria vulgaris and M. orontium).
Modeling rates of turnover of repetitive DNA among organellar genomes
-
As the plastid IR is highly conserved in size and position among Digitalideae species (Fig. 1d), it is apparent that these repeats have experienced very little turnover; in fact, the IR has remained quite stable in most land plants over 500 million years of evolution[5]. In stark contrast, the six Digitalideae mitogenomes share little to no large DNA repeats, indicating that these repeats turn over at a fast rate (Fig. 2c). Thus, angiosperm organellar genomes exhibit substantial differences in the rate of turnover of repetitive DNA: The plastomic IRs are essentially static, whereas the mitogenomic large repeats are mostly to fully turned over in Digitalideae. The two scenarios, either no turnover (i.e., stasis) or complete turnover, represent the extremities of our expectations regarding the amount of repetitive DNA (R) shared between any two genomes.
To assess the level of turnover of repeat content between genomes, the actual R (RActual) was quantified and statistically compared with the alternative expectations of R under a stasis model (RStatic) or a full turnover model (RTurnover). Plastomic RActual (mean = 25,746 bp; median = 25,757 bp) was not significantly different (p > 0.05) from RStatic (mean = 25,621 bp; median = 25,739 bp) but was significantly different (p << 0.05) from RTurnover (mean = 4,317 bp; median = 4,334 bp) according to one-tailed Wilcoxon signed-rank tests (Supplementary Table S9), consistent with qualitative observations of a lack of turnover of the IR in Digitalideae plastomes (Fig. 1d). In contrast, the qualitatively fast rate of mitogenomic repeat turnover (Fig. 2d) was corroborated by the finding that mitochondrial RActual (mean = 1,431 bp; median = 0 bp) was significantly different (p << 0.05) from RStatic (mean = 15,547 bp; median = 11,292 bp) but not significantly different (p > 0.05) from RTurnover (mean = 589 bp; median = 258 bp) in one-tailed Wilcoxon signed-rank tests (Supplementary Table S10). Thus, large repeats turn over at significantly faster rates in Digitalideae mitogenomes compared with the plastomic IR.
Phylogenetic congruence among Digitalideae genomes
-
Phylogenetic relationships were examined for six Digitalideae species and four related Plantaginaceae species using datasets comprising whole plastomes, a mitochondrial concatenation of 35 genes and 20 introns, or the nuclear rDNA cluster (Fig. 4). All three datasets generated identical tree topologies, and most branches received maximal bootstrap support. As seen in the initial phylogenetic analyses (Supplementary Fig. S1), the sister grouping of D. lutea URI10 with D. ferruginea rather than D. grandiflora is unexpected, but all other Digitalideae relationships are consistent with previous phylogenetic studies based on fewer loci[26,29]. Digitalideae is more closely related to Plantagineae than to Antirrhineae, as expected on the basis of previous multigene[25,34] and whole plastome[51−53] analyses of Plantaginaceae relationships, but further sampling of the nuclear rDNA and mitogenomes from other Plantaginaceae tribes is needed to evaluate the phylogenetic congruence among genomes across Plantaginaceae.
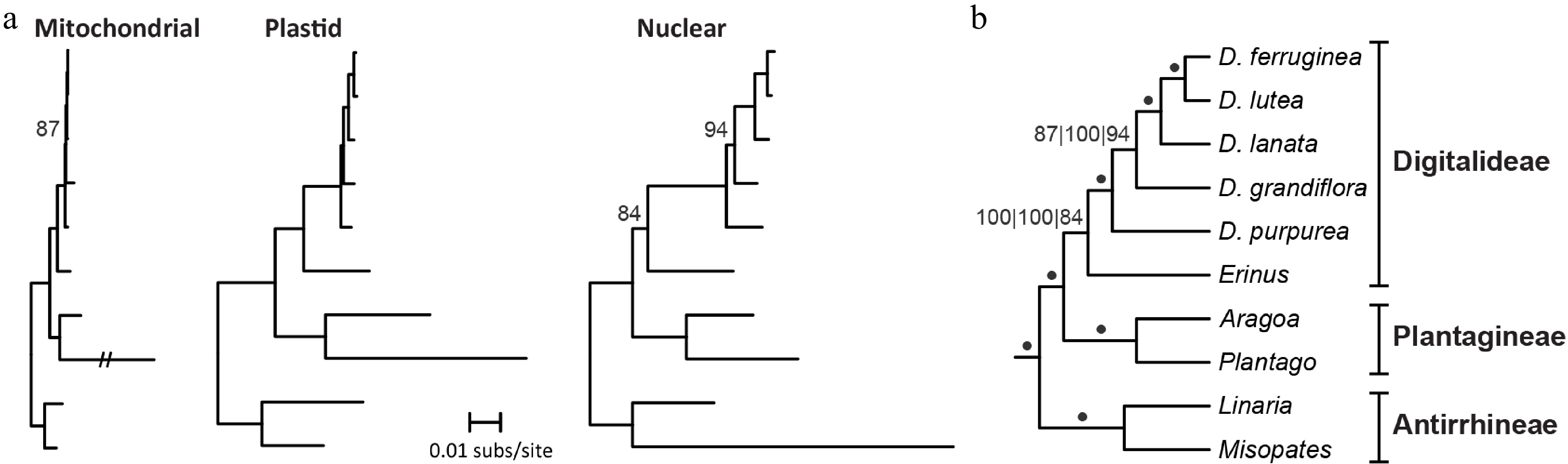
Figure 4.
Phylogenetic signal among Digitalideae genomes. (a) Phylograms of the concatenation of 32 mitochondrial CDSs, 3 rRNAs, and 20 introns present in all species (left); entire plastomes (excluding one copy of the IR) (middle); and nuclear rDNA (right). A long mitochondrial branch for Plantago ovata was reduced to 20% of the original length. All bootstrap values < 100% are listed. Outgroup taxa from Lamiales were used to root the tree but are not shown. (b) Cladogram summarizing the results from the three phylogenetic analyses. A dot indicates 100% bootstrap support from all three genome analyses. Bootstrap values for each analysis (left: mitochondrial; middle: plastome; right: nuclear rDNA) are shown if any analysis has < 100% bootstrap support. Section groupings are shown.
-
To date, thousands of plastomes and hundreds of mitogenomes have been sequenced from angiosperms, and comparisons of their genomic characteristics[3−5,7,10,24] have established that the plastome evolves substantially more slowly in structure and content in comparison with the mitogenome. This pattern is mirrored in Digitalideae. Functional content and gene order are identical, and size and structure are nearly so, among Digitalideae plastomes (Fig. 1). This is consistent with the general conservation of plastomic structure in other Plantaginaceae, except for the more structurally variable plastomes from Plantagineae[33,51,52,54], and is in line with expectations for a conservatively evolving plastome in most angiosperms[5].
Compared with their plastomes, Digitalideae mitogenomes are more variable in size, structure, and content (Table 1; Figs. 2 and 3; Supplementary Fig. S2). Mitogenomic sizes vary by less than twofold among Digitalideae species, which is large compared with the plastome but unexceptional compared with the much larger mitogenomic size ranges in other groups of closely related angiosperms[20,22,55−58]. Gene content differs among Digitalideae through the presence/absence of rps10 and some tRNAs (Fig. 3; Supplementary Table S8), and it varies more so among Lamiales as a result of additional gene losses at different evolutionary timepoints. The loss of rps2 and rps11 occurred early in eudicot history and are thus expectedly absent from all Lamiales, whereas the loss of rps1, rps7, and rps19 occurred multiple times across Lamiales[59]. Several Digitalideae mitogenomes have a duplicated rrn26, which may have some benefit to achieve the high expression of ribosomal RNAs, as suggested for plastid rRNAs[5]. Intriguingly, most Digitalis mitogenomes contain duplicate copies of the very short (22 bp) and doubly trans-spliced nad5 Exon 3, which perhaps might relieve some bottleneck during transcriptional processing. However, neither rrn26 nor nad5 Exon 3 is universally duplicated in angiosperm mitochondria, suggesting that any potential benefit is unlikely to be strong.
In terms of Digitalideae MIPT content (Supplementary Table S7), the overlapping plastomic positions of origin for most MIPTs suggest that most were acquired early in Digitalideae evolution or were even present in the Digitalideae crown ancestor. Of the 11 MIPT blocks, four are partially or fully shared between Erinus and most Digitalis species, indicating their ancestral presence in the tribe. Another four MIPTs are shared among most or all Digitalis species, also indicating their origin in an early Digitalis ancestor. Only three MIPTs were found to be uniquely present in a single species, indicating either a recent transfer event, or possibly an earlier event coupled with loss from other species. These large MIPTs have introduced copies of plastid-derived rps7 and trnV-GAC, which are otherwise absent from all sequenced Plantaginaceae mitogenomes. This raises the possibility of the potential functional recapture of these lost mitochondrial genes. However, we failed to find any RNA-seq reads that support the expression of these MIPT copies of rps7 or trnV-GAC, suggesting they are not expressed and instead represent nonfunctional transfer events from the plastome.
Large repeats share few homologous segments among Digitalideae mitogenomes (Fig. 2d), which is consistent with the repeat patterns inferred from several studies of other groups of close relatives: maize[60], beet[61], strawberry[14], Oenothera[62], and Fagaceae[63]. However, a few mitochondrial studies have found interspecific retention of some repeats, such as in Brassica[11,24] and lettuce[64]. Unfortunately, in most comparative studies of angiosperm mitogenomes, large repeat positions in plant mitogenomes have neither been visualized nor compared, so the degree of repeat retention and turnover is often not inferable. Regardless, the results from this study and inferable evidence from others confirm that large repeats turn over at a fast pace in angiosperm mitochondria, roughly at the timescale of speciation or close to it, whereas the plastome has retained its large IR for ~500 million years in most land plant lineages. Indeed, our statistical analysis of repeat turnover has shown that the sharing of repeat fragments among Digitalideae mitogenomes is consistent with a model of complete turnover, although at least a few of the mitochondrial repeats share endpoints and may represent the partial retention of an ancestral repeat (Supplementary Table S10). In contrast, the essentially full retention of the plastomic IR is consistent with a repeat stasis model (Supplementary Table S9). Additional mitogenomic studies from closely related species are needed to quantify how often large repeats are retained across species boundaries, as seen in Brassica and lettuce. In particular, the use of the repeat modeling equations will allow for a statistical evaluation of the lability of large repeats in other lineages.
Finally, our phylogenomic analysis supported the grouping of Erinus and Digitalis in Digitalideae and several sectional relationships within Digitalis (Fig. 4). Sampling included a representative for both Digitalideae species and three of the five proposed sections of Digitalis: E. alpinus as the sole member of Erinus, D. purpurea from section Digitalis, D. grandiflora and D. lutea from section Macranthae, and D. ferruginea and D. lanata from section Globiflorae. Unexpectedly, in our analysis, D. lutea URI10 grouped with D. ferruginea within section Globiflorae. However, in a previous study of D. lutea, most individuals grouped in section Macranthae, but an Italian individual grouped instead with D. subalpina[26], a species that is unassigned to any section.
Overall, even though the monophyly of D. lutea is in question, our finding of D. lutea URI10 in section Globiflorae is inconsistent with the phylogenetic placement of all other published D. lutea samples. A comparison of the trnL/trnF and ITS regions from our URI10 individual with the Digitalis markers available in GenBank showed >99.8% identity to D. ferruginea subsp. schischkinii but <96% identity to any of the other D. lutea markers in GenBank. This unexpected result indicates that the SRA samples published under D. lutea URI10 (biosample SAMN20054101) are potentially misidentified, although the nonmonophyly among individuals from D. ferruginea[26] compounds the uncertainty, tempering any strong conclusions at this stage. Clearly, more taxonomic effort is needed at a phylogenomic level within Digitalideae to sample species exhibiting nonmonophyly (D. lutea and D. ferruginea), species from unsampled sections (Frutescentes, Isoplexis), and species currently unplaced within any section (e.g., D. subalpina).
-
In this study, we have expanded the availability of mitogenomes, plastomes, and nuclear rDNA for six Digitalideae species (D. ferruginea, D. grandiflora, D. lanata, D. lutea, D. purpurea, and E. alpinus). Comparative analyses of these genomic resources from Digitalideae and other Lamiales demonstrate that the plastomes are virtually identical among species, whereas the mitogenomes are more variable in size, structure, arrangement, repeat content, gene content, and intron content. In particular, the evolutionary stability of large repeats is significantly different between organellar genomes, in which mitogenomic repeats have essentially turned over fully among species, whereas the plastomic IR content has remained remarkably stable. Finally, phylogenetic analyses of entire plastomes, mitochondrial genes and introns, and nuclear rDNA exhibit congruent phylogenetic signals among Digitalideae and other Plantaginaceae genera, encouraging future systematic analyses to resolve the remaining taxonomic uncertainties regarding Digitalideae systematics and species circumscription.
The authors wish to thank Jeff Palmer and the Joint Genome Institute for sharing the unpublished Sanger sequencing reads from Digitalis purpurea; and Sara Handy, Robert Literman, and Elizabeth Hunter for discussing the Illumina sequencing reads from Digitalis. This research was supported in part by the Complex Biosystems program at the University of Nebraska–Lincoln (graduate stipend to Rasel Ahmed), the US National Science Foundation (award MCB 2212075 to Jeffrey P. Mower), and the National Natural Science Foundation of China (Grant No. 32170238 to Zhiqiang Wu).
-
Not applicable. No animal experimentation, human subjects, or collection of wild species were involved in this research.
-
The authors confirm their contributions to the paper as follows: study conception and design: Mower JP, Wu Z; data collection: Mower JP, Wang J; analysis and interpretation of results: Mower JP, Wang J, Ahmed R, Wu Z; draft manuscript preparation: Mower JP. All authors reviewed the results and approved the final version of the manuscript.
-
The Sanger sequencing reads for Digitalis purpurea cannot be deposited in the sequence archives but are available upon reasonable request from the corresponding author JP Mower. All other data analyzed during this study are included in this published article, and the sources are listed in the supplementary information files.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Jeffrey P. Mower, Jie Wang
- Supplementary Table S1 Nucleotide data sets for Digitalideae assembly
- Supplementary Table S2 GenBank accessions for Digitalideae marker phylogenetics.
- Supplementary Table S3 GenBank accessions for phylogenomics.
- Supplementary Table S4 Homologous segments between pairs of Digitalideae mitogenomes.
- Supplementary Table S5 Repeats (min 100 bp) in Digitalideae mitogenomes.
- Supplementary Table S6 Homologous segments between pairs of Digitalideae mitogenomic repeats.
- Supplementary Table S7 Mitochondrial DNA of plastid origin (MIPTs) in Digitalideae mitogenomes.
- Supplementary Table S8 Ribosomal and transfer RNAs in Digitalideae mitogenomes.
- Supplementary Table S9 Wilcoxon signed-rank tests for large plastomic repeats.
- Supplementary Table S10 Wilcoxon signed-rank tests for large mitogenomic repeats.
- Supplementary Fig. S1 Phylogenetic analysis of marker data sets.
- Supplementary Fig. S2 Depth of read coverage for Digitalideae mitochondrial genomes.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Mower JP, Wang J, Ahmed R, Wu Z. 2026. Rapid turnover of mitogenomic repeats and congruent phylogenomic signal among Digitalideae genomes. Genomics Communications 3: e004 doi: 10.48130/gcomm-0026-0003 

Rapid turnover of mitogenomic repeats and congruent phylogenomic signal among Digitalideae genomes
- Received: 11 December 2025
- Revised: 23 January 2026
- Accepted: 28 January 2026
- Published online: 04 March 2026
Abstract: Digitalideae (Plantaginaceae) comprises ~25 species of foxgloves in two genera (Digitalis and the monotypic Erinus). Several species are notable as garden ornamentals or for their medical benefits and toxic side-effects, yet their genomic resources are limited to plastid genomes from two species. We assembled sequencing data from six Digitalideae species to evaluate the structural and functional evolution of their organellar genomes and to evaluate the phylogenetic signal of these organellar genomes and nuclear rDNA. Digitalideae plastomes are virtually identical among species, exhibiting invariant gene and intron content, a collinear gene order, and very similar sizes and repeat structures. The mitochondrial genomes are nearly identical in terms of gene and intron content but exhibit variation in size, substantial structural rearrangements, and extensive turnover of large repeat content. Comparison of large mitogenomic repeats is consistent with a turnover model, whereas plastomic inverted repeat content is consistent with a retention model, demonstrating distinctly different rates of repeat turnover between the organellar genomes. Phylogenetic trees constructed from entire plastomes, mitochondrial genes and introns, or nuclear rDNA are fully congruent among Digitalideae and other Plantaginaceae, and the results reveal some taxonomic uncertainties regarding Digitalideae systematics and possible species misidentifications in the existing data.
-
Key words:
- Digitalideae /
- Mitochondrial genome /
- Plastid genome /
- Repeat evolution /
- Phylogenetics