









-
WRKY transcription factors are a class of key gene regulatory proteins unique to plants, widely distributed across the plant kingdom, and constituting one of the largest families of transcription factors in plants[1]. The cDNA of WRKY was first cloned from sweet potato (Ipomoea batatas; SPF1) and wild oat (Avena fatua; ABF1, 2), among others[2,3]. Rushton et al.[4] identified WRKY1, WRKY2, and WRKY3 from parsley, named them WRKY, and first demonstrated that WRKY proteins play an important role in regulating plant responses to pathogens. Based on the number of WRKY domains and the type of zinc-finger structure, WRKY proteins are classified into three major groups[5]. WRKYGQK is the most typical sequence; variants such as WRRY, WSKY, and WKRY exist in a few proteins. The zinc-finger structure at the C-terminus of the domain is crucial for stabilizing the three-dimensional conformation and DNA-binding ability of the WRKY domain[6]. Extensive studies have revealed that WRKY proteins can function either as transcriptional activators or repressors, with their specific roles depending on the target genes they bind to and the protein interaction environment in which they are situated.
WRKY transcription factors are extensively involved in various aspects of plant growth and development, such as seed germination, flowering time regulation, and organ morphogenesis. Research on AtWRKY41 has further elucidated the signaling pathway through which WRKY proteins regulate seed dormancy. The results indicate that silencing of AtWRKY41 shortens seed dormancy and downregulates the transcription of AtABI3 in both mature and imbibed seeds[7]. In rice, however, loss-of-function mutation of OsWRKY29 enhances seed dormancy. Moreover, in the absence of ABA, OsWRKY29 can bind to the promoters of the ABA-responsive gene OsVP1 and the abscisic acid response element binding factor OsABF1 to downregulate their expression, thereby shortening seed dormancy[8]. WRKY proteins also play important roles in photomorphogenesis during the seedling stage. For example, AtWRKY36 directly binds to the promoter region of AtHY5 to downregulate its transcription under white light, thereby promoting hypocotyl elongation in seedlings. In contrast, UVB-activated AtUVR8 can interact with AtWRKY36 to inhibit its binding to the AtHY5 promoter, thus preventing the suppression of AtHY5 transcription by this transcription factor[9]. Unlike AtWRKY36, loss of function of AtWRKY32 leads to hypocotyl elongation, suggesting that this transcription factor promotes photomorphogenesis. Under dark conditions, AtCOP1 suppresses AtWRKY32, resulting in hypocotyl elongation[10]. After accumulating sufficient energy and nutrients during the vegetative growth stage, plants enter the reproductive stage, during which they precisely regulate flowering time in response to endogenous and exogenous signals. Recent studies have shown that members of the WRKY family play significant roles in the flowering process. In the vernalization pathway of Arabidopsis, cold-induced AtWRKY34 can bind to the AtCUL3A promoter to activate its expression. The E3 ubiquitin ligase AtCUL3A subsequently promotes the degradation of AtFRI, leading to reduced H3K4me3 levels in the chromatin of the AtFLC gene and thereby inhibiting AtFLC expression. This ultimately induces the expression of flowering-promoting genes and accelerates flowering. In another study, it was found that WRKY63 suppresses flowering under non-vernalization conditions by activating AtFLC, but under vernalization conditions, it promotes flowering by activating the repressors of AtFLC, COOLAIR, and COLDAIR[11]. In the GA (gibberellin)-mediated flowering pathway, DELLA proteins interact with AtWRKY12 and its homolog AtWRKY13, altering their transcriptional regulation of the downstream gene AtFUL and thereby affecting flowering. Besides participating in the GA pathway, AtWRKY12 and AtWRKY13 also influence flowering in the age-dependent pathway by regulating the expression of miR172b[12].
In tea plants, WRKY transcription factors play a crucial role in regulating stress resistance, growth and development, as well as the formation of tea quality. CsWRKY29 is induced by low temperature and ABA, enhances freezing tolerance by binding to the CsABI5 promoter and activating cold-responsive and sugar metabolism genes[13]. CsWRKY15 is induced by ethylene, drought, low temperature, and GA, and enhances broad-spectrum stress resistance by activating antioxidant enzymes and the GA signaling pathway[14]. Overexpression of CsWRKY51 leads to dwarfing, leaf curling, increased branching and flowering, reduced chlorophyll content, and significantly elevated accumulation of amino acids such as glutamine[15]. Another study revealed that the CsWRKY12-CsVQ4L module regulates the accumulation of ester-type catechins in tea plants. Overexpression of CsWRKY12 promotes the expression of CsSCPL4 and CsSCPL5, resulting in increased levels of EGCG and ECG. Furthermore, the VQ motif-containing protein CsVQ4L interacts with CsWRKY12; specifically, CsVQ4L binds directly to CsWRKY12 and further enhances CsWRKY12-mediated activation of CsSCPL4 and CsSCPL5, synergistically promoting the accumulation of ester-type catechins in young tea leaves[16]. Studies confirmed that the transcription factor CsWRKY40 directly binds to and positively regulates CsFBXL13, thereby participating in low-temperature response and the regulation of spring bud flush[17]. CsWRKY71 is a key negative regulator of theanine biosynthesis, inhibiting the expression of the major gene CsTSI. The gibberellin signaling pathway can relieve this inhibition, thereby promoting theanine accumulation[18]. In summary, WRKY transcription factors play important roles in various aspects of tea plant growth and development. The pangenome can overcome the limitations of a single reference genome by revealing gene presence/absence variations (gPAVs) and copy-number variations (CNVs), and by classifying genes into core and variable categories, thereby providing a more comprehensive reflection of population genetic diversity and offering critical insights for understanding plant evolutionary history and environmental adaptation[19].
To gain a deeper understanding of the evolution and function of WRKY gene family members during the growth and development of tea plants, this study conducted a comprehensive analysis of WRKY genes in tea plants using genomic data from 22 tea varieties. Based on the identification of WRKY members across these 22 tea varieties, this research systematically investigated their physicochemical properties, constructed a phylogenetic tree, identified core WRKY members in tea plants, calculated the Ka/Ks ratios among WRKY members, and analyzed the expression profiles of WRKY genes in different tissues and at different stem nodes of tea plants. This work provides a holistic perspective on the evolution and functional diversification of WRKY genes across diverse tea varieties.
-
A total of 22 tea plant genomes, representing three Camellia sinensis varieties (var. sinensis, var. assamica, and var. pubilimba) and major tea-producing regions in China, were obtained from the Tea Graph Pangenome Database. These high-quality genome assemblies were generated using Illumina and PacBio sequencing platforms, with detailed cultivar information and assembly statistics provided in Supplementary Table S1. The 22 tea plant genomes used in this study were all downloaded from the Tea Graph Pangenome Database (
www.tea-pangenome.cn )[20]. After obtaining all genomic data, custom scripts were used to detect gene alternative splicing forms, and the longest transcript for each gene was retained. The protein sequences of 72 WRKY gene family members from Arabidopsis thaliana were downloaded from The Arabidopsis Information Resource (TAIR) database. Using A. thaliana WRKY members as seed sequences, a BLASTP analysis was performed against the 22 tea plant genomes with stringent parameters (E-value ≤ 1e-20)[21]. All candidate genes were annotated for conserved domains using the Pfam and PANTHER databases within the InterProScan software[22]. Meanwhile, the physicochemical properties of WRKY proteins from the 22 tea plant varieties were analyzed using the Protein Parameter Calc plugin in TBtools software[23]. Meanwhile, we counted the number of introns in WRKY members across all varieties and performed significance analysis using ANOVA, followed by Fisher's LSD post hoc test.To investigate the phylogenetic relationships of WRKY proteins among the 22 tea plant varieties, multiple sequence alignment of all identified WRKY members was conducted using MAFFT v7.505 with parameters -auto[24], and the aligned sequences were subsequently trimmed with automated parameters using TrimAl v1.4.rev15 with parameters -automated1[25]. A phylogenetic tree was constructed for the WRKY members using IQ-TREE v2.0 with the parameters -m MF -T 20[26]. Finally, the phylogenetic tree was visualized using the online website iTOL (
https://itol.embl.de )[27].Clustering and Ka/Ks analysis of the tea plant WRKY gene family
-
Gene family clustering of protein sequences from the 22 tea plant varieties was performed using OrthoFinder software[28]. Subsequently, the results for WRKY gene family members were extracted from the clustering outcomes. Furthermore, syntenic gene pairs, both among the 22 tea plant varieties and within each variety, were identified using JCVI (a Python implementation of MCScan)[29], and gene pairs belonging to the WRKY gene family were specifically extracted. The obtained gene pairs were subjected to sequence alignment using ParaAT with default parameters[30], followed by format conversion and calculation of their Ka/Ks values using KaKs_Calculator 3.0 software with parameters-m YN[31]. ANOVA combined with Fisher's LSD method was used for significance analysis.
Expression profiling of WRKY gene family members
-
To investigate the expression patterns of WRKY genes across diverse tea plant genetic backgrounds, we selected four representative cultivars from the 22 accessions for transcriptome analysis: Huangdan (HD), Longjing43 (LJ43), Shuchazao (SCZ), and Tieguanyin (TGY). These cultivars were chosen based on the following criteria: (1) taxonomic representativeness—all belong to C. sinensis var. sinensis, the most widely cultivated variety; (2) geographical diversity—they originate from major tea-producing regions in China, including Fujian (HD, TGY) and Zhejiang/Sichuan (LJ43, SCZ); (3) processing suitability—HD and TGY are typical oolong tea cultivars, while LJ43 and SCZ are representative green tea cultivars, allowing comparison between distinct quality trait priorities; (4) phenological variation—they exhibit differences in spring bud flush timing (early in LJ43 and SCZ vs. moderate to late in HD and TGY); and (5) genomic data availability—high-quality reference genomes and corresponding transcriptome data are available for these cultivars in the Tea Plant Information Archive (TPIA). These selections enable a comprehensive assessment of WRKY gene expression in relation to cultivar-specific developmental and metabolic characteristics. The transcriptomic datasets used included those from different tea plant tissues and leaves at different stem node positions. All expression data were visualized using TBtools, with row-wise comparison and Zero-to-One normalization applied[32].
-
In this study, WRKY protein members across 22 tea plant genomes were identified using BLASTP and InterProScan. A total of 1,699 WRKY members were identified from the 22 tea cultivars. The cultivar GH3H contained the fewest members (65), while JGY contained the most (84) (Supplementary Table S1). The physicochemical properties of all WRKY members were further analyzed. Overall, no significant differences in the physicochemical properties of WRKY proteins were observed among the different cultivars (Fig. 1a−d). Their molecular weights (MWs) ranged from 10,163.41 (LJ107062) to 130,277.88 (LTDC003505), and their isoelectric points (pI) varied from 4.77 (e.g., MSBH156683, DX5H099312, GH3H064613, HD1100446, HJY092440, F7121467, JMZ062949, LTDC025752, F6098493, CSS0013893.1, F5113173, YH9H028196, F8101164, ZYQ051037) to 10.48 (F5048378). The instability index ranged from 33.66 (MSBH130915) to 81.25 (HD.13G0016890, F5102968). The grand average of hydropathy (GRAVY) values ranged from −1.469 (QL10H092552) to 0.394 (JS046202, QL10H009067) (Supplementary Table S2).
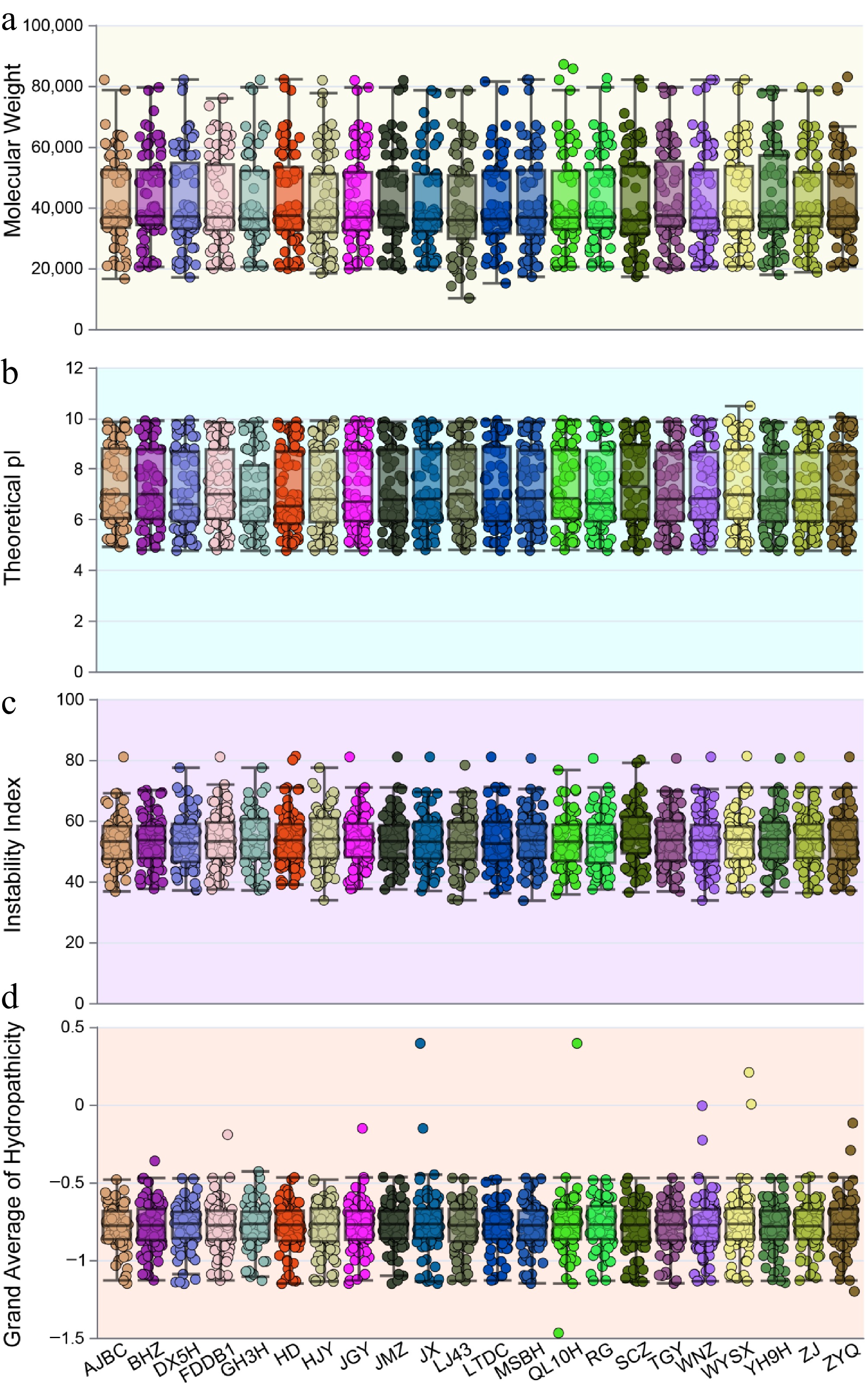
Figure 1.
Statistical analysis of the physicochemical properties of WRKY proteins in 22 tea plant cultivars. (a) Molecular weight, (b) isoelectric point, (c) instability index, (d) hydropathicity.
Phylogenetic tree and cluster analysis of WRKY in tea plants
-
To investigate the phylogenetic relationships of WRKY proteins in tea plants, a phylogenetic tree was constructed using WRKY protein sequences from the 22 tea cultivars. The tree comprised 1,699 WRKY proteins from the 22 tea plants and 74 WRKY members from Arabidopsis thaliana. As shown in Fig. 2a, the WRKY proteins from tea plants could be divided into 11 clades. The largest clade, Clade I, contained 373 WRKY members, while the smallest, Clade II, contained 36 WRKY members. The distribution of WRKY members from the 22 tea cultivars across these clades was further analyzed. WRKY genes from most cultivars were present across all 11 clades. However, cultivars GH3H, SCZ, and QL10H lacked WRKY members in Clade II (which includes Arabidopsis WRKY23/43/56) (Fig. 2b). Furthermore, the number of WRKY members within the same clade varied among different tea cultivars. For instance, within Clade I, SCZ had 20 members, whereas QL10H had only 16. This suggests a significant numerical variation in WRKY members among different tea plant germplasms (or accessions), which may reflect functional differentiation or adaptive divergence of this gene family across distinct genetic backgrounds.
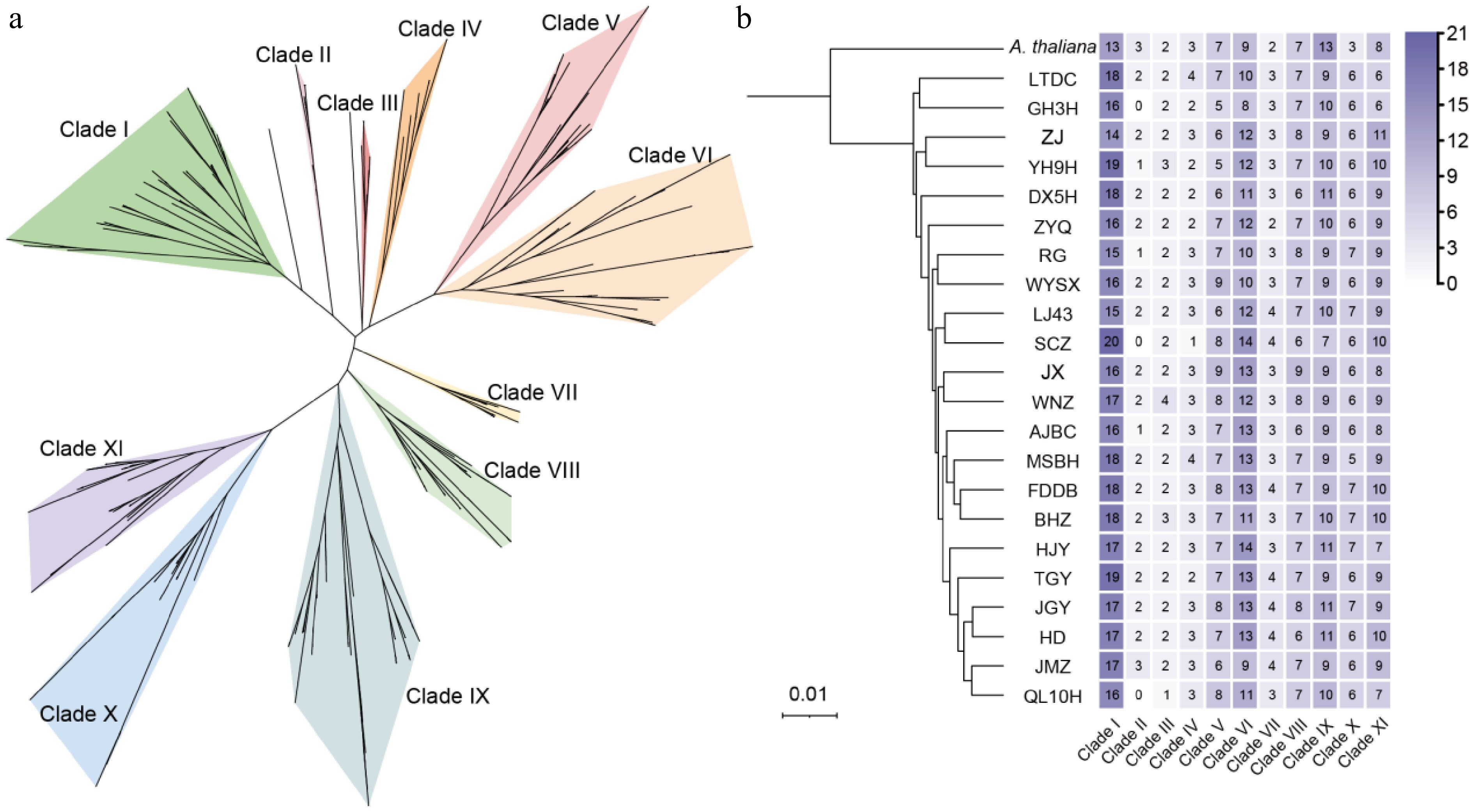
Figure 2.
Phylogenetic tree and quantitative statistics of WRKY members in 22 tea plant genomes. (a) The background colors in the phylogenetic tree are used to distinguish different evolutionary clades. (b) The phylogenetic tree is derived from previously published literature. The numbers in the heatmap represent the count of members from each cultivar within each clade, with color intensity ranging from light to dark indicating the corresponding member count.
Furthermore, the WRKY protein sequences from the 22 tea plant cultivars were subjected to clustering analysis. OrthoFinder clustered the 1,697 WRKY proteins from tea plants into 69 orthologous groups (OGs) (Fig. 3). This study further classified these 69 OGs, identifying 24 core OGs, 32 soft-core OGs, and 12 shell OGs. Additionally, the clustering results showed that, among the 24 core OGs, only 16 clustered with Arabidopsis thaliana, while 15 of the soft-core OGs and 7 of the shell OGs clustered with Arabidopsis, respectively (Supplementary Table S3). These results indicate the diversification of WRKY genes at the tea plant species level as well as the varietal diversification of WRKY members among different cultivars.
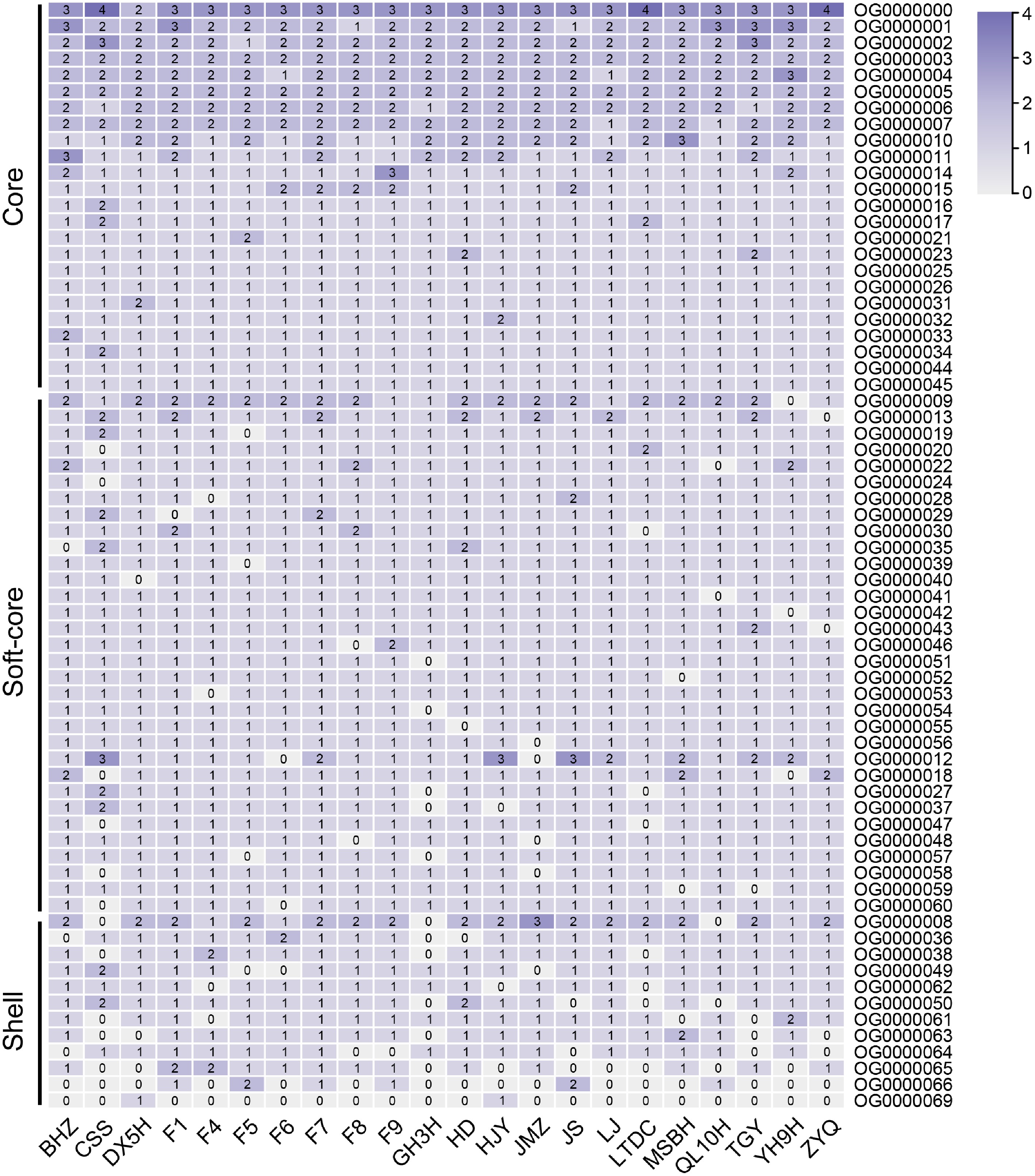
Figure 3.
Cluster analysis of tea plant WRKY members based on OrthoFinder results. "Core" represents gene clusters where members are present in all 22 tea plant cultivars. "Soft-core" represents gene clusters where members are present in at least 90% of the tea plant cultivars. "Shell" represents gene clusters where members are present in less than 90% of the tea plant cultivars.
Analysis of selection pressure and intron number statistics of WRKY genes within tea plant cultivars
-
To investigate the selection pressure acting on the WRKY gene family during tea plant evolution, we calculated the Ka/Ks values for WRKY members across the 22 cultivars. The results showed that the Ka/Ks values among WRKY gene family members were largely consistent across the 22 tea cultivars, primarily distributed between 0.2 and 0.6 (Fig. 4a). Furthermore, only WRKY members in cultivar F7 exhibited Ka/Ks values greater than 1.0. These findings indicate that WRKY members within tea plant cultivars are generally under purifying selection. Additionally, as introns are key factors influencing gene expression efficiency and genetic load, we further analyzed the intron numbers of WRKY members across different cultivars (Fig. 4b). The intron numbers of WRKY members in the 22 tea cultivars were statistically analyzed. The results revealed that most WRKY members in tea plants possess three introns, with the maximum number observed being 15. Furthermore, except for cultivars JMZ and QL10H, all other cultivars contained WRKY members that were intron-less genes. This suggests potential variability in the gene structure of this family, which may be associated with its expression regulation and functional diversification.
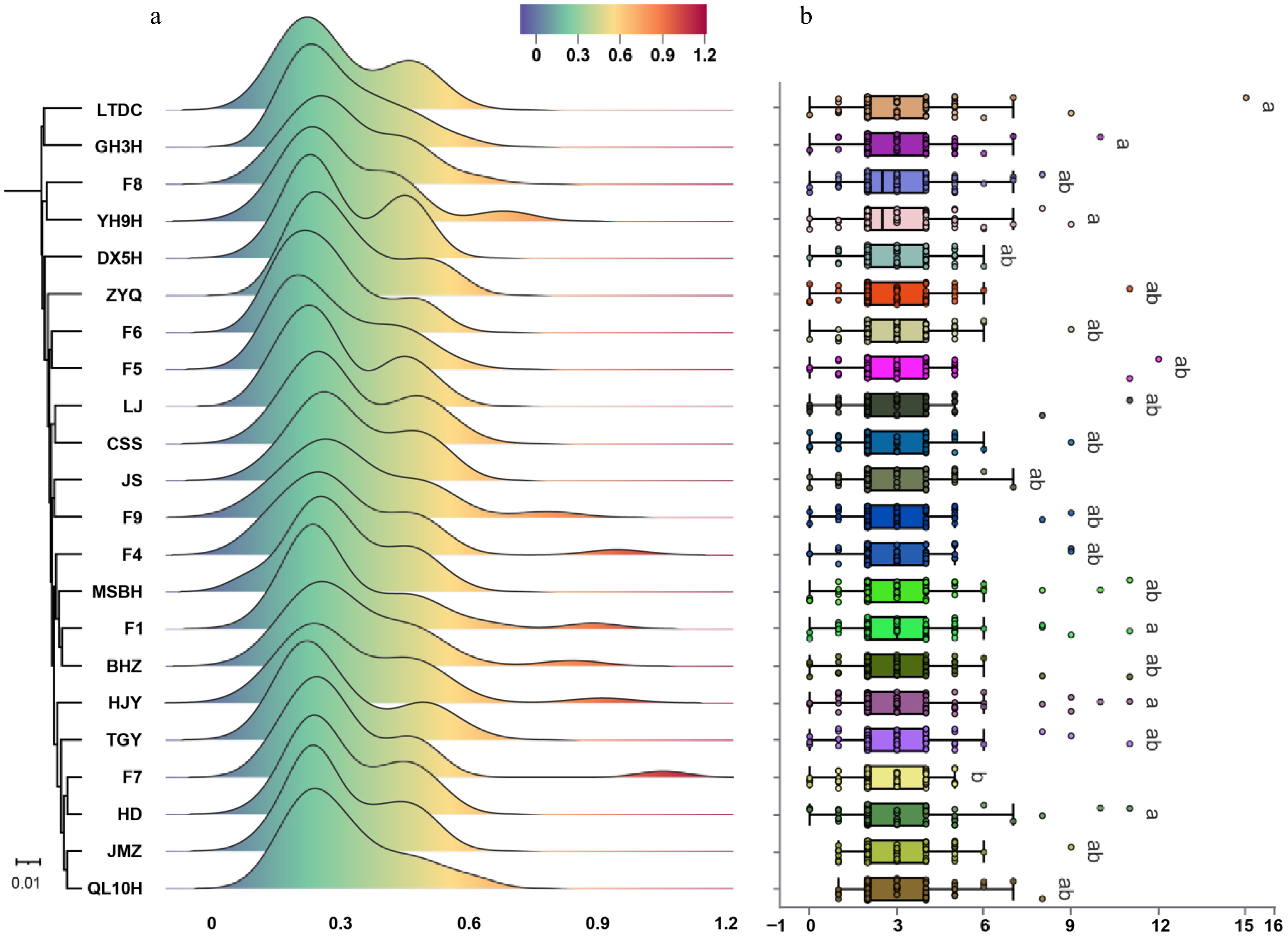
Figure 4.
Ka/Ks analysis and intron number identification of WRKY members in 22 tea plant cultivars. (a) Distribution of Ka/Ks values among WRKY members in 22 tea plant cultivars. (b) Statistics of intron numbers in WRKY members across 22 tea plant cultivars. Significance analysis was performed using ANOVA, followed by Fisher's LSD post hoc test.
Analysis of selection pressure and intron number statistics within phylogenetic tree clades
-
Syntenic gene pairs formed by WRKY members from the 22 cultivars were filtered based on their distribution in the phylogenetic tree. We found significant differences in the Ka/Ks values of WRKY gene pairs among different evolutionary clades. For example, clades I, II, IV, V, VI, and IX exhibited higher Ka/Ks values compared to the other clades (Fig. 5a). This suggests that a substantial number of WRKY members within these clades have experienced elevated evolutionary pressure during evolution, potentially driving functional innovation or adaptive evolution. Further statistical analysis of intron numbers in WRKY members across different clades revealed that clades with fewer introns (e.g., Clades I, II, VI, and IX) generally showed higher Ka/Ks values. Furthermore, intron-less genes were present within these clades, implying that the simplification of gene structure (intron loss) may act synergistically with a stronger, relatively higher selection pressure, accelerating the functional differentiation of certain clades (Fig. 5b). In summary, against an overall conservative background, the tea plant WRKY gene family likely achieved functional diversification and specialization in specific evolutionary lineages through the synergistic effects of relatively higher selection pressure and gene structure variation (e.g., intron loss).
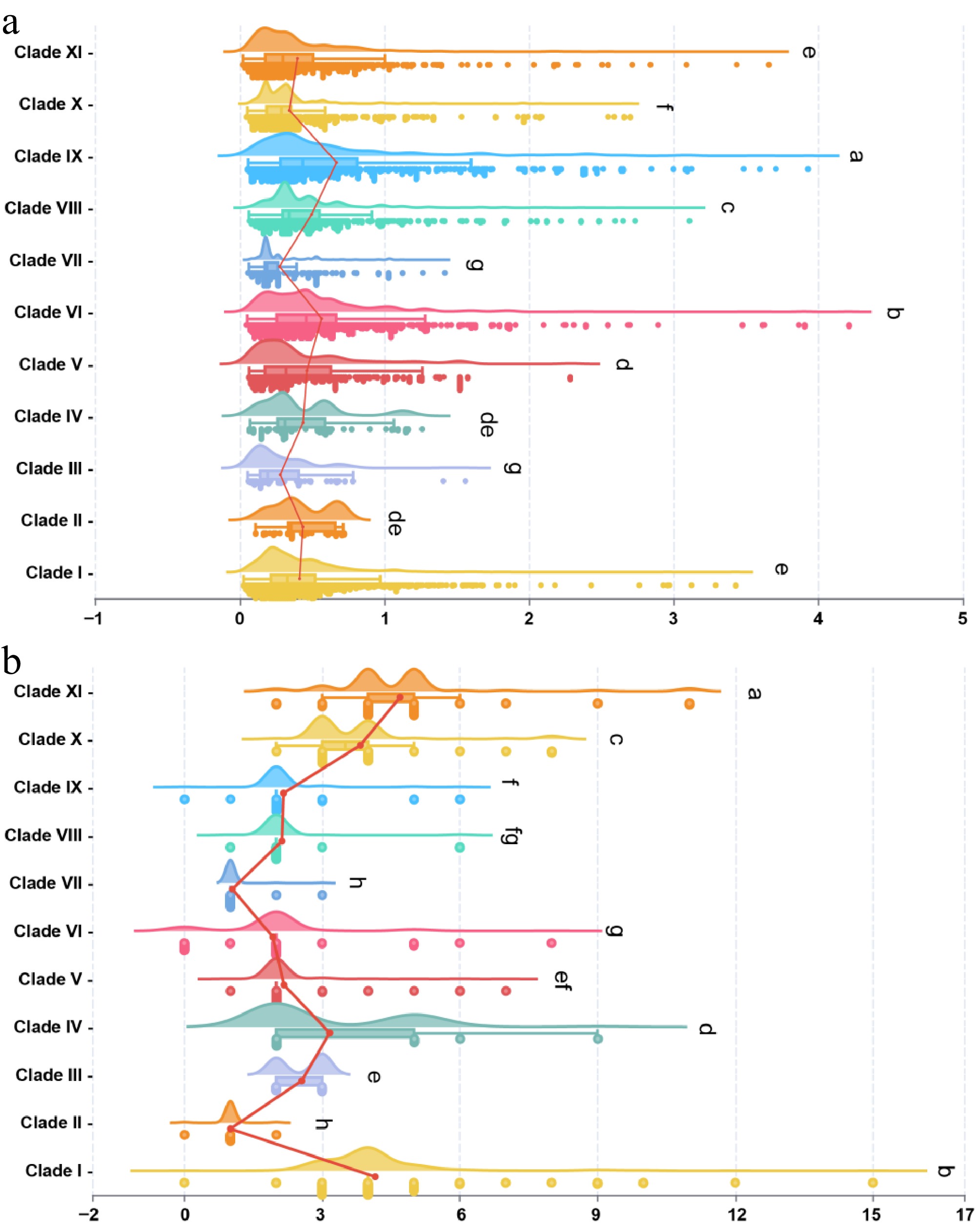
Figure 5.
Ka/Ks analysis and intron number statistics of WRKY members across 11 clades. (a) Distribution of Ka/Ks values among WRKY members in the 11 clades. (b) Statistics of intron numbers in WRKY members across the 11 clades. Significance analysis was performed using ANOVA, followed by Fisher's LSD post hoc test.
Root-predominant and tissue/clade-specific expression of WRKY genes in tea plants
-
To understand the functions of the WRKY gene family in tea plants, this study extracted transcriptomic data for four tea cultivars (which correspond to varieties included in the 22-tea pangenome used in this study) from a tea plant genomic database. A systematic comparison was conducted on the expression levels of their WRKY members in different tissues (apical bud, flower, fruit, young leaf, mature leaf, old leaf, root, stem) (Fig. 6). The results showed that WRKY genes in all four cultivars exhibited significantly high expression in roots, suggesting that the WRKY family may play a core regulatory role in the biological processes of the tea plant root system, such as root development, stress response, or nutrient absorption. Furthermore, some WRKY genes also showed relatively high expression in flowers and fruits, indicating their potential function in reproductive development or secondary metabolite accumulation. From the perspective of evolutionary clades (Clade I–XI), distinct expression patterns were observed among different clades. Clade I showed significant expression in young and mature leaves. Clade VIII and Clade IX exhibited relatively high expression in apical buds and leaves, while Clade VII was specifically and highly expressed in fruits. In summary, WRKY genes in tea plants demonstrate clear tissue-specific expression patterns and functional differentiation across evolutionary clades.

Figure 6.
Analysis of WRKY gene expression in different tissues of tea plants.
Expression of WRKY genes in tea plants is closely associated with leaf development
-
Expression of WRKY genes in tea plants exhibits a strong association with the early stages of leaf development, particularly in apical buds and the first leaf, where transcript levels progressively decrease in lower leaf positions. This expression gradient, combined with clade-specific patterns, suggests that WRKY members are likely involved in key processes such as leaf morphogenesis, cell differentiation, and the accumulation of secondary metabolites (e.g., catechins and theanine) that determine the quality of tender tea shoots. The distinct expression profiles of Clade V, VI, IX, and XI in buds and young leaves further imply functional specialization among WRKY subclades in regulating early leaf growth and flavor-related metabolic pathways. To investigate the expression dynamics of WRKY genes during tea leaf development, this study further analyzed the expression patterns of WRKY members in apical buds and leaves at different node positions (first to fourth leaves) across four tea cultivars (Fig. 7). The results showed that WRKY genes were generally highly expressed in apical buds and the first leaf. As the leaf node position descended, the expression of most WRKY genes exhibited a gradual declining trend, with some showing minimal or no expression. This indicates that WRKY genes play a significant role in the early stages of tea leaf development, contributing to the formation of tea quality. From an evolutionary clade perspective, Clades V and XI were highly expressed in buds and the first leaf. Clades VI and IX were specifically highly expressed in the first leaf. Although Clade I was also highly expressed in buds and leaves, a substantial number of its members remained highly expressed in the second to fourth leaves. In summary, WRKY genes displayed distinct stage-specific expression patterns across different leaf node positions in tea plants, with particularly active expression in apical buds and young leaves. This suggests their important functions in leaf morphogenesis and early development.

Figure 7.
Expression analysis of WRKY genes at different leaf developmental stages in tea plants.
-
The development of plant genomic technologies has provided valuable genetic information resources for the study of genes and gene families. Previous research has predominantly focused on the characterization of individual gene families within a single or a limited number of plant accessions. However, this approach has inherent limitations, making it difficult to reveal the presence/absence variation and evolutionary dynamics of gene families across different cultivars within a species. By employing a pan-genomic approach based on 22 tea plant genomes, the present study overcomes these limitations and provides a comprehensive view of the WRKY gene family at the population level. Consequently, systematic analyses of single gene families or all gene families within specific pathways based on large-scale genomic data[33,34], as well as investigations into the evolutionary dynamics of gene families within populations utilizing plant pangenome sequencing technologies, can effectively address the shortcomings of these traditional methods[35,36]. Building upon prior analyses of the WRKY gene family in tea plants, this study further employs a tea pangenomic perspective to perform a comprehensive genome-wide systematic analysis, thereby providing an in-depth functional elucidation of WRKY gene family members[20].
Gene presence/absence variation represents one of the most common forms of genomic variation. Based on the tea pangenome, this study systematically analyzed the composition and distribution of WRKY gene family members across 22 cultivars, identifying a total of 24 core OGs, 32 soft-core OGs, and 12 shell OGs. Compared to previous studies that focused on identifying the WRKY gene family, performing phylogenetic analysis, and profiling expression patterns in single tea cultivars[37,38], this work clarifies the conservation, specificity, and dynamic evolution of this family throughout tea plant evolution. These distinct OG categories reflect both the conservation and diversity of the WRKY gene family among tea cultivars: core OGs likely perform indispensable functions in fundamental growth and developmental processes; soft-core OGs may be involved in cultivar-specific adaptive regulation; while shell OGs might be associated with localized evolutionary events.
This study revealed significant differences in the selection pressure acting on WRKY genes across distinct evolutionary clades. Notably, clades such as I, II, and IV exhibited higher Ka/Ks ratios, suggesting they may have experienced elevated evolutionary pressure during evolution. Genes within these clades were often associated with a reduced number or even a complete loss of introns, implying a close link between gene structure simplification and functional innovation. This observation raises the possibility that intron loss and elevated selection pressure may synergistically contribute to functional divergence within certain WRKY clades. Such a hypothesis is consistent with previous findings in other gene families, where retroposition-mediated intron loss, coupled with relaxed or positive selection, has been implicated in the emergence of novel biological functions[39]. However, direct evidence linking intron loss to functional divergence in WRKY genes remains to be established, and this interpretation should be considered speculative at this stage. Concurrently, expression analyses demonstrated that these clades exhibited highly specific expression patterns in different tea plant tissues (e.g., roots, young leaves, apical buds). For instance, Clade I was highly expressed in young and mature leaves, while Clade VII showed fruit-specific expression. Together, these results indicate that during tea plant evolution, the WRKY gene family likely underwent relatively higher selection pressure, which drove the optimization of gene structure and functional differentiation, thereby enabling its specific regulatory roles in different developmental stages and tissues of the tea plant.
Notably, this study demonstrates that WRKY genes exhibited markedly higher expression in the apical buds and first leaves of tea plants, with expression gradually decreasing in lower leaf positions. This pattern suggests a crucial role for this gene family in the early development of new tea shoots. As the primary source of harvested tea leaves, the developmental state of new shoots directly impacts both tea yield and quality[40]. The high expression of WRKY genes in young leaves may be closely linked to their regulation of leaf morphogenesis, the accumulation of secondary metabolites (such as tea polyphenols and caffeine), and stress response pathways[37,41]. In this study, we observed that WRKY gene expression profiles varied across different tea cultivars, with HD and TGY showing similar patterns, as did LJ43 and SCZ (Fig. 7). These groupings correspond to known genetic backgrounds, phenological traits, and quality characteristics of the four cultivars. HD and TGY are both C. sinensis var. sinensis cultivars originating from Fujian, typically characterized by moderate to late bud flush and high ester-type catechin accumulation, making them suitable for oolong tea production. In contrast, LJ43 and SCZ are typical green tea cultivars from the Jiangnan region, known for early bud break and high amino acid content. The expression similarity within each pair suggests that WRKY gene expression patterns are not merely cultivar-specific but may reflect conserved regulatory mechanisms associated with distinct developmental strategies and metabolic priorities. These observations further support the notion that WRKY genes contribute to functional diversification underlying tea plant adaptation and quality formation. In this study, we selected four representative tea cultivars—HD, TGY, LJ43, and SCZ—for transcriptome analysis. These cultivars differ in their geographical origins, phenological traits, and processing suitability. Notably, HD and TGY are both oolong tea cultivars originating from Fujian, characterized by moderate to late bud flush and high ester-type catechin accumulation, whereas LJ43 and SCZ are typical green tea cultivars from the Jiangnan region, known for early bud break and high amino acid content. The expression patterns of WRKY genes clustered these cultivars into two corresponding pairs (HD/TGY and LJ43/SCZ) (Fig. 7), suggesting that WRKY expression profiles are not merely cultivar-specific but may reflect conserved regulatory modules associated with distinct developmental strategies and metabolic priorities. This observation further supports the notion that WRKY transcription factors contribute to functional diversification underlying tea plant adaptation and quality formation. Furthermore, the expression specificity of different evolutionary clades in buds and leaves (e.g., high expression of Clade V and XI in buds) further implies functional diversification of WRKY genes in the formation of tea quality traits. These findings provide potential gene targets for future molecular breeding strategies aimed at modulating shoot development and enhancing tea quality.
Based on the tea pangenome, this study systematically elucidated the composition, evolution, and expression characteristics of the WRKY gene family, providing valuable resources for the genetic improvement of tea plants. The identification of core OGs suggests that these genes may be indispensable for fundamental biological processes in tea plants and could be prioritized as conserved targets in breeding programs. In contrast, the soft-core and shell OGs reflect genetic diversity among cultivars, potentially associated with cultivar-specific traits such as stress resistance and metabolite accumulation. Furthermore, the widespread high expression of WRKY genes in roots implies their potential functions in root development and stress responses, offering candidate genes for breeding tea cultivars with enhanced stress tolerance and nutrient efficiency[42−44]. In the future, functional validation and breeding applications targeting key WRKY genes can be advanced by integrating technologies such as gene editing and molecular marker-assisted selection, thereby promoting the precision improvement of tea plant varieties.
This research was funded by National Key R & D Program of China (2020YFE0202900), the National Natural Science Foundation of China (31972460), General Program for Basic Science (Natural Science) Research in Universities of Jiangsu Province (25KJD210002), and the Science and Technology Project of Jiangsu Province for Industry-Academia-Research Collaboration (HX2025293).
-
The authors confirm their contributions to the paper as follows: conceived and designed the experiments: Yang B, Zhao F; analyzed the data, wrote, and revised the manuscript: Huang W, Shen M, Xiao Y, Fang W, Jiang J. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Bin Yang, Fei Zhao
- Supplementary Table S1 Information on 22 tea plant cultivars and statistics of WRKY gene family count.
- Supplementary Table S2 Statistics on physicochemical properties of WRKY protein members.
- Supplementary Table S3 Clustering analysis of WRKY transcription factors in 22 tea plant cultivars.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yang B, Zhao F, Xiao Y, Huang W, Shen M, et al. 2026. Pangenome analysis of the WRKY gene family unveils its functional roles in the growth and development of Camellia sinensis. Genomics Communications 3: e009 doi: 10.48130/gcomm-0026-0008 

Pangenome analysis of the WRKY gene family unveils its functional roles in the growth and development of Camellia sinensis
- Received: 22 January 2026
- Revised: 02 April 2026
- Accepted: 17 April 2026
- Published online: 19 May 2026
Abstract: WRKY transcription factors are crucial regulators in plants, extensively involved in growth, development, stress responses, and secondary metabolism. However, their evolutionary dynamics and functional diversification across tea plant cultivars have not been systematically elucidated. In this study, we performed a pan-genome-wide analysis of the WRKY gene family using genomic data from 22 tea cultivars. A total of 1,699 WRKY members were identified and classified into 24 core, 32 soft-core, and 12 shell homologous groups. Evolutionary analysis indicated that certain clades (e.g., Clade I, II, and IV) experienced elevated evolutionary pressure, which was associated with a reduction in intron numbers. Expression profiling revealed that WRKY genes were highly expressed in roots, apical buds, and young leaves, exhibiting clear tissue and developmental stage-specific patterns. These results elucidate the functional diversification of the WRKY gene family during tea plant evolution and highlight their potential regulatory roles in the growth and development of young shoots. These candidate genes and positively selected clades provide valuable molecular targets for marker-assisted breeding and functional genomics studies aimed at improving tea quality.
-
Key words:
- WRKY /
- Camellia sinensis /
- Pan-gene family /
- Growth and development /
- Phylogeny