









-
Cell death is a core biological process that helps organisms maintain homeostasis, regulate their development, and respond to pathological damage. Depending on the mechanisms of death, morphological characteristics, and immunogenicity, cell death can be classified into two main categories: regulated cell death (RCD) and accidental cell death (ACD). RCD is an actively regulated process mediated by specific molecular pathways that have clear physiological or pathological significance. Over time, the study of cell death has expanded beyond apoptosis, autophagy, pyroptosis, and necroptosis, with the mechanisms of ferroptosis, cuproptosis, and other forms of cell death being discovered and refined. The following sections elaborate on the types of cell death in chronological order of their discovery.
Apoptosis, as a genetically programmed cell death mechanism, executes orderly cellular demise through activation of the caspase protease cascade via the mitochondrial pathway or the death receptor pathway. This process maintains tissue homeostasis by eliminating damaged DNA and infected cells, and through tumor suppression, characterized by cytoplasmic shrinkage, chromatin condensation, and apoptotic body formation[1].
Autophagy-dependent cell death represents a distinct regulated death modality initiated by excessive autophagic degradation of essential organelles, contrasting with classical apoptosis. Marked by cytoplasmic vacuolization, this paradoxical process demonstrates dual functionality in cellular protection and destruction, depending on the context[2].
Pyroptosis, unlike autophagy, constitutes an inflammatory type of RCD mediated by gasdermin family pore-forming proteins. Activated through inflammasome-mediated Caspase-1/4/5 cleavage, it eliminates intracellular pathogens while potentially triggering sepsis or autoimmune disorders when hyperactivated[3]. This lytic process features cellular swelling preceding membrane rupture, chromatin degradation, and the release of proinflammatory cytokines, accompanied by nuclear condensation and DNA fragmentation[4].
Necroptosis emerges as a caspase-independent type of regulated necrosis characterized by plasma membrane permeabilization and the release of damage-associated molecular patterns (DAMPs). This death modality participates in pathophysiological processes including ischemia–reperfusion injury and chronic inflammation, and serves as a backup death mechanism if apoptosis is deficient[5].
Ferroptosis, distinct from classical death pathways, represents an iron-dependent regulated type of necrosis triggered by lethal lipid peroxidation accumulation. Initiated through inactivation of glutathione peroxidase 4 (GPX4) or inhibition of the cystine-glutamate antiporter (System Xc-), it features a reduction in mitochondrial cristae and membrane densification. This process modulates drug resistance in tumors and contributes to neurodegenerative disorders like Parkinson's disease[6].
Cuproptosis, mechanistically analogous to ferroptosis, constitutes a copper-induced cell death mechanism mediated through copper-binding-induced aggregation of lipoylated mitochondrial proteins and subsequent respiratory chain collapse. Dependent on copper transporter SLC31A1, this copper homeostasis-related death pathway holds therapeutic potential for Wilson's disease and offers novel strategies for copper metabolism-targeted cancer therapies[7].
Distinct from the aforementioned RCD pathways, ACD represents a passive demise triggered by extreme physicochemical insults in the absence of orchestrated molecular regulation. Characterized by cellular swelling, rapid plasma membrane disintegration, and massive cytoplasmic content leakage, ACD elicits robust inflammatory responses through the release of DAMPs. This nonprogrammed death modality mediates initiation signals for repairing tissue damage while pathologically exacerbating organs' dysfunction under diseased conditions.
Cell death in normal physiological conditions is a highly regulated biological process. However, in patients with gastrointestinal (GI) tumors, cancer cells can influence the immune microenvironment through distinct modes of cell death. For example, autophagy suppresses immunogenic cell death (ICD) and immune cells while enhancing antigen presentation. Ferroptosis releases DAMPs and inhibits immune T-cells. Cuproptosis involves mitochondrial stress and the release of DAMPs, suppressing immune T-cells, among others, thereby impacting the efficacy of cancer immunotherapy and patients' prognosis. Immunotherapy, which activates or enhances the body's antitumor immune response, has become a critical strategy in treating gastrointestinal cancer. Major approaches include immune checkpoint inhibitors (ICIs), CAR-T-cell therapy, and cancer vaccines. As a first-line treatment of advanced gastric cancer, pembrolizumab demonstrated better overall survival (OS) than chemotherapy in programmed death ligand-1 (PD-L1)-positive (CPS ≥ 1) patients (10.6 vs 11.1 months; hazad ratio (HR), 0.90; 95% confidence interval (CI), 0.75–1.08). However, in the CPS ≥ 10 subgroup, immunotherapy showed a superior OS over chemotherapy alone (17.4 vs 10.8 months; HR, 0.62; 95% CI, 0.45–0.86). Another study revealed that immunotherapy combined with chemotherapy achieved a prolonged OS (12.9 vs 11.5 months; HR, 0.78; 95% CI, 0.70–0.87) and progression-free survival (PFS) (6.9 vs 5.6 months; HR, 0.76; 95% CI, 0.67–0.85) compared with standard chemotherapy[8]. Similarly, nivolumab combined with chemotherapy as a first-line treatment for PD-L1-positive (CPS ≥ 5) patients improved both OS (14.4 vs 11.1 months; HR 0.71; 98.4% CI, 0.59–0.86) and PFS (7.7 vs 6.05 months; HR 0.68; 98% CI, 0.56–0.81), establishing this regimen as a recommended first-line treatment option. Studies have confirmed that ICI therapy has become the standard first-line treatment for microsatellite instability-high (MSI-H) colorectal cancer. In MSI-H colorectal cancer patients treated with pembrolizumab versus chemotherapy alone, with a median follow-up of 32.4 months, immunotherapy demonstrated a superior PFS (16.5 vs 8.2 months)[9]. In esophageal cancer, pembrolizumab combined with chemotherapy as first-line treatment for esophageal squamous cell carcinoma (ESCC) demonstrated a clinically meaningful improvement in OS for patients with PD-L1 (CPS ≥ 10) of 13.9 vs 8.8 months. Nivolumab combined with chemotherapy or ipilimumab (dual immunotherapy) as a first-line treatment for ESCC resulted in a median OS of 13.7 months in PD-L1 ≥ 1% patients[10]. CAR-T-cell therapies targeting Claudin18.2, Human epidermal growth factor receptor 2, carcinoembryonic antigen, guanylyl cyclase C, and epidermal growth factor receptors have entered clinical trials, with the current data showing partial responses[11]. Clinical trials investigating personalized cancer vaccines to activate specific T-cell responses have also been initiated in surgically treated pancreatic cancer patients.
Despite the promising prospects of immunotherapy, its clinical translation still faces critical challenges, such as resistance to immunotherapy mediated by the tumor microenvironment (TME) and immune-related adverse events (irAEs). For instance, regulatory T-cells (Tregs) exert immunosuppressive effects by secreting Interleukin (IL)-10 and transforming growth factor (TGF)-β to inhibit the activity of effector T-cells and by expressing CTLA-4 to competitively deplete CD80/CD86 ligands, thus weakening the efficacy of ICIs[12]. Additionally, cancer-associated fibroblasts secrete collagen and hyaluronic acid to form physical barriers, which hinder T-cells' infiltration and contribute to the 'immune desert-like' TME[13]. Tumor cells, by consuming large amounts of glucose and tryptophan, lead to T-cell dysfunction as a result of energy depletion and the accumulation of tryptophan metabolites[14]. Moreover, sustained antigen stimulation results in epigenetic reprogramming of T-cells, forming an irreversible exhaustion phenotype that depends on thymocyte selection-associated HMG BOX protein (TOX)[15]. These reactions are essentially aberrant immune responses to self-antigens. The occurrence of irAEs is often associated with patients’ pre-existing autoimmune diseases and the dosage of ICIs. Conducting comprehensive pretreatment screening can help reduce their incidence, and thus, this topic will not be explored further.
The efficacy of immunotherapy is closely linked to the TME, and the occurrence of cell death is also inseparable from the TME. Recent studies have revealed that autophagy, by regulating iron/copper metabolism, oxidative stress, and other processes in the TME, plays a significant role in the progression of newly discovered ferroptosis and cuprotosis[16,17]. Therefore, this review summarizes the mechanisms of these three types of cell death and how they regulate immune responses in the TME, aiming to provide a theoretical foundation for combination therapies as a GI tumor treatment.
-
Autophagy is an evolutionarily conserved intracellular degradation process in which double-membrane vesicles called autophagosomes form with the help of autophagy-related (ATG) proteins. These vesicles engulf cellular components and deliver them to the lysosomes to be broken down by hydrolytic enzymes. The process involves several key stages: initiation, nucleation, elongation, closure, fusion, and degradation (Fig. 1).
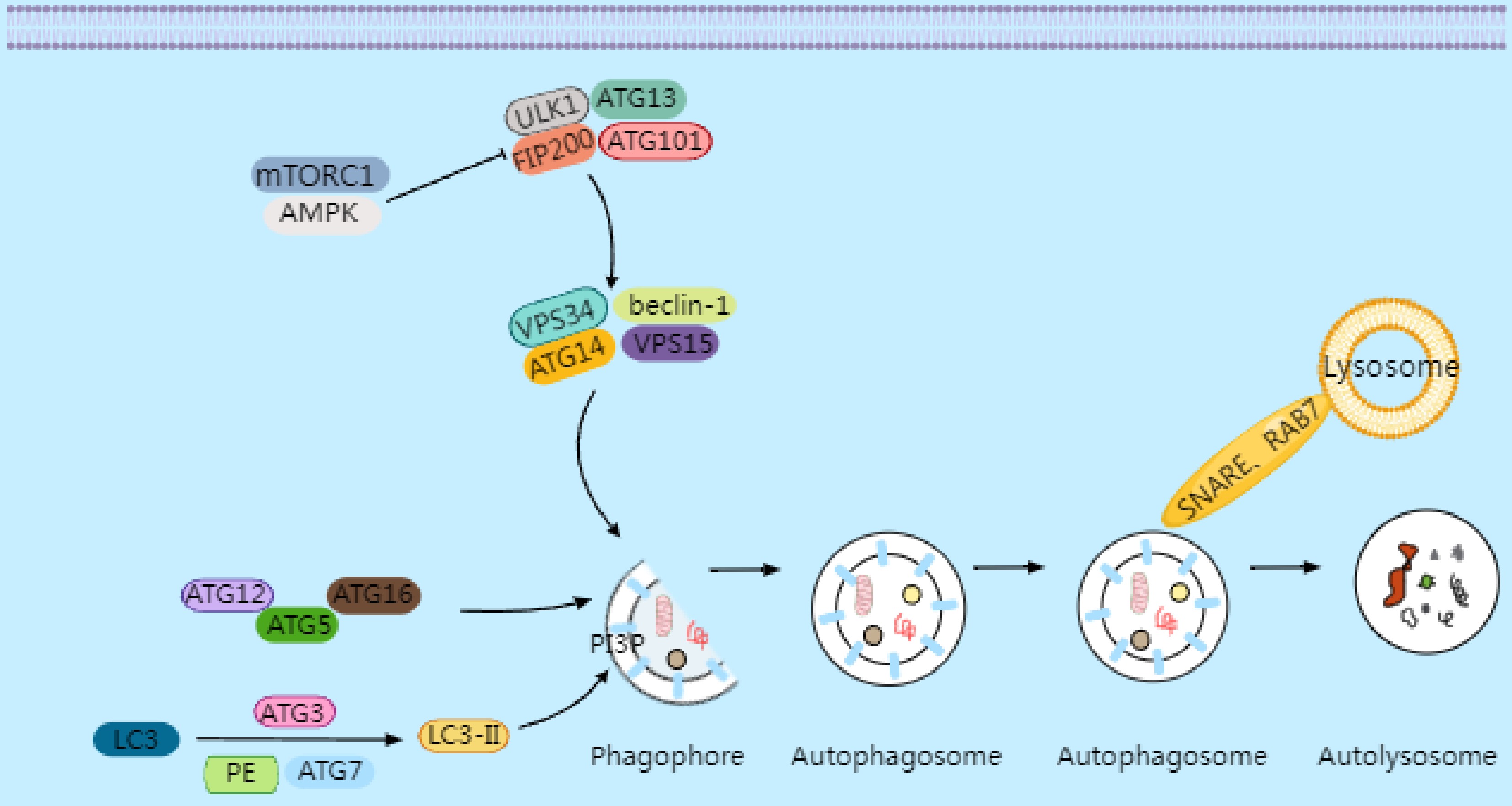
Figure 1.
Under conditions of cellular stress, nutrient deprivation, or inactivation of mammalian target of rapamycin complex 1 (mTORC1), the inhibition of the ULK1 complex is relieved, initiating the formation of the phagophore. Downstream from the ULK complex is the autophagy-specific VPS34 complex I, which catalyzes the production of phosphatidylinositol 3-phosphate (PI3P) on autophagic membranes, thereby promoting the expansion of autophagosome membranes. PI3P triggers the recruitment of autophagy's conjugation machinery, including the ATG16–ATG5-ATG12 complex, ATG3, and ATG7. These proteins facilitate the conjugation of microtubule-associated protein 1A/1B light chain 3 (LC3) to phosphatidylethanolamine (PE), converting it into LC3-II (LC3's lipidated form). LC3-II binds to newly formed autophagosome membranes and remains associated until the autophagosome fuses with lysosomes. The fusion process between autophagosomes and lysosomes requires Rab GTPase family members and soluble NSF attachment protein receptor (SNARE)proteins. Rab7 collaborates with SNARE proteins to drive membrane fusion between autophagosomes and lysosomes, forming autolysosomes. Following the degradation of the contents by hydrolases, small-molecule nutrients are released and recycled back to the cytoplasm.
Under nutrient deprivation or stress, activation of adenosine monophosphate-activated protein kinase (AMPK) or inhibition of mammalian target of rapamycin complex 1 (mTORC1) triggers the Unc-51 like kinase 1 (ULK1) complex (ULK1, ATG13, focal adhesion kinase interacting protein of 200 kDa [FIP200], and ATG101), initiating the formation of phagophores[18]. The VPS34 complex (vacuolar protein sorting 34 [VPS34], Beclin-1, ATG14, and VPS15) then produces phosphatidylinositol 3-phosphate (PI3P), promoting membrane expansion. PI3P recruits conjugation systems such as ATG16L1–ATG5–ATG12, ATG3, and ATG7, which lipidate light chain 3 (LC3) to form LC3-II[19]. LC3-II embeds in the autophagosome membrane and guides fusion with lysosomes via Rab GTPases (e.g., Rab7) and soluble NSF attachment protein receptor (SNARE) proteins, forming autolysosomes where the cargo is degraded and nutrients are recycled[20].
Autophagy is a highly regulated intracellular degradation process, with its core signaling network integrating various environmental stimuli and intracellular signaling pathways to ensure cellular homeostasis under stress conditions. The key signaling networks regulating autophagy can be broadly categorized as the mTORC1 pathway, the AMPK pathway, and stress signaling pathways.
mTORC1 is the major negative regulator of autophagy, suppressing its initiation by integrating signals from growth factors, amino acids, and energy levels[21]. Under nutrient-rich conditions, amino acids activate mTORC1 via Rag GTPases, whereas growth factors like insulin act through the phosphatidylinositol 3-kinase (PI3K)–A k strain transforming (AKT)–tuberous sclerosis complex 1 and 2 (TSC1/2) pathway. Active mTORC1 phosphorylates ULK1 and ATG13, inhibiting the assembly of the ULK1 complex and suppressing autophagy[22,23]. Under nutrient deprivation, mTORC1 inactivation leads to ULK1 dephosphorylation, triggering the formation of phagophores and activating autophagy[24].
AMPK, a cellular energy sensor, activates autophagy under low ATP conditions[25]. Energy stress increases the adenosine monophosphate (AMP)/adenosine triphosphate (ATP) ratio, activating AMPK. AMPK promotes autophagy directly by phosphorylating ULK1 and indirectly by inhibiting mTORC1[26]. Additionally, AMPK activates the tuberous sclerosis complex 2 (TSC2) to suppress mTORC1 and phosphorylates Beclin-1 to enhance interaction with VPS34, promoting theproduction of PI3P and autophagosome nucleation[27].
Under stress, reactive oxygen species (ROS) accumulation activates AMPK, inhibits mTORC1, and induces autophagy[26,28]. ROS oxidizes Kelch-like ECH-associated protein 1 (Keap1), releasing nuclear factor erythroid 2-related factor 2 (Nrf2) to translocate into the nucleus and upregulating autophagy-related genes[29]. Furthermore, ROS activates the mitochondrial proteins BCL-2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3) and Nip3-like protein X (NIX), which recruit autophagy-related proteins and promote mitophagy[30].
Regulation of tumor immunity by autophagy
-
Autophagy plays a complex and multifaceted role in the tumor immune microenvironment (TIME) by regulating the metabolism and signaling of tumor cells, immune cells, and stromal cells.
Autophagy releases glucose and fatty acids through glycophagy and lipophagy, maintaining cellular energy homeostasis. Under nutrient-deprived conditions, the AMPK/mTORC1 signaling axis activates autophagy, promoting the recycling of metabolic substrates to sustain ATP levels while depriving T-cells of their metabolic substrates[31]. Tumor cells, through their high expression of glutamine transporters and metabolic enzymes, convert glutamine into α-ketoglutarate, which enters the tricarboxylic acid (TCA) cycle. Under conditions of glutamine deficiency, autophagy is activated to recycle intracellular components and regulate metabolic adaptation via the AMPK/mTOR pathway. T-cell activation relies on glutamine metabolism to produce nucleotides and energy, but glutamine depletion in the tumor microenvironment directly suppresses T-cells' proliferation and effector functions. Additionally, in dendritic cells (DCs), glutamine regulates the activity of the transcription factor EB (TFEB) via the folliculin (FLCN) protein. When tumor cells deplete glutamine through the FLCN–TFEB signaling axis, FLCN becomes inactivated, leading to the impaired antigen-presenting capacity of DCs and their inability to effectively activate CD8+ T-cells[32].
The stability of PD-L1 protein is regulated by its intracellular transport and degradation pathways. Studies have shown that ubiquitination of PD-L1 is a key signal for its degradation via autophagy. Certain members of the tripartite motif (TRIM) family, such as TRIM21 and TRIM25, function as E3 ubiquitin ligases, directly catalyzing PD-L1's K48- or K63-linked ubiquitination. This modification enhances PD-L1's interaction with Sequestosome 1 (SQSTM1/p62), which bridges ubiquitinated PD-L1 with autophagosomes through its UBA (ubiquitin-binding) domain and LIR (LC3-interacting region) motif. The PD-L1–SQSTM1 complex is integrated into the autophagosomal membrane by LC3-II and is subsequently degraded following fusion with the lysosomes[33,34]. The degradation of PD-L1 directly alleviates its inhibitory signals on the PD-1 receptor on T-cells, restoring T-cell activation[35]. This process also reduces the recruitment of Tregs and myeloid-derived suppressor cells (MDSCs), as well as the secretion of immunosuppressive factors like IL-10 and TGF-β.
The TME is often characterized by hypoxia, nutrient deprivation, and oxidative stress. Autophagy supports the metabolic adaptability of tumor cells by degrading damaged organelles and redundant proteins, recycling metabolic substrates, and providing energy and biosynthetic precursors. ICIs activate T-cells to kill tumor cells; However, this process is accompanied by a high consumption of glucose and amino acids within the TME by the T-cells. Through its mechanism, autophagy assists tumor cells in surviving metabolic competition. Autophagy inhibitors, such as hydroxychloroquine, block lysosomal acidification and inhibit autophagy, leading to the accumulation of metabolic waste in tumor cells and disruption of the energy supply. This weakens the ability of tumor cells to adapt to the metabolic stress induced by PD-1/PD-L1 inhibitors, thereby reversing resistance to treatment[36].
Autophagy enhances the immunogenicity of tumor cells by promoting the release of ATP, calreticulin externalization, and high mobility group box 1 secretion, activating DCs and CD8+ T-cells, and inducing ICD in tumor cells[37]. Chemotherapy- or radiotherapy-induced autophagy-dependent ICD can enhance antitumor immune responses[38]. Autophagy also influences antigens' presentation: by degrading intracellular proteins to generate antigenic peptides, autophagy facilitates their presentation to CD8+ T-cells[39] Deficient autophagy reduces antigen presentation efficiency[40], thereby impairing T-cell-mediated tumor-killing capabilities[41].
The regulatory effects of autophagy on immunosuppressive cells are primarily evident in Tregs and tumor-associated macrophages (TAMs). Autophagy maintains the metabolic adaptability and functional stability of Tregs, promoting their immunosuppressive activities. Autophagy inhibitors can impair Tregs' functions, thereby enhancing antitumor immunity[42]. Autophagy also promotes the polarization of M2-like TAMs, which suppress T-cells' functions by secreting IL-10 and vascular endothelial growth factor (VEGF). Autophagy-dependent mitochondrial quality control is essential for maintaining the immunosuppressive phenotype of TAMs[43].
In summary, the role of autophagy in tumor-related immunity is dynamic and paradoxical. On one hand, autophagy maintains genomic stability, suppresses inflammation, and activates immune surveillance. On the other hand, autophagy supports metabolic adaptation, promotes metastasis, and mediates therapy resistance. For clinical treatment, it is necessary to develop individualized strategies based on the tumor's type, stage, and microenvironment characteristics to balance the double-edged sword of autophagy.
An autophagy-targeting combined immunotherapy strategy for GI tumors
-
During tumor initiation, autophagy primarily acts as a tumor suppressor. By eliminating dysfunctional mitochondria, autophagy reduces ROS-induced DNA damage and genomic instability. It also suppresses early tumorigenesis by activating antitumor immunity through immunogenic cell death. In the progression phase, autophagy exhibits dual roles, both promoting and suppressing cancer. It can expose antigens to activate immune responses, but also maintains tumor cells' self-renewal capacity, enhances chemotherapeutic drug efflux (leading to treatment resistance), and influences antigen presentation to modulate tumors' immunity. During the advanced stages of cancer, autophagy predominantly functions as a tumor promoter. It facilitates the formation of an immunosuppressive microenvironment and promotes tumor cells' adaptation to metabolic stress and immune escape.
Autophagy inhibitors suppress lysosomal acidification, reducing the secretion of exosomal PD-L1 while increasing tumor cells' sensitivity to PD-1/PD-L1 inhibitors. Combined with PD-1/PD-L1 blockade, they decrease exosomal PD-L1 secretion, enhance T-cell recognition, and inhibit tumorigenesis. Several clinical trials are currently evaluating the efficacy of combining autophagy inhibitors with ICIs in advanced gastroesophageal junction cancer and Epstein–Barr virus-positive metastatic gastric cancer (Table 1). Preliminary results show prolonged PFS in combination therapy groups compared with monotherapy groups[44].
Table 1. Summary of clinical research on targeting cell death pathways in cancer immunotherapy.
Cell death
typeCombination strategy Mechanism of action Indication/target Clinical stage/
evidence levelKey findings Autophagy Autophagy inhibitors chloroquine/hydroxychloroquine (CQ/HCQ) + ICIs Blocks lysosomal acidification, reduces exosomal PD-L1 secretion, and enhances tumor sensitivity to PD-1/PD-L1 inhibitors Advanced gastroesophageal junction cancer, Epstein–Barr virus (EBV) gastric cancer Phase II trials (NCT04132505, NCT03755440) Combination significantly prolonged median progPFS vs monotherapy group. Rab27a/nSMase2 inhibitors + anti-vascular endothelial growth factor (VEGF) Inhibits exosomal PD-L1 packaging and release, blocks pre-metastatic niche formation Colorectal cancer metastasis models Preclinical studies Significantly reduced liver metastases when combined with bevacizumab. MAPK/ERK kinase (MEK) inhibitor (trametinib) + autophagy inhibitor Blocks autophagy-dependent metabolic reprogramming Colorectal cancer liver metastasis Preclinical studies Synergistically inhibited liver metastasis progression. Ferroptosis Ferroptosis inducers (erastin/RSL3) + PD-1 inhibitor CD8+ T-cell secreted IFN-γ inhibits SLC7A11, enhances ferroptosis sensitivity, creates a positive immune feedback loop Gastric cancer mouse model Preclinical studies Increased tumor-infiltrating T-cells (three- to five fold) and enhanced IFN-γ secretion. VPS34 inhibitor (SAR405) + Atezolizumab Blocks autophagosome formation to induce ferroptosis, synergistically enhances anti-PD-L1 efficacy Advanced colorectal cancer, pancreatic cancer Preclinical studies Significantly suppressed tumor growth; clinical trials needed to confirm mechanism. Ferroptosis nanoparticles + STING agonist Co-delivers ferroptosis inducer and ADU-S100, activates DCs and inhibits PD-L1 Colon cancer mouse model Preclinical studies Reversed immunosuppressive microenvironment, achieved significant tumor suppression. Ferroptosis induction + photothermal therapy (PTT) + anti-PD-1 PTT directly kills cells and releases tumor-associated antigens (TAAs), ferroptosis enhances immunogenic cell death (ICD), reverses T-cell exhaustion Gastrointestinal tumors (theoretical model) Preclinical studies Triple synergy provides a 'heat-sensitization + immune activation' strategy. Copper ionophore (Elesclomol) + immune checkpoint inhibitors (ICIs) Copper accumulation triggers lipoylated protein aggregation, inducing ICD; concurrently downregulates PD-L1 transcriptional activity Advanced gastric adenocarcinoma/gastroesophageal junction cancer Phase III Trial (GEMSTONE-303) Sugemalimab (anti-PD-L1) combined with antibody-dependent phagocytosis significantly improved survival. Cuproptosis Cuproptosis inducer + anti-V-domain Ig suppressor of T cell activation (VISTA) antibody Blocks VISTA-mediated immunosuppression by myeloid-derived suppressor cells (MDSCs), synergizes with cuproptosis to activate immune response Adenomatous polyposis coli (APC)-mutant colorectal cancer mouse model Preclinical studies Significantly suppressed tumor progression. Beyond autophagy inhibitors, inhibiting Rab family proteins (e.g., Rab27a) or neutral sphingomyelinase 2 (nSMase2) specifically blocks the exosomal packaging of PD-L1. Early-stage tumor cells systemically suppress T-cells' activation in the lymph nodes via exosomal PD-L1. Inhibiting Rab27a or nSMase2 reduces exosomal PD-L1 release, enhancing CD8+ T-cells' antitumor activity. Exosomal PD-L1 establishes premetastatic niches by inducing immunosuppressive macrophage polarization in the target organs of metastasis (e.g., the liver and lung). Inhibiting nSMase2 reduces exosome-dependent M2 macrophage activation, delaying metastasis. In advanced tumors, exosomal PD-L1 binds PD-1 on peripheral T-cells, causing systemic T-cell exhaustion. Inhibition of Rab27a or nSMase2 can restore T-cells' function and enhance the response to chemotherapy or immunotherapy. In colorectal cancer models, Rab27a inhibitors combined with anti-VEGF agents simultaneously suppress angiogenesis and immune evasion, significantly reducing metastatic lesions[45]. Beyond targeting autophagy-related pathways, combination strategies with multitarget kinase inhibitors have also shown promise. Studies have revealed that mitogen-activated protein kinase (MAPK) inhibitors (e.g., trametinib) combined with autophagy inhibitors markedly inhibit liver metastasis in colorectal cancer by disrupting autophagy-dependent metabolic reprogramming[46].
The autophagy-targeting combined immunotherapy strategy for GI tumors provides a novel direction to overcome immunotherapy resistance by modulating tumor metabolism-immune crosstalk. Future research should further investigate the dynamic role of autophagy in immune editing and advance the clinical translation of precision treatment regimens through interdisciplinary technologies.
-
Ferroptosis is a form of iron-dependent programmed cell death triggered by excessive accumulation of lipid peroxidation. It is driven by dysregulated iron metabolism, the buildup of lipid peroxides, and the failure of the antioxidant defense system (Fig. 2).
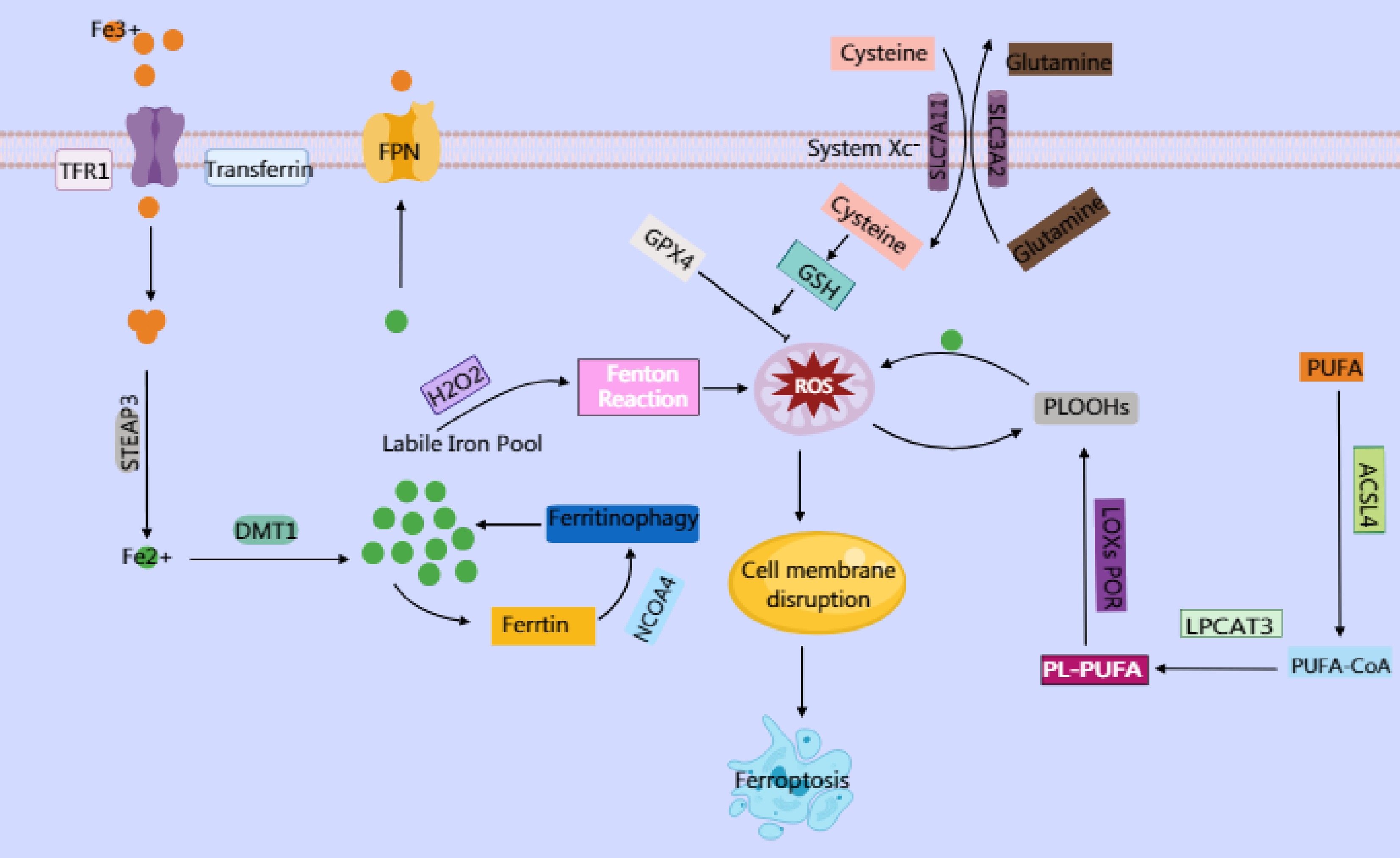
Figure 2.
Ferroptosis is an iron-dependent cell death characterized by lipid peroxidation. Elevated intracellular iron imported via transferrin and transferrin receptor (TFR1) is reduced by six-transmembrane epithelial antigen of prostate 3 (STEAP3) to its ferrous form (Fe2+), which is then transported by divalent metal transporter 1 (DMT1) into the labile iron pool. This labile iron contributes to the generation of reactive oxygen species (ROS), particularly hydroxyl radicals (·OH), through the Fenton reaction, where Fe2+ catalyzes the conversion of hydrogen peroxide (H2O2) into highly reactive ROS. In turn, ROS trigger oxidative damage, including the peroxidation of polyunsaturated phospholipids (PUFA-PLs), which serve as critical substrates for ferroptosis. PUFA-PLs are formed through the incorporation of polyunsaturated fatty acids (PUFAs) into membrane phospholipids by Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), followed by their oxidation with the involvement of lipoxygenases (LOXs) and cytochrome P450 oxidoreductase (POR). Under normal physiological conditions, the body utilizes GPX4 to reduce toxic lipid hydroperoxides (PLOOHs) into nontoxic lipid alcohols (PLOHs) by consuming GSH, thereby maintaining redox homeostasis. GPX4 mediates cystine uptake through the cystine–glutamate antiporter (System Xc−), a heterodimeric transmembrane transporter composed of SLC7A11 and SLC3A2. Intracellular cystine is reduced to cysteine, whereas Glu is exported. Cysteine serves as a critical precursor for synthesizing GSH, which is an essential cofactor for GPX4. However, when GPX4 function is impaired or GSH synthesis is disrupted, lipid peroxides accumulate.
Molecular mechanisms of ferroptosis
-
Intracellular iron homeostasis is critical for ferroptosis. Iron enters the cells via transferrin receptor 1 (TFR1) and is reduced to Fe2+ by six-transmembrane epithelial antigen of prostate 3 (STEAP3). Fe2+ is transported into the cytoplasm via DMT1 or exported by ferroportin (FPN). This balance can be modulated by iron chelators (e.g., deferoxamine [DFO]) or ferroptosis inhibitors, thereby influencing ferroptosis[47].
Disruption of iron homeostasis prompts nuclear receptor coactivator 4 (NCOA4)-mediated recognition and autophagic degradation of ferritin (ferritinophagy), releasing free iron and expanding the labile iron pool. Excess Fe2+ catalyzes hydroxyl radical formation via the Fenton reaction, generating ROS[48].
Elevated ROS induces oxidative damage, particularly peroxidation of polyunsaturated fatty acid-containing phospholipids (PUFA-PLs), whose synthesis is facilitated by Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3). This peroxidation is further accelerated by lipoxygenases (LOXs) and cytochrome P450 oxidoreductase (POR)[49,50].
Ordinarily, GPX4 maintains redox homeostasis by reducing lipid hydroperoxides (PLOOHs) to nontoxic alcohols (PLOHs), using glutathione (GSH) as a cofactor. GSH synthesis depends on cystine uptake via System Xc− (composed of SLC7A11 and SLC3A2). Inhibition of System Xc− (e.g., by erastin) or direct GPX4 blockade (e.g., by RSL3) leads to lethal lipid peroxide accumulation and ferroptosis[51,52].
Notably, certain tumor cells evade ferroptosis despite low GPX4 expression via ferroptosis suppressor protein 1 (FSP1) or dihydroorotate dehydrogenase (DHODH), which reduce ubiquinone (CoQ10) to ubiquinol (CoQH2). CoQH2 quenches lipid peroxyl radicals, halting peroxidation propagation[53].
The accumulation of lipid peroxides reduces membrane fluidity, increases permeability, and leads to the formation of hydrophilic pores, causing ion imbalance and leakage of cellular contents. Oxidative cleavage products further damage proteins and nucleic acids, ultimately leading to cell death.
The System Xc-–GPX4–GSH axis constitutes a central ferroptosis defense mechanism. Here, p53 transcriptionally represses SLC7A11, impairing antioxidant capacity and promoting ferroptosis. It also modulates ferroptosis via regulation of H2B monoubiquitination (H2Bub1) and USP7-mediated nuclear translocation[54,55]. Conversely, Nrf2 enhances ferroptosis resistance by activating GPX4 and SLC7A11[56]. The Hippo pathway effectors YAP and TAZ upregulate ACSL4 and TFR1, promoting lipid peroxidation and iron accumulation to sensitize cells to ferroptosis[57].
Regulation of tumor immunity by ferroptosis
-
Ferroptosis, as a form of iron-dependent, lipid peroxidation-driven cell death, has recently become a hot topic in tumor immunotherapy research. Its impact on immunotherapy exhibits complex bidirectional regulatory characteristics.
On one hand, ferroptosis can enhance tumors' immunogenicity to promote immune responses. It does so by releasing DAMPs, which activate the antigen-presenting function of DCs and promote T-cell infiltration. For instance, CD8+ T-cells can secrete IFN-γ to suppress the expression of SLC7A11 in tumor cells, thereby increasing tumor sensitivity to ferroptosis and forming a positive immune feedback loop[58]. Additionally, ferroptosis leads to the release of tumor cell contents, increasing the antigen load on DCs and activating CD8+ T-cells. Ferroptosis can also promote the polarization of TAMs toward the pro-inflammatory M1 phenotype and eliminate immunosuppressive populations such as MDSCs, thereby relieving their suppression of CD8+ T-cells and natural killer cells and enhancing the efficacy of immunotherapy.
On the other hand, ferroptosis can suppress anti-tumor immunity. Ferroptotic cells may release oxidized phospholipids (e.g., PE-OOH), which inhibit DCs' maturation and antigen-presenting functions, leading to immune evasion. Additionally, cancer-associated fibroblasts can secrete glutamine to enhance the antioxidant capacity of tumor cells, weakening the immune-activating effects of ferroptosis. In the TME, the fatty acid transporter CD36 is highly expressed and promotes the uptake of PUFAs by CD8+ T-cells, resulting in the peroxidation and accumulation of lipids and ultimately triggering ferroptosis. Although the CD36-mediated regulation of T-cell ferroptosis has been observed in multiple mouse models, including lung cancer, colorectal cancer, and melanoma, most studies are based on single mouse strains. In humans, T-cells exhibit significant heterogeneity. For example, tissue-resident T-cells can survive for a considerably long time in the TME of solid tumors and maintain antitumor activity. However, the current evidence only shows high CD36 expression in tumor-infiltrating CD8+ T-cells[59], whereas the expression level of CD36 and ferroptosis sensitivity in tissue-resident T-cells lack support from human data. Exhausted T-cells, which exhibit functional exhaustion because of persistent antigen stimulation, may exacerbate ferroptosis through CD36-dependent pathways through metabolic reprogramming, such as excessive fatty acid uptake[60].
Combination therapy strategy of ferroptosis and immunotherapy for GI tumors
-
Ferroptosis induced by erastin and RSL3 promotes the proliferation of CD8+ T-cells. Preclinical studies showed that in mouse models of gastric cancer, the combination of ferroptosis inducers with PD-1 inhibitors increased tumor-infiltrating T-cells by three- to fivefold and markedly enhanced effector functions, such as IFN-γ secretion[61]. Currently, both ferroptosis inducers and immunotherapy have demonstrated antitumor efficacy in esophageal cancer. Although the efficacy of combination therapy has not been reported, the combination of ferroptosis and immunotherapy is expected to become a novel therapeutic approach for esophageal cancer[62]. In some gastric cancers, cells exhibit elevated intracellular oxidative stress by mediating GSH efflux, thereby increasing susceptibility to ferroptosis. Dietary restriction of amino acids combined with GPX4 inhibitors significantly suppresses tumors' growth. Additionally, ferroptosis upregulates PD-L1 expression via the STAT3 signaling pathway[63], suggesting that ferroptosis may enhance immunotherapy responses by remodeling the TME. Preclinical studies in advanced colorectal and pancreatic cancer models revealed that SAR405 (a VPS34 inhibitor) combined with atezolizumab blocks autophagosome formation, induces ferroptosis, and synergizes with PD-L1 inhibitors to amplify immune responses[64]. However, clinical validation of these mechanisms is still required.
Nanoparticles achieve drug enrichment at tumor sites through surface modifications. In colorectal cancer, ferroptosis-related gene-based nanoparticles can specifically deliver iron ions or small-molecule inhibitors to tumor tissues, directly acting on cancer cells and indirectly enhancing the efficacy of immunotherapy by modulating immune cells' activity and the TME's status[65].
By co-delivering erastin/RSL3 with stimulator of interferon genes (STING) agonists, nanoparticles induce ferroptosis while activating the STING pathway in DCs. This dual action promotes DCs' maturation and antigen presentation, suppresses PD-L1 expression, disrupts immune checkpoint-mediated tolerance, and reverses the immunosuppressive microenvironment. In subcutaneous mouse models, this strategy achieved significant tumor suppression[66].
Photothermal therapy (PTT) directly kills tumor cells via localized hyperthermia while releasing tumor-associated antigens, forming dual immune-activating signals with ferroptosis-induced ICD[67]. Combining PTT with anti-PD-1/PD-L1 antibodies reverses the exhaustion of T-cells induced by ferroptosis and hyperthermia, enhancing CD8+ T-cells' infiltration. The integration of these modalities enables multidimensional regulation for precision oncology[68].
GI tumors often develop resistance to conventional therapies through inactivation of apoptosis pathways or epithelial–mesenchymal transition. Ferroptosis selectively kills cancer cells with a mesenchymal phenotype[69], whereas PTT directly disrupts drug-resistant cells through thermal ablation. Beyond thermal damage, PTT also triggers ICD to activate antitumor immunity[70]. Thus, the synergy between ferroptosis and PTT offers a strategy for GI malignancies.
Currently, research on ferroptosis in GI tumor immunotherapy remains limited. This review summarizes preclinical findings and proposes actionable strategies to advance the clinical translation of modulating ferroptosis in gastric cancer immunotherapy.
-
Cuproptosis is a newly proposed form of cell death characterized by the abnormal accumulation of intracellular copper ions (Cu2+), which triggers cell death through specific molecular mechanisms. Unlike autophagy and ferroptosis, cuproptosis relies on the direct cytotoxic effects of copper ions and their interference with critical metabolic pathways, particularly in cells that are dependent on mitochondrial respiration (Fig. 3).
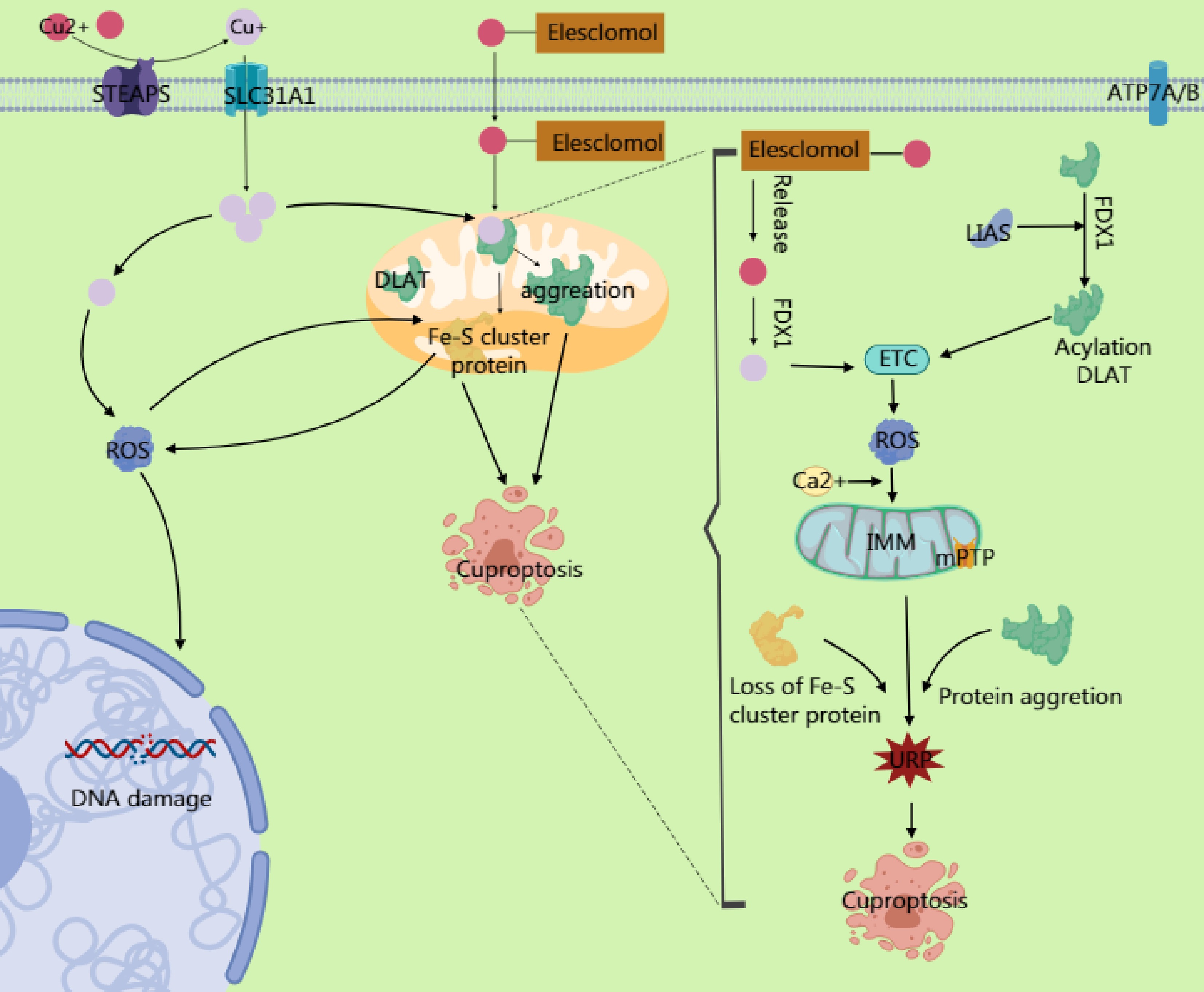
Figure 3.
Copper ions are reduced to Cu+ by six-transmembrane epithelial antigen of prostate 3 (STEAP) family proteins, and their dynamic balance is maintained through the transport proteins SLC31A1 and ATP7A/B. Excess Cu2+ can be converted to Cu+ via ferredoxin 1 (FDX1). The generated Cu+ induces reactive oxygen species (ROS) production through the Fenton reaction, leading to DNA damage and other effects. Simultaneously, it inhibits Fe-S cluster protein synthesis. Excess Cu2+ enters the mitochondrial matrix through transporters on the mitochondrial membrane, targeting enzymes modified with lipoic acid. Cu2+ binds to the lipoylation site of the TCA cycle enzyme dihydrolipoamide S-acetyltransferase (DLAT), causing the dsyfunction in the pyruvate dehydrogenase (PDH) complex and impairing electron transport chain (ETC) activity. This ETC impairment increases electron leakage and massive ROS generation. The ROS further oxidizes cysteine residues in Fe–S cluster proteins, forming a positive feedback loop that exacerbates Fe–S cluster degradation. The accumulated ROS from ETC dysfunction, combined with Ca2+ overload in the mitochondrial matrix (caused by copper-induced disruption of mitochondrial Ca2+ homeostasis), directly activates the components of the mitochondrial permeability transition pore (mPTP), prompting its opening. The mPTP, a high-conductance channel in the mitochondria, compromising the integrity of the inner mitochondrial membrane (IMM) and releasing proapoptotic factors. Additionally, the accumulation of mitochondrial ROS and degradation of the Fe–S cluster may activate endoplasmic reticulum stress through calcium signaling or ROS diffusion, triggering the unfolded protein response (UPR). This upregulates proapoptotic factors, ultimately driving the cell toward death.
Molecular mechanisms of cuproptosis
-
Cuproptosis is driven by the dysregulation of copper homeostasis, mitochondrial metabolic collapse, and proteotoxic stress. Key mechanisms include the following.
Intracellular Cu2+ homeostasis is maintained by reduction via STEAP proteins, uptake through SLC31A1, and efflux via ATP7A/B[7]. Pathological or carrier-induced (e.g., elesclomol) copper overload leads to mitochondrial Cu2+ accumulation, targeting lipoylated tricarboxylic acid (TCA) cycle proteins, causing aggregation and cell death. ATP7B mutation impairs copper export, leading to copper accumulation and neurodegenerative disorders such as Wilson’s disease[71].
Elevated mitochondrial Cu2+ is reduced to Cu+ by ferredoxin 1 (FDX1). Cu+ promotes ROS generation via the Fenton reaction and inhibits Fe–S cluster synthesis. Cu2+ binds lipoylated dihydrolipoamide S-acetyltransferase (DLAT), inducing oligomerization, disrupting the production of acetyl-CoA, lowering NADH/FADH2, and impairing the function of the electron transport chain (ETC)[72,73].
ETC dysfunction increases electron leakage and ROS, oxidizing Fe–S cluster proteins and forming a damaging feedback loop. ROS- and Cu2+-induced Ca2+ overload activate mitochondrial permeability transition pores (mPTPs), reducing the membrane potential (ΔΨm) and triggering the release of cytochrome C. ROS also induce endoplasmic reticulum (ER) stress and the unfolded protein response, upregulating proapoptotic factors[7,74].
As mentioned above, the occurrence of cuproptosis is driven by three key axes: dysregulation of copper homeostasis, mitochondrial metabolic collapse, and proteotoxic stress. Targeting these mechanisms by modulating copper homeostasis, mitochondrial metabolic rescue, and alleviating proteotoxicity may offer therapeutic strategies for regulating cuproptosis.
Nrf2 activates antioxidant genes (e.g., for GSH synthesis) to quench Cu+ toxicity and may stabilize Fe–S clusters to inhibit FDX1. It also cooperates with hypoxia-inducible factor-1α (HIF-1α) under hypoxia to enhance glycolysis, reducing mitochondrial dependence and cuproptosis sensitivity[75]. Here, p53 affects FDX1 via Fe–S cluster biogenesis; loss of p53 impairs Fe–S synthesis and reduces copper toxicity[76]. The p53-induced circFRMD4A inhibits PKM2, forcing reliance on TCA and increasing cuproptosis sensitivity. Moreover, p53 upregulates lipoylation genes, enhancing the binding of copper to TCA enzymes.
Trimethylation of lysine 27 on histone H3 (H3K27me3) silences copper transporters (SLC31A1, ATP7A/B) and TCA-related genes, disrupting copper homeostasis and reducing lipoylated protein production. Its downregulation alleviates copper toxicity. H3K27me3 also suppresses GSH synthesis, enhancing copper ions' toxicity[77]. miR-137 targets SLC31A1 to reduce copper uptake and affects GSH synthesis, collectively increasing copper's toxicity[78].
Glycolysis and glutamine metabolism supply reduced nicotinamide adenine dinucleotide phosphate (NADPH) to maintain reduced GSH, neutralizing copper-induced ROS. α-Ketoglutarate (α-KG) supports the ETC function via TCA-derived NADH/FADH2, reducing ROS. α-KG also stabilizes HIF-1α to modulate copper efflux. GSH binds Cu2+ for export via multidrug resistance-associated proteins[79,80].
Cu2+ catalyzes the Fenton reaction, producing hydroxyl radicals. FAO-derived ROS worsen oxidative damage, directly oxidizing lipoylated proteins and Fe–S clusters, disrupting the ETC and energy metabolism. Impaired Fe–S clusters also reduce FDX1 activity. In addition, p53 deficiency disrupts Fe–S biogenesis, dampening FDX1 function and cuproptosis. Enhanced Food and Agriculture Organization of the United Nations (FAO) increases mitochondrial copper accumulation and acetyl-CoA production; the α-KG thus derived epigenetically dysregulates copper transporters, accelerating cuproptosis[81,82].
Regulation of tumor immunity by cuproptosis
-
On one hand, cuproptosis remodels the immune microenvironment by inducing ICD. The opening of the mPTP and rupture of the plasma membrane triggered by cuproptosis release DAMPs, tumor-associated antigens, and HSP70. These molecules activate DCs via Toll-like receptors and the STING pathway, promoting DCs' maturation and enhancing antigen-presenting capacity.
On the other hand, cuproptosis downregulates the expression of indoleamine 2,3-dioxygenase 1 (IDO1), reducing the conversion of tryptophan to kynurenine (Kyn). This suppresses Tregs' recruitment and restores cytotoxic T lymphocytes' activity. Tryptophan metabolism via IDO1 generates Kyn, which activates the aryl hydrocarbon receptor to inhibit CD8+ T-cells' proliferation and promotes Tregs' infiltration of the TME, thereby dampening antitumor immunity. DAMPs released during cuproptosis activate DCs and cytotoxic T-lymphocytes, which secrete cytokines like IFN-γ to further suppress IDO1's expression.
Cuproptosis relies on mitochondrial copper accumulation, leading to the lipoylation of TCA cycle-related enzymes and disruption of iron–sulfur cluster proteins. This metabolic dysregulation not only directly induces cell death but also activates immunogenic signaling through glutathione depletion and ROS burst.
By integrating ICD induction, immune checkpoint modulation, and metabolic reprogramming, cuproptosis offers novel perspectives for reshaping the immune microenvironment. However, its mechanistic complexity—including its cell-type dependency and dose-dependent effects—requires further interdisciplinary investigation to fully elucidate these pathways.
Synergistic cuproptosis–immunotherapy strategies for GI tumors
-
Copper ions regulate PD-L1's stability by modulating its ubiquitination-mediated degradation[83]. A high-copper microenvironment promotes the expression of PD-L1, whereas copper chelators or ionophores (e.g., elesclomol) enhance the efficacy of PD-1 inhibitors by restoring copper homeostasis. In the Phase III GEMSTONE-303 trial for advanced or metastatic gastric adenocarcinoma or gastroesophageal junction adenocarcinoma, sugemalimab not only blocks the PD-1–PD-L1 interaction but also enhances immunotherapy's efficacy by mediating antibody-dependent cellular phagocytosis of PD-L1-positive tumor cells and TAMs without compromising effector T-cells[84].
In colorectal cancer, truncating mutations in the APC gene upregulate the immune checkpoint VISTA via the METTL3–HIF-1α signaling axis, promoting MDSCs' infiltration and establishing an immunosuppressive microenvironment. In murine colon adenocarcinoma models, combining cuproptosis inducers with VISTA monoclonal antibodies simultaneously activates cuproptosis pathways and blocks VISTA-mediated immune evasion, significantly suppressing tumor progression[85].
Copper-based nanoplatforms not only induce cuproptosis but also increase membrane-associated PD-L1 expression. Consequently, when copper-based nanoplatforms are co-administered with anti-PD-L1 antibodies, they significantly inhibit lung metastases (including metastatic colorectal cancer) by coupling ICD induction with PD-1/PD-L1 blockade, achieving enhanced antitumor immunity[86].
Radiotherapy upregulates the expression of PD-L1 via DNA damage and IFN-γ secretion, exacerbating T-cell exhaustion. Cuproptosis nanomaterials counteract this by reducing the expression of PD-L1 in tumor cells while increasing tumor-infiltrating DCs, thereby reversing radiotherapy-induced immune escape[87,88]. Biomimetic nanodrugs dissociate through glutathione-responsive processes and exhibit comprehensive antitumor capabilities, including copper-induced cell death and TIME reversal. Furthermore, cuproptosis nanodrugs can co-deliver navoximod to directly inhibit IDO1 activity, lower Kyn levels, and reduce Treg recruitment, mitigating immune evasion. Thus, copper-induced cuproptosis combined with TIME modulation represents a promising therapeutic strategy.
Beyond nanomaterials, the MetaCell system (a neutrophil-encapsulated Fe–Cu metal–organic framework) releases Cu2+ via photothermal effects, concurrently activating cuproptosis and ferroptosis. This system has demonstrated significant tumor suppression and prevented recurrence in multiple solid tumor models[89]. However, its efficacy in GI tumors requires further validation.
-
Autophagy not only facilitates the degradation of ferritin via the receptor NCOA4, releasing free Fe2+ to promote the Fenton reaction (generating ROS and accelerating lipid peroxidation), but also degrades lipid droplets to release PUFAs, providing substrates for lipid peroxidation. Furthermore, it selectively degrades GPX4, further weakening cellular antioxidant capacity and thereby promoting the ferroptosis process (Fig. 4).
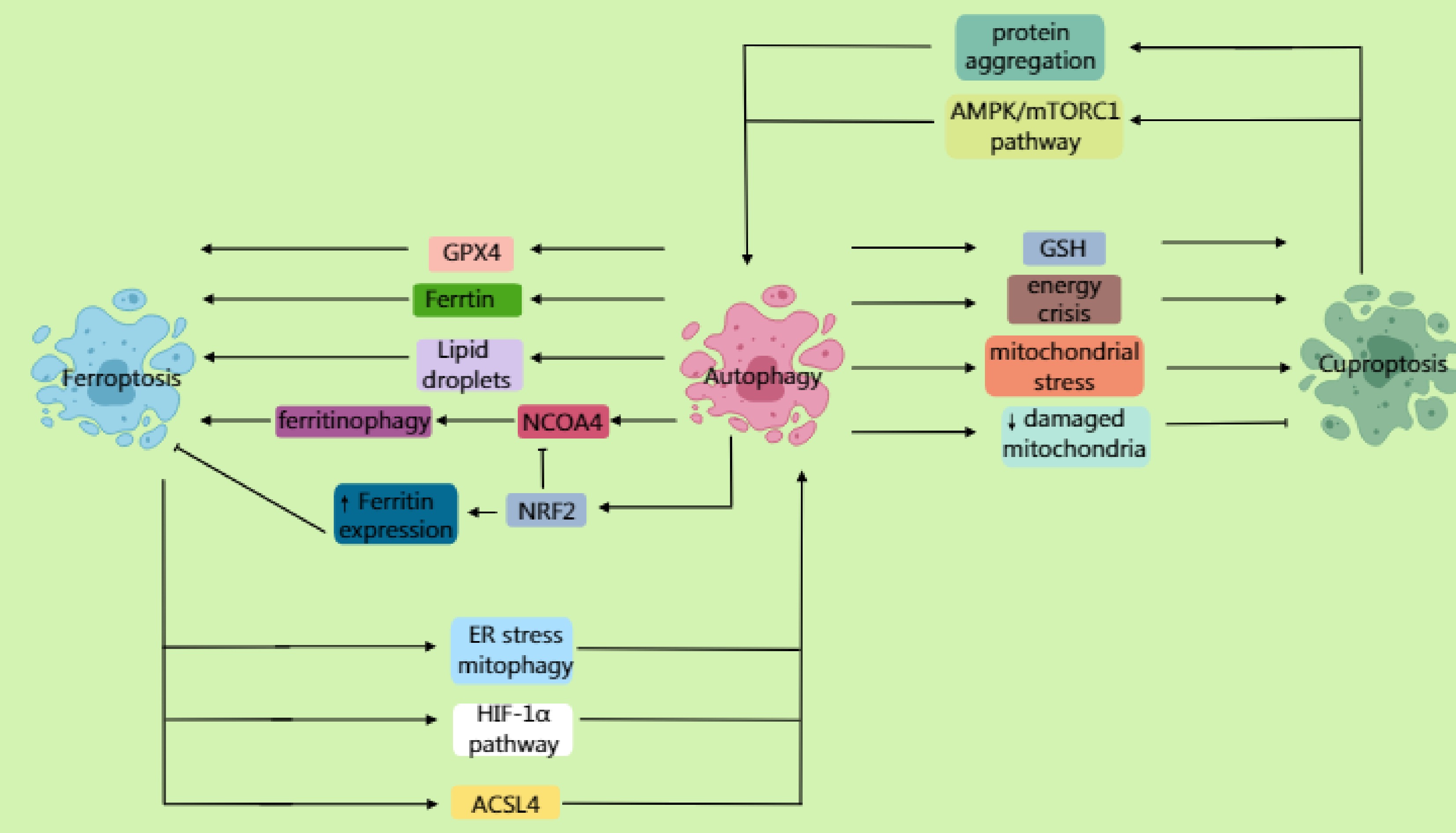
Figure 4.
Autophagy not only mediates the lysosomal degradation of ferritin through the receptor NCOA4, promoting the Fenton reaction to generate reactive oxygen species (ROS) and accelerate lipid peroxidation, but also degrades lipid droplets, releasing polyunsaturated fatty acids (PUFAs). Furthermore, by selectively degrading GPX4, it promotes the ferroptosis process. NRF2 antagonizes ferroptosis by upregulating the expression of ferritin to promote iron storage while inhibiting NCOA4-mediated ferritinophagy. Inhibition of the mTOR pathway activates autophagy, enhancing susceptibility to ferroptosis via the ferritinophagy or GPX4 degradation pathways. Ferroptosis can further induce autophagy; ferroptosis-dependent lipid ROS damage the endoplasmic reticulum (ER) and mitochondria, triggering ER stress and mitophagy. It also activates pathways such as HIF-1α, upregulating autophagy genes. ACSL4-mediated membrane phospholipid peroxidation products can directly activate autophagosome formation. In cuproptosis, autophagy may degrade important intracellular copper chelators (such as glutathione and metallothioneins), leading to elevated levels of free copper ions. Moreover, under specific conditions, excessive autophagy may exacerbate a cellular energy crisis and mitochondrial stress, sensitizing cells to cuproptosis. Additionally, autophagy (specifically mitophagy) clears the damaged mitochondria caused by copper overload, reducing cellular sensitivity to cuproptosis. Beyond promoting cell death, autophagy may delay cuproptosis by clearing damaged mitochondria or abnormally aggregated lipoylated proteins. Cuproptosis can similarly induce autophagy. Copper ions bind to mitochondrial tricarboxylic acid (TCA) cycle enzymes, inducing abnormal protein aggregation and activating mitophagy. Copper overload increases mitochondrial ROS, inducing autophagy through AMPK/mTORC1 imbalance.
Beyond the interplay at the molecular mechanism level, signaling pathways also exhibit cross-regulation. NRF2 counteracts ferroptosis by upregulating the expression of ferritin to enhance iron storage while suppressing NCOA4-mediated ferritinophagy. Conversely, iron overload competitively binds divalent iron to NCOA4, accelerating its degradation and inhibiting ferritinophagy. When mTOR signaling is inhibited, it activates autophagy, enhancing ferroptosis sensitivity through either the ferritinophagy pathway or GPX4 degradation.
Furthermore, ferroptosis can further induce autophagy. Ferroptosis-dependent lipid ROS damage the ER and mitochondria, triggering ER stress and mitophagy. It also activates pathways such as HIF-1α, upregulating autophagy genes. The key molecule p62/SQSTM1 accumulates during ferroptosis, competitively binding Keap1 to release Nrf2 and promote antioxidant genea' expression. ACSL4-mediated membrane phospholipid peroxidation products can directly activate autophagosome formation. Autophagy induced by ferroptosis is also dynamic and paradoxical: it activates DCs and enhances T-cell responses, but also inhibits PD-L1 degradation and the release of DAMPs. Metabolically, autophagy recycles amino acids/iron ions from ferroptotic cell debris, supporting glutaminolysis in the neighboring tumor cells. Additionally, autophagy maintains the NADPH pool, balancing ferroptosis-associated oxidative stress. In terms of genomic stability, autophagy clears ROS-damaged mitochondria, reducing nuclear DNA oxidative damage. It can also provide dNTP precursors to support homologous recombination repair. However, excessive autophagy can deplete ribosomes, inducing the risk of replication stress.
In contrast to ferroptosis, during cuproptosis, autophagy may degrade important intracellular copper chelators, leading to elevated levels of free copper ions. Moreover, under specific conditions, excessive autophagy may exacerbate a cellular energy crisis and mitochondrial stress, sensitizing cells to cuproptosis. Additionally, autophagy (specifically mitophagy) clears damaged mitochondria caused by copper overload, potentially reducing cellular sensitivity to cuproptosis. Besides its pro-death role, autophagy might delay cuproptosis by clearing damaged mitochondria or aberrantly aggregated lipoylated proteins.
Cuproptosis can also induce autophagy. Copper ions bind to mitochondrial TCA cycle enzymes, inducing abnormal protein aggregation and activating mitophagy. Copper overload increases mitochondrial ROS, inducing autophagy via an imbalance in AMPK/mTORC1 signaling. Autophagy induced by cuproptosis is similarly dynamic and paradoxical: it not only induces the release of mitochondrial DNA, activating the relative pathway, but also inhibits PD-L1 degradation and the release of DAMPs. Metabolically, cuproptosis-triggered mitophagy forces cells to shift towards glycolysis. Regarding genomic stability, autophagy clears ROS-damaged mitochondria, reducing oxidative damage to nuclear DNA. It can also provide dNTP precursors to support homologous recombination repair. However, the mitochondrial DNA leakage induced by cuproptosis may potentially be integrated into the nuclear genome.
-
This review synthesizes the pivotal roles of RCD pathways, namely autophagy, ferroptosis, and cuproptosis, in reshaping the immunometabolic landscape of GI tumors and their synergistic potential with immunotherapy. Key mechanistic insights reveal that autophagy modulates immune evasion through PD-L1 degradation and metabolic substrate competition, ferroptosis enhances immunogenicity via lipid peroxidation-driven antigen release, and cuproptosis triggers mitochondrial proteotoxic stress to disrupt immunosuppressive checkpoints. The triangular crosstalk among these pathways further underscores their interconnected roles in driving tumor progression or therapy resistance, exemplified by autophagy-mediated ferritinophagy fueling ferroptosis and copper-induced oxidative stress amplifying cuproptosis. Emerging therapeutic strategies, such as autophagy inhibitors combined with immune checkpoint blockade, iron/copper-targeted nanomedicines integrated with PTT, and metabolic reprogramming interventions, demonstrate promising preclinical efficacy in overcoming drug resistance and reversing TIME immunosuppression. However, their clinical translation faces significant hurdles, including tumors' heterogeneity, the dose-limiting toxicity of metal ionophores, and limited human data on immune cell-specific RCD effects. Future research must prioritize multidisciplinary approaches, leveraging artificial intelligence-driven biomarker discovery to predict the activation of RCD pathways, advancing spatiotemporally controlled nanodelivery systems for precision targeting, and employing single-cell multi-omics to decipher the dynamic interplay between RCD and immune cell subsets. By bridging mechanistic discoveries with translational innovation, this work lays a foundation for developing next-generation immunotherapies that harness the dual power of cell death and immune activation, ultimately transforming the therapeutic paradigm for GI cancers.
The study was funded by the Natural Science Foundation of Jiangsu Province (BK20231252), Key Medical Research Projects of Jiangsu Provincial Health Commission (K2023026), the Science and Technology Planning Social Development Project of Zhenjiang City (SH2022047), and Medical Education Collaborative Innovation Fund from Jiangsu University (JDY2022003).
-
This article is a literature review and did not directly involve human participants or animal experiments. All content presented herein is based on previously published research literature. Throughout the writing process, we have endeavored to ensure the objectivity and accuracy of the cited studies. The authors declare no conflicts of interest.
-
The authors confirm contribution to the paper as follows: draft manuscript preparation, figure design: Han M, Jiang Y; manuscript revision for intellectual content and comment: Dong K; writing – review & editing: Li X, Wang D. All authors reviewed the results and approved the final version of the manuscript.
-
The data that support the findings of this study are available in the PubMed database.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Miao Han, Kebin Dong, Yuchun Jiang, Xiaoqin Li
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Han M, Dong K, Jiang Y, Li X, Wang D. 2026. Synergistic cell death pathways in gastrointestinal tumor immunotherapy: molecular mechanisms, metabolic reprogramming, and clinical translation. Gastrointestinal Tumors 13: e001 doi: 10.48130/git-0025-0020 

Synergistic cell death pathways in gastrointestinal tumor immunotherapy: molecular mechanisms, metabolic reprogramming, and clinical translation
- Received: 27 April 2025
- Revised: 14 November 2025
- Accepted: 03 December 2025
- Published online: 09 January 2026
Abstract: This review elucidates the intricate interplay between regulated cell death pathways—autophagy, ferroptosis, and cuproptosis—and their synergistic potential for immunotherapy in gastrointestinal tumors. By systematically dissecting the molecular mechanisms of these pathways, the authors highlight their dynamic regulation of the tumor immune microenvironment, including antigen presentation, metabolic competition, and immune evasion. Key insights include autophagy-mediated programmed death ligand-1 degradation, ferroptosis-driven immunogenic feedback via lipid peroxidation, and cuproptosis-induced mitochondrial collapse through copper-dependent proteotoxic stress. The review critically evaluates emerging combination strategies, such as autophagy inhibitors with immune checkpoint inhibitors, iron/copper-targeted nanomedicines coupled with photothermal therapy, and metabolic reprogramming interventions. These approaches demonstrate enhanced efficacy in overcoming drug resistance and reversing the immunosuppressive features of the tumor immune microenvironment. Despite promising preclinical advances, challenges such as tumor heterogeneity, spatiotemporal regulation of cell death–immune crosstalk, and clinical translation barriers are underscored. The authors advocate for multidisciplinary integration of nanotechnology, artificial intelligence, and multi-omics to optimize precision therapies. This work provides a robust framework for leveraging regulated cell death pathways to refine immunotherapy paradigms in gastrointestinal tumors, from mechanistic discoveries to actionable clinical strategies.
-
Key words:
- Death /
- Pathways /
- Impacts /
- Immunotherapy