










-
Chimeric Antigen Receptor T (CAR-T) cell immunotherapy has yielded groundbreaking progress in the management of hematologic malignancies; nonetheless, its clinical pertinence and efficacy in the management of solid tumors remain severely circumscribed by inherent biological barriers and tumor microenvironment constraints. In spite of these successes, the translational application of CAR-T cell immunotherapy to the realm of solid neoplasms has proven substantially suboptimal[1−3]. Multiple barriers contribute to this limitation, including inadequate trafficking of infused T cells into tumour tissues, an immunosuppressive tumour microenvironment, and insufficient persistence of CAR-T cells following infusion[1,4,5]. Mitigating these recalcitrant challenges is imperative for extending the therapeutic dividends of CAR-T cell immunotherapy to patients afflicted with solid neoplasms.
Claudin18.2 has emerged as a propitious therapeutic target for gastrointestinal cancers, particularly gastric cancer (GC) and pancreatic adenocarcinoma (PAAD), owing to its selective overexpression in malignant tissues and minimal expression in normal healthy epithelia, rendering it a salient candidate for targeted immunotherapeutic interventions. Its appeal stems from a tumor-restricted expression profile, with high prevalence in primary and metastatic lesions but minimal presence in normal tissues, thereby mitigating the risk of on-target, off-tumor toxicity—a significant hurdle in CAR-T therapy[6]. The clinical validation of Claudin18.2 targeting was demonstrated by zolbetuximab, a monoclonal antibody that met its primary endpoint in a Phase III trial for advanced gastric cancer (GC) and gastroesophageal junction cancer (GEJ). This success has catalyzed the development of Claudin18.2-directed CAR-T cells[7]. Given its favorable expression pattern, Claudin18.2 also presents a favorable candidate for CAR-T therapy development, especially as it may reduce the potential for the 'on-target off-tumor' effect that is a common concern with CAR-T treatments. As a result, several Claudin18.2-targeting CAR-T products are under preclinical or clinical investigation[6]. Among these, the second-generation product CT041 has yielded an encouraging 48.6% overall response rate in clinical trials[8,9]. Nonetheless, the durability of responses remains a challenge, as indicated by a median progression-free survival of only 4.2 months in GC patients. This limitation underscores the critical need for nascent approaches to potentiate the persistence and efficacy of Claudin18.2-targeting CAR-T therapies.
A promising approach to improving CAR-T cell treatments in solid tumours is the use of armoring strategies involving cytokines, chemokines, and/or their receptors[10]. Several cytokines, including interleukin-2 (IL-2), interleukin-15 (IL-15), and interleukin-7 (IL-7), have been shown to improve the expansion and persistence of CAR-T cells in vivo[11−14]. A clinical study based on this armored CAR-T technology is currently underway. Among these, interleukin-9 (IL-9) has recently gained attention as a potential therapeutic cytokine for enhancing T-cell function. IL-9 is a T-helper 9 (Th9)-derived cytokine that plays an important role in T-cell differentiation, survival, and proliferation[15]. Additionally, IL-9 has been shown to influence the tumor microenvironment, enhancing immune cell infiltration, and potentially overcoming immune suppression in solid tumors[15−17].
In the present investigation, we engineered murine CAR-T cells that selectively target Claudin18.2, endowed with constitutive IL-9 expression. Subsequently, we appraised their anti-tumor potency via in vitro and in vivo assays, and strived to disentangle the underpinning molecular mechanisms by leveraging immunocompetent murine models.
-
The murine pancreatic cancer cell line Panc02 and hepatocellular carcinoma cell line Hepa1-6 were kept in our lab. Mouse melanoma cell lines B16F10 and 293T were purchased from the Cancer Institute of Xuzhou Medical University (Xuzhou, China). All the tumor cell lines are from C57BL/6 mice. To achieve Claudin18.2 positive target cells, we infected Murine Claudin18.2 lentiviral vectors into Panc02, Hepa1-6, and B16F10 cells. Randomly, all cells were cultured in DMEM (GIBCO), and 10 % FBS (GIBCO) was added as standard.
CAR-T cell production
-
A second-generation CAR was created with Claudin18.2-specific scfv (clone 69H9) coupled to murine cd8-hinged domain, transmembrane domain, and intracellular signaling region of 4-1BB and CD3ζ. IL-9 expressing CAR-T cells were made by joining the murine IL-9 (mIL-9) coding sequence to the CAR construct via a cleavage F2A peptide. Retroviral particles were packaged from 293T with pCL-Eco and CAR vector using 293T. Viral supernatant was obtained at 48, and 72 h post-transfection, and PEG8000 concentration was performed, then stored at −80 °C before use.
Spleens from female C57BL/6 mice (CD45.1 congenic) were isolated after CO2 euthanasia and cervical dislocation. Splenocytes were filtered into single-cell suspensions, purified using RoboSep™ kit, and resuspended at 1 × 108 cells/ml. T cells were activated in 6-well plates pre-coated with 1 µg/ml anti-mouse CD3 (Thermofisher), then cultured in TCM supplemented with 4 µg/ml anti-mouse CD28 (eBioscience) at 3 × 106 cells/ml in 5% CO2 at 37 °C for 24 h. Activated T cells were then transduced with CAR or IL-9 CAR retrovirus using RetroNectin-coated plates (Takara). The untransduced T cells were used as a control. All T cells were cultured in RPMI-1640 medium supplemented with 10% FBS, 100 U/mL penicillin, and recombinant human IL-2 (50 IU/mL).
Cytokine production assay
-
Cytokine production assessment: Murine CAR-T cells were cocultured with tumor cells at an E : T ratio of 1:1 for 24 h. Supernatants of the culture were made, and assay on the secretion of IL-9, IFN-γ, IL-2, and granzyme B was carried out with ELISA kits (Thermo Fisher) as per the manufacturer's guidelines.
In vitro cytotoxicity assays
-
The cytolytic activity of CAR-T cells was evaluated using a luciferase-based assay. Target tumor cells (Panc02, B16F10, and Hepa1-6) were cocultured with CAR-T cells at various E : T ratios (3:1, 1:1, and 1:3) for 24 h in triplicate wells. Following coculture, specific tumor cell lysis was determined using the Bright-Glo™ Luciferase Assay System (Promega), and cytotoxicity was calculated according to standard protocols.
Flow cytometry
-
Briefly, T cells were stained under the standard protocol and flow cytometry techniques. Fluorophore-conjugated anti-mouse monoclonal antibodies including CD45.1 (APC/Cyanine7, clone A20), CD45.2 (PE, clone 104), CD3 (FITC/PE, clone 17A2), CD4 (BUV395, clone RM4-5), CD8α (BUV805, clone 53-6.7), CD44 (Brilliant Violet 650™, clone IM7), and CD62L (Brilliant Violet 510™, clone MLE-14) were purchased from BioLegend (San Diego, CA, USA), or BD Biosciences (Milpitas, CA, USA). All data were acquired by BD FACSCantoll, then analyzed using FlowJo software.
In vivo therapeutic potency of CAR-T cells
-
All the animal experiments were performed at Xuzhou Medical University's animal facility. All methods strictly followed the guidelines and regulations of experimental animals. No animals died before meeting the criteria for euthanasia. Panc02-18.2 (4 × 106), B16F10-18.2 (5 × 105), and Hepa1-6-18.2 (1 × 107) suspension cells were subcutaneously inoculated into the right flank of CD45.2+ C57BL/6 recipient mice at the age of 8 weeks. One day before the CAR-T cells injection, cyclophosphamide (150 mg/kg) was administered intraperitoneally (i.p.). On Day 0, a dose of 5 × 106 CAR-T cells, made from splenic T cells of CD45.1+ congenic C57BL/6 mice, were injected intravenously.
IHC analysis
-
Tumor specimens were harvested from euthanized mice, fixed in 10% neutral buffered formalin, and embedded in paraffin. Sections (4 μm) were stained with antibodies against mouse CD3 (Cell Signaling Technology), and Claudin18.2 (Abcam). Images were captured using an Olympus microscope, and quantitative analysis of positive staining was performed using Image-Pro Plus 7.0 software.
Statistical analysis
-
Statistical analysis was carried out using GraphPad Prism 10 software. An unpaired two-tailed t-test was used for comparisons between two groups. The results were presented as the mean ± SEM. The one-way ANOVA or two-way ANOVA was performed to analyzed comparisons between two or more groups. Bonferroni's multiple-comparison was used for further correction. The p-values with statistical significance were defined as * p < 0.05; ** p< 0.01; *** p < 0.001; **** p < 0.0001.
-
We created a second-generation CAR construct harboring a Claudin18.2-specific single-chain variable fragment (scFv), followed by 4-1BB and CD3ζ intracellular signaling motifs (CAR). The mIL-9 sequence was directly fused with the CAR sequence through an F2A self-cleavage sequence so as to generate the IL-9 CAR (Fig. 1a). CD3+CD45.1+ T cells were isolated from the spleen of CD45.1 congenic C57BL/6 mice. Following activation with αCD3/CD28 antibodies, murine T cells were infected with the CAR or IL-9 CAR to produce CAR-Ts and IL-9 CAR-Ts, respectively. The untransduced T cells (UNT) served as the control group. The CAR-positive frequency was quantified via flow cytometry (Fig. 1b). The positive frequency of CAR-T surpassed 57%, while that of IL-9 CAR-T exceeded 52% (Fig. 1c, d). The fundamental phenotype (CD4/CD8) of IL-9 CAR-T cells showed no significant differences compared to conventional CAR-T cells and untransduced T cells (Fig. 1e, f).
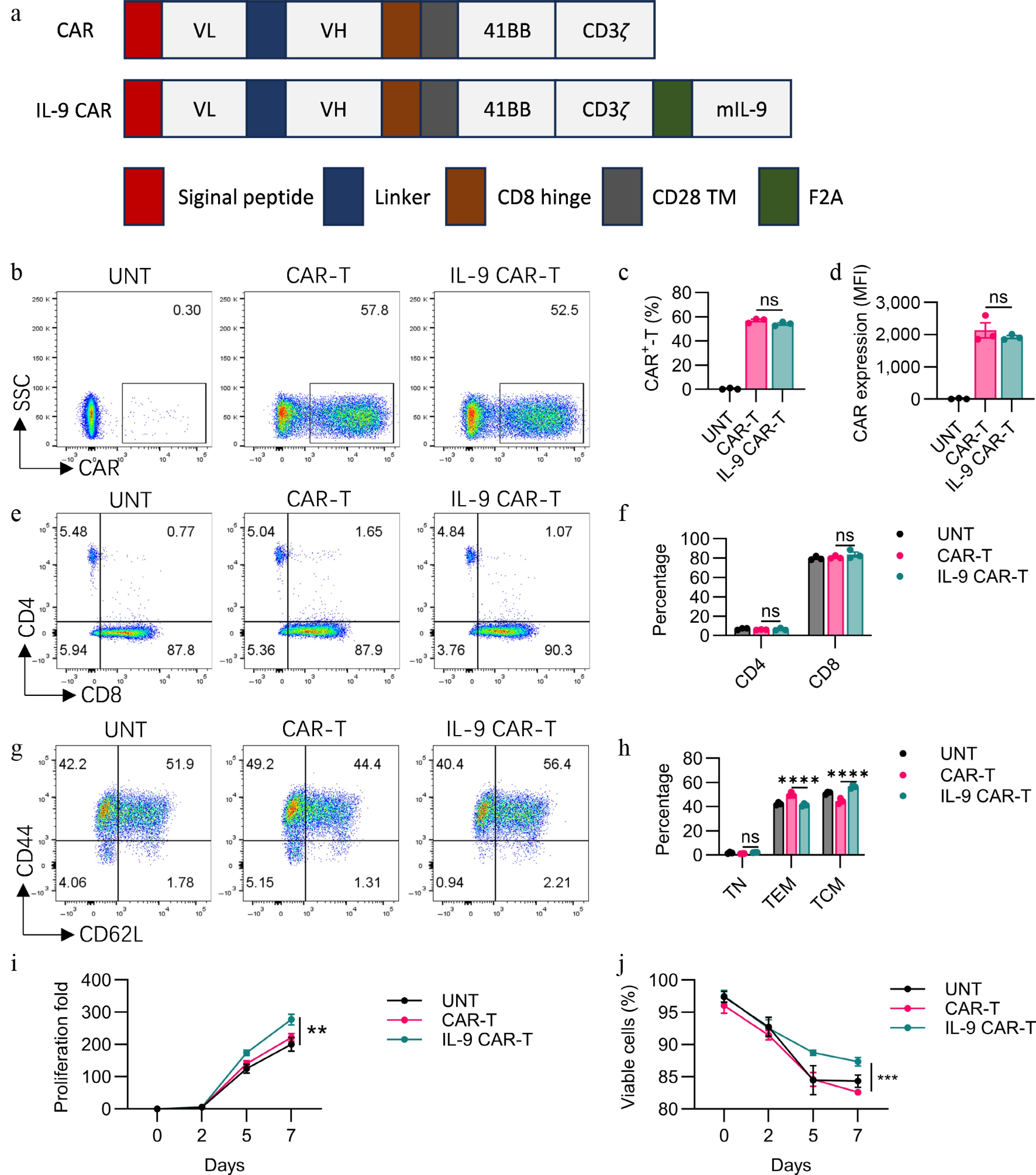
Figure 1.
Functional characterization of engineered IL-9-expressing anti-Claudin18.2 CAR-T cells. (a) Architecture of the conventional and cytokine-incorporated CAR retroviral constructs. CAR expression was validated by (b) flow cytometry, with the (c) transduction efficiency, and (d) expression level (MFI) quantified. The CD4/CD8 ratio within the CAR-T cell products was (e) analyzed, and (f) summarized. The immunophenotype defined by CD44 and CD62L expression is displayed in (g) representative plots, and (h) a cumulative bar graph. Proliferative capacity, (i) fold expansion, and (j) cell viability were tracked over time in culture. Results are representative of three independent experiments performed in triplicate (mean ± SEM; n = 3 biologically independent samples). Statistical notations: ns, p > 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
IL-9 CAR-T cells showed enhanced proliferation capacity in vitro
-
The growth of CAR-T cells was tracked. We noted that IL-9 CAR-T cells exhibited a markedly elevated proliferative potential relative to conventional CAR-T cells (Fig. 1g). Additionally, IL-9 CAR-T cells exhibited higher viability relative to conventional CAR-T cells and UNT cells (Fig. 1h).
In vitro cytokine production and cytotoxicity of CAR-T cells improved by expressing IL-9
-
Claudin18.2 expressions on different types of cells were determined with the help of flow cytometry as depicted in Supplementary Fig. S1a. The tumoricidal potential of CAR-T cells towards Panc02, B16F10, and Hepa1-6 cells expressing claudin18.2 (Panc02-18.2, B16F10-18.2, and Hepa1-6-18.2), or wildtype (Panc02, B16F10, and Hepa1-6) were also assessed in vitro. Compared to UN-T cells, CAR-T and IL-9 CAR-T displayed strong cytotoxicity against Panc02-18.2, B16F10-18.2, and Hepa1-6-18.2 (Fig. 2d–f), but not against Panc02, B16F10, and Hepa1-6 (Fig. 2a, b). However, the enhanced killing activity of IL-9 CAR-T cells compared to conventional CAR-T cells in this short-term killing assay in vitro was not observed (Fig. 2d–f). Cytotoxicity tests showed that all of the CAR cells were able to lyse the Claudin18.2' target cells in the same way as each other in vitro.
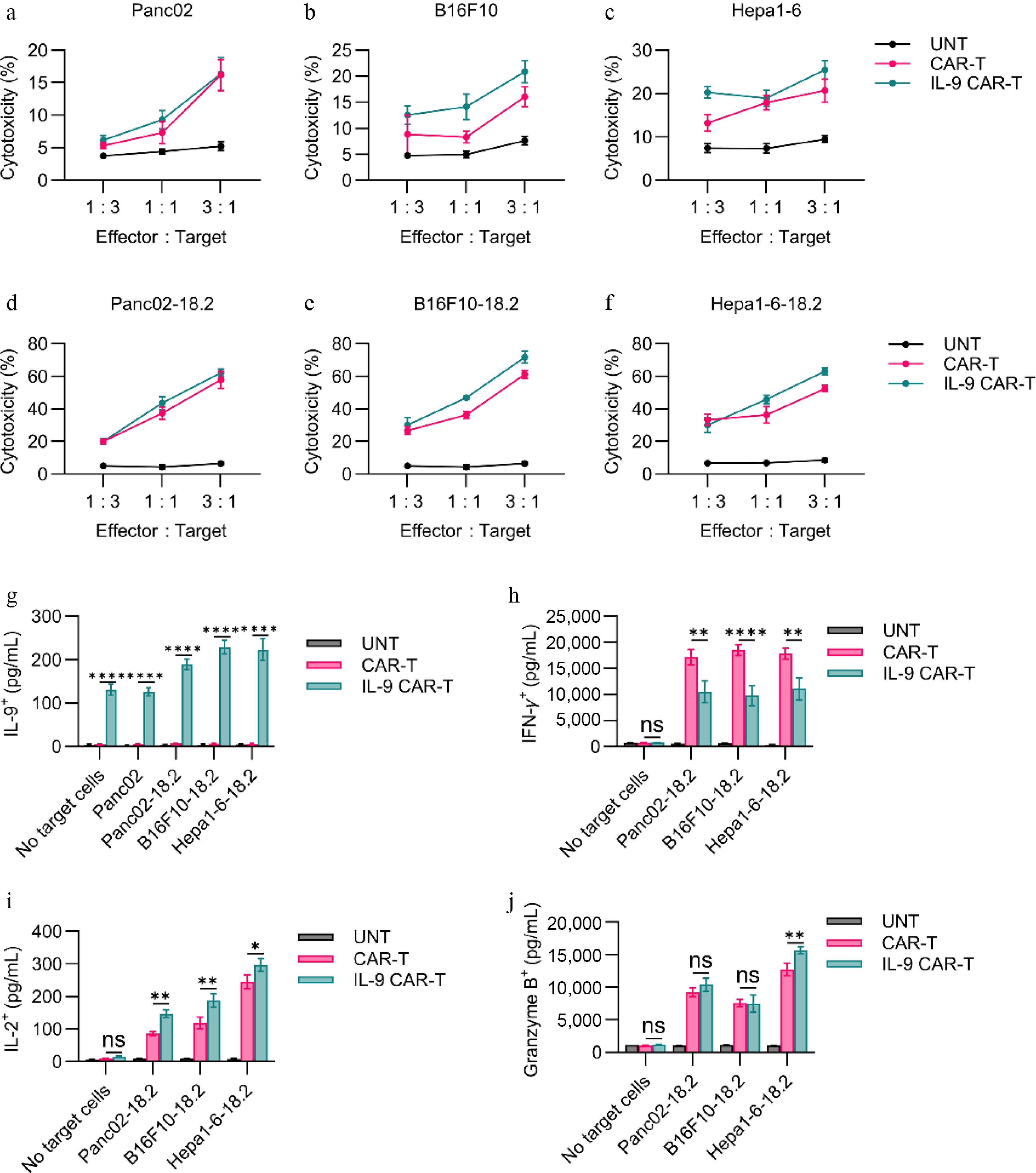
Figure 2.
In vitro functional assessment of CAR-T cells. (a)–(f) Cytotoxic activity of CAR-T cells against Claudin18.2-negative or Claudin18.2-overexpressing target cells was evaluated after 24 h of co-culture at various effector-to-target (E : T) ratios, using a standard non-radioactive cytotoxicity assay. (g)–(j) Production of IL-9, IFN-γ, Granzyme B, and IL-2 by CAR-T cells was measured by ELISA following 24-h co-culture with target cells at a 1:1 E : T ratio. Data are representative of at least three independent experiments performed in triplicate (n = 3 biologically independent samples), and presented as mean ± SEM. Statistical significance: ns, not significant (p > 0.05); * p < 0.05; ** p < 0.01; **** p < 0.0001.
The cytokine elaborative capacity of CAR-T cells in vitro was checked by ELISA. To determine cytokine release, CAR-T cells were cocultured with antigen-expressing tumor cells (Panc02-18.2, B16F10-18.2, and Hepa1-6-18.2). For IL-9 secretion, it was found that exposure to tumor cells greatly improved the production of IL-9 by IL-9 CAR-T cells compared to CAR-Ts or UNT cells with or without antigen-expressing (Fig. 2g). For IFN-γ secretion, after coculturing with Claudin18.2-positive tumor cells, conventional CAR-T cells secreted more IFN-γ than IL-9 CAR-T cells (Fig. 2h). For IL-2 secretion, IL-9 CAR-T cells secreted more IL-2 than CAR-T cells after coculturing with Panc02-18.2, B16F10-18.2, or Hepa1-6-18.2 tumor cells (Fig. 2i). For granzyme B, IL-9 CAR-T cells secreted more granzyme B than CAR-T cells after coculturing with Hepa1-6-18.2 tumor cells, but no significant difference was found when coculturing with Panc02-18.2 or B16F10-18.2 tumor cells (Fig. 2j).
IL-9-engineered CAR-T cells exhibited augmented antitumor capacity in diverse solid tumor models
-
To verify the different antitumor effects of each CAR-T cell in vivo, we established an in vivo Panc02-18.2 tumor model in C57BL/6 mice (Fig. 3a). From Fig. 3b and c, we can see that the IL-9 CAR-T cells inhibit the growth of the tumor more strongly, by 94.48% (Fig. 3d). On the contrary, the CAR-T cells have only a slight inhibitory effect on the tumour, with a growth inhibition effect of 57.38%. The antitumor effect experiments were then carried out for IL-9 CAR-T cells on B16F10-18.2 and Hepa1-6-18.2 mouse solid tumors. For the B16F10-18.2 model (Fig. 3e), there was a more significant antitumor effect of the IL-9 CAR-T cells, compared to normal CAR-T cells according to the tumor volume and weight (Fig. 3f, g). The tumor growth inhibition rate of IL-9 CAR-T cells was 72.5%, and the tumor growth inhibition rate of usual CAR-T cells was 47.93% (Fig. 3h). In addition to the Hepa1-6-18.2 tumor model in Fig. 3i, IL-9 CAR-T has a more efficient antitumor effect than common CAR-T cells (Fig. 3j, k), and the tumor inhibition rate of the IL-9 CAR-T cell group can reach 99.58%, which is much higher than the 62.79% of the control group of the general CAR-T (Fig. 3l). It can therefore be deduced from the above results that the IL-9 CAR-T cells still achieved the most significant efficacy advantage compared to the ordinary CAR-T cells, in all three different solid tumor models.
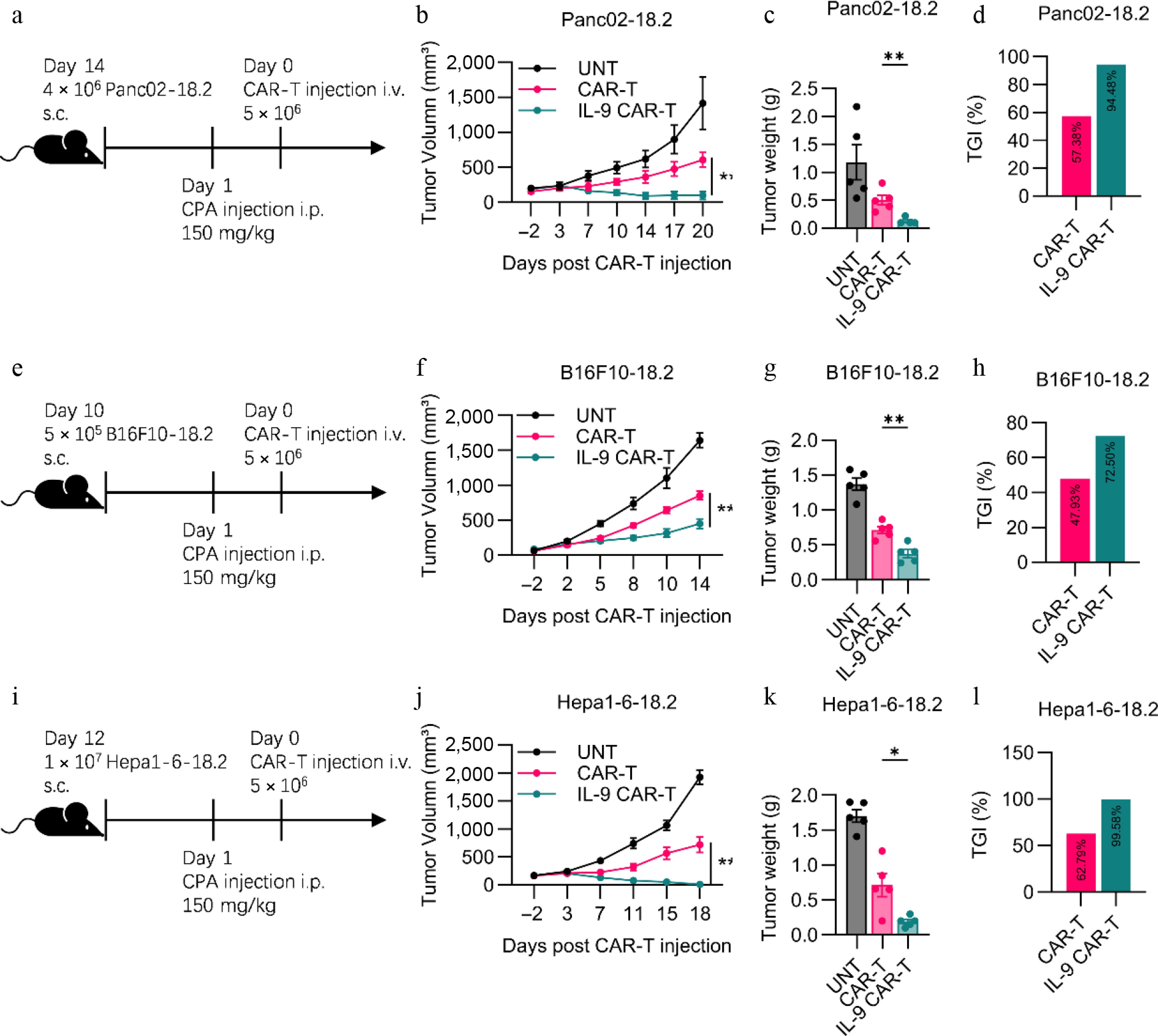
Figure 3.
Enhanced antitumor efficacy of IL-9-expressing CAR-T cells in immunocompetent solid tumor models. (a) Experimental timeline for the Panc02-hClaudin18.2 pancreatic cancer model. Endpoint (b) tumor volume, (c) tumor weight, and (d) tumor growth inhibition (TGI) for the pancreatic cancer model. (e) Experimental timeline for the B16F10-hClaudin18.2 melanoma model. Endpoint (f) tumor volume, (g) tumor weight, and (h) TGI for the melanoma model. (i) Experimental timeline for the Hepa1-6-hClaudin18.2 liver cancer model. Experimental scheme of Hepa1-6-18.2 mouse liver solid tumor model in vivo antitumor experiment. (j) Histogram of the volume of tumors at the endpoint of the animal experiment. (k) Histogram of tumor weight at the endpoint of the animal experiment. (l) Histogram showed tumor growth inhibition of CAR-T cell treatment. All animal experiments were repeated more than twice with similar results. The results are expressed as the mean ± SEM. Statistical significance was defined as follows: p > 0.05; * p < 0.05; ** p < 0.01; *** p < 0.001.
IL-9 CAR-T cells possessed enhanced expansion and T-cell infiltration in vivo
-
To further disentangle the underpinning mechanisms by which transgenic IL-9 expression potentiates the in vivo antitumor capacity of CAR-T cells, we appraised the in vivo expansion kinetics of these CAR-T cells. An identical experimental regimen was conducted as illustrated in Fig. 3a. Seven days subsequent to CAR-T cell administration, mice from each cohort were euthanized for the harvesting of blood, spleen, and solid tumor specimens. Relative to the conventional CAR-T cell cohort, IL-9-engineered CAR-T cells exhibited a superior in vivo proliferative capacity of adoptively transferred CAR-T cells, encompassing their total T-cell proportions, and absolute counts in peripheral blood and splenic tissues (Fig. 4a, b). Concurrently, we assessed the Ki67 expression levels within adoptively transferred CAR-T cells in the spleen. The IL-9 treatment markedly elevated the frequencies of Ki67+ CAR-T cells among the total T-cell pool, signifying that IL-9 supplementation strongly augmented the in vivo proliferative potential of adoptive CAR-T cells (Fig. 4c, d). Next, we analyzed tumor tissues by IHC staining with antigen expression, including the CD3 and claudin18.2 biomarkers for the illustration of intratumoral infiltration of T-cells, and the killing effect of tumor-specific antigens. Compared with conventional CAR-T cells, IL-9 CAR-T cells significantly enhanced T cell infiltration on Day 7 after CAR-T injection (Fig. 4e, f). Simultaneously, IL-9 CAR-T cells markedly eradicated the target antigen Claudin18.2 within tumor tissues, demonstrating that IL-9 expression facilitated the clearance of antigen-positive tumor cells by CAR-T cells (Fig. 4g, h).
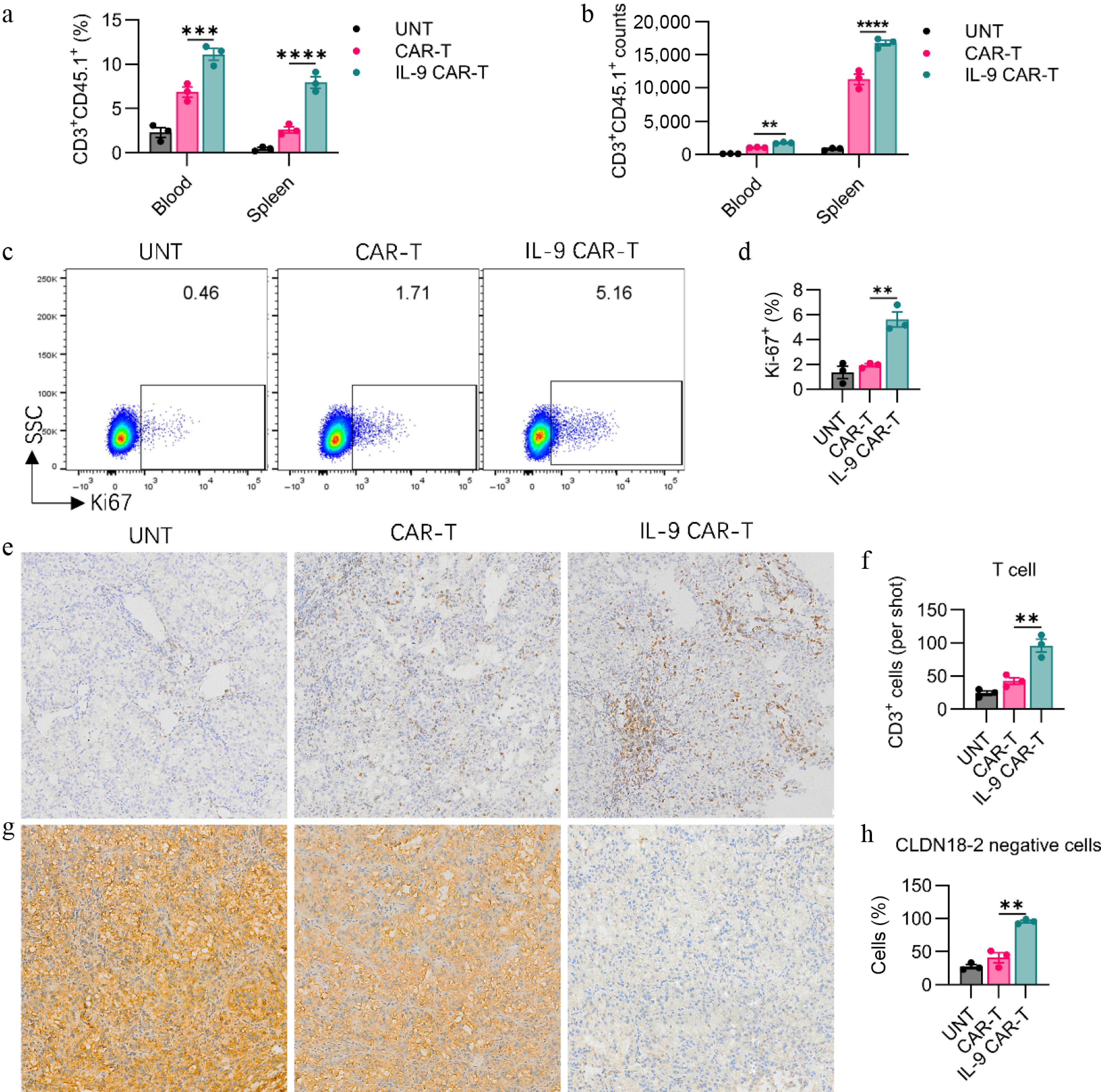
Figure 4.
IL-9 expression enhances CAR-T cell expansion, persistence, and tumor infiltration in vivo. The (a) percentage, and (b) absolute number of transferred CD3+CD45.1+ CAR-T cells in the blood and spleen of treated mice on day 7 post-infusion. Representative (c) flow cytometry plots and (d) quantification of Ki67 expression in CD3+CD45.1+ CAR-T cells from the spleen, indicating proliferating cells. Representative (e) immunohistochemistry (IHC) images (200× magnification), and (f) quantitative analysis of CD3+ T cell infiltration in tumor tissues on day 7. Representative (g) IHC images (200× magnification), and (h) quantification of Claudin 18.2 (Claudin18.2) expression in tumor tissues. Data in (a), (b), (d), (f), and (h) represent mean ± SEM (n = 3 mice per group). Significance: ns, not significant; ** p < 0.01; *** p < 0.001; ****p < 0.0001.
IL-9 CAR-T treatment developed the memory phenotype in endogenous T cells
-
Next, we analyzed T-cell phenotypic profiles in the spleens of mice 7 d post CAR-T cell infusion. Relative to conventional CAR-T cells, IL-9 CAR-T cells drove a greater proportion of endogenous T cells to differentiate into CD62L+CD44+ central memory T cells in the spleens of mice on Day 7 (Fig. 5c, d), whereas no differences in central memory subsets or effector memory subsets were observed within CD45.1+ T cells (Fig. 5a, b). These outcomes suggested that IL-9 facilitated endogenous T cells to transition toward a memory phenotype, which could potentially contribute to sustained long-term antitumor activity.
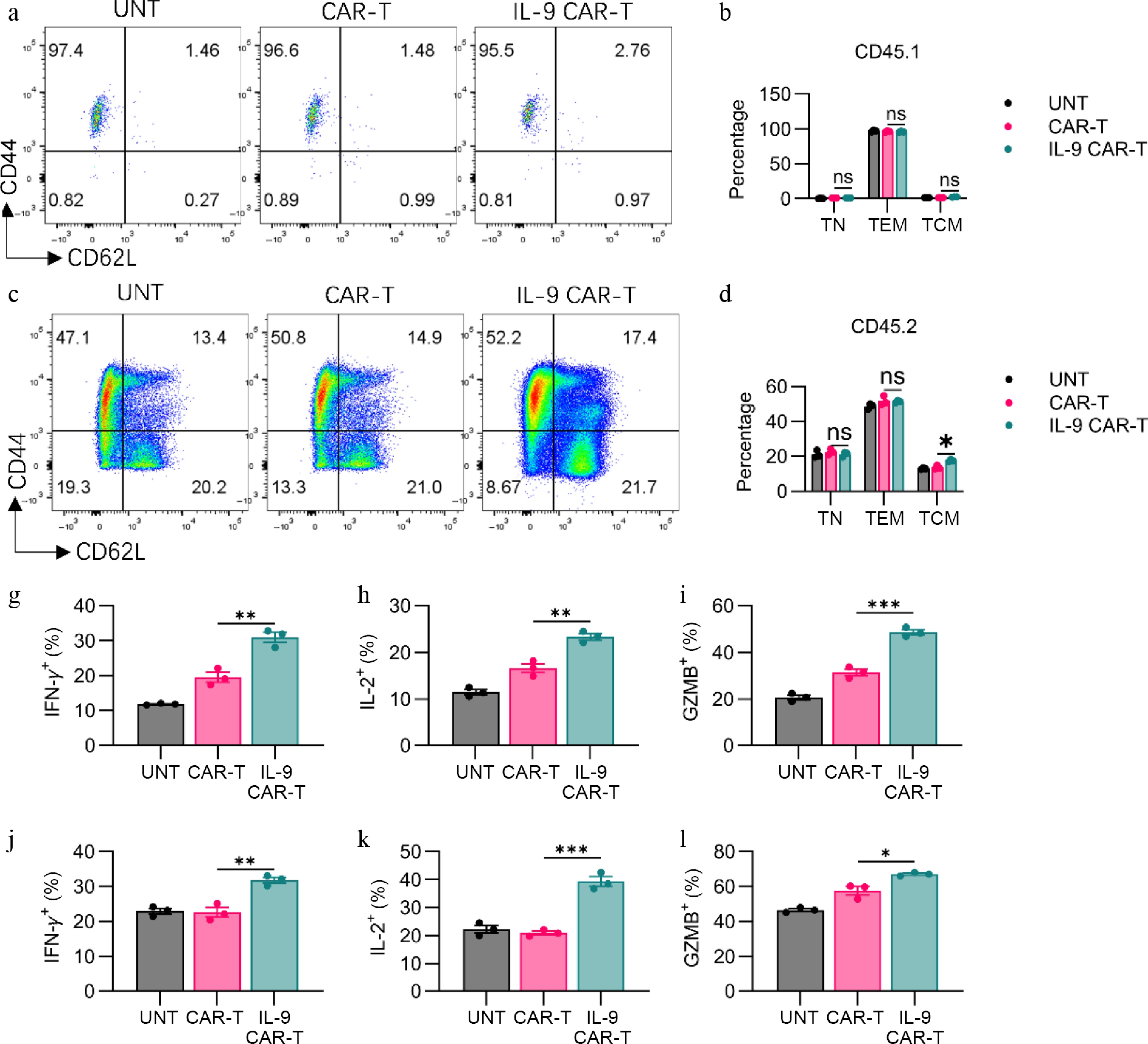
Figure 5.
IL-9 expression promoted the shift to memory phenotype and the anti-tumor efficacy of CAR-T cells. (a) Representative flow plots showed CD44 or CD62L expression on CD3+CD45.1+ T cells. (b) Histogram showing percentage of CD44 or CD62L expression on CD3+CD45.1+ T cells. (c) Representative flow plots showing CD44 or CD62L expression on CAR-T cells. (d) Histogram showing percentage of CD44 or CD62L expression on CD3+CD45.2+ T cells. Histogram showing percentage of (g) IFN-γ, (h) IL-2, and (i) granzyme B expression on CD3+CD45.1+ T cells. Histogram showing percentage of (j) IFN-γ, (k) IL-2, and (l) granzyme B expression on CD3+CD45.2+ T cells. The results are expressed as the mean ± SEM. Statistical significance was defined as follows: ns, not significant, p > 0.05; * p < 0.05; ** p < 0.01; *** p < 0.001.
IL-9 CAR-T treatment boosted the effector molecules expression of CAR-T and endogenous T cells
-
Furthermore, we quantified the expression levels of effector molecules within CAR-T cells on Day 7 post CAR-T cell infusion. Relative to conventional CAR-T cells, IL-9 CAR-T cells generated higher levels of IFN-γ on Day 7, contrasting with their in vitro behavior (Fig. 5g, Supplementary Fig. S2a). In addition, the frequencies of IL-2+ and granzyme B+ CAR-T cells from the spleen were also markedly elevated following IL-9 expression (Fig. 5h, i, Supplementary Fig. S2b, S2c). Concurrently, we assessed the expression profiles of effector molecules on CD45.2+ endogenous T cells, revealing that IL-9 expression promoted endogenous T cells to generate greater quantities of IFN-γ, IL-2, and granzyme B, relative to conventional CAR-T cells (Fig. 5j–l, Supplementary Fig. S3a–S3c). These results illustrate that IL-9 markedly augmented the in vivo antitumor capabilities of both CAR-T cells and endogenous T cells, simultaneously.
-
The advancement of chimeric antigen receptor T (CAR-T) cells has transmuted cancer immunotherapy, with impressive clinical responses observed in hematologic malignancies. However, the therapeutic potential of CAR-T cells in solid tumors remains limited by a spectrum of challenges, inclusive of tumor microenvironment (TME)-elicited immunosuppression, inadequate T-cell infiltration, and subpar persistence of infused CAR-T cells[18−20]. One of the major limitations of CAR-T therapy is the insufficient expansion of CAR-T cells after infusion[21]. Ectopic expression of cytokines or chemokines has been engineered into CAR-T cells in prior investigations, intended to enhance the infiltration and survival of adoptively transferred CAR-T cells[13,14,22]. In our case, we have explored a novel strategy of enhancing CAR-T cell function by incorporating interleukin-9 (IL-9) expression into the CAR construct, thus generating IL-9-expressing Claudin18.2 CAR-T cells (IL-9 CAR-T). Prior investigations have demonstrated that polarizing human T cells to secrete IL-9 enhanced the antitumor activity and proliferative capacity[23,24]. In our study, IL-9 CAR-T cells exhibited significantly higher proliferative capacity in vitro and in vivo compared to conventional CAR-T cells, which might contribute to the improved in vivo antitumor effects in three solid tumor models.
Another hallmark of an effective CAR-T therapy is the ability to produce pro-inflammatory cytokines and exhibit cytotoxicity against target tumor cells[25]. In our study, although the secretion of IFN-γ was lower compared with conventional CAR-T cells in vitro, IL-9 CAR-T cells produced more IFN-γ in vivo, which may be due to the complicated tumor micro environment (TME) giving more stimulation to the CAR-T cells. Meanwhile, IL-9 CAR-T cells produced more IL-2 and granzyme B in vivo, which may also lead to the tumor cytotoxicity and CAR-T cell expansion.
Possessing the ability to rapidly generate a secondary wave of effector T cells, central memory T cells (Tcm) serve as a subset of antigen-experienced lymphocytes that constitutively express the surface markers CD62L and CD44, and they are pivotal to providing protection against systemic challenges. Additionally, they have a tendency to migrate preferentially to secondary lymphoid organs, which are pivotal in mediating robust therapeutic antitumor immune responses against established cancers[26−28]. Prior investigations have demonstrated that IL-9-secreting T cells contributed to a central memory phenotype, characterized by reduced exhaustion, hyperproliferation, and prolonged survival in vivo. In our study, we also found a similar situation where IL-9 promoted CAR-T cells to transition toward a central memory phenotype, both in vitro and in vivo[15]. Tumor-specific memory cells play a critical role in sustaining long-term tumor control during immunotherapy. Rechallenge experiments should be conducted for further exploration.
-
In summary, our findings substantiate that enforced IL-9 expression within CAR-T cells significantly augments their in vivo antitumor potency, proliferative capacity, intratumoral infiltration, and cytokine secretion profiles, thereby yielding superior therapeutic outcomes across multiple solid tumor models. Moreover, the potential of IL-9 CAR-T cells to elicit sustained memory responses and long-term antitumor immunity against established cancers, further underscores the clinical significance of this strategy. These observations lay a robust foundation for forthcoming clinical explorations of IL-9 CAR-T cell regimens, which may represent a transformative strategy to enhance the clinical efficacy of CAR-T cell therapy in solid tumor settings. Subsequent preclinical investigations will be imperative to precisely optimize the therapeutic dosage, administration timeline, and potential combinatorial approaches for IL-9 CAR-T cells, thereby maximizing their therapeutic potential in the field of cancer immunotherapy.
This article was supported by Jiangsu Province Basic Research Program Natural Science Foundation (BK20231156).
-
The study was conducted in accordance with the Declaration of Helsinki, and all procedures were approved by the Xuzhou Med Univ's IACUC, identification number: 2025051678, approval date: 2025.04.06.
-
The authors confirm their contributions to the paper as follows: study conception and design: Bu X; data collection: Han L, Liu W; analysis and interpretation of results: Song Y, Li R; draft manuscript preparation: Song Y. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Yipeng Song, Rantian Li
- Supplementary Fig. S1 (A) Representative flow cytometry plots of claudin18.2 expression on Panc02-18.2, B16F10-18.2 and Hepa1-6-18.2 and their corresponding wild-type counterparts.
- Supplementary Fig. S2 (A) Representative flow plots showed IFN-γ expression on CD3+CD45.1+ T cells in spleen. (B) Representative flow plots showed IL-2 expression on CD3+CD45.1+ T cells in spleen. (C) Representative flow plots showed granzyme B expression on CD3+CD45.1+ T cells in spleen.
- Supplementary Fig. S3 (A) Representative flow plots showed IFN-γ expression on CD3+CD45.2+ T cells in spleen. (B) Representative flow plots showed IL-2 expression on CD3+CD45.2+ T cells in spleen. (C) Representative flow plots showed granzyme B expression on CD3+CD45.2+ T cells in spleen.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Song Y, Li R, Han L, Liu W, Bu X. 2026. Augmentation of Claudin18.2 CAR-T cell anti-tumor activity by IL-9 expression in mouse solid tumor models. Gastrointestinal Tumors 13: e007 doi: 10.48130/git-0026-0007 

Augmentation of Claudin18.2 CAR-T cell anti-tumor activity by IL-9 expression in mouse solid tumor models
- Received: 01 December 2025
- Revised: 07 February 2026
- Accepted: 25 March 2026
- Published online: 28 April 2026
Abstract: Chimeric Antigen Receptor T (CAR-T) Cell Therapy has been successful in curing hematologic malignancy but has not shown any results on solid tumors. One big obstacle is lack of dispersal coupled with CAR-T cells remaining undetected in the sick tissue environment. IL-9 (interleukin-9) retains T cells survival and function to improve antitumor immune reactions. This study developed Claudin18.2-targeted CAR-T cells that constantly expressed IL-9. It studied their ability to fight tumors in mice with working immune systems suffering from tumors. In vitro, it was found that the proliferative and chemotactic abilities of IL-9 expressing CAR-T cells are strong, but their cytotoxicity against Claudin18.2+ tumor cells is the same as that of regular CAR-T cells. In vivo, in treatment with IL-9, CAR-T cells showed a much stronger effect on tumor growth with the pancreatic model, melanoma model, and hepatocellular carcinoma model than in vitro, with the therapeutic effects improving, with the number of CAR-T cells getting bigger. With better results, more CAR-T cells are formed with a higher infiltration into the tumor, therefore more IFN-γ, IL-2, and granzyme B. IL-9 expression gives rise to internal T cell development, which become a central storage, which could potentially retain anti-tumor immunity. IL-9 also has some pretty good results with improving efficiency and half-life of Claudin18.2-specific CAR-T cells, it might also be a good way to make solid cancer CAR-T therapies more effective.
-
Key words:
- CAR-T cell therapy /
- Interleukin-9 /
- Claudin 18.2 /
- Solid tumors /
- Tumor microenvironment