










-
Because of hepatitis B virus (HBV) infection, China bears the highest global burden of hepatocellular carcinoma (HCC), accounting for 42.5% of new cases and 41.8% of deaths worldwide[1,2]. Given that most patients are diagnosed at advanced stages, systemic therapy is the primary treatment option, yet the 5-year survival rate is just 14.4%[3]. Despite immune checkpoint inhibitor-based combinations reshaping the treatment landscape, over 60% of patients are unresponsive[4−7], emphasizing the urgent need for more effective and tolerable agents.
Icaritin, a flavonoid monomer derived from Epimedium, is a first-in-class natural agent targeting STAT3[8,9]. Early clinical studies demonstrated its efficacy and safety in patients with HCC[9,10], leading to its approval in China in January 2022 following positive Phase III trial results. Our previous work identified its first lncRNA target, linking its mechanism to suppressed tumor glycolysis[11]. With its potent efficacy and favorable safety profile, icaritin represents a unique therapeutic option for advanced HCC. Previous studies revealed that STAT3 had the effect on promoting glycolysis[12−14]. In current study, we attempt to elucidate the mechanism by which icaritin regulates STAT3 expression through the glycolytic pathway.
-
The human HCC cell line HepG2 was obtained from the American Type Culture Collection (ATCC). Cells were cultured in Dulbecco's Modified Eagle Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin–streptomycin (Gibco) at 37 °C in a humidified atmosphere containing 5% CO2. To mimic hypoxia, cells were pretreated with 20 μM cobalt chloride (CoCl2) for 24 h. Icaritin (purity > 98%, Shenogene Pharma Group) was dissolved in dimethyl sulfoxide (DMSO) to prepare a stock solution, which was stored at −20 °C. The final concentration of DMSO in all experiments did not exceed 0.1%.
Plasmid construction and cell transfection
-
The full-length human aldolase B (ALDOB) coding sequence was amplified by polymerase chain reaction (PCR) and cloned into the pcDNA3.1(+) vector (Invitrogen) to generate the ALDOB overexpression plasmid (pcDNA3.1-ALDOB). An empty pcDNA3.1(+) vector was used as a negative control. HepG2 cells were seeded into six-well plates and cultured overnight to reach 70%–80% confluence prior to transfection. Transfection was performed using Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer's instructions. Briefly, for each well, 2.5 μg of plasmid DNA (pcDNA3.1-ALDOB or the empty pcDNA3.1[+] vector) was diluted in 125 μL of Opti-MEM medium, and 5 μL of Lipofectamine 3000 reagent was diluted in another 125 μL of Opti-MEM medium. The diluted DNA was then mixed with the diluted Lipofectamine 3000 reagent at a 1:1 ratio and incubated for 15 min to form DNA–lipid complexes. The mixture was subsequently added dropwise to the cells. After 6 h of incubation, the culture medium was replaced with fresh DMEM containing 10% FBS to reduce potential cytotoxicity. Cells were harvested 48 h after transfection for subsequent experiments, including quantitative real-time PCR (qPCR) and Western blot analysis.
RNA sequencing and data analysis
-
Total RNA was extracted from HepG2 cells treated with or without icaritin (10 μM, 48 h) using TRIzol reagent (Invitrogen). The RNA's integrity was assessed on an Agilent 2100 Bioanalyzer, and library construction was performed, followed by sequencing on an Illumina NovaSeq 6000 platform (Novogene). Raw reads were aligned to the human reference genome (GRCh38) using HISAT2, and differential expression analysis was conducted with the DESeq2 R package. Genes with an adjusted p-value < 0.05 and |log2(fold change)| > 1 were considered to be significantly differentially expressed.
RNA extraction and qPCR
-
Total RNA was extracted from cultured cells using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. cDNA was synthesized from 1 μg of total RNA using the PrimeScript RT reagent Kit (Takara). qPCR was performed using SYBR Green Premix Pro Taq HS (Accurate Biotechnology) on a QuantStudio 5 Real-Time PCR System (Applied Biosystems). Relative mRNA expression levels were calculated using the 2−ΔΔCᴛ method and normalized to the expression of Actin. The primer sequences used are listed in Supplementary Table S1.
Western blot analysis
-
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Beyotime Biotechnology) containing protease and phosphatase inhibitors (Roche). Protein concentrations were determined using a BCA protein assay kit (Beyotime). Equal amounts of protein were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinyl fluoride (PVDF) membranes (Millipore). The membranes were blocked with 5% nonfat milk and then incubated overnight at 4 °C with primary antibodies against ALDOB (1:1000, Proteintech, 66862-1-Ig), STAT3 (1:1,000, Cell Signaling Technology, 12640S), phospho-STAT3 (Tyr705) (1:2,000, Cell Signaling Technology, 9145S), and Actin (1:5,000, Proteintech, 60004-1-Ig). Following incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature, protein signals were detected using an electrochemiluminescence (ECL) system (Tanon). Band intensities were quantified by densitometric analysis using ImageJ software and normalized to the levels of Actin.
Cell proliferation assays
-
Cell proliferation was assessed using three complementary assays. For the CCK-8 assay, transfected cells seeded in 96-well plates were treated with icaritin for 48 h, followed by incubation with 10 μL of a CCK-8 solution (Beyotime) for 2 h. Absorbance at 450 nm was measured using a microplate reader. For colony formation, 1,000 cells per well were seeded into six-well plates, treated with icaritin for 10–14 days, then fixed with methanol, stained with 0.1% crystal violet, and counted. Incorporation of 5-ethynyl-2'-deoxyuridine (EdU) was evaluated using the Cell-Light EdU Apollo567 In Vitro Kit (RiboBio) as per the manufacturer's protocol, with stained cells imaged under a fluorescence microscope (Nikon).
Measurement of glycolysis
-
Glucose consumption and lactate production were determined using commercial colorimetric assays (Glucose Uptake Assay Kit, ab136955; Lactate Assay Kit, ab65331; Abcam) following the manufacturer's guidelines. To account for variations in cell proliferation, the results were normalized to cell numbers enumerated from parallel wells using a hemocytometer. Glycolytic activity was monitored in real time with a Seahorse XFe96 Analyzer using the Glycolysis Stress Test Kit (Agilent Technologies). In brief, cells cultured in XF96 microplates were equilibrated in extracellular flux (XF) assay medium, and the extracellular acidification rate (ECAR) and the oxygen consumption rate (OCR) were measured after the addition of glucose, oligomycin, and 2-deoxy-D-glucose (2-DG).
Luciferase reporter assay
-
A fragment of the human STAT3 gene promoter was cloned into the pGL3-Basic vector (Promega) to generate the STAT3 promoter–luciferase reporter construct. HepG2 cells were co-transfected with the reporter plasmid and the Renilla luciferase control vector (pRL-TK, Promega) using Lipofectamine 3000. After 24 h, cells were treated with icaritin for an additional 24 h. Luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega) on a GloMax Navigator Microplate Luminometer (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity.
Chromatin immunoprecipitation assay
-
Chromatin immunoprecipitation (ChIP) was carried out using the SimpleChIP® Plus Sonication Chromatin IP Kit (Cell Signaling Technology, #56383) according to the manufacturer's instructions. Briefly, HepG2 cells were cross-linked, and chromatin was sonicated to generate DNA fragments averaging 20–500 bp. Immunoprecipitation was performed overnight at 4 °C with antibodies targeting H3K18la (PTM Biolabs, PTM-1406RM) or p300 (Cell Signaling Technology, #54062); normal rabbit immunoglobulin G (IgG) served as a negative control. Following purification, precipitated DNA was subjected to qPCR analysis with primers specific to the STAT3 promoter region. The enrichment was calculated as a percentage of the input chromatin.
Statistical analysis
-
All results are expressed as mean ± standard deviation (SD) of at least three independent experiments. Statistical analyses were carried out using GraphPad Prism 9.0. The unpaired two-tailed Student's t-test was used for comparisons between two groups, and one-way or two-way analysis of variance (ANOVA) followed by Tukey's post hoc test was applied for multiple group comparisons. Statistical significance was set at p < 0.05.
-
To identify the underlying target of icaritin in HCC cells, we performed RNA sequencing (RNA-seq). The analysis compared the gene expression profiles between cells treated with or without icaritin (Fig. 1a). Differentially expressed genes (DEGs) were filtered, yielding 25 upregulated and 49 downregulated mRNAs (Fig. 1b). Our previous study demonstrated that icaritin exerts pharmacological effects by inhibiting glycolysis in HCC cells. To identify glycolysis-related targets of icaritin, we focused on 118 glucose metabolism-related genes curated from the PathCards database, which integrates multiple pathways including glycolysis, gluconeogenesis, and the pentose phosphate pathway. ALDOB was identified as a potential target (Fig. 1c). Subsequent qPCR analysis confirmed that icaritin treatment significantly inhibited ALDOB expression (Fig. 1d). To further investigate its function, we overexpressed ALDOB in HCC cells, as validated by qPCR (Fig. 1e). In summary, we identified ALDOB as a candidate molecule through which icaritin inhibits glycolytic activity in HCC cells.
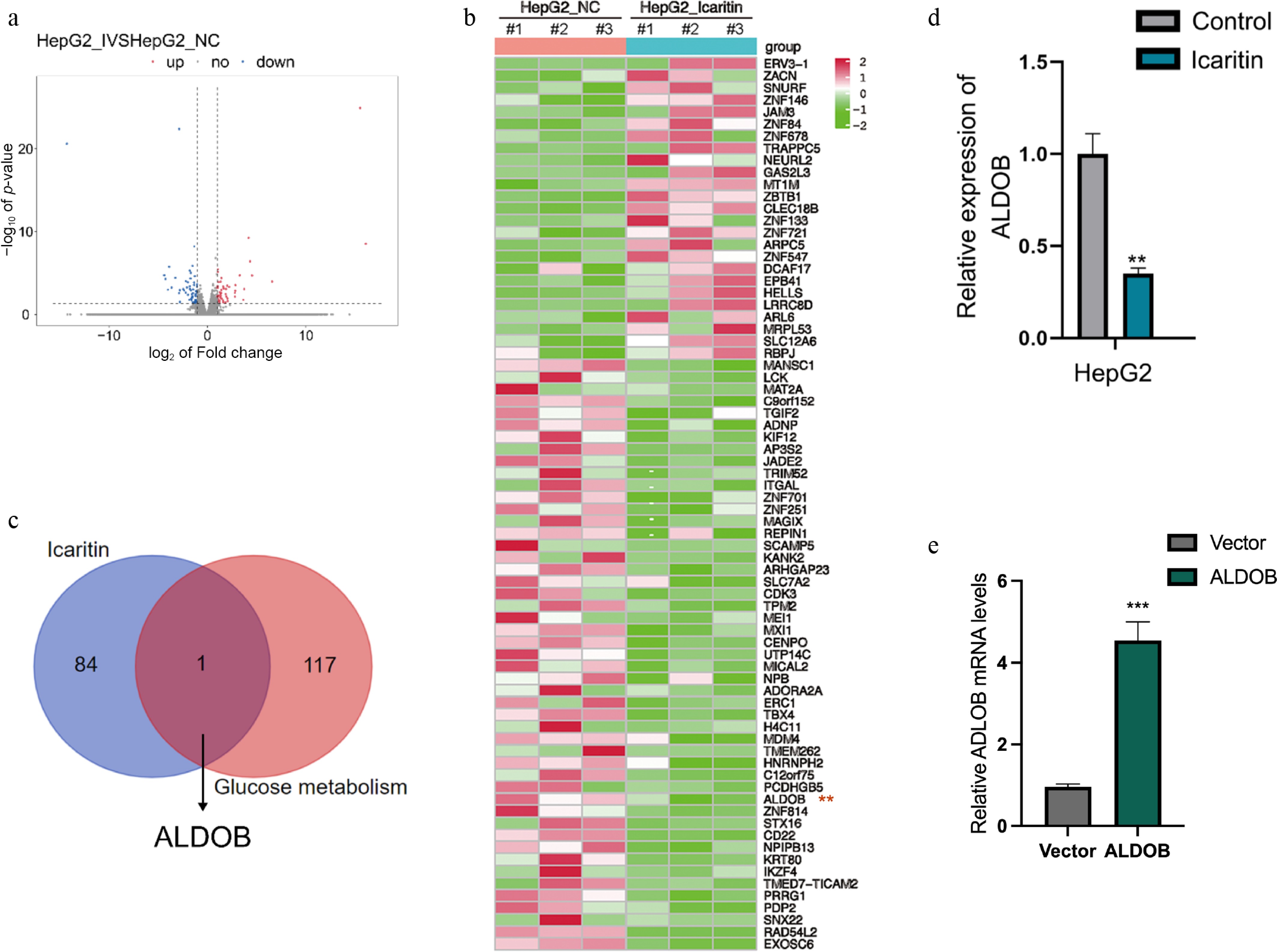
Figure 1.
Identification of ALDOB as a glycolysis-related target downregulated by icaritin in HCC cells. (a) RNA-seq identification of differentially expressed genes (DEGs) in HCC cells treated with or without icaritin (b) Identification of DEGs, with 49 downregulated and 25 upregulated genes. (c) Screening of downregulated DEGs for glycolysis-associated genes identified ALDOB. (d) Icaritin significantly reduced ALDOB's mRNA expression as measured by qPCR. (e) Successful overexpression of ALDOB in HCC cells was verified by qPCR. * p < 0.05, ** p < 0.01, *** p < 0.001; n = 3.
Overexpression of ALDOB rescues icaritin-induced inhibition of HCC cell proliferation
-
We next determined whether ALDOB mediates the anti-proliferative effect of icaritin. Firstly, CCK-8 assays showed that the proliferation inhibition caused by icaritin was compromised by ALDOB overexpression (Fig. 2a). We next performed colony formation assays and found that ALDOB overexpression similarly restored the clonogenic potential of icaritin-treated cells (Fig. 2b). Finally, EdU incorporation assays directly quantified DNA synthesis and confirmed that ALDOB overexpression reversed the icaritin-induced suppression of cell proliferation (Fig. 2c). In conclusion, these functional gains-of-function experiments establish that icaritin inhibits HCC cells' proliferation via ALDOB.
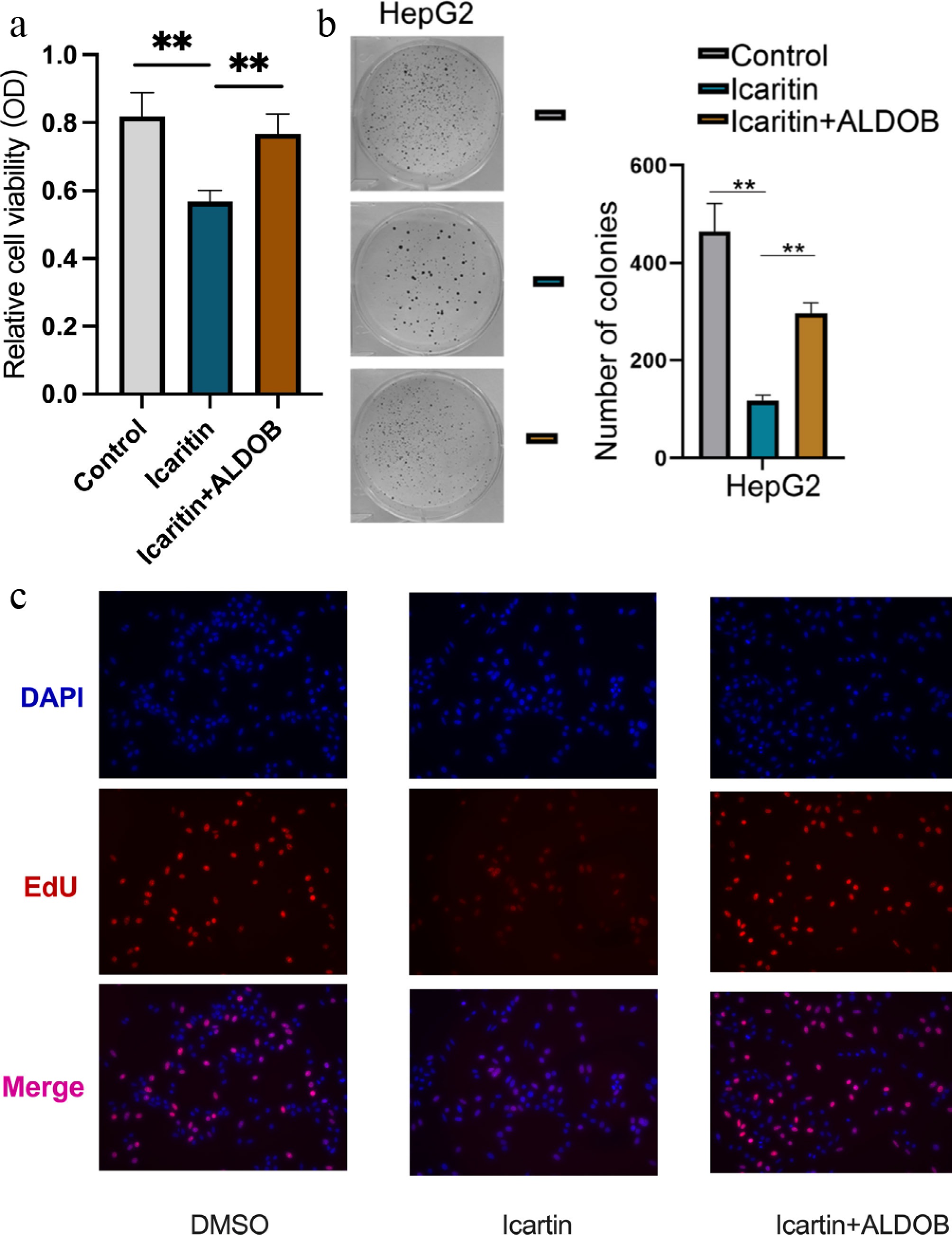
Figure 2.
Overexpression of ALDOB rescues icaritin-induced inhibition of HCC cell proliferation. (a) Cell viability was measured by an CCK-8 assay in HCC cells with or without ALDOB overexpression following treatment with the indicated concentrations of icaritin. (b) Representative images (left) and quantification (right) of colony formation assays under the same conditions. (c) The percentage of proliferating cells was assessed by an EdU assay. * p < 0.05, ** p < 0.01, *** p < 0.001; n = 6.
ALDOB overexpression reverses the icaritin-induced suppression of glycolysis in HCC cells
-
We further investigated whether icaritin modulates glycolytic metabolism via ALDOB. Icaritin treatment significantly reduced glucose uptake and lactate production in HCC cells, both hallmarks of glycolysis. Notably, these inhibitory effects were effectively reversed by ALDOB overexpression (Fig. 3a, b). To directly assess glycolytic flux, we used the Seahorse XF Analyzer. Consistent with these findings, icaritin potently suppressed the extracellular acidification rate (ECAR), an indicator of glycolytic lactate generation, and this suppression was abolished in ALDOB-overexpressing cells (Fig. 3c, d). Collectively, these data demonstrate that icaritin impairs glycolytic metabolism in HCC cells through ALDOB.
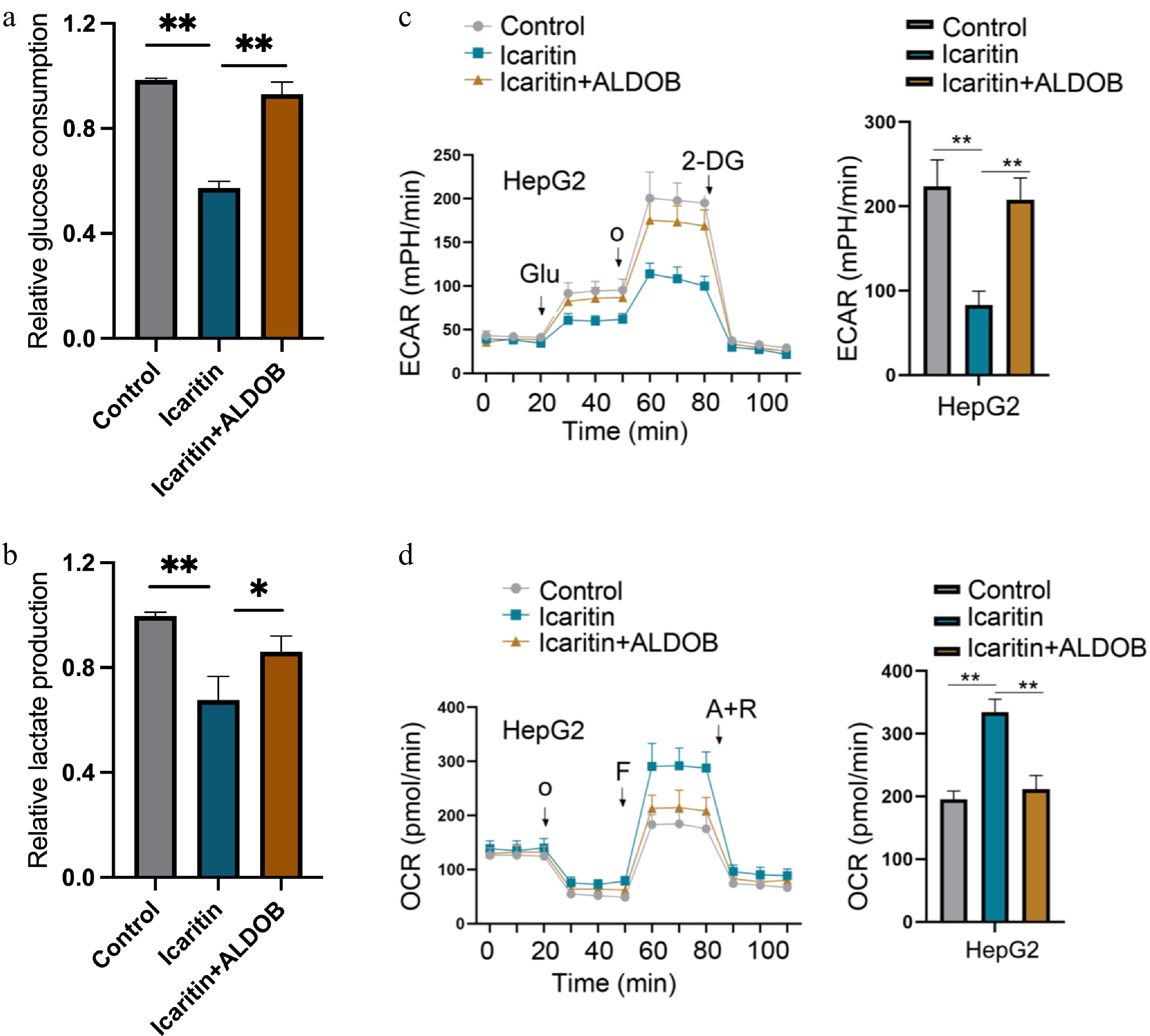
Figure 3.
ALDOB overexpression reverses the icaritin-induced suppression of glycolysis in HCC cells. (a) Glucose uptake was measured in control and ALDOB-overexpressing HCC cells treated with or without icaritin. (b) Intracellular lactate production was assessed under the same conditions. (c) Glycolytic function was analyzed by measuring the extracellular acidification rate (ECAR) using the Seahorse XF Analyzer. (d) Mitochondrial respiration was assessed by measuring the oxygen consumption rate (OCR) under the same conditions. * p < 0.05, ** p < 0.01, *** p < 0.001; n = 4.
Icaritin suppresses STAT3 signaling in an ALDOB-dependent manner
-
To elucidate the underlying molecular mechanism, we examined the status of STAT3, a known target of icaritin. qPCR analysis showed that icaritin suppressed mRNA expression of STAT3, and this effect was reversed by ALDOB overexpression (Fig. 4a). Western blot analysis further demonstrated that icaritin downregulated the protein expression of both ALDOB and STAT3, while also inhibiting STAT3 phosphorylation. Crucially, all these inhibitory effects were abolished upon ALDOB overexpression (Fig. 4b, c). These findings indicate that icaritin transcriptionally represses STAT3 in an ALDOB-dependent manner.
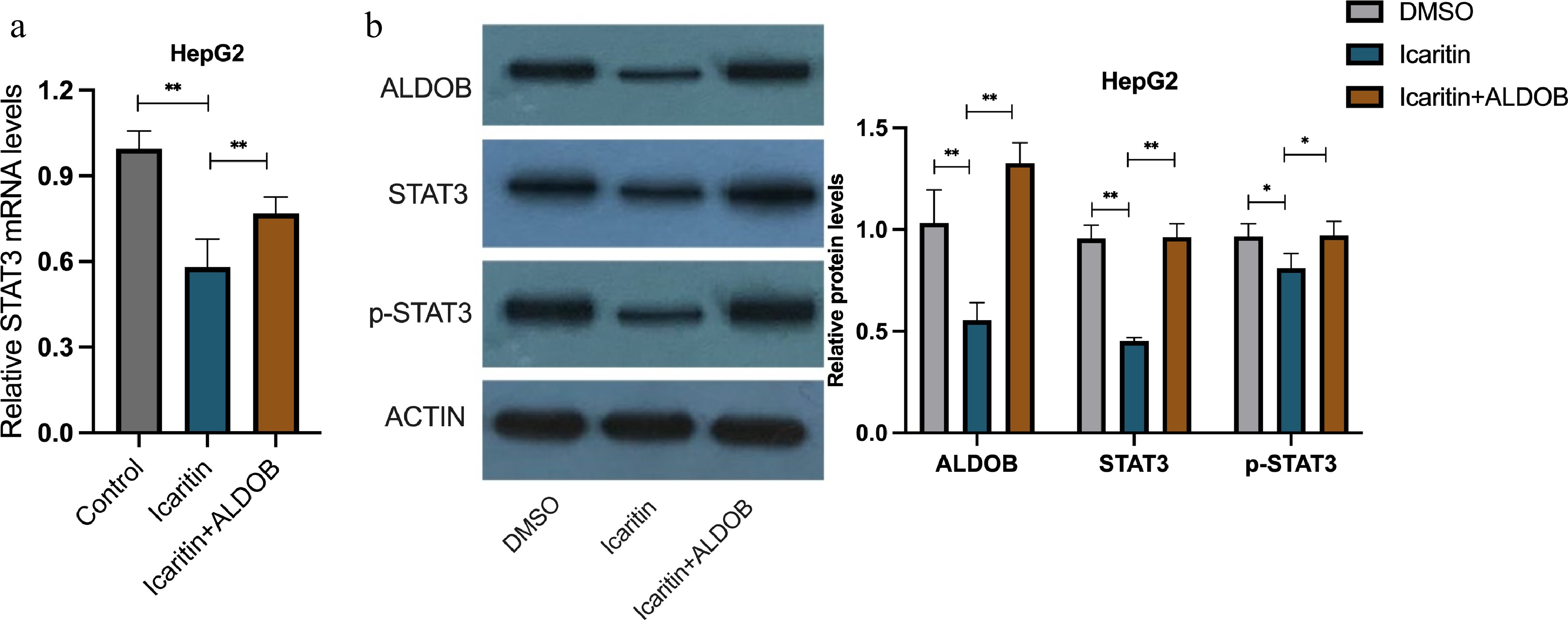
Figure 4.
Icaritin suppresses STAT3 signaling in an ALDOB-dependent manner. (a) The mRNA expression level of STAT3 was determined by qPCR in HCC cells with or without ALDOB overexpression following icaritin treatment. (b) Representative Western blot images showing the protein levels of ALDOB, STAT3, phosphorylated STAT3 (p-STAT3), and the loading control Actin under the same conditions. * p < 0.05, ** p < 0.01, *** p < 0.001; n = 3.
Icaritin represses STAT3 transcription by modulating the H3K18la modification on its promoter
-
To delineate the mechanism by which icaritin suppresses STAT3 transcription, we investigated epigenetic regulation at its promoter. The luciferase reporter assay confirmed that icaritin inhibits the STAT3 promoter's activity (Fig. 5a). Given that lactate serves as a substrate for histone lactylation, we performed ChIP assays. Icaritin treatment significantly reduced the enrichment of H3K18la on the STAT3 promoter, an effect that was rescued by the addition of exogenous lactate (Fig. 5b, c). Furthermore, we found that the histone acetyltransferase p300, a known reader of lactylation, binds to the STAT3 promoter. Icaritin diminished both the recruitment of p300 and the concomitant H3K18la modification at the promoter (Fig. 5d). Collectively, these results unveil a novel epigenetic pathway whereby icaritin, by reducing lactate production, attenuates p300-mediated H3K18la modification on the STAT3 promoter, thereby repressing its transcription.
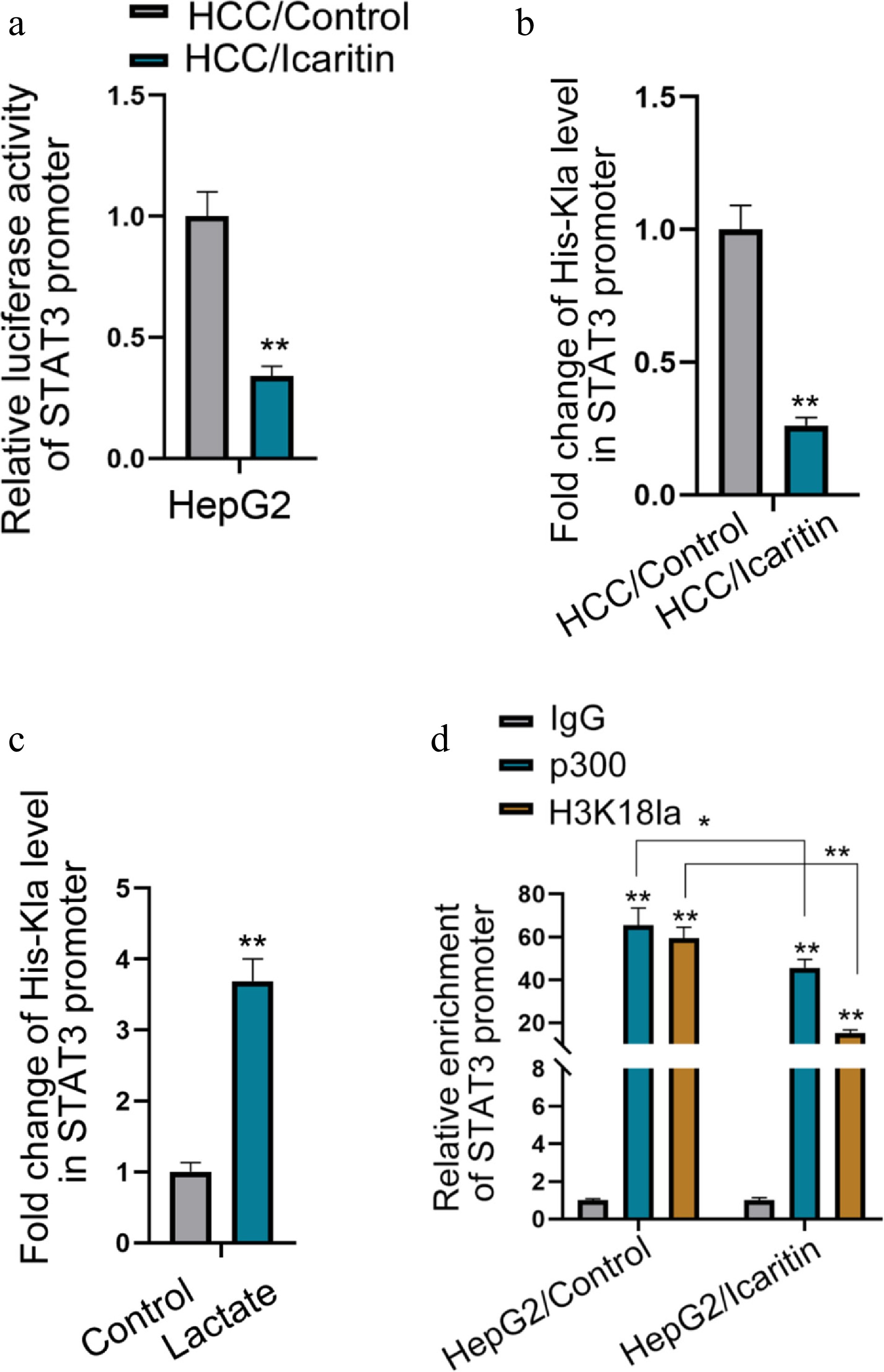
Figure 5.
Icaritin represses STAT3 transcription by modulating H3K18la modification on its promoter. (a) STAT3 promoter activity was measured by a dual-luciferase reporter assay in HCC cells treated with icaritin. (b) ChIP assay quantifying the enrichment of H3K18la on the STAT3 promoter after icaritin treatment. (c) ChIP assay showing the enrichment of H3K18la on the STAT3 promoter in cells treated with icaritin in the presence or absence of exogenous sodium lactate. (d) ChIP assays analyzing the binding of p300 and the level of H3K18la on the STAT3 promoter under icaritin treatment. * p < 0.05, ** p < 0.01; n = 3.
-
This study systematically elucidates a novel mechanism of action for the natural compound icaritin in HCC: It targets the glycolytic enzyme ALDOB, thereby modulating a metabolism–epigenetics axis that leads to the inhibition of the oncogenic transcription factor STAT3. Our findings reveal a previously unknown mechanism by which icaritin influences epigenetic regulation through modulating glucose metabolism in HCC, providing new insights into the metabolic–epigenetic crosstalk in cancer.
Our research is the first to identify ALDOB as a key downstream target of icaritin. ALDOB, a key enzyme in fructose metabolism, catalyzes the cleavage of fructose-1,6-bisphosphate into dihydroxyacetone-3-phosphate and glyceraldehyde-3-phosphate, and this action plays a critical role in promoting glycolysis. However, ALDOB has recently gained attention for its tumor-suppressive role in HCC[15]. Studies have shown that ALDOB expression is downregulated in HCC tissues, with its low expression correlating with poor patient prognosis, whereas its overexpression can inhibit HCC cell proliferation and tumorigenesis[16]. These results indicate that ALDOB exerts inhibitory effects on glycolysis and tumorigenesis, which are not consistent with the current results. We propose that ALDOB acts as a multifunctional molecule in regulating glycolysis and HCC's progression, with its role being context-dependent, potentially influenced by microenvironmental conditions such as hypoxia or nutrient availability. Our study was conducted under hypoxic conditions. Under hypoxia, HCC cells undergo metabolic reprogramming toward enhanced glycolysis to adapt to low oxygen availability. In this specific context, ALDOB, despite its tumor-suppressive functions, can be co-opted to support glycolytic flux.
Using CCK-8, colony formation, and EdU assays, we conclusively demonstrated that ALDOB overexpression rescues HCC cells from icaritin-induced proliferation arrest. These results indicate that the anti-proliferative effect of icaritin is highly dependent on its regulation of ALDOB. We subsequently linked ALDOB function directly to the glycolytic process through glucose uptake, lactate production, and Seahorse extracellular flux analyses. The results suggest that icaritin, via ALDOB, significantly suppresses glycolytic flux in HCC cells. HCC is a malignancy highly dependent on aerobic glycolysis[17]. Rapid glycolysis provides not only energy but also abundant biosynthetic precursors for rapidly proliferating tumor cells[18]. Therefore, by inhibiting glycolysis through ALDOB, icaritin effectively cripples the "energy and material" supply of HCC cells, providing a solid metabolic basis for its potent anti-proliferative effect.
The most intriguing finding of this study lies in connecting the icaritin–ALDOB–glycolysis axis to an epigenetic mechanism for transcriptional repression of STAT3. STAT3 is a crucial oncogenic transcription factor in HCC, whose constitutive activation promotes cell proliferation, survival, angiogenesis, and immune evasion[19,20]. Although icaritin has been reported as a STAT3 inhibitor[8], the precise mechanism, particularly at the transcriptional level, has remained unclear.
Icaritin regulates STAT3 at the transcriptional level, as evidenced by the ALDOB-dependent reduction in STAT3 mRNA, thereby connecting glycolytic activity to transcriptional control. In recent years, the role of metabolites as substrates or cofactors for epigenetic modifications has become increasingly prominent[21]. Lactate, as a precursor for histone lactylation, is a key molecule linking metabolism to epigenetics[22]. Seminal work by Zhang et al. demonstrated that lactate-derived H3K18la modification can promote gene transcription, directly coupling glycolysis to epigenetic regulation[23]. Moreover, p300 has been established as a bona fide lactyltransferase that catalyzes H3K18la using lactyl-CoA derived from lactate[23]. Our data show that icaritin reduces both p300 recruitment and H3K18la enrichment at the STAT3 promoter. Importantly, exogenous lactate rescues H3K18la levels, demonstrating that icaritin acts by limiting the availability of the substrate (lactyl-CoA) rather than directly inhibiting p300 expression or its binding stability. The decreased p300 enrichment detected by ChIP likely reflects reduced catalytic engagement under low-lactate conditions, rather than impaired recruitment per se. Our results demonstrate that icaritin suppresses STAT3 transcription by reducing its promoter's H3K18la modification. This effect is reversed by exogenous lactate, confirming that icaritin acts through inhibiting glycolysis and reducing lactate to diminish STAT3 lactylation and silence its transcription.
This study reveals a novel positive feedback loop in HCC whereby STAT3 activation drives glycolysis[24,25], whose lactate product then fuels further STAT3 activation via lactylation. Icaritin breaks this cycle by targeting ALDOB, showcasing its advantage in multitarget, network-based regulation.
We uncovered a novel signaling axis from a metabolic enzyme (ALDOB) to an oncogenic transcription factor (STAT3), mediated by histone lactylation, deepening the understanding of the interplay between metabolic abnormalities and epigenetic dysregulation in HCC.
Our findings provide a solid scientific basis for the clinical application of icaritin. ALDOB expression levels or serum lactate levels might serve as biomarkers for predicting icaritin's efficacy. Furthermore, this mechanism suggests that icaritin could synergize with other therapies, such as immune checkpoint inhibitors, since lactate accumulation is a key factor in the immunosuppressive tumor microenvironment[26]. It validates that targeting the metabolism–epigenetics interface is a viable anti-cancer strategy, providing new ideas for developing similar drugs.
Naturally, there are some limitations in the current research. Primarily, the research is based on cell models; further validation of the ALDOB–lactate–STAT3 axis in animal models and clinical samples is necessary. Secondly, the precise mechanism by which icaritin regulates ALDOB expression at the transcriptional or post-transcriptional level remains to be fully elucidated. Thirdly, while our data demonstrate that icaritin reduces p300 recruitment to the STAT3 promoter, the precise molecular mechanism by which lactate availability influences p300 chromatin binding—whether through direct regulation of its lactyltransferase activity, conformational changes, or interactions with other cofactors—requires further investigation. Additionally, whether other histone lactylation sites or additional lactate-sensitive genes contribute to icaritin's anti-tumor effects remains to be explored.
In summary, this study delineates the multistep molecular mechanism by which icaritin, through downregulating ALDOB, inhibits glycolysis, reduces lactate production, diminishes H3K18la modification and p300 recruitment on the STAT3 promoter, and ultimately suppresses STAT3 transcription. This work not only comprehensively reveals the mechanism of action of icaritin but, more importantly, discovers a novel pathway mediated by lactylation that connects cancer metabolism with epigenetic regulation, providing a new theoretical foundation and potential targets for the targeted therapy of HCC.
This study was supported by the National Natural Science Foundation of China (Grant No. 82204800) and the National Major Science and Technology Project Fund (Grant No. 2024ZD0520405). Shenogene Pharma Group, Beijing, China.
-
This study involved only in vitro experiments using commercially available human hepatocellular carcinoma cell lines (HepG2), which were obtained from the American Type Culture Collection (ATCC). No human participants, human tissue samples, or animal subjects were used. Therefore, ethical approval was not required for this study.
-
The authors confirm their contributions to the paper as follows: conceptualization: Zheng X, Xun C; methodology: Zheng X, Qu W; investigation, formal analysis, writing – original draft, visualization: Zheng X; validation, resources: Qu W; writing – review and editing, funding acquisition: Qu W, Xun C; supervision, project administration: Xun C. All authors reviewed the results and approved the final version of the manuscript.
-
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
-
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
- Supplementary Table S1 Primers and overexpression sequences used in this study.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zheng X, Qu W, Xun C. 2026. Icaritin Targets ALDOB-dependent Glycolysis to Attenuate STAT3 Transcription via Histone Lactylation under Hypoxia in Hepatocellular Carcinoma. Gastrointestinal Tumors 13: e009 doi: 10.48130/git-0026-0009 

Icaritin Targets ALDOB-dependent Glycolysis to Attenuate STAT3 Transcription via Histone Lactylation under Hypoxia in Hepatocellular Carcinoma
- Received: 28 September 2025
- Revised: 02 April 2026
- Accepted: 09 April 2026
- Published online: 26 May 2026
Abstract: Icaritin, a natural anti-cancer agent, is known to target STAT3 and inhibit glycolysis in hepatocellular carcinoma (HCC), but the connecting mechanism has been elusive. In this study, we identified the glycolytic enzyme aldolase B (ALDOB) as a key target of icaritin by transcriptome sequencing of icaritin-treated hypoxic HepG2 cells. Icaritin significantly inhibited HCC cell proliferation and glycolysis, and these effects were substantially reversed by overexpression of ALDOB. At the molecular level, icaritin suppressed STAT3 promoter activity and mRNA expression. Crucially, a chromatin immunoprecipitation (ChIP) assay revealed that icaritin reduced the enrichment of histone H3K18 lactylation (H3K18la) and its associated enrichment of p300 on the STAT3 promoter. This suppressive effect was rescued by the addition of exogenous lactate, directly linking lactate's availability to the epigenetic activation of STAT3. These findings revealed that icaritin suppresses STAT3 transcription in HCC through inhibiting ALDOB-mediated glycolysis, ultimately reducing the glycolysis-dependent H3K18la modification on the STAT3 promoter. This work identifies a critical metabolism–epigenetics pathway, providing a foundational mechanism for icaritin's anti-tumor effect.
-
Key words:
- Hepatocellular carcinoma /
- Icaritin /
- Glycolysis /
- Lactylation /
- STAT3