









-
Lonicera japonica Thunb. is an important medicinal herb widely used in both traditional Chinese medicine and modern preparations[1,2]. Due to its widespread cultivation in many countries[3], the chemical composition of L. japonica has been extensively studied, with chlorogenic acid and luteoloside identified as major active ingredients[4,5]. The active constituents of L. japonica provide a wide range of pharmacological properties, including antibacterial, anti-inflammatory, and antiviral effects[6,7]. Although L. japonica is now cultivated widely[3], it still faces numerous challenges in agricultural production, the most prominent being the significant decrease in main active compounds as flowers bloom, which severely reduces the plant's utilization efficiency[8−10]. To address this issue, research has increasingly focused on studying the synthesis and regulatory mechanisms of active ingredients during the flower development stage of L. japonica. However, there remains considerable room for exploration regarding the underlying regulatory mechanisms[10,11].
Research has shown that fluctuations in endogenous hormones such as abscisic acid (ABA), salicylic, and jasmonic acid are linked to the growth and development of Lonicera japonica, thereby affecting its secondary metabolism[9]. Guan et al. demonstrated that certain members of the MYB, AP2/ERF, bHLH, and NAC transcription factor families are closely associated with the biosynthesis of chlorogenic acid and luteolin in L. japonica[12]. Despite significant advances in understanding the regulatory effects of hormones and transcription factors on L. japonica, research at the post-transcriptional level remains limited, leaving room for further exploration to fully elucidate the mechanisms underlying the active components of L. japonica. Recently, Xia et al. identified numerous miRNAs along with their target genes in L. japonica, providing a comprehensive miRNA expression profile and valuable insights for functional genomics research[13]. Additionally, miRNAs have been reported to regulate both flower development and secondary metabolism in L. japonica[14]. AS is also a critical post-transcriptional regulatory mechanism, allowing a single gene to significantly increase transcriptome and proteome diversity by producing multiple mRNA isoforms[15−17]. In Arabidopsis thaliana, AS has been shown to be closely related to flower development[18]. The FLOWERING LOCUS T (FT) protein is a key component of the 'florigen' signal, which is crucial for transitioning from vegetative to reproductive growth. In Platanus acerifolia, levels of various AS_PaFT isoforms correlate with different stages of flower development[19]. In Brachypodium distachyon, age-dependent AS of FT2 produces the FT2β isoform, which forms heterodimers with FT2α and FT1, disrupting the florigen-mediated flowering initiation complex, and delaying flowering[20]. Moreover, studies have shown that AS regulates the flowering transition by generating diverse transcripts of the FLOWERING LOCUS C (FLC) gene in A. thaliana[21]. In an early flowering mutant of trifoliate orange (Poncirus trifoliata), five alternatively spliced transcripts of PtFLC were identified, with their abundances varying between juvenile and adult stages[22]. These findings suggest that AS of the FLC gene may be a target of natural selection for flowering regulation under natural conditions.
It has been shown that AS has a strong connection to plant secondary metabolism[23]. In Catharanthus roseus, AS of strictosidine β-D-glucosidase (SGD) generates a truncated form known as shSGD which interacts with SGD to influence the relevant reactions and regulate monoterpene indole alkaloid biosynthesis[24]. In plants, allantoin biosynthesis is dependent on transthyretin-like proteins, while in Arabidopsis thaliana, the two transthyretin-like isoforms are due to the expression of AS[25]. JASMONATE ZIM-domain (JAZ) repressor, a key inhibitor of flavonoid biosynthesis[26], is regulated by AS in tea plants resulting in the creation of three JAZ splice isoforms[27]. CsJAZ1-1 and CsJAZ1-2 (rather than CsJAZ1-3), in the absence of jasmonates, competitively bind to CsMYC2, a positive regulator of flavonoid biosynthesis[28], thereby repressing flavonoid biosynthetic genes. In rapeseed (Brassica napus L.), BnaPAP2 has been demonstrated as a MYB transcription factor essential for regulating anthocyanin biosynthesis[29]. AS gives rise to different isoforms of BnaPAP2.A7 with opposite functions. The complete BnaPAP2.A7 harbors crucial MYB domains and could interact with a bHLH protein in vitro. In contrast, the two truncated splice isoforms of BnaPAP2.A7 lack the ability to interact with bHLH proteins[29]. Accordingly, AS emerges as a crucial mechanism for regulating anthocyanin biosynthesis in rapeseed.
In this study, we combined previous transcriptome data with the genome data of L. japonica to investigate differential AS events across different growth periods. On this basis, further key secondary metabolic genes for the occurrence of AS were screened by analysis. Molecular biology techniques were used to verify gene expression before and after splicing at different developmental stages, elucidating the impact of AS events on L. japonica quality. Our study further expands the current understanding of AS and provides a new theoretical basis for the growth and quality improvement of L. japonica.
-
The L. japonica plants used in this experiment were grown in the Pingyi outdoor cultivation base (Shandong Province, China) without exposure to extreme drought, plant diseases, and insect pests. Flowers of L. japonica were collected at five different developmental stages, named juvenile bud stage (JBS), third green stage (TGS), complete white stage (CWS), silver flowering stage (SFS), and gold flowering stage (GFS). Flowers of the same development stage from at least 10 plants were combined to form a single biological replicate. Collected plant materials were frozen in liquid nitrogen and stored at −80 °C for further analysis.
RNA extraction
-
RNA was extracted from the flowers of L. japonica and transcriptome analysis was performed following a previously described protocol[8]. Initially, 100 mg of each flower sample was ground to powder in liquid nitrogen. Total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen, Hilden, Germany) and the quantity and quality were measured using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). RNA was then reverse-transcribed using a cDNA synthesis kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. The obtained cDNA was used for the following PCR verification experiment.
Transcriptome data acquisition and annotation
-
The raw data of transcriptome sequences from five flowering stages of L. japonica were obtained from NCBI Sequence Read Archive under project PRJNA507904 (
www.ncbi.nlm.nih.gov/sra/?term=PRJNA507904 ) deposited by our previous publication[9]. Clean reads were aligned with the reference genome via HISAT2[30]. Single-gene sequences were compared with Viridiplantae database (taxid: 33090) from the UniProt using BLASTX, with a cutoff E-value of 10−5. Single-gene sequences that could not be aligned with the database were scanned for coding region nucleotide (5'-3') and amino acid sequences using ESTScan[31]. The value of fragments per kilobase of transcript per million mapped reads (FPKM) was calculated using cufflinks[32] to represent the expression level of genes.Identification of alternative splicing events
-
A paired comparison of five flowering stages of RNA transcriptome data was performed to identify AS events. The reference genome of L. japonica (PRJCA001719) was obtained from China National Center for Bioinformation (
https://bigd.big.ac.cn/gwh/Assembly/660/show ). rMATS (http://rnaseq-mats.sourceforge.net/index.html ) software was used for identifying the differential AS events with the default parameters (adjusted p < 0.05 and |ΔPSI| > 10). All AS events were matched with the corresponding five major types of AS events.Function annotation and enrichment
-
Gene functions were analyzed by using MapMan bin codes[33]. Predictions of identified genes from L. japonica were performed by converting annotations to the Arabidopsis genome and considering orthologous genes. Pathway mapping of identified genes was conducted using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (
www.genome.jp/kegg )[34].Validation of alternative splicing events by PCR
-
PCR reactions were performed for the amplification of gene fragments by the designed primers (listed in Supplementary Table S1) using the PCR Reaction Kit (Accurate) and Instrument (Applied Biosystems, CA, USA), in which samples at the SFS stage were used. For further validating the alternative splicing events, the target fragments were purified and recovered using a Gel Extraction Kit (Accurate) and then recombined into the pET28 vector. PCR amplification reaction was performed again using the primers and the following sequencing analysis was conducted.
Quantitative real-time PCR analysis
-
To verify the expression of selected genes at different flowering stages, Primer 5.0 software was used to design primers (Supplemental Table S1) and qRT-PCR was conducted using ABI 7500 fluorescence quantitative PCR instrument (Applied Biosystems, CA, USA). The SYBR® Green Pro Taq HS qPCR Kit (Accurate Biotechnology Co., Ltd, Hunan, China) was used, and β-actin as the housekeeping gene[35]. Three biological replicates were performed on each group sample and the relative expression levels were calculated based on the 2−ΔΔCᴛ method[36].
Statistical analysis
-
The SPSS statistical software (version 22.0; IBM, Armonk, NY, USA) was used for statistical evaluation. Statistical significance was evaluated by the student's t-test when only two groups were compared or one-way ANOVA followed by Tukey's test when multiple groups were compared. A p-value < 0.05 was considered as the statistical significance. Three independent biological replicates per sample were tested in this study.
-
The RNA-seq data obtained consisted of 15 libraries divided into five groups, each representing a different flowering stage, with three biological replicates per group (Table 1). Most raw reads exceeded 30 million, except for sample GFS_1, which had 27 million reads. The highest read count was observed in sample SFS_3, with 50 million reads. The majority of total raw bases exceeded 4 gigabases (G), with sample CWS_3 having the fewest bases at 3.9 G, and sample GFS_2 having the most at 7.4 G. The average read length was over 120 bases; samples in the GFS stage had the longest reads at 148 bases, while reads from other stages ranged from 120 to 130 bases.
Table 1. Quality data of samples from each of the five stages of the L. japonica transcriptome.
Sample Raw reads (M) Raw bases (G) Raw Q20 (G) Raw Q30 (G) Clean reads (M) Clean bases (G) Clean Q20 (G) Clean Q30 (G) Average
length (bp)JBS_1 32.623 4.226 3.981 (94.2%) 3.725 (88.1%) 29.040 (89.0%) 3.654 (86.5%) 3.560 (97.4%) 3.419 (93.6%) 125.8 JBS_2 49.964 6.245 5.884 (94.2%) 5.528 (88.5%) 44.648 (89.4%) 5.431 (87.0%) 5.285 (97.3%) 5.087 (93.7%) 121.6 JBS_3 40.982 5.123 4.830 (94.3%) 4.533 (88.5%) 36.754 (89.7%) 4.464 (87.2%) 4.344 (97.3%) 4.175 (93.5%) 121.5 TGS_1 48.764 6.095 5.741 (94.2%) 5.379 (88.2%) 43.324 (88.8%) 5.281 (86.6%) 5.141 (97.4%) 4.941 (93.6%) 121.9 TGS_2 48.817 6.102 5.768 (94.5%) 5.434 (89.1%) 44.174 (90.5%) 5.377 (88.1%) 5.233 (97.3%) 5.037 (93.7%) 121.7 TGS_3 36.493 4.728 4.483 (94.8%) 4.231 (89.5%) 32.980 (90.4%) 4.176 (88.3%) 4.075 (97.6%) 3.929 (94.1%) 126.6 CWS_1 34.153 4.316 4.050 (93.8%) 3.794 (87.9%) 30.309 (88.7%) 3.702 (85.8%) 3.599 (97.2%) 3.459 (93.4%) 122.1 CWS_2 40.121 5.110 4.829 (94.5%) 4.536 (88.8%) 35.785 (89.2%) 4.425 (86.6%) 4.317 (97.6%) 4.156 (93.9%) 123.7 CWS_3 30.453 3.917 3.693 (94.3%) 3.462 (88.4%) 27.201 (89.3%) 3.407 (87.0%) 3.317 (97.4%) 3.187 (93.6%) 125.2 SFS_1 48.938 6.117 5.757 (94.1%) 5.393 (88.2%) 43.576 (89.0%) 5.295 (86.6%) 5.152 (97.3%) 4.950 (93.5%) 121.5 SFS_2 46.323 5.790 5.469 (94.5%) 5.137 (88.7%) 41.419 (89.4%) 5.054 (87.3%) 4.925 (97.4%) 4.738 (93.7%) 122 SFS_3 50.913 6.364 5.930 (93.2%) 5.519 (86.7%) 44.178 (86.8%) 5.344 (84.0%) 5.189 (97.1%) 4.978 (93.1%) 121 GFS_1 27.133 4.070 3.977 (97.7%) 3.853 (94.7%) 25.913 (95.5%) 3.834 (94.2%) 3.798 (99.0%) 3.721 (97.0%) 148 GFS_2 49.948 7.492 7.324 (97.8%) 7.099 (94.8%) 47.727 (95.6%) 7.066 (94.3%) 7.001 (99.1%) 6.861(97.1%) 148 GFS_3 38.352 5.753 5.620 (97.7%) 5.444 (94.6%) 36.600 (95.4%) 5.425 (94.3%) 5.374 (99.1%) 5.265 (97.0%) 148.2 Differential alternative splicing events occurred among different developmental stages
-
The HISAT2 software was used to map clean reads to the reference genome. The average number of clean reads for each flowering stage was as follows: JBS (36,814,019), TGS (40,159,280), CWS (31,098,199), SFS (43,057,608), and GFS (36,746,742) (Table 1). As shown in Fig. 1, the average total mapping rate for most samples exceeded 97%. The unique mapping rates for each stage were 68.61% for JBS, 77.05% for TGS, 74.50% for CWS, 62.09% for SFS, and 86.95% for GFS (Table 2). Additionally, rMAT was used to compare splicing patterns across four developmental stages (TGS, CWS, SFS, and GFS) relative to JBS, identifying all AS events. In total, 31,739 AS events (20,916 AS genes) were detected at TGS, 30,572 AS events (20,501 AS genes) at CWS, 31,147 AS events (20,632 AS genes) at SFS, and 32,071 AS events (21,020 AS genes) at GFS, compared to JBS (Fig. 2).
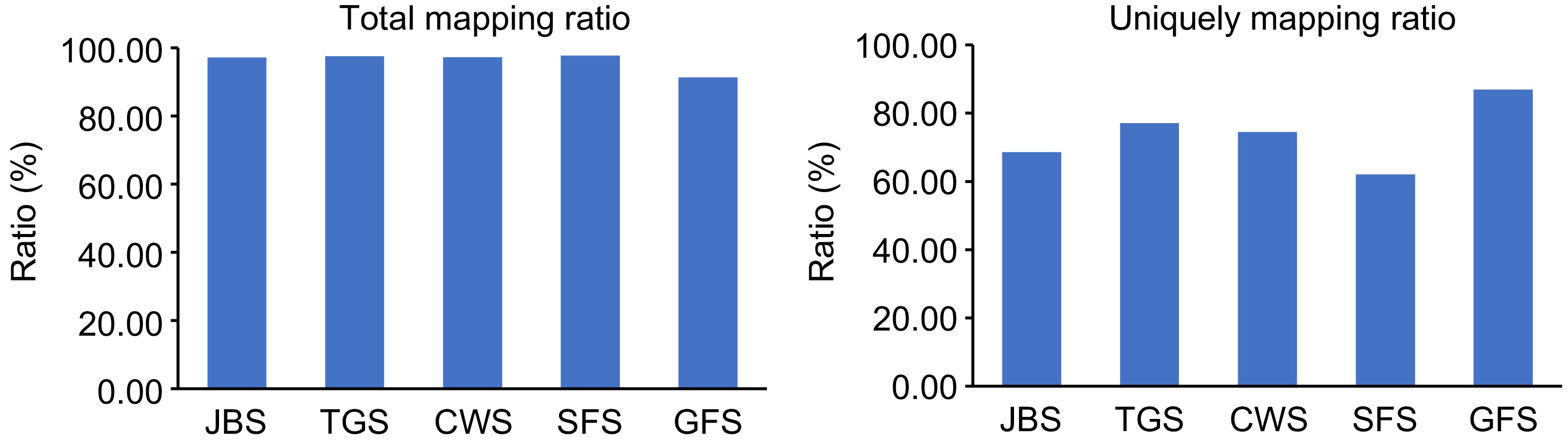
Figure 1.
Mapping of L. japonica transcriptome to the reference genome. 'Total mapping ratio' refers to the proportion of clean reads that align with the reference genome, while 'Uniquely mapping ratio' represents the proportion of clean reads that align uniquely to a specific location on the reference genome.
Table 2. Mapping of the L. japonica transcriptome to the reference genome.
Sample Total mapping ratio Uniquely mapping ratio JBS_1 96.85% 56.37% JBS_2 97.19% 81.70% JBS_3 97.32% 67.75% TGS_1 98.50% 53.61% TGS_2 96.94% 88.34% TGS_3 96.97% 89.20% CWS_1 96.66% 92.97% CWS_2 97.32% 58.13% CWS_3 97.53% 72.41% SFS_1 97.54% 62.00% SFS_2 98.39% 55.27% SFS_3 96.94% 68.99% GFS_1 91.43% 87.21% GFS_2 91.17% 86.69% GFS_3 91.25% 86.94% 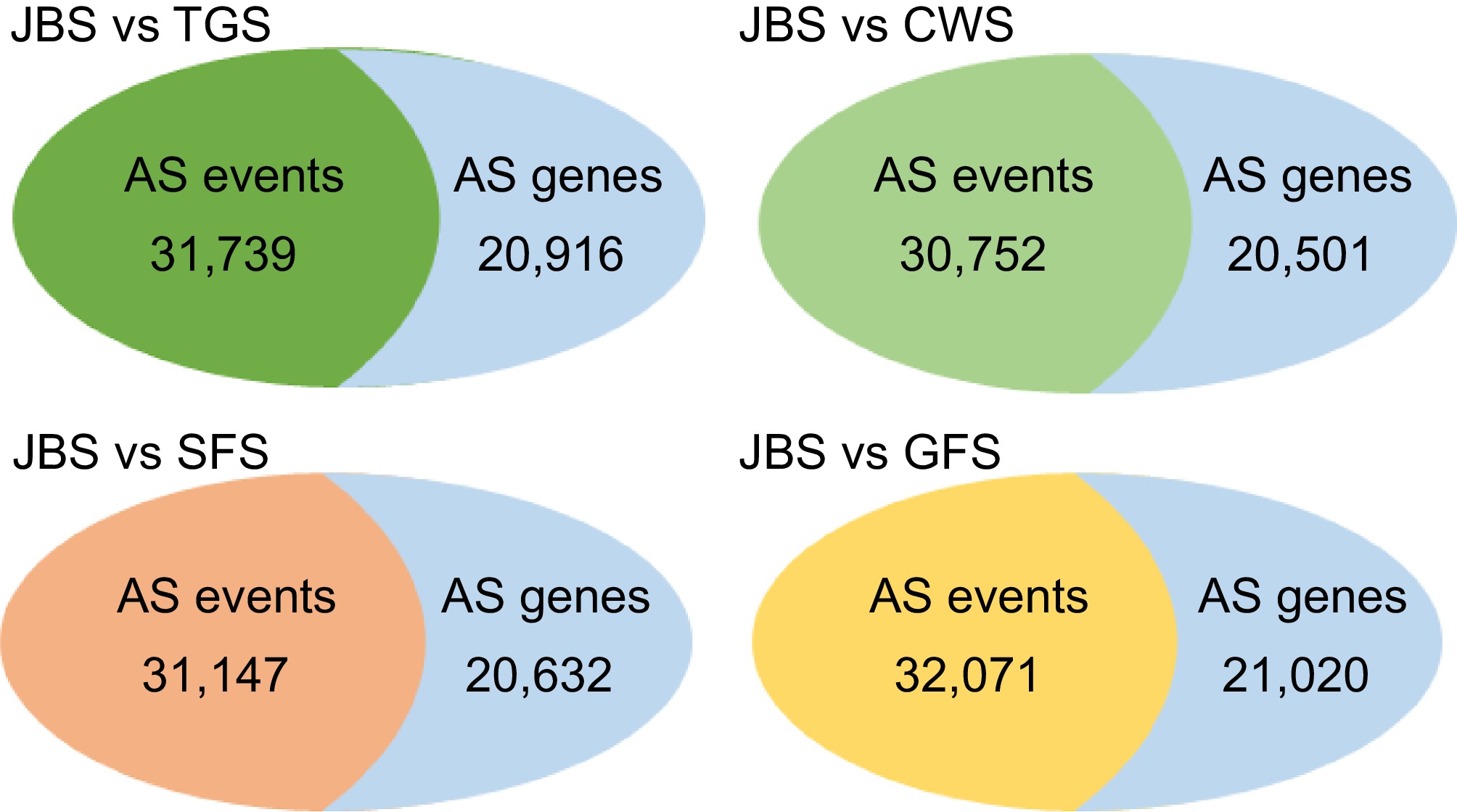
Figure 2.
Overview of the number of alternative splicing events and related genes between different stages. All of the AS events and related genes were identified through the comparison of four stages including TGS, CWS, SFS, and GFS with the first stage of JBS. rMATS software was used for identifying the differential alternative splicing events.
Distribution of AS events at different flowering stages
-
Based on the observed number of AS events, five major types of AS events were identified: exon skipping (ES), mutually exclusive exons (MXE), alternative 5' donor sites (A5SS), alternative 3' acceptor sites (A3SS), and intron retention (IR) (Fig. 3). The distribution of AS events across different flowering stages showed distinct patterns for each AS type. A total of 14,976, 15,654, 15,548, and 14,692 ES events were observed, respectively, across the different stages, making it the most prevalent type of AS event. However, the number of MXE events identified was 996, 1,136, 1,045, and 940, respectively, representing a smaller proportion of the overall AS events. A5SS events were observed 5,755, 5,799, 5,734, and 5,739 times across the stages, making this one of the more frequent types of splicing events. And the A3SS events occurred 5,617, 5,673, 5,609, and 5,585 times, respectively, showing a similar distribution to the A5SS events. From this distribution, it is evident that ES is the most prevalent AS type, representing 40% of all events. This is followed by A3SS, A5SS, IR, and finally MXE, with MXE accounting for less than 5% of total AS events (Fig. 3).

Figure 3.
Number of identified alternative splicing events at different periods. The left side represents a schematic of the five AS types identified, with the white boxes representing the constitutive regions, and the grey representing the alternative regions. The four coloured blocks on the right represent the number of events in which several types of AS occurred
MXE and ES are important at the TGS and SFS stages
-
To gain deeper insights, a comparative analysis of differential AS events was performed between JBS vs TGS and JBS vs SFS, revealing distinct patterns (Fig. 4). In the JBS vs TGS comparison, 177, 150, 292, 18, and 2,222 differential AS events were identified for A3SS, A5SS, MXE, IR, and ES, respectively. In the JBS vs SFS comparison, these numbers were 185, 171, 243, 18, and 1,650 for the same AS types. Notable variations in the discrepancy proportion among the different AS types were observed. MXE exhibited the highest discrepancy proportion, approximately 25%, reaching 27.94% in JBS vs TGS, and 24.40% in JBS vs SFS. ES displayed the second-highest discrepancy proportion, ranging from 10%–15%, with values of 14.29% in JBS vs TGS, and 11.40% in JBS vs SFS. A3SS and A5SS showed a more moderate discrepancy proportion, around 3.50% and 2.90%, respectively; A3SS reached 3.16% in JBS vs TGS, and 3.29% in JBS vs SFS, while A5SS peaked at 2.62% in JBS vs TGS, and 2.92% in JBS vs SFS. IR presented the lowest discrepancy proportion, around 0.47% in both comparisons. Based on these results, ES and MXE were selected for further analysis due to their higher frequency of AS events and greater discrepancy proportion, with MXE showing the largest difference proportion among all AS types.
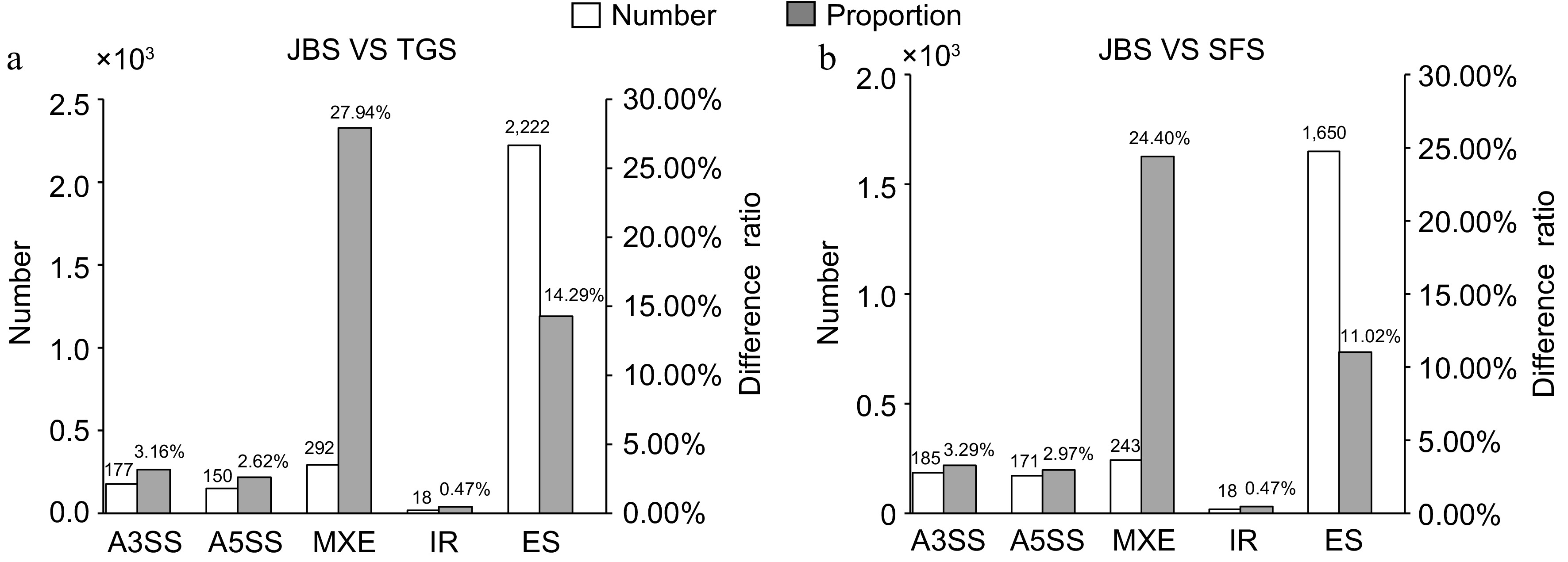
Figure 4.
Distribution of differential alternative splicing events at different flowering stages. The number and proportion of differential alternative splicing events from JBS vs TGS, and JBS vs SFS were calculated. The left vertical axis represents the number of differential alternative splicing events, while the right vertical axis represents the proportion of differential alternative splicing events.
Function of MXE
-
To explore the impact of AS on physiological metabolism in L. japonica, we conducted a statistical screening of genes exhibiting variable splicing across the five flowering stages. The analysis focused intensively on genes undergoing ES and MXE splicing events. Genes with MXE splicing events were identified specifically in TGS and SFS when compared to JBS. Functional analysis of these genes revealed distinct patterns of enrichment. Genes with MXE splicing events (GMXEs) identified in TGS were predominantly involved in protein metabolism, RNA metabolism, transport, DNA metabolism, and development. In contrast, GMXEs identified in SFS were primarily associated with protein metabolism, lipid metabolism, RNA metabolism, secondary metabolism, and signaling/stress/amino acid metabolism. Notably, genes related to lipid metabolism and secondary metabolism were more abundant in JBS than in SFS. Additionally, GMXEs specifically involved in DNA metabolism, cell wall formation, major carbohydrate metabolism, photosynthesis, and mitochondrial electron transport were uniquely identified in TGS (Fig. 5).
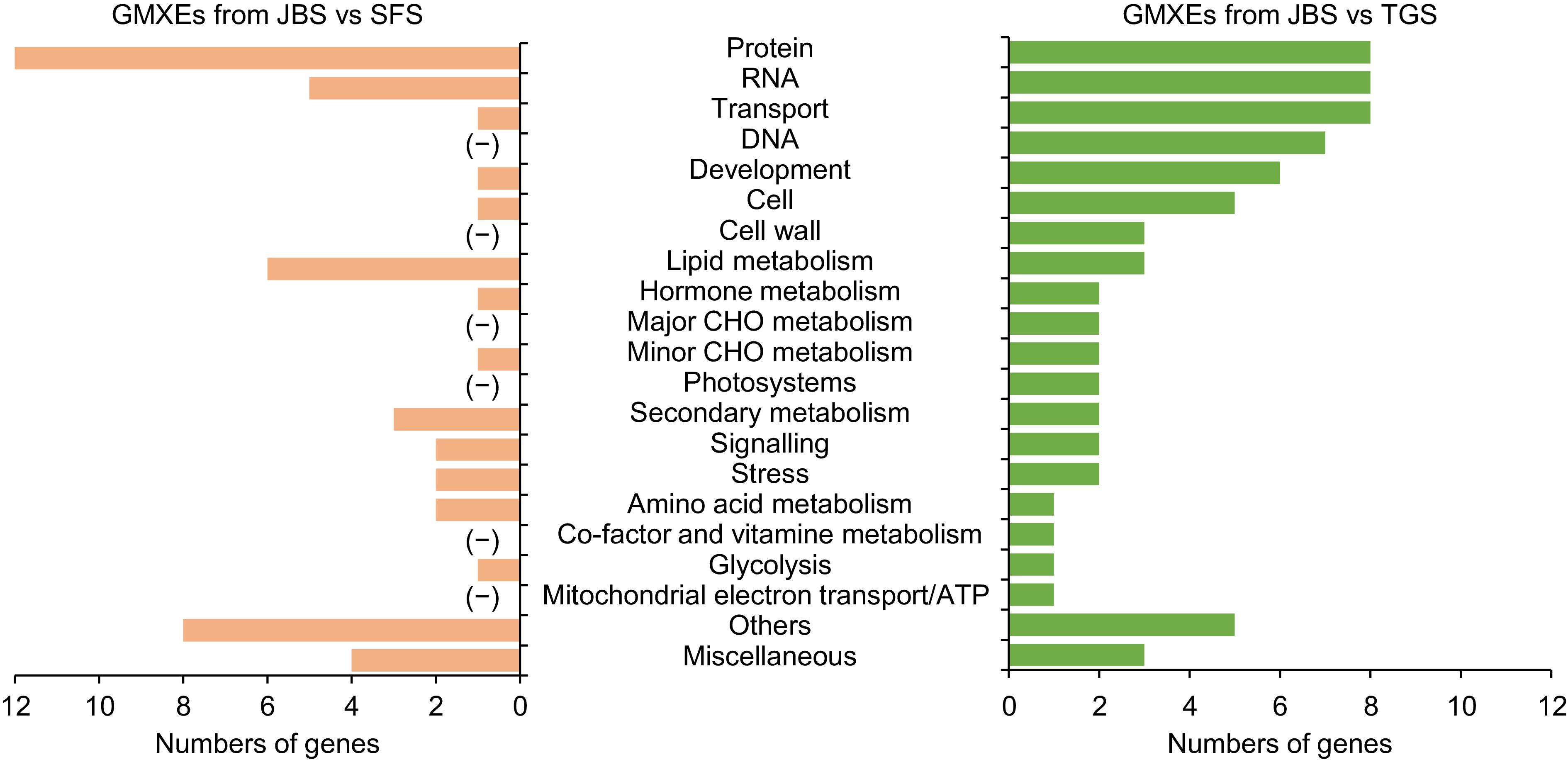
Figure 5.
The function of genes involved in mutually exclusive exon events. Gene functions were predicted and categorized using MapMan bin codes. '(−)' indicates that no gene was categorized. Abbreviations: CHO, carbohydrate.
Function of ES
-
To investigate the impact of ES events on physiological metabolism, genes modified by ES events (GES) were functionally categorized. GESs identified in TGS and SFS were primarily enriched in protein metabolism, RNA metabolism, cell metabolism, transport, and signaling pathways. Additionally, the number of GESs associated with development, DNA metabolism, lipid metabolism, amino acid metabolism, and major carbohydrate metabolism were significantly lower in SFS compared to TGS. Secondary metabolism played a notable role, with 14 genes associated with secondary metabolic functions in the JBS vs TGS comparison, and 18 genes in the JBS vs SFS comparison (Fig. 6). The gene annotations were submitted to the KEGG database for enzyme identification. Four key enzymes closely related to secondary metabolism were identified: tyrosine decarboxylase (TYDC), chalcone isomerase (CHI), strictosidine synthase (STR), and 2'-hydroxyisoflavone reductase (IFR). Interestingly, TYDC and CHI were found in the TGS group but not in the SFS group, suggesting that ES events may occur specifically in TGS, involving metabolic pathways related to aromatic amino acid metabolism, isoquinoline alkaloid biosynthesis, and flavonoid biosynthesis. Conversely, STR and IFR were present in the SFS group but absent in the TGS group, indicating that ES events likely occur only in SFS, affecting metabolic pathways associated with indole alkaloid biosynthesis and isoflavonoid biosynthesis.
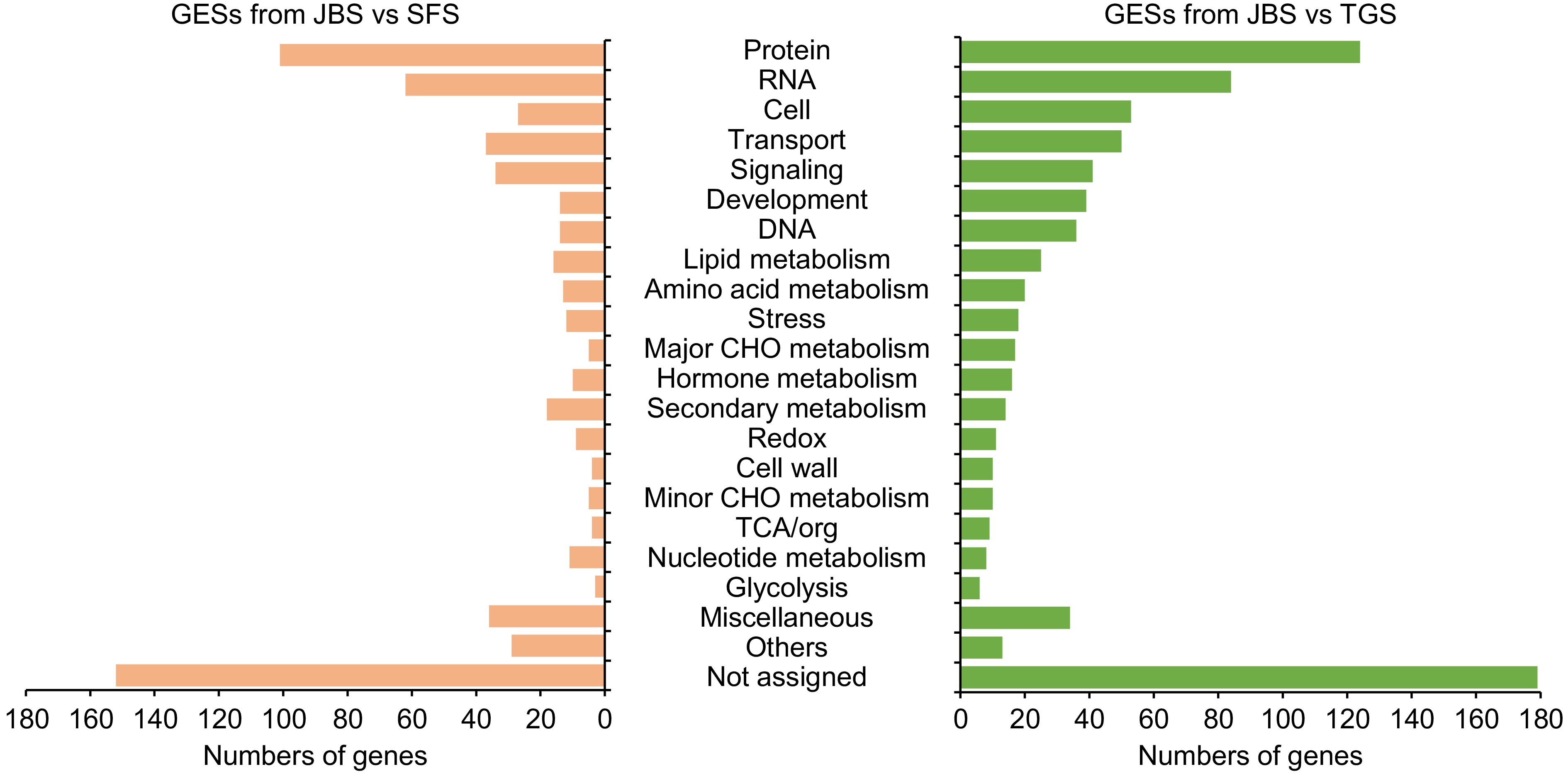
Figure 6.
The function of genes involved in exon skipping events. Gene functions were predicted and categorized using MapMan bin codes. Abbreviations: TCA, tricarboxylic acid.
Metabolic pathway analysis
-
The metabolic pathways involving the four key genes were summarized and analyzed, revealing two main pathways: flavonoid metabolism and alkaloid metabolism (Fig. 7). In the flavonoid metabolic pathway, the process begins with phenylalanine, which is converted into the intermediates cinnamoyl-CoA and p-coumaroyl-CoA to isoliquiritigenin. This compound is then catalyzed by CHI to form liquiritigenin. Through a series of complex biochemical reactions, liquiritigenin is further transformed into daidzein and 2'-hydroformononetin, which are subsequently synthesized into (−)-vestitone, catalyzed by IFR, thus completing the flavonoid metabolic pathway. In the alkaloid metabolic pathway, both tyrosine and tryptophan contribute to the alkaloid formation, with tyrosine and phenylalanine capable of interconversion. Tyrosine is catalyzed by TYDC to form tyramine, which subsequently leads to dopamine and progresses further along the alkaloid pathway. Meanwhile, tryptophan is converted to tryptamine, which is then transformed into 3-alpha(S)-strictosidine by STR, entering the downstream steps of the alkaloid biosynthesis pathway.
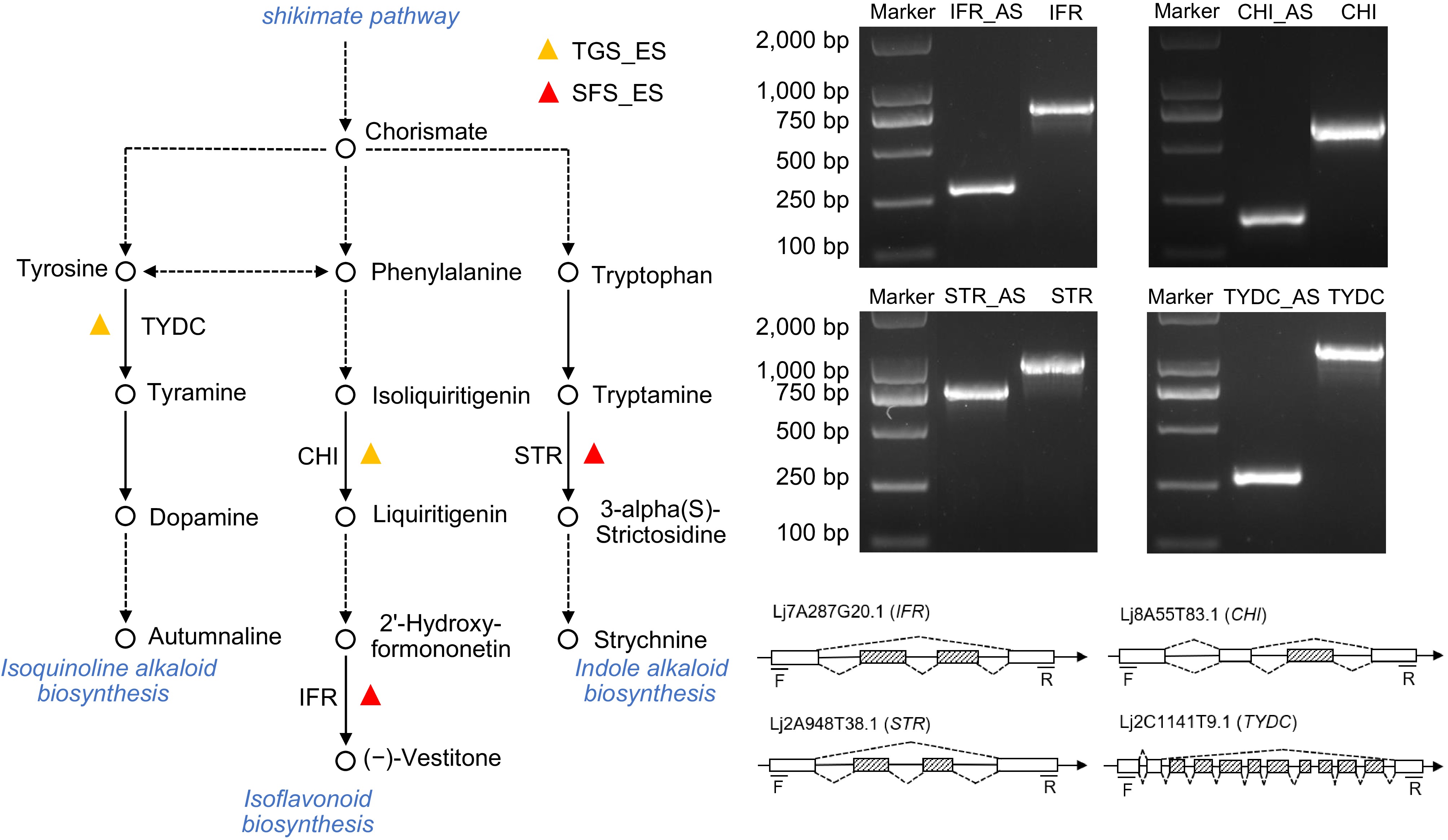
Figure 7.
The metabolic pathway mapping and structure validation of exon skipping event genes. The ES genes were mapped to the metabolic pathways using the KEGG database. Hollow circles represent compounds in the pathway, while solid black lines represent a single reaction step and dotted lines represent multiple steps. Gene structure was analyzed by PCR and sequencing to identify splicing variants. Boxes represent the exons, boxes filled with dashed lines represent the splicing exons, solid lines represent the introns, and dotted lines indicate the splicing sites of introns.
AS event validation
-
In the previous analysis, four genes were screened: IFR, CHI, STR, and TYDC. The first two are associated with flavonoid metabolism, while the latter two are linked to alkaloid metabolism. To confirm the accuracy of these findings, the sequence structures of these genes were validated. Primers were designed to amplify potential variants based on the identified AS types in these genes. Due to the presence of ES events, a homologous recombination strategy was employed, connecting the target gene with or without the corresponding spliceosome sequence in a vector. Differences in the length of PCR products from the same gene were used to assess the occurrence of AS events (Fig. 7). Sequencing technologies were then utilized to confirm the produced isoforms and the AS types of these genes. PCR analysis of the IFR gene with the same primer pair produced two products, approximately 250 and 750 bp in length. Sequencing results confirmed that the IFR gene underwent a two-ES splicing event. Additionally, PCR and sequencing analyses validated the occurrence of ES events during the transcription of the other three genes, CHI, STR, and TYDC (Fig. 7, Supplemental Figs S1 & S2).
qRT-PCR of several AS genes
-
To further investigate the relationship of AS events of the selected genes with their potential functions, the expression levels of AS-modified transcripts of the IFR, CHI, STR, and TYDC genes were analyzed using qRT-PCR (Fig. 8). The IFR_AS transcript exhibited low expression levels during the first three stages, followed by a significant increase in the SFS and GFS stages. CHI_AS expression was significantly higher during the TGS, CWS, and GFS stages compared to the JBS and SFS stages. The STR_AS gene showed an initial slight decrease in expression, followed by a rapid increase, reaching its peak during the GFS stage. In contrast, TYDC_AS had the highest expression levels at the early flowering stage, followed by a steady decline throughout the later flowering stages.
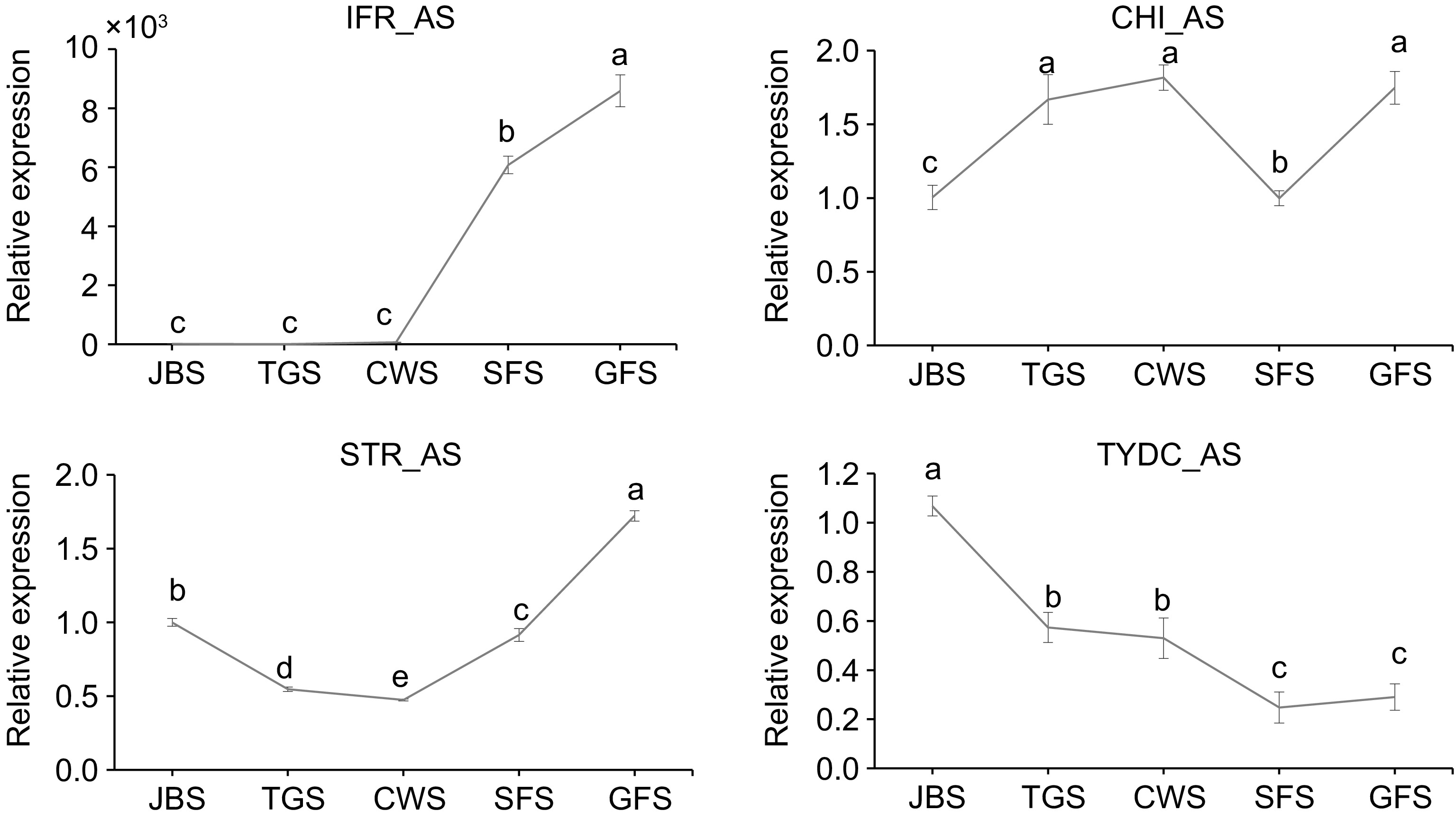
Figure 8.
Expression levels of genes involved in AS events at different flowering stages. RNAs were extracted and the expression of genes was measured by qRT-PCR. The data are presented as the mean ± SD from three independent biological replicates. Means with the same letter were not significantly different among floral stages, while different letters indicate that the change is significant according to Tukey's multiple comparison (p < 0.05).
-
The flowers of L. japonica, the traditional Chinese medicinal plant under investigation, are of particular interest due to their medicinal properties. AS is recognized as a critical mechanism for post-transcriptional regulation of floral development[37]. In plants, IR is the most prevalent type of AS, while MXE splicing events are less frequent[38]. Studies have shown that approximately 60% of introns in Arabidopsis undergo variable splicing, with IR accounting for about 40% of AS events[39,40]. However, IR is not always the most predominant type. Sometimes plants may favor ES for precise protein isoform regulation under stress[41] or rapid developmental transitions[42]. Notably, a high frequency of AS events occur in flowers further suggesting a key role for AS in regulating the development in Arabidopsis[38]. In our study, we analyzed the AS events across different flowering stages of L. japonica by combining de novo sequencing with genomic data. Specifically, the results indicated that ES events were the most prevalent, constituting 40% of the total AS events observed (Fig. 3). A comparison between normal temperature incubation (22 °C) with cold stress (4 °C) in Arabidopsis thaliana exhibited a higher occurrence of IR events, with a lesser proportion of ES events[43]. Jabre et al. also arrived at similar results, but it is notable that for ES events alternative regions with higher inclusion levels (higher PSI values) display more nucleosome occupancy across the splice sites[44]. CRISPR/Cas9 technology was employed to induce ES in rice betaine aldehyde dehydrogenase (BADH), resulting in the creation of BADH-2, which enhances rice quality by imparting flavor during its maturation[45]. Additionally, under drought conditions, the third exon of the proline synthesis enzyme in Arabidopsis thaliana undergoes ES, leading to proline accumulation and indicating the significance of ES as a pivotal mechanism in plant response to abiotic stressors[46]. These results above indicate that IR and ES play crucial roles in plant development, under both normal growth conditions and under stress-induced scenarios.
Dynamic roles of AS in secondary metabolism
-
It has been reported that AS serves as a significant mechanism in the regulation of secondary metabolism[23]. This is particularly evident in medicinal plants where the abundance of bioactive constituents is indicative of their quality. In Actinidia chinensis, several structural genes for anthocyanin biosynthesis, including chalcone synthase, dihydroflavonol-4-reductase, and anthocyanidin synthase are regulated by AS during fruit development[47]. In Ginkgo biloba, selective splicing regulates flavonoid biosynthesis genes and their transcriptional regulators in different tissues, thereby affecting secondary metabolism[48]. In L. japonica, the presence of flavonoids and phenolic acids is various during floral development, while some of indicative components such as chlorogenic acid and luteoloside, were highest in CWS, while lower in other developmental stages[9]. We screened variable splicing genes related to flavonoid synthesis, CHI, and IFR, and their expression trends also changed with the flowering stage. The expression level of IFR_AS was relatively low at CWS and higher in SFS and GFS, which was opposite to the changing pattern of chlorogenic acid and luteoloside. The potential interplay between AS events and the accumulation of secondary metabolites may exist, warranting further in-depth investigation.
Studies have shown variations in CHI expression within the same plant species. Notably, the inner petals exhibit higher CHI expression compared to the outer petals[49]. In Fagopyrum dibotrys, CHI expression is most prominent in flowers and least pronounced in the stems[50]. CHI has been verified its distinctive function as a unique enhancer within the flavonoids pathway[51]. Our research has unveiled differing levels of CHI expression across various stages of flowering. The expression of CHI_AS exhibited a higher value at CWS and GFS than at JBS and SFS. IFR is a key enzyme in the isoflavone synthesis pathway, controlling the synthesis of isoflavone in plants[52]. The importance of IFR has been highlighted in previous publications with regard to its significant role in plants' secondary metabolism and defense mechanisms against stressors. In soybeans, exposure to ABA triggers the upregulation of isoflavone reductase expression, aiding in the mitigation of ABA-induced stress through the synthesis of secondary metabolites[53]. During periods of flooding stress, the upregulation of genes associated with isoflavone is notably observed, suggesting a significant involvement in responding to flooding stress[54]. The present study specifically delves into the variations in IFR levels throughout the floral development of L. japonica. During the initial three stages (JBS, TGS, CWS), there was minimal disparity in the expression levels of IFR_AS. However, in the SFS and GFS stages, IFR_AS consistently exhibited higher expression levels. Furthermore, in accordance with the research conducted by Yang et al.[55], we can infer that CHI_AS and IFR_AS play an essential role in the biosynthesis of flavonoids during the flower development of L. japonica. While investigating flavonoids, we also examined genes undergoing variable splicing with the alkaloid biosynthetic pathway. The genes STR and TYDC, critical components located upstream of indole alkaloids and isoquinoline alkaloids, are subject to variable splicing. Consequently, it is plausible to postulate that AS is capable of influencing the secondary metabolism of L. japonica.
-
The objective of this study was to identify the specific genes engaged in AS and the corresponding AS events that transpire during the flowering phase of L. japonica flowering utilizing the plant's transcriptome and genome. Comparative analysis between the first flowering stage with the other four stages revealed 20,916, 20,501, 20,632, and 21,020 genes undergoing AS, accompanied by 31,739, 30,752, 31,147, and 32,071 AS events. TGS and SFS were selected for further examination based on variations in active components across different flowering stages. Upon comparing these stages, the AS type exhibiting the greatest disparity in occurrence (ES) and the highest ratio of differences in AS events incidence (MXE) was singled out for examination. Subsequent gene function analysis and categorization of genes in these AS forms led to the selection of genes linked with secondary metabolism for detailed investigation. Our findings imply that AS may be essential for regulating secondary metabolism at distinct developmental phases of L. japonica. Our study elucidates the correlation between alterations in secondary metabolism and AS events during L. japonica flowering. These results not only enrich our comprehension of the intricacies of L. japonica in secondary metabolism regulation but also establish a platform for further investigation into the regulatory mechanisms governing active component synthesis.
This work was supported by the Zhejiang Province Key R&D Program Project (Grant No. 2023C04019), the Research Initiation Funding of Zhejiang Sci-Tech University (Grant No. 23042215-Y), Zhejiang Province Basic Public Welfare Research Program (Grant No. LGC22H200012), and Zhejiang Xinmiao Talents Program (Grant No. 2023R406037).
-
The authors confirm contribution to the paper as follows: study conception and designed: Zhang L, Yang B; writing - draft manuscript: Chen J, Tian X, Yasmeen F; writing - manuscript revision and editing: Zhang L, Zhang N; language polishing: Yasmeen F; ES events identification: Tian X; transcriptomic and genomic data analysis: Ni H; bioinfomatic analysis: Yang H; experiments performed: Chen J, Tian X. All of the authors have read and agreed to the final published version of the manuscript.
-
The data used and analyzed in the current study are available from the corresponding author on reasonable request. The sequences used for de novo assembly have been deposited in the NCBI Sequence Read Archive under project PRJNA507904 in our previous work (www.ncbi.nlm.nih.gov/sra/?term=PRJNA507904).
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Jie Chen, Xu Tian
- Supplementary Table S1 The sequence of primers used in this study.
- Supplementary Fig. S1 Gel electrophoresis analysis of PCR products for validating the alternative splicing events in genes.
- Supplementary Fig. S2 Sequence alignment of the amplified fragments of the alternative splicing isoforms.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Chen J, Tian X, Weng X, Ni H, Yang H, et al. 2025. Transcriptomic analysis to understand the alternative splicing event on regulating secondary metabolism during flowering stages of Lonicera japonica. Medicinal Plant Biology 4: e014 doi: 10.48130/mpb-0025-0011 

Transcriptomic analysis to understand the alternative splicing event on regulating secondary metabolism during flowering stages of Lonicera japonica
- Received: 13 February 2025
- Revised: 15 March 2025
- Accepted: 20 March 2025
- Published online: 06 May 2025
Abstract: Lonicera japonica Thunb. is an important traditional Chinese medicinal resource. The synthetic pathways, as well as the functional genes involved in the biosynthesis of phenolic acids and flavonoids, have been intensively studied; however, the mechanism of post-transcriptional regulation during the flowering stages remains largely unexplained. In this study, the L. japonica transcriptome and genome were used to identify the alternative splicing (AS) events that occur during L. japonica flowering and the relevant genes. A total of 31,739, 30,752, 31,147, and 32,071 AS events were identified based on a comparison of flowering stages with the first flowering stage, and 20,916, 20,501, 20,632, and 21,020 genes were involved in the AS events. Exon skipping (ES) has been identified as the most frequent type of alternative splicing event, whereas mutually exclusive exons (MXE) are typically among the least frequently occurring types. Functional analysis of genes underlying the two types of AS events showed these genes were mainly related to protein metabolism, RNA metabolism, cell cycle, transport, signaling, and secondary metabolism. Further pathway mapping analysis demonstrated the occurrence of AS event in chalcone isomerase (CHI), 2'-hydroxyisoflavone reductase (IFR), tyrosine decarboxylase (TYDC), and strictosidine synthase (STR) genes, which are responsible for the production of liquiritigenin, vestitone, tyramine, and strychnine, respectively. PCR and sequencing confirmed these AS events, further validating our findings. Our study elucidates the correlation between alterations in secondary metabolism and alternative splicing events during L. japonica flowering, which enhances our understanding of the complex regulation of its secondary metabolism and provides a basis for subsequent in-depth exploration of the regulatory mechanisms of active ingredient synthesis in L. japonica.
-
Key words:
- L. japonica /
- Alternative splicing /
- Exon skipping /
- Mutually exclusive exons /
- Secondary metabolism