









-
Natural products are chemical compounds synthesized through primary and secondary metabolic pathways in microorganisms, plants, and animals[1]. These compounds, especially plant secondary metabolites (SMs), play critical roles in complex biological interactions and have promising applications in medicine, pharmaceuticals, food, health care products, and the spice industy[2]. Within plants, SMs exhibit extensive diversity and can be grouped into phenolic compounds, terpenes, and nitrogen-containing compounds based on their metabolic pathways[3]. Among these, flavonoids are one of the most widespread and structurally diverse groups of phenolic compounds in plants. They are synthesized through the phenylpropanoid pathway and play essential roles in plant growth, development, and defense mechanisms[4]. Flavonoids contribute to UV protection, pathogen resistance, and symbiotic interactions, in addition to their roles in plant pigmentation and signaling pathways[5]. They are particularly notable for their applications in anti-inflammatory, antibacterial, antiallergic, anticancer, and disease prevention and treatment scenarios[6−8]. Anthocyanins, as a subgroup of flavonoids, are widely present in plants and are crucial in plant pigmentation and stress responses. They also act as antioxidants, promoting human health[7]. Over the past decade, anthocyanin biosynthesis has garnered significant attention. However, pathway regulation and artificial intervention in medicinal plants remain at an early stage. Understanding the dynamic changes and metabolic networks of SMs in plants provides a theoretical basis for plant genetic improvement and SM production applications.
Cornus wilsoniana is a deciduous shrub belonging to the genus Cornus, native to the northern temperate regions of China[9]. It blooms in clusters of white flowers in spring and bears purple-black drupes in winter. C. wilsoniana boasts distinctive growth traits and a wide array of morphological features that render it invaluable for numerous applications. Its smooth, mottled bark, with patches that peel away, along with uniquely shaped leaves, flowers, and fruits, makes it a standout choice for landscape ornamentation, ecological afforestation, and various economic pursuits such as oil extraction and timber processing. Furthermore, its exceptional adaptability allows it to flourish in a range of soil conditions, including alkaline, neutral, slightly acidic, and mildly saline-alkaline, thus supporting its extensive cultivation. Most notably, the tree's remarkable longevity and continuous growth provide a stable, long-term resource for economic use while also positioning it as a highly promising candidate for both ecological afforestation and medicinal cultivation. C. wilsoniana is a highly valued medicinal plant abundant in lipids, flavonoids, organic acids, and terpenes. Its high concentrations of unsaturated fatty acids have been demonstrated to play significant roles in lipid-lowering and fatty acid supplementation[10]. However, current research on C. wilsoniana primarily focuses on its oil content[9], leaving the dynamic changes of SMs and their regulatory networks during its developmental processes insufficiently understood.
The biosynthesis of SMs is typically confined to specific plant tissues and occurs at distinct developmental stages[11]. Floral organs provide an excellent model system for studying plant development and metabolism, as they harbor a diverse array of bioactive metabolites with significant health benefits. Notably, in addition to their fundamental role in reproductive success, floral organs serve as dynamic hubs for the synthesis and accumulation of SMs, such as anthocyanins, which contribute to pigmentation, pollinator attraction, and defense responses[12]. In recent years, increasing attention has been directed toward understanding the genomic and metabolic regulation of SM biosynthesis during flower development. Studies have demonstrated that genes involved in the biosynthesis of SMs, particularly flavonoids, terpenoids, phenolics, and polysaccharides, are highly active throughout floral development[13−15]. These metabolic pathways are tightly regulated by complex molecular networks, including transcription factors, epigenetic modifications, and hormonal crosstalk, which collectively orchestrate the spatiotemporal expression of key biosynthetic genes.
Moreover, floral organ development is influenced by a range of intrinsic and environmental factors, such as petal movement, reversible expansion and contraction of petal cells, and external cues like temperature and light availability[16]. Variations in fruit yield and quality traits have been linked to differential gene expression patterns established during floral organogenesis[17]. Additionally, metabolomic and transcriptomic analyses indicate that different plant genotypes exhibit distinct SM expression profiles during flower development, highlighting the role of genetic background in shaping metabolic diversity[18]. For instance, short-term heat stress (2 h at 38°C) in Solanum lycopersicum pollen led to a substantial increase in flavonoid content, including Kaempferol dihexoside, Quercetin-3-O-rutinoside, and Kaempferol-3-O-rutinoside, suggesting that environmental stressors can modulate SM biosynthesis in a genotype-dependent manner.
Collectively, investigating the dynamic regulation of SM biosynthetic pathways and their associated regulatory networks during floral bud development, particularly in medicinal plant species, holds promise for uncovering novel strategies to enhance their pharmacological potential and adaptive resilience. The rapid advancement of genomic sequencing technologies has significantly enhanced our ability to investigate species evolution and unravel complex biological regulatory mechanisms[19]. Meanwhile, mass spectrometry provides critical chemical evidence to decode metabolic networks[20]. The integration of transcriptomics and metabolomics offers a powerful approach to elucidate the intricate relationships between genes and metabolites, shedding new light on the regulatory dynamics of secondary metabolism. To elucidate the metabolic mechanisms underlying flavonoid biosynthesis in C. wilsoniana, we conducted multi-omics analyses across different genotypes during flower bud development. By integrating transcriptomic and metabolomic data, we visualized the flavonoid metabolic network in flower bud tissues of C. wilsoniana and identified key genes involved in flavonoid biosynthesis and floral organ development.
Our findings identified 17 genes potentially involved in the complete anthocyanin and proanthocyanidin biosynthesis pathways of C. wilsoniana. This work provides theoretical support for developing the medicinal and health-promoting potential of C. wilsoniana and offers new insights into the study of SMs and functional genomics in the Cornaceae family.
-
The biological samples utilized in this study were sourced from C. wilsoniana tissue materials, which were procured from the cultivation field of the seed resource bank at Guangdong Academy of Forestry, Guangdong, China (113°38′0″ E, 23°20′0″ N). The study encompassed floral buds at distinct developmental stages: the calyx differentiation stage (H1 and L1), the pistil differentiation stage (H2 and L2), and the stamen differentiation stage (H3 and L3). These stages were examined in two genotypes characterized by high yield (H) and low yield (L) under the same growing conditions. Specifically, after fruiting, we classified plants with the highest fruit production as high-yield, while those that did not bear fruit were designated as low-yield. A total of three independent biological replicates were collected for each sample. All gathered materials were rapidly frozen using liquid nitrogen and subsequently stored at −80 °C until they underwent RNA sequencing and metabolite extraction procedures.
Transcriptome sequencing and analysis
-
Samples were collected and subjected to RNA isolation and purification using the pBIzol kit (BIOFLUX, Hangzhou Bori Technology, Hangzhou, China). Subsequently, cDNA libraries were constructed and subjected to sequencing. Quality assessment of the extracted RNA samples was a critical step in the pipeline. This was realized through a combination of techniques. A NanoDrop ultraviolet spectrophotometer (Thermo, Waltham, MA, USA) facilitated the quantification of RNA, while the integrity of the samples was assessed using the Bioanalyzer 2100 System (Agilent, Santa Clara, CA, USA). This comprehensive evaluation assured the suitability of the RNA for downstream analysis. For the construction of cDNA libraries, we integrated ~3 μg of the pristine RNA samples. These libraries, meticulously prepared, were the foundation for subsequent sequencing endeavors. Leveraging the capabilities of the Illumina HiSeq 4000 platform, we embarked on high-throughput sequencing, generating paired-end reads spanning 150 bp. This sequencing was entrusted to the expertise of Allwegene Biotechnology Co., Ltd. (Beijing, China).
The raw sequencing reads were preprocessed using Trimmomatic v0.39 to remove adapters and low-quality sequences[21]. This quality-centric processing ensured that the downstream analysis would be based on high-fidelity sequences. To establish an intricate alignment with the reference genome of C. wilsoniana, we leveraged the STAR v2.7.10b[22]. This alignment process facilitated the accurate mapping of the processed reads, paving the way for a granular exploration of gene expression patterns. To quantify gene expression, we used RSEM 1.3.3[23] to calculate Fragments Per Kilobase of transcript per Million mapped reads (FPKM). DESeq2 v1.34[24] was used to identify differentially expressed genes with p-value ≤ 0.05 and |log2(Fold Change)| > 2.
Metabolome analysis
-
Floral bud samples at the calyx differentiation stage, pistil differentiation stage, and stamen differentiation stage were used for their metabolome analysis. Metabolites were extracted following a standardized protocol. Briefly, freeze-dried samples were ground using a mixer mill (MM 400, Retsch) at 30 Hz for 1.5 min, and 100 mg of powdered tissue was extracted in 1.2 mL of 70% aqueous methanol. The samples were incubated overnight at 4 °C, centrifuged at 12,000 rpm for 10 min, and the resulting supernatant was filtered using a 0.22-μm membrane filter (SCAA-104, ANPEL, Shanghai, China) before analysis by Ultra Performance Liquid Chromatography-Mass Spectrometry (UPLC-MS/MS). The metabolite extracts were analyzed using a UPLC-ESI-MS/MS system consisting of a UPLC system (Nexera X2, Shimadzu) coupled with a QTRAP 4500 mass spectrometer (Applied Biosystems). Chromatographic separation was achieved using an Agilent SB-C18 column (1.8 μm, 2.1 mm × 100 mm) with a mobile phase of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). A gradient elution was applied, starting with 5% B at 0 min, linearly increasing to 95% B within 9 min, maintained for 1 min, then returned to 5% B over 1.1 min, followed by equilibration for 2.9 min. The flow rate was set at 0.35 mL/min, the column temperature at 40 °C, and the injection volume was 4 μL. The QTRAP 4500 operated in multiple reaction monitoring (MRM) mode with both positive and negative ionization; the electrospray ionization (ESI) parameters were a source temperature of 550 °C, ion spray voltages of 5,500 V (positive) and −4,500 V (negative), a curtain gas at 25 psi, and a high setting for the collision gas.
Data acquisition and peak integration were performed using Analyst v1.6.3 software (AB Sciex). Metabolite identification was conducted using an in-house database together with the KEGG Compound database (
www.genome.jp/kegg )[25], matching peaks based on retention time, molecular weight, and fragment ions, while quantification was performed in MRM mode by integrating and normalizing peak areas. Raw LC-MS data were processed using Analyst v1.6.3 (AB Sciex), with metabolite intensities normalized by unit variance scaling (UV scaling) to ensure comparability, and quality control (QC) samples were analyzed at regular intervals to assess reproducibility and instrument stability. To conduct the Uniform Manifold Approximation and Projection (UMAP) analysis, we used the DESeq2[24] v1.34 package to identify significantly changed metabolites, with a threshold of |log2(Fold Change)| ≥ 2 and p-value ≤ 0.05.Weighted Gene Co-Expression Network Analysis
-
We constructed a gene co-expression network for the flower bud developmental stage of C. wilsoniana using WGCNA v1.73[26]. We calculated the Pearson correlation coefficients between the target genes based on the count matrix of the genes by converting the correlation matrix into a neighbor-joining matrix. Hierarchical clustering and dynamic tree cut function were used for module detection to cluster all differentially expressed genes. For high reliability of the results, the minimum number of genes was set to 30 and the sensitivity was set to 4.0. Gene significance and module affiliation were calculated to correlate the individual modules with the phenotypic data (assay content of SMs) and to explore the core genes. Information on the corresponding module genes that were highly correlated with flavonoids was extracted, and linear gene pairs with weights ≥ 0.45 were filtered for connectivity statistics and network construction.
Genes involved in anthocyanin biosynthesis and analysis
-
To elucidate the genetic components controlling the flavonoids, we performed a meticulous search and comparative analysis of genes in the relevant modules. The basis for this analysis was the comprehensive UniProt Reference Clustering (UniRef) database (
www.uniprot.org/help/downloads ), which contains comprehensive biosynthesis-related genes in flavonoid secondary metabolism. We used DIAMOND[27] to establish the linkage between the C. wilsoniana proteome and the reference genes using stringent criteria (e-value ≤ 1e-5, comparative identity ≥ 70%, and comparative coverage ≥ 50%). After identifying the homologous genes, the expression signals of these homologs were further examined to screen for genes that are expressed in at least one stage during bud development. Using these genes, we designed the pathway for anthocyanin biosynthesis. -
The development of flower buds in C. wilsoniana marks a critical phase for metabolite accumulation, with buds at different developmental stages serving as key tissues for elucidating the tree's metabolic mechanisms. In this study, transcriptomic analysis was performed on 18 flower bud samples (three biological replicates per stage) across distinct developmental phases: sepal differentiation stage (H1 and L1), pistil differentiation stage (H2 and L2), and stamen differentiation stage (H3 and L3).
UMAP analysis indicates that biological replicates from samples at the same blooming stage exhibited high consistency (Fig. 1a), while noticeable heterogeneity was observed between the two genotypes (high yield (H) and low yield (L)) within the same period. This observation lays a foundational basis for subsequent research endeavors. Differential expression analysis identified a total of 2,013 differentially expressed genes (DEGs) across the three developmental stages. Hierarchical clustering based on the expression levels of all 2,013 DEGs uncovered dynamic expression patterns across the six groups (Fig. 1b). To elucidate the roles of DEGs in regulating biological pathways, we conducted KEGG enrichment analysis (Fig. 1c & Supplementary Table S1). These pathways suggest critical involvement in several essential processes, such as SM biosynthesis, regulation of plant development and adaptation, the structural integrity of the plant cuticle, and nitrogen utilization for metabolic efficiency. Collectively, these findings underscore the multifaceted regulatory roles of DEGs in coordinating developmental and environmental responses.
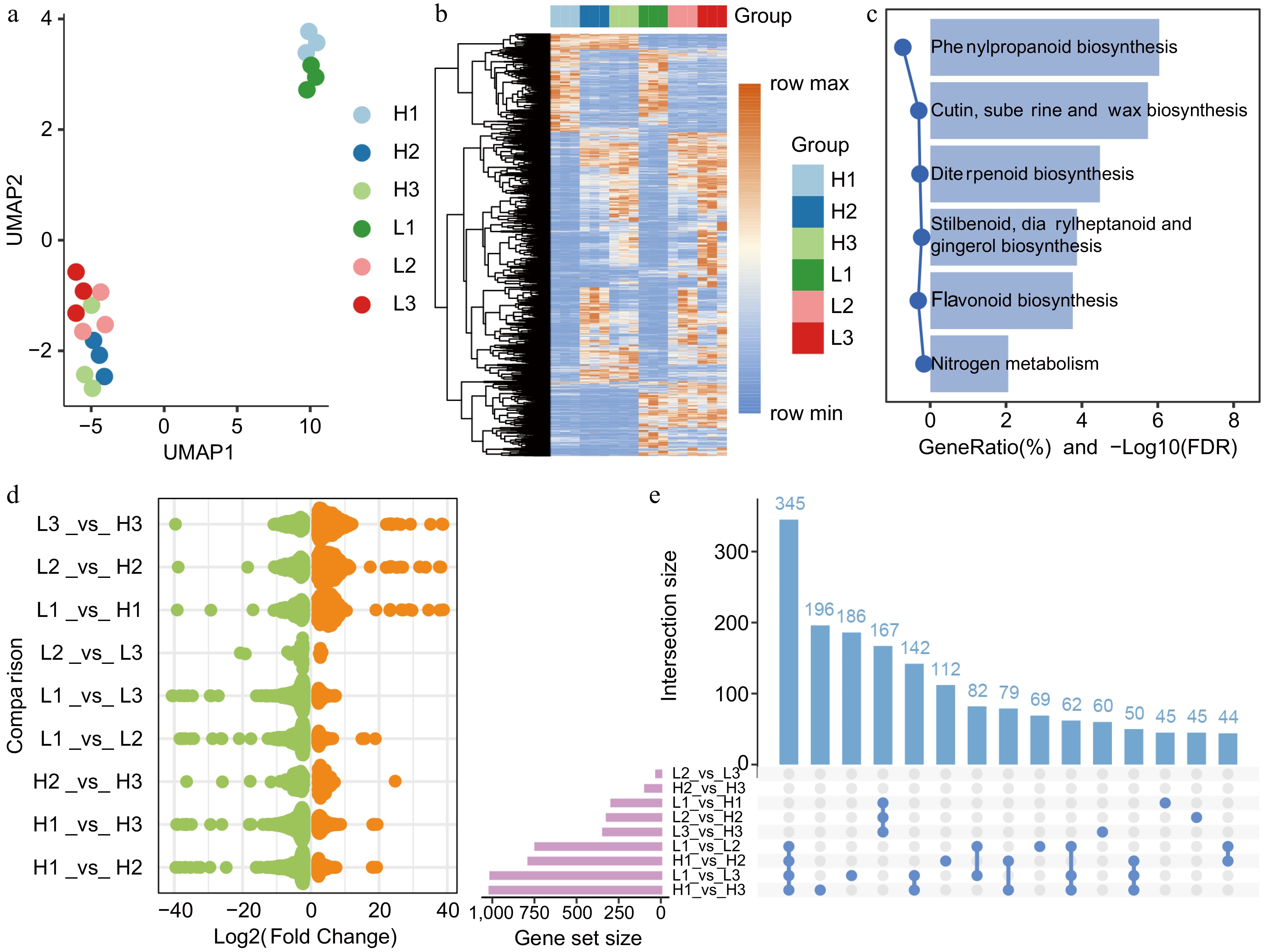
Figure 1.
Transcriptomic characteristics of flower bud developmental stages in C. wilsoniana. (a) UMAP scatterplot of flower bud samples. (b) Hierarchical clustering heatmap of the relative abundance of 2,013 DEGs. (c) KEGG enrichment analysis of 2,013 DEGs. (d) Log2(Fold Change) distribution of upregulated (orange, adjusted p-value < 0.05) and downregulated (green, adjusted p-value < 0.05) genes in different comparison groups. (e) Bar chart of DEG counts across different comparison groups.
To further examine the dynamic changes in DEGs across different groups, we performed independent DEG analyses for various developmental stage comparisons (Fig. 1d; Supplementary Fig. S1). Our analysis reveals that the differential expression profiles vary significantly across different flower bud developmental stages. Notably, a substantial number of genes are upregulated in the genotype comparison (L vs H), suggesting that genotype-specific activation of key regulatory pathways may contribute to yield differences. In contrast, most DEGs between consecutive developmental stages are downregulated, indicating a reprogramming of gene expression as the flower bud progresses from one stage to the next. The most substantial number of DEGs was observed between the sepal and stamen differentiation stages, despite differing yield phenotypes, as evidenced by the comparisons of H1_vs_H3 and L1_vs_L3 (Fig. 1e). This was followed by the largest number of DEGs observed between the sepal and pistil differentiation stages, as indicated by the comparisons of H1_vs_H2 and L1_vs_L2. Previous studies have shown that the development of floral organs is accompanied by dramatic metabolic changes, particularly in the biosynthesis of SM and lipid metabolism. DEGs from early flower bud development to full bloom are often enriched in the flavonoid biosynthesis pathway[12,15].To further characterize the distinct features of DEGs across different groups during various flower developmental stages and between different genotypes, we conducted KEGG enrichment analysis for each group and their overlapping gene sets (Fig. 1e). The results revealed that DEGs from these comparisons (H1_vs_H2, H1_vs_H3, L1_vs_L2, and L1_vs_L3) played the most significant role in SM biosynthesis pathways. (Supplementary Fig. S2 & Supplementary Table S2). Further intersection analysis of DEGs identified from H1_vs_H2, H1_vs_H3, L1_vs_L2, and L1_vs_L3 revealed 345 overlapping DEGs, which were also significantly enriched in these KEGG pathways (Supplementary Fig. S2 & Supplementary Table S3). These findings suggest that SM biosynthesis pathways play a crucial role in flower bud differentiation and development, potentially contributing to the structural and functional maturation of floral organs. The enrichment of DEGs in phenylpropanoid and flavonoid biosynthesis pathways highlights their involvement in the synthesis of key bioactive compounds, such as lignin, anthocyanins, and flavonols, which are essential for cell wall reinforcement, pigmentation, and defense responses.
Additionally, DEGs identified from L1_vs_L2 and L2_vs_L3 were also associated with 'Fatty acid elongation' and 'Pentose and glucuronate interconversions', distinguishing them from the profiles observed in H1_vs_H2 and H2_vs_H3. The distinct enrichment patterns observed between different genotypes indicate genotype-specific regulatory mechanisms governing metabolic transitions. The association of DEGs from L1_vs_L2 and L2_vs_L3 with fatty acid elongation and pentose/glucuronate interconversions suggests their potential roles in lipid metabolism and cell wall remodeling, processes that are critical for floral organ expansion, cuticle formation, and overall developmental plasticity.
Metabolomic characteristics of flower bud developmental stages in C. wilsoniana
-
To investigate the differences in metabolite composition during the flower bud development of C. wilsoniana, a differential metabolomic analysis was conducted using the same nine comparison groups employed in the transcriptomic analysis. UMAP analysis of the three biological replicates also confirmed the high reliability and reproducibility of the metabolomic data obtained from C. wilsoniana samples (Fig. 2a). Mass spectrometry analysis identified a total of 701 metabolites across all stages of flower bud development (Supplementary Table S4).
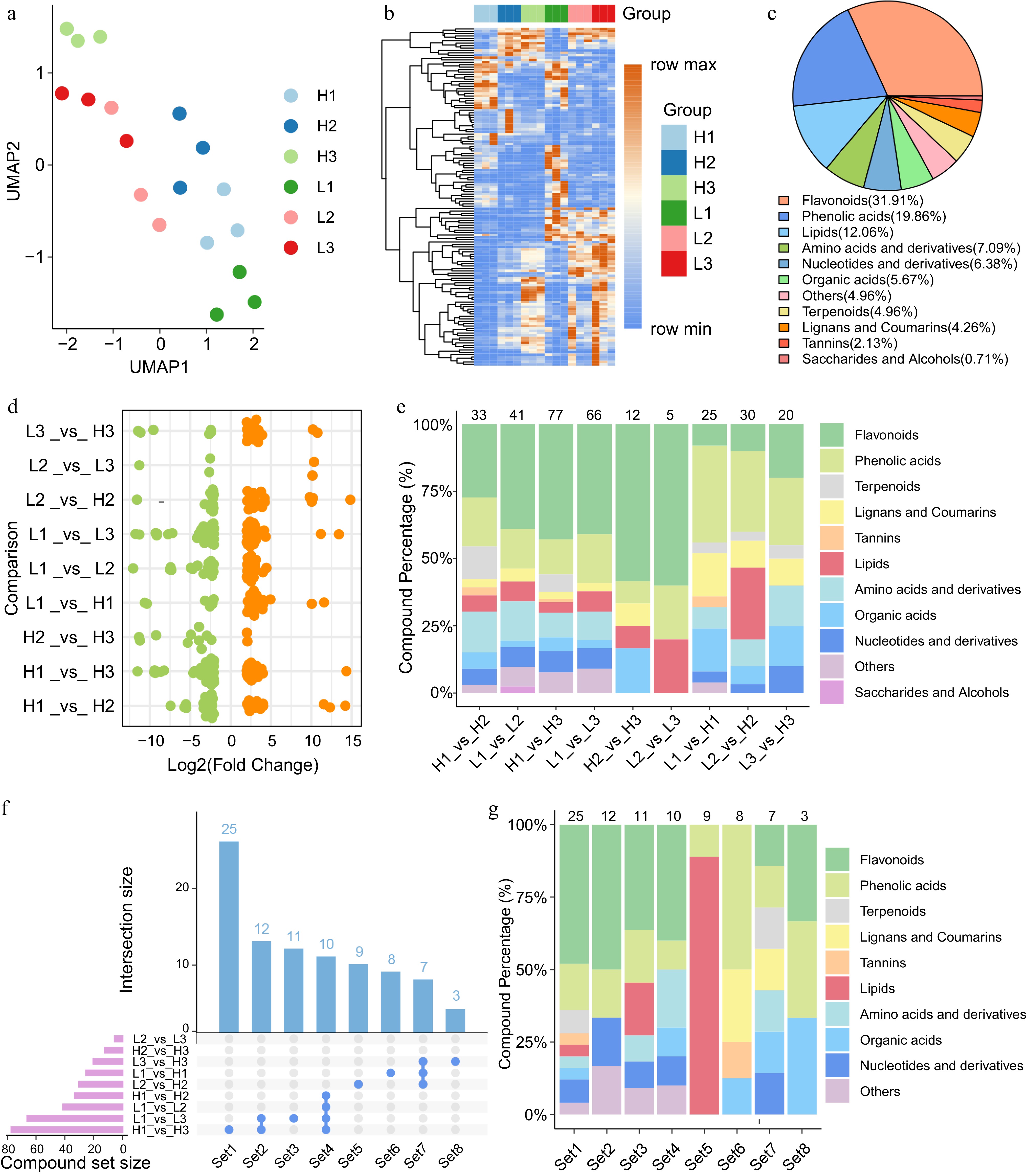
Figure 2.
Metabolomic characteristics of flower bud developmental stages in C. wilsoniana. (a) UMAP scatterplot of flower bud samples. (b) Hierarchical clustering heatmap of the relative abundance of 141 DEMs. (c) Classification of the 141 DEMs detected during flower bud developmental stages in C. wilsoniana. (d) Log2(Fold Change) distribution of upregulated (orange, Adjusted p-value < 0.05) and downregulated (green, Adjusted p-value < 0.05) metabolites across different comparison groups. (e) Trends in the proportional changes of DEM categories across the comparison groups. (f) Bar chart of DEM counts across different comparison groups. (g) Class composition of different sets of DEMs. Supplementary Table S5 lists the information for Set1−Set8.
To further identify key metabolites involved in the flower bud development of C. wilsoniana, stringent significance thresholds were applied, resulting in the identification of 141 metabolites with significantly altered abundance during this process. A heatmap of the relative abundance of these metabolites revealed significant differences in concentrations across different stages, corroborating the UMAP results (Fig. 2b). These differentially expressed metabolites (DEMs) predominantly consisted of SMs which play crucial roles in regulating developmental and physiological processes. Specifically, the DEMs included 45 flavonoids, 28 phenolic acids, seven terpenoids, six lignans and coumarins, three tannins, and a variety of other metabolites (Fig. 2c). Flavonoids and phenolic acids represented the largest groups, reflecting their critical roles in stress responses, floral pigmentation, and reproductive development. Terpenoids, lignans, and coumarins are known to contribute to signaling pathways and structural integrity[28,29].
In parallel with our transcriptomic analysis, we conducted independent analyses of DEMs across nine comparison groups (Fig. 2d, e & Supplementary Fig. S3). Notably, the comparisons between the sepal differentiation stage (H1 and L1) and the stamen differentiation stage (H3 and L3) yielded the largest number of DEMs, while the fewest DEMs were observed between the stamen (H3 and L3) and pistil (H2 and L2) differentiation stages, mirroring the trends observed in transcriptomic differential analysis. These findings imply that the most significant metabolic reprogramming occurs during the transitions from sepal differentiation to either pistil or stamen differentiation stages, likely reflecting the heightened metabolic demands and functional specialization required for floral organ development. In contrast, the minimal changes observed between the pistil and stamen differentiation stages suggest that these stages share more similar metabolic profiles, possibly due to overlapping roles in late-stage floral development. Moreover, flavonoids, phenolic acids, and terpenoids were significantly enriched among DEMs in comparisons H1_vs_H2, H1_vs_H3, L1_vs_L2, and L1_vs_L3, whereas comparison L2_vs_H2 exhibited a notable increase in the proportion of lipids.
We further analyzed the unique and shared DEMs, along with their expression profiles, across the comparison groups to uncover the underlying biological differences (Fig. 2f, g). A comprehensive analysis of the flavonoid profiles enriched in Set1 (H1_vs_H3) and Set3 (L1_vs_L3) revealed that the high-yield genotype exhibits a greater diversity of flavonoid species and a wider range of chemical modifications (Supplementary Table S5). In particular, anthocyanins and flavonoid derivatives, such as Cyanidin-3-O-(6″-O-p-Coumaroyl)glucoside and Luteolin-7-O-neohesperidoside, displayed high metabolic activity. This diversity may confer enhanced free radical scavenging ability, antioxidant capacity, defensive functions, and cell wall strengthening, thereby promoting the differentiation and development of floral organs[30]. For example, Kaempferol-3-O-neohesperidoside, uniquely present in Set1, has been reported to participate in regulating cell expansion and floral organ formation[31].
In contrast, Set3 is dominated by anthocyanins and catechin-type compounds with overall lower diversity and more uniform glycosylation and acylation modifications. This limited diversity may restrict their roles in signal transduction and defense responses, reflecting an insufficient or less specific activation of key branches of flavonoid metabolism during flower bud development in the low-yield genotype. Previous studies have shown that glycosylation and acylation modifications of flavonoids play critical roles in plant growth regulation and environmental adaptation[32]. Thus, these metabolic differences may serve as key regulatory mechanisms in floral organ differentiation, stress defense, and energy allocation, ultimately affecting the reproductive efficiency and yield performance of different genotypes.
Furthermore, analysis of the DEM expression profiles revealed that, in both high- and low-yield genotypes, the abundance of flavonoid compounds significantly increased from the sepal stage to the stamen stage (Supplementary Fig. S4), consistent with previous reports on the role of flavonoids in floral organ development[33]. Interestingly, the overall expression levels of flavonoid compounds during flower bud development were significantly higher in the low-yield genotype compared to the high-yield genotype. This phenomenon may indicate that the low-yield genotype exhibits a compensatory or adaptive response during flower bud development by increasing flavonoid accumulation to counterbalance the insufficient activity in other key metabolic pathways[32]. However, this compensatory effect appears inadequate to fully overcome deficits in cell differentiation and organ formation, ultimately leading to reduced reproductive efficiency and yield.
Identification of gene modules highly correlated with SMs
-
To identify genes associated with SMs, we performed Weighted Gene Co-expression Network Analysis (WGCNA) on 2,013 DEGs across different genotypes and time points of C. wilsoniana flower bud development. To minimize noise from lowly expressed genes, we set a minimum module size of 30 genes and a sensitivity threshold of 4.0 for module detection. This analysis grouped the genes into five co-expression modules (Supplementary Table S6), with pairwise correlation evaluation performed among them. Each highly correlated gene group corresponds to a branch of the hierarchical clustering tree (Fig. 3a), and genes within the same module tend to exhibit significant topological overlap.
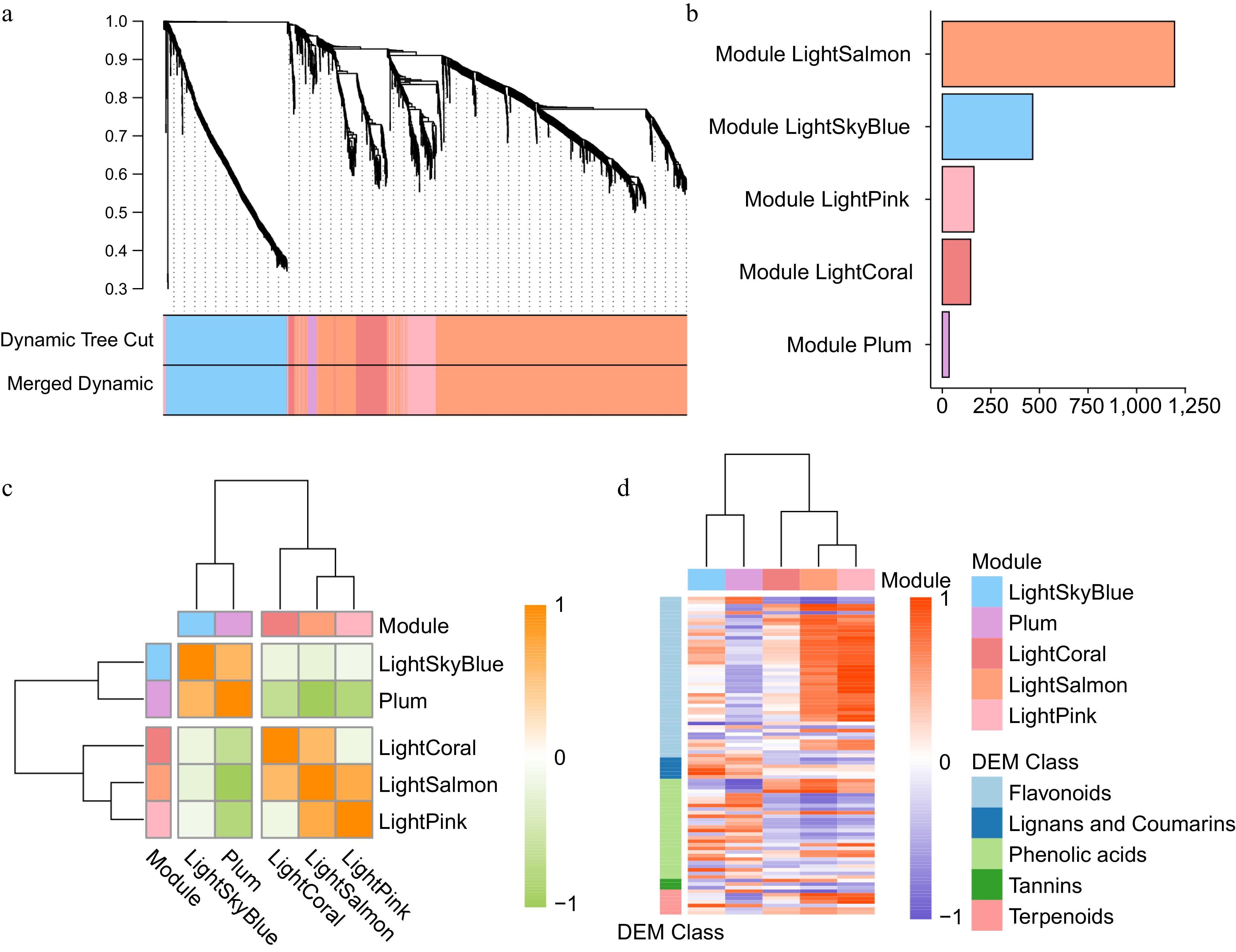
Figure 3.
WGCNA reveals key modules associated with secondary metabolism. (a) Hierarchical clustering dendrogram of DEGs based on topological overlap dissimilarity, with module identification using dynamic tree cut and merged dynamic methods. (b) Distribution of the gene number in each module. (c) Heatmap illustrating Pearson correlation among module genes within co-expression modules. (d) Heatmap showing the expression correlation between modules and different categories of differentially expressed SMs.
Among the identified modules, the LightSalmon module contained the largest number of genes (1,195), followed by the LightSkyBlue module (465 genes) and the LightPink module (163 genes), whereas the Plum module comprised only 35 genes (Fig. 3b). Furthermore, hierarchical clustering grouped these five modules into two highly interconnected clusters (Fig. 3c), indicating distinct yet coordinated expression patterns during flower bud development.
To further elucidate the roles of these modules in secondary metabolism, we correlated them with DEMs known to participate in secondary metabolic pathways during C. wilsoniana flower bud development (Fig. 3d). Our analysis revealed that the LightSalmon and LightPink modules were highly associated with the biosynthesis of flavonoids and terpenoids, while the LightSkyBlue and Plum modules showed a stronger correlation with lignin and coumarin pathways (Fig. 3d). These findings suggest that genes within these modules likely contribute to the early regulatory steps of secondary metabolism and may play critical roles in the plant’s environmental response mechanisms during flower bud development.
Flavonoid-associated module gene expression patterns and enrichment analysis
-
To elucidate the functions of 1,358 genes within the LightSalmon and LightPink modules, we performed clustering analyses on the genes in these modules. As shown in Fig. 4a, the genes in the LightSalmon module can be divided into two groups. The majority exhibit high expression during the pistil differentiation stage (H2 and L2) and stamen differentiation stage (H3 and L3) but low expression during the calyx differentiation stage (H1 and L1). The other group shows the opposite trend, with higher expression during the calyx differentiation stage (H1 and L1) and lower expression during the pistil and stamen differentiation stages (H2, L2, H3, and L3). These results indicate that the expression of DEGs in the LightSalmon module undergoes significant changes as C. wilsoniana enters the pistil differentiation stage. Similarly, clustering analysis of the LightPink module genes reveals that these genes exhibit expression patterns closely associated with the differentiation of floral organs (Fig. 4b). The majority show relatively low expression during the sepal differentiation stage (H1 and L1) but gradually increase during the differentiation of the reproductive organs (gynoecium and androecium; H2, L2, H3, and L3), suggesting that they play important roles in the transition from early structural components to functional reproductive organs. A very small subset of genes displays the opposite trend, with high expression during sepal differentiation and reduced expression during the differentiation of the reproductive organs. When the clustering results of both the LightSalmon and LightPink modules are combined, it can be inferred that the significant expression changes observed during the different stages of floral organ differentiation are closely related to the dynamic regulation of flavonoids and their derivatives during the morphological establishment and functional transition of C. wilsoniana flowers.
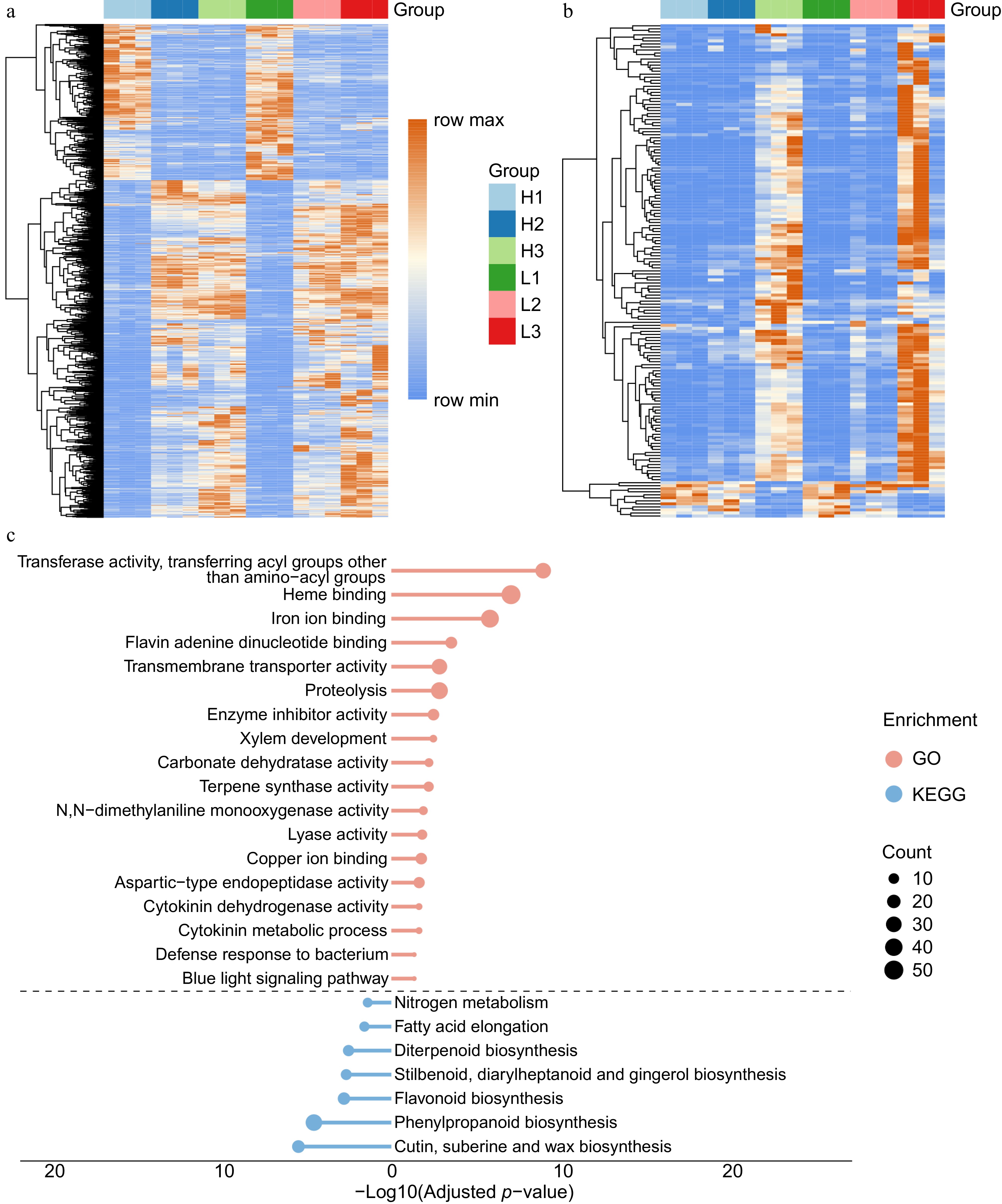
Figure 4.
Expression patterns of DEGs in different modules related to flavonoids. (a) Expression patterns of DEGs in the LightSalmon module. (b) Expression patterns of DEGs in the LightPink module. (c) Typical significantly enriched GO terms for the DEGs in the LightSalmon and LightPink modules.
We then performed GO enrichment and KEGG pathway enrichment analysis on all the flavonoid-related DEGs in these two modules. The results revealed 18 significantly enriched GO terms and seven KEGG pathway terms (Supplementary Table S7).
Notably, our GO enrichment analysis revealed that terms such as 'Transferase activity, transferring acyl groups other than amino-acyl groups' (GO:0016747) and 'Heme binding' (GO:0020037) were highly over-represented (p < 1 × 10−10). Studies have demonstrated that acyl modifications are critical for modulating the solubility, stability, and bioactivity of flavonoid intermediates[34,35]. This suggests that acyl transfer and redox reactions are central to the regulation of enzymes driving flavonoid and terpenoid biosynthesis. Moreover, the significant enrichment of terms such as 'Iron ion binding' (GO:0005506) and 'Flavin adenine dinucleotide binding' (GO:0050660) underscores the role of metalloenzymes in facilitating electron transport and oxidative reactions, which are essential for the structural diversification and catalytic activity within these secondary metabolic pathways[36].
Complementary KEGG pathway analysis further corroborated these findings, revealing robust enrichment in 'Phenylpropanoid biosynthesis' (ko00940) and 'Flavonoid biosynthesis' (ko00941). The high-rich factors and fold enrichment values associated with these pathways reflect a pronounced upregulation of key biosynthetic genes orchestrating the formation of flavonoid backbones and their derivatives. In parallel, significant enrichment in pathways related to terpenoid metabolism, including 'Diterpenoid biosynthesis' (ko00904) and 'Cutin, suberine, and wax biosynthesis' (ko00073), highlights a coordinated regulatory network that balances the synthesis of structurally and functionally diverse SMs.
Furthermore, the enrichment of GO terms associated with the 'Cytokinin metabolic process' and the 'Blue light signaling pathway' points to an intricate interplay between hormonal cues and light-responsive mechanisms in modulating these biosynthetic pathways[37,38]. The detection of terms related to defense responses (e.g., 'Defense response to bacterium') and 'Transmembrane transporter activity' further implies that these gene modules contribute not only to flavonoid biosynthesis but also to cellular processes involved in stress adaptation and the compartmentalization of SMs[39].
Collectively, these enrichment analyses revealed that the DEGs within the LightSalmon and LightPink modules integrate a spectrum of biological processes, from redox homeostasis and acyl transfer to hormonal signaling and stress defense, that fine tune flavonoid and terpenoid metabolism during plant development. This complex regulatory network in C. wilsoniana likely underpins the dynamic production of bioactive compounds, thereby influencing plant growth, defense, and ultimately, yield.
Construction of the co-expression network related to flavonoids.
-
To comprehensively assess the key genes in the co-expression network, we filtered the feature gene pairs from two modules related to flavonoid compounds. To obtain high-confidence results, a threshold of weight ≥ 0.45 was set, resulting in a total of 679 linear gene pairs (Supplementary Table S8). Notably, among these pairs, 11 hub genes exhibited connectivity degrees greater than 10, and three of these surpassed a connectivity degree of 100, underscoring their potential as central regulators in the network. For visualization, the filtered gene pairs were imported into Cytoscape v3.9.1[40]. As shown in Fig. 5 and Supplementary Table S9, the co-expression network included 17 transcription factors (TFs) and 191 structural genes. Within the network, TFs predominantly belong to the bHLH (5 genes), bZIP (3 genes), and G2-like (3 genes) families (Fig. 5, Supplementary Table S10).
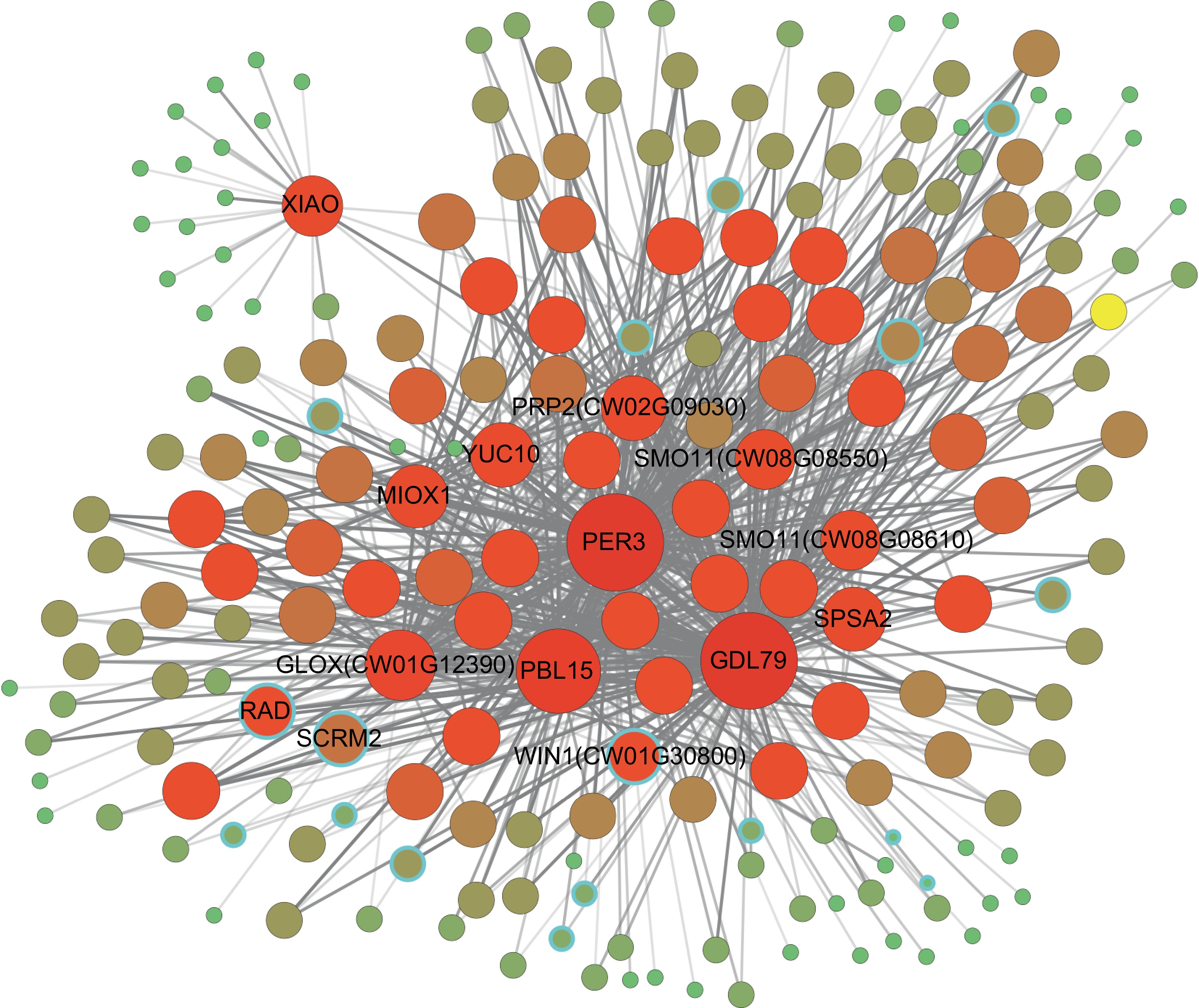
Figure 5.
Co-expression network of flavonoid-related genes. The size and color depth of the nodes represent the connectivity of the nodes. Nodes with a blue border represent TFs. Supplementary Table S9 lists the information for G1−G12. The depth of the colored edges represents the weight values. The distant region and elliptical region indicate two highly interactive gene clusters.
TFs such as WIN1 (CW01G30800), RAD, and SCRM2 demonstrate extensive regulatory interactions (tandem degree ≥ 5), suggesting that they coordinate the expression of multiple downstream genes. Notably, the TFs identified in this regulatory network are predominantly associated with floral development and stress responses. For instance, MYB26 (CW01G15170, CW01G15160) is closely linked to another development and is involved in regulating the thickening of the inner anther wall[41,42]. This suggests that MYB26 may play a critical role in early flower development, particularly by modulating cell wall assembly and secondary metabolism within the flavonoid biosynthetic pathway.
Conversely, the ERF family member WIN1 (CW01G24940 and CW01G30800) not only promotes cuticle formation by inducing the expression of wax biosynthetic enzymes, thereby ensuring the integrity of floral organ surfaces, but also enhances drought tolerance under stress conditions[43,44]. WIN1 may regulate flavonoid biosynthesis by activating a suite of stress-responsive genes, thus bolstering the plant's ability to cope with environmental challenges.
Furthermore, bHLH family members RAD (CW02G09830) and SCRM2 (CW01G35230) play pivotal roles in floral morphogenesis and stomatal differentiation, respectively—processes that are essential for proper organ development and function. Additionally, other TFs such as NFYA2 (CW02G02970) and TGA10 (CW09G27040) are involved in modulating stress responses and hormone signaling during flower development, and may indirectly influence the flavonoid biosynthetic pathway. Collectively, these TFs underscore pivotal roles in orchestrating floral development and secondary metabolism, particularly flavonoid synthesis, indicating that these gene clusters represent the core regulatory network driving flavonoid biosynthesis in C. wilsoniana.
In addition to transcriptional regulation, several metabolic enzymes and structural regulators further refine this network by modulating substrate availability and cellular architecture. For example, PALY (CW03G17800, connectivity 6), a phenylalanine ammonia-lyase-like protein, directly participates in the phenylpropanoid pathway and serves as an important precursor for flavonoid biosynthesis[45]. YUC10 (CW11G10330, connectivity 37) plays a pivotal role in auxin biosynthesis, regulating auxin gradients that are critical for floral organ morphogenesis and may indirectly influence flavonoid synthesis[46]. Moreover, PBL15 (CW11G28970, connectivity 117) functions as a receptor-like cytoplasmic kinase involved in developmental signal transduction and stress responses[47]. Metabolic enzymes such as SPSA2 (CW06G06650) and MIOX1 (CW08G03400, connectivity 31) are key players in carbohydrate and cell wall metabolism, providing the necessary substrates and energy for flavonoid production[48]. Genes like XIAO (CW06G00450, connectivity 21), which regulates organ size and cell proliferation[49], are potentially involved in sterol metabolism, further suggesting that modifications in cellular membranes and wall composition are integral to both the structural integrity of flowers and the compartmentalization of flavonoid biosynthesis. These genes are likely to serve as key switches in the biosynthesis and metabolic regulation network of flavonoids in C. wilsoniana.
Identification of key genes in flavonoid biosynthesis
-
To elucidate the key genes involved in flavonoid biosynthesis in C. wilsoniana, a comparative genomics analysis was performed. We manually inspected the proteome of C. wilsoniana and matched it against the UniProt reference clustering (UniRef) database. As a result, a total of 17 genes were identified, constituting a complete anthocyanin and proanthocyanidin biosynthesis pathway (Fig. 6a), thus enriching the KEGG pathway for flavonoid biosynthesis. Among these, eight key genes were found in the LightSalmon and LightPink modules, which are highly associated with flavonoid biosynthesis and metabolism. Additionally, we identified a total of 24 key compounds from this pathway in differential expression modules (Fig. 6b). Understanding the expression diversity of these metabolic genes and metabolites during flower bud development is crucial for anthocyanin biosynthesis and metabolism, particularly in light of their antioxidant and free radical scavenging activities.
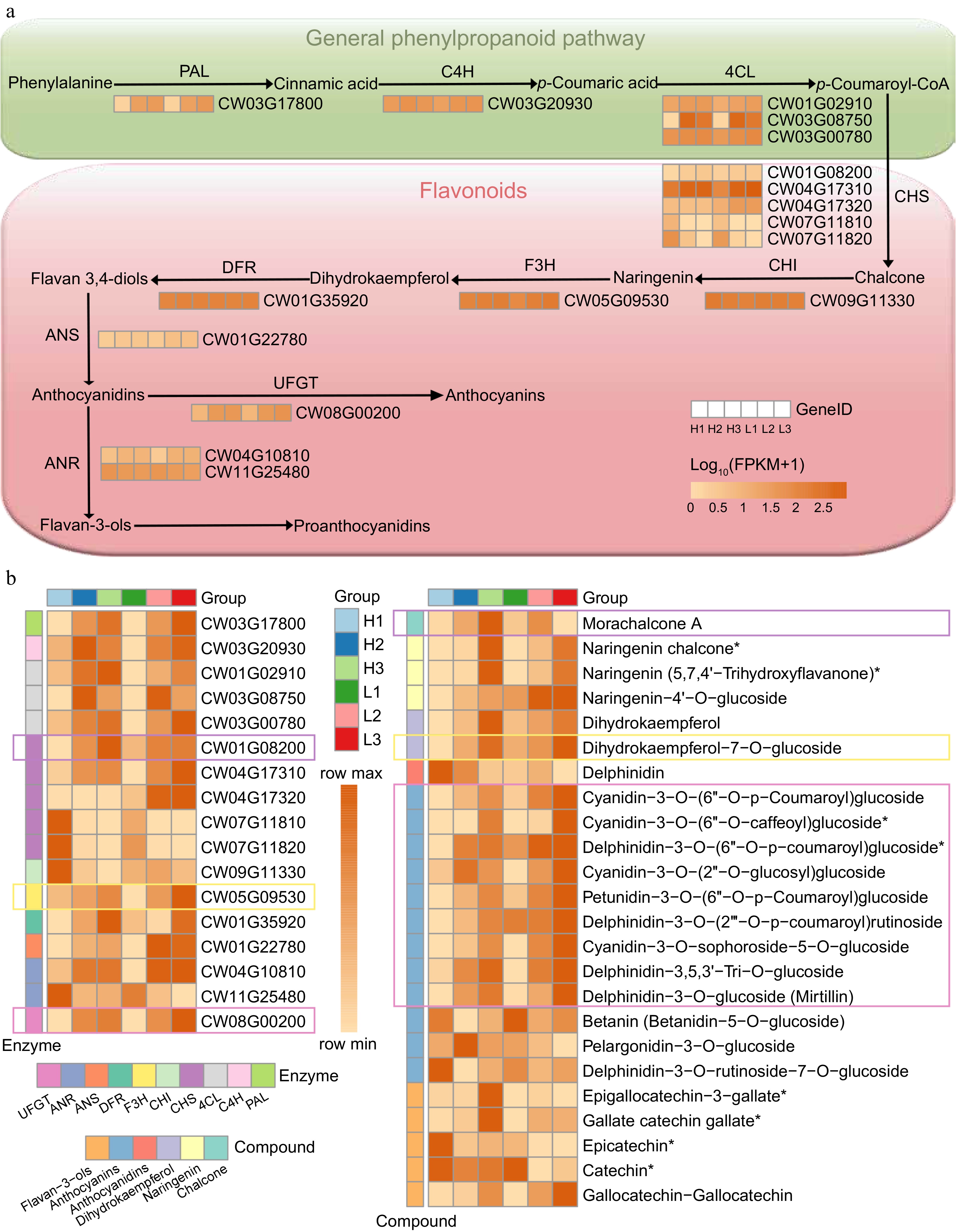
Figure 6.
Genes involved in flavonoid biosynthesis. (a) A simplified representation of the flavonoid biosynthesis pathway, where the expression levels of each gene were estimated based on FPKM from RNA sequencing of flower bud tissue from high-yield and low-yield genotypes. (b) A heatmap of the expression abundances of key enzymes and metabolites in the anthocyanin pathway during the flower bud development stages, with expression levels standardized by the Max-Min method. Rectangles of different colors represent enzymes and metabolites with similar expression patterns. * indicates metabolites with possible structural isomers.
The results indicate significant changes in the expression levels of key enzymes and metabolites involved in the anthocyanin pathway at different developmental stages. Several compounds, such as naringenin, dihydrokaempferol, and anthocyanins tended to show low or no expression during the sepal differentiation stage while exhibiting higher expression during the stamen differentiation stage. These results align with previous reports suggesting that the accumulation of intermediates and end-products of the flavonoid pathway can fluctuate markedly with developmental cues and environmental conditions[50]. Moreover, it suggested that later stages of flower bud development may favor the upregulation of genes involved in anthocyanin production[51].
Notably, the abundance of anthocyanins showed significant differences between H3 and L3, with the L3 consistently exhibiting higher anthocyanin levels than the H3. This pattern contrasts with the broader diversity of flavonoid species observed in the high-yield genotype and is supported by our metabolomic analysis, which indicates that Set3 (L1_vs_L3) is dominated by anthocyanins and catechin-type compounds but displays less chemical diversity. Such an accumulation of anthocyanins in L3 may represent a compensatory or adaptive strategy to bolster antioxidant capacity and stress defense[32], given that the low-yield genotype exhibits less extensive activation of other key metabolic branches. These findings are in line with previous reports demonstrating that anthocyanin biosynthesis can be upregulated under certain developmental or stress conditions[51]. However, the relatively narrow range of flavonoid modifications in L3 suggests that this genotype might be limited in its ability to fine-tune specific regulatory or defensive functions. Indeed, while enhanced anthocyanin accumulation may partially mitigate deficiencies in other metabolic pathways, it does not appear sufficient to improve the overall reproductive efficiency and yield performance in the low-yield genotype. Consequently, this genotype-specific metabolic reprogramming underscores the intricate balance between flavonoid diversity, stress tolerance, and floral organ differentiation, providing a framework for future research aimed at improving yield through targeted manipulation of flavonoid biosynthesis pathways.
Furthermore, robust causal associations were observed between specific enzymes and metabolites in the flavonoid biosynthesis pathway, as evidenced by comprehensive correlation analyses (Supplementary Table S11). Notably, the transcript abundance of F3H (CW05G09530) exhibited a strong positive correlation with Dihydrokaempferol-7-O-glucoside, with both parameters showing a marked increase during the stamen differentiation stage. This finding underscores the critical importance of enzyme-substrate specificity and the temporal regulation of gene expression in modulating anthocyanin biosynthesis. In addition, UFGT, a key enzyme responsible for the glycosylation of anthocyanidins into stable anthocyanins, displayed a strong positive correlation with anthocyanin levels. UFGT expression peaked concurrently with anthocyanin accumulation in the stamen differentiation stage, suggesting that its activity plays a critical role in converting unstable anthocyanidin intermediates into mature anthocyanins. Notably, correlation analysis revealed that UFGT expression was significantly associated with the accumulation of specific anthocyanin compounds, particularly Cyanidin-3-O-(6″-O-p-Coumaroyl)glucoside and Delphinidin-3-O-(6″-O-p-coumaroyl)glucoside. This observation underscores the pivotal role of UFGT-mediated glycosylation in stabilizing these pigments, thereby contributing not only to the vibrant coloration during stamen development but also to enhanced antioxidative capacity and overall floral defense.
-
Flavonoids play multifaceted roles in plant development, serving as key regulators of pigmentation, signaling molecules in plant-microbe interactions, and mediators of stress responses. In this study, we employed an integrative transcriptomic and metabolomic approach to elucidate the molecular framework governing flavonoid biosynthesis during C. wilsoniana flower bud development. By comparing high-yield (H) and low-yield (L) genotypes across developmental stages, we uncovered a genotype-specific metabolic reprogramming that fine-tunes secondary metabolism in response to reproductive and environmental cues.
Our transcriptomic analysis identified 2,013 DEGs, with significant enrichment in the phenylpropanoid and flavonoid biosynthetic pathways. The dynamic shifts in gene expression between developmental stages highlight the intricate coordination between floral organ differentiation and SM accumulation. Specifically, genes associated with anthocyanin and flavonol biosynthesis exhibited marked upregulation during the transition from sepal differentiation to reproductive stages, suggesting their pivotal role in modulating pigmentation and reproductive fitness[50,51]. Parallel metabolomic profiling further revealed a diverse array of 141 DEMs, among which flavonoids and phenolic acids were predominant. Notably, the high-yield genotype exhibited a broader spectrum of flavonoid species with extensive glycosylation and acylation modifications, whereas the low-yield genotype was characterized by an increased accumulation of anthocyanins and catechins. This suggests that the low-yield genotype prioritizes antioxidant defense at the expense of broader metabolic diversity, which may inadvertently affect reproductive efficiency[30,33]. The differential accumulation of flavonoid intermediates and end-products between the two genotypes underscores a genotype-specific metabolic specialization. In the high-yield genotype, a more diverse array of flavonoid modifications likely enhances biological versatility, supporting multiple functional roles such as stress defense, floral pigmentation, and reproductive signaling[32]. In contrast, the low-yield genotype’s reliance on enhanced anthocyanin accumulation may serve as a compensatory mechanism to reinforce antioxidant defenses but may come at a trade-off by limiting metabolic flexibility and reproductive output[34,51].
WGCNA provided further insights into the regulatory architecture underlying flavonoid biosynthesis. The LightSalmon and LightPink modules, which exhibited strong associations with flavonoid metabolism, contained key structural genes such as PAL, CHS, ANR, and UFGT, as well as critical TFs belonging to the MYB, bHLH, and ERF families. MYB26, a known regulator of another development, was identified as a potential upstream regulator of flavonoid biosynthetic genes, whereas WIN1, a TF involved in cuticle formation, may influence metabolic flux toward specialized flavonoid branches[50,51]. These findings reinforce the notion that transcriptional regulation plays a crucial role in orchestrating the metabolic shifts required for floral organogenesis and adaptive stress responses.
The development and morphogenesis of floral organs in plants are governed by highly complex and intricate regulatory mechanisms. Specifically, genes that are highly expressed during gynoecium and androecium differentiation are often involved in the biochemical pathways required for reproductive organ development, gametogenesis, and fertilization[52,53]. In contrast, genes with elevated expression during sepal differentiation are likely implicated in the formation of early protective structures, such as sepals, and in defense mechanisms against environmental stresses[54]. Flavonoids not only serve as key contributors to flower pigmentation but also play critical roles in antioxidation, pathogen defense, signal transduction, and protection against ultraviolet radiation. Studies have shown that the spatiotemporal expression of flavonoids in floral organs is often tightly linked to their specific functional roles or adaptive responses to environmental conditions[4,55]. From the perspective of module expression profiles, the dynamic changes in flavonoid-related gene expression underscore their regulatory roles in floral organ development. The differential expression of genes in the LightSalmon and LightPink modules during sepal and reproductive organ differentiation suggest that core structural genes or regulatory factors within the flavonoid biosynthetic pathway may be crucial in mediating the transition from 'protective structures' to 'reproductive functions' in floral organs. Specifically, genes upregulated during reproductive organ differentiation may promote the synthesis of anthocyanins or flavonols, thereby providing protective or signaling functions for pollen and pistils[56,57], whereas flavonoid biosynthesis-related genes highly expressed during sepal differentiation may primarily contribute to early color formation and stress defense[58,59].
Comparative genomics analysis further refined our understanding of the evolutionary trajectory of the flavonoid biosynthetic pathway, identifying 17 candidate genes that collectively constitute a complete anthocyanin and proanthocyanidin biosynthetic network. Among these, eight key genes were embedded within the LightSalmon and LightPink co-expression modules, underscoring their pivotal roles in flavonoid metabolism. Additionally, several core metabolic enzymes, including PAL, F3H, and UFGT, emerged as critical regulatory nodes within the network. Notably, the expression of F3H (CW05G09530) exhibited a strong positive correlation with Dihydrokaempferol-7-O-glucoside levels during stamen differentiation, emphasizing the importance of enzyme-substrate specificity and the spatiotemporal regulation of anthocyanin biosynthesis[50,51]. Similarly, UFGT demonstrated a robust association with anthocyanin accumulation, particularly in the glycosylation of Cyanidin-3-O-(6″-O-p-coumaroyl)glucoside and Delphinidin-3-O-(6″-O-p-coumaroyl)glucoside, highlighting its essential role in stabilizing anthocyanidins and modulating pigment composition. These findings reinforce the intricate coordination between genetic regulation and metabolic flux in fine-tuning flavonoid biosynthesis, ultimately shaping floral pigmentation, stress responses, and reproductive success.
Taken together, our findings reveal a complex regulatory network that dynamically integrates developmental cues, metabolic flux, and environmental responsiveness to modulate flavonoid biosynthesis in C. wilsoniana. This work provides valuable insights into the genetic and metabolic underpinnings of floral organ development and highlights potential targets for genetic improvement. Future studies leveraging genome editing and metabolic engineering approaches could further elucidate the functional roles of these key regulatory nodes, ultimately paving the way for enhanced yield, stress resilience, and bioactive compound production in medicinal and horticultural crops.
We thank the members of Dijun Chen's laboratory for providing useful discussions and valuable comments. The authors also acknowledge the High Performance Computing Center of Nanjing University for providing high performance computing (HPC) resources. This work was supported by the National Natural Sciences Foundation of China (32070656, 32270709), and the National Key Research and Development Program of China (SQ2022YFE012895).
-
The authors confirm contribution to the paper as follows: sample collection, methodology, data analysis, organization, visualization, draft manuscript preparation: Cai J; conceptualization: Cai J, Chen M, Chen D; study guidance: Chao H, Chen M, Chen D; manuscript revision: Chao H. All authors reviewed the results and approved the final version of the manuscript.
-
Raw transcriptome sequencing data of C. wilsoniana have been deposited in the Genome Sequence Archive (https://ngdc.cncb.ac.cn/gsa: Accession No. CRA010376)[60]. The raw metabolome data have been deposited in OMIX (https://ngdc.cncb.ac.cn/omix/releaseList: Accession No. OMIX003453). The reference genome of C. wilsoniana was downloaded from GWH (https://ngdc.cncb.ac.cn/gwh: Accession No. GWHCAYR00000000).
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Jiaqi Cai, Haoyu Chao
- Supplementary Table S1 KEGG pathway enrichment analysis of DEGs across three developmental stages.
- Supplementary Table S2 KEGG pathway enrichment analysis of DEGs across independent developmental stage comparisons.
- Supplementary Table S3 Overlapping DEGs and their KEGG pathway enrichment across key developmental stage comparisons.
- Supplementary Table S4 Metabolite abundance at the bud development stage.
- Supplementary Table S5 Representation and class characterization of different DEM sets.
- Supplementary Table S6 DEMs in different modules based on WGCNA identification.
- Supplementary Table S7 GO and KEGG pathway enrichment analysis of flavonoid-related genes in two key modules.
- Supplementary Table S8 Linear pairs of modular genes with Weight ≥ 0.45 in the co-expression network.
- Supplementary Table S9 The tagging of each gene in Figure 5 corresponds to the ID.
- Supplementary Table S10 The connectivity of different nodes is calculated by Cytoscape in Figure 5.
- Supplementary Table S11 Correlation analysis of key enzymes and metabolites involved in the anthocyanin pathway.
- Supplementary Fig. S1 Log2(Fold Change) for DEGs with the most significant differences across comparison groups.
- Supplementary Fig. S2 DEGs in some groups were significantly enriched in multiple SM biosynthetic pathways.
- Supplementary Fig. S3 Log2(Fold Change) for DEMs with the most significant differences across comparison groups.
- Supplementary Fig. S4 Expression profiles of different DEM sets.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Cai J, Chao H, Chen M, Chen D. 2025. Integration of metabolome and transcriptome reveals flavonoid metabolic network during the developmental stage of flower buds in Cornus wilsoniana. Medicinal Plant Biology 4: e017 doi: 10.48130/mpb-0025-0015 

Integration of metabolome and transcriptome reveals flavonoid metabolic network during the developmental stage of flower buds in Cornus wilsoniana
- Received: 10 January 2025
- Revised: 18 April 2025
- Accepted: 22 April 2025
- Published online: 23 May 2025
Abstract: Secondary metabolites (SMs) are crucial for plant adaptation and human health, yet the regulatory mechanisms underlying their biosynthesis remain incompletely understood. Cornus wilsoniana, a woody oil crop and medicinal plant, is renowned for its rich flavonoid content, yet the genetic and metabolic basis of its secondary metabolism remains largely unexplored. Here, we integrate transcriptomic and metabolomic analyses to dissect the regulatory landscape governing flavonoid biosynthesis during flower bud development. Comparative analyses between high-yield and low-yield genotypes across distinct developmental stages reveal a coordinated reprogramming of flavonoid pathways, with significant shifts in gene expression and metabolite accumulation. WGCNA links critical biosynthetic genes to co-expression modules enriched in hormone signaling, redox homeostasis, and light perception, underscoring the interplay between developmental and environmental cues in shaping metabolic fluxes. Furthermore, comparative genomics reveals 17 candidate genes forming a complete anthocyanin and proanthocyanidin biosynthetic network. Our findings illuminate the molecular framework underlying flavonoid metabolism in C. wilsoniana, providing a foundation for metabolic engineering and genetic improvement strategies aimed at enhancing bioactive compound production for medicinal and agricultural applications.
-
Key words:
- Cornus wilsoniana /
- Transcriptome /
- Metabolome /
- Flower /
- Flavonoid