









-
Rheum officinale is a traditional Chinese medicinal herb whose dry rhizomes and roots are rich in bioactive compounds such as anthraquinones, flavonoids, stilbenes, and tannins. These constituents exhibit a range of pharmacological activities, including antiviral, anti-inflammatory, antibacterial, antioxidant, and anti-tumor effects[1,2]. It is widely used in the treatment of constipation, abdominal pain, diarrhoea, jaundice and inflammation of some organs[3,4]. The increasing market demand for Rheum officinale (rhubarb), coupled with the depletion of wild resources, has shifted the primary supply to cultivated sources. However, challenges such as fragmented cultivation practices, extensive farming management, and outdated planting and processing techniques in production areas have resulted in significant quality inconsistencies in commercially available rhubarb. Therefore, implementing standardized cultivation practices and advanced processing technologies is crucial for enhancing both the yield and quality of rhubarb[5].
At present, R. officinale is experiencing increasing pressure due to decline in wild populations driven by rising market demand, necessitating artificial cultivation as the primary solution[5]. However, the absence of standardized harvesting and processing methods has resulted in significant variability in the quality of medicinal rhubarb[4,6,7]. Recognizing the significance of optimal harvest timing, in accordance with the growth rhythm of medicinal rhubarb, is imperative. Given its perennial nature, the composition of medicinal constituents in R. officinale fluctuates annually. Therefore, evaluating active ingredients across different growth years is of utmost importance for determining the optimal harvest period. Previous studies on Rheum species and other perennial medicinal plant, such as Panax ginseng, have shown that secondary metabolites like anthraquinones accumulate progressively during early growth stages (2−4 years), peaking at maturity[8,9]. This evidence informed the focus on 2- to 4-year-old R. officinale to document dynamic metabolite shifts and identify the optimal harvest timing. While studies on harvest periods exist for a various medicinal species[10], similar investigations into R. officinale are scarce. Furthermore, although the rhizomes and roots serve as the main reservoirs of chemical constituents in R. officinale, the alterations in their composition with maturation remain largely unexplored[11].
Understanding the key genes involved in the biosynthesis of secondary metabolites in medicinal plants is crucial for enhancing their medicinal quality. Recent studies have investigated differentially expressed genes (DEGs) associated with secondary metabolite pathways across various developmental stages. For instance, in Dendrobrium officinale[12], several DEGs were identified to be implicated in the metabolism of water-soluble polysaccharides, flavonoid, and phosphoserine, respectively. A comparative transcriptomic analysis in Ficus carica revealed 1,274 upregulated and 813 downregulated genes between tree-ripe and commercially ripe fruits[13]. Similarly, transcriptomic analyses in yellowhorn (Xanthoceras sorbifolium) unveiled correlations between MYB and WD40 transcription factors and anthocyanin accumulation[14]. In Dendrobium moniliforme, Weighted Gene Co-expression Network Analysis (WGCNA) highlighted the significance of genes like FLS, CCR, and MYB in flavonoid biosynthesis[15]. Despite these advances, a comprehensive comparison of rhizome and root transcriptomes at different harvest times remains unexplored in R. officinale, impeding the identification of key genes associated with anthraquinone biosynthesis.
In this study, the grown R. officinale was divided into three ages (two, three, and four years), to better analyze the content of bioactive metabolites in each age. Changes in bioactive compounds among the three ages groups were assessed by transcriptomic and High-performance liquid chromatography (HPLC) analyses. Short Time-series Expression Miner (STEM) and Weighted Gene Co-expression Network Analysis (WGCNA) were employed to identify genes exhibiting high correlations with bioactive compounds. The outcomes of this study will facilitate the effective exploration and utilization of harvested tissues for both academic research and commercial applications as a traditional Chinese medicinal resource. Furthermore, some key genes identified during the transcriptomic analysis will be applied in the genetic breeding of R. officinale.
-
The samples were collected from Taoshuwan Village, Guanyin Town, Zhenba County, Shaanxi Province, China (108°06′26″ E, 32°24′04″ N; altitude 1,226 m). This region experiences a subtropical monsoon climate, with a mean annual temperature of 14–16 °C and precipitation ranging from 900 to 1,200 mm. The soil is loamy and well-drained. R. officinale plants were cultivated following local practices: organic manure (7.5 tons/ha) was applied annually, with manual weeding ensuring organic standards. The R. officinale plants were identified to be an herbaceous plant of Rheum L. in Polygonaceae by Professor Yong-gang Yan of Shaanxi University of Chinese Medicine. A voucher specimen (No. RO_901) has been deposited in the Key Laboratory for Research and Development of Qin Medicine, Shaanxi University of Chinese Medicine. On September 1st of each year (2020–2023), three healthy plants aged two, three, and four years were randomly selected per age group (three biological replicates each; n = 6 total per group). Roots and rhizomes were separated, washed, and transported on ice to the artificial climate room (20 ± 2 °C; 60% ± 5% relative humidity) at Shaanxi University of Chinese Medicine. Tissues were sliced into 3.0 cm segments, sun-dried for 7 d, and further dried at 40 °C in an electric oven to constant weight.
Bioactive components measurement by HPLC Assays
-
The R. officinale samples underwent drying in an electric blast drying oven, followed by grinding and sieving through the fourth sieve. Subsequently, 0.1000 g of the samples was precisely weighed and transferred to a 50 mL stopper triangle bottle, to which 4.5 mL of methanol was added and weighed. Ultrasonic treatment was applied for 30 min (power: 500 W, frequency: 40 kHz). After equilibration at room temperature, the mixture was centrifuged at 10,000 rpm for 10 min, and the supernatant was filtered through a 0.22 μm microporous membrane. The resulting filtrate was subjected to bioactive components content analysis using high-performance liquid chromatography (HPLC)[16].
HPLC analysis was conducted using an Acchrom S3000 system equipped with a 3410 UV Detector. Acchrom Unitary-C18(2) analytical column (250 mm × 4.6 mm, particle size: 5 μm) was employed for compound separation. The mobile phase consisted of methanol (A) and 0.2% phosphoric acid aqueous solution (B). The elution program began with 10% eluent A and maintained a flow rate of 1.0 mL/min. The linear gradient elution was performed as follows: 15%−30% mobile phase A (0−5 min), 30%−50% A (5−10 min), 50%−58% A (10−15 min), 58%−60% A (15−25 min), 60%−90% A (25−36 min), 90%−100% A (36−40 min). This was followed by isocratic elution with 100% A (40−45 min). Column re-equilibration was achieved with 10% A for 10 min prior to subsequent runs. The entire analysis was conducted at 30 °C with detection wavelength set at 260 nm[17].
Reference substance preparation involved the accurate determination of gallic acid, catechin, sennoside B, chrysophanol-8-O-glucoside, emodin-8-O-glucoside, aloe-emodin, rhein, emodin, chrysophanol, and physcion (Dexter Biotechnology Co., Ltd). These reference substances were diluted with methanol in a 10 mL volumetric flask to prepare a reference reserve solution. Subsequently, 1.0 mL of each reference stock solution was accurately measured and diluted tenfold with methanol to obtain a mixed reference stock solution at the corresponding mass concentration, stored at 4 °C for subsequent use (Supplementary Table S1).
Transcriptomic analysis
-
Roots and rhizomes of Rheum officinale root were pulverized into powder, and three biological replicates were conducted. Total RNA extraction was carried out utilizing the RNeasy Plant Mini Kit (Qiagen, Germany), followed by the elimination of genomic DNA using RNase-free DNase I (Qiagen, Germany). High-quality RNA specimens were employed for sequencing library preparation. RNA purification, reverse transcription, library construction, and sequencing were performed by Kidio Biotechnology Co., Ltd. (Guangzhou, China). Each sample utilized 1 μg of total RNA for library construction, following the manufacturer's guidelines. Subsequently, the RNA-seq library underwent sequencing using an Illumina NovaSeq 6000 system. The raw data were processed using AdapterRemoval v2 for adapter trimming[18]. The RSEM software was employed to analyze the expression levels of transcripts[19]. Genes meeting the criteria of an absolute value of log2 ratio of ≥ 1 and a false discovery rate (FDR) significance score of < 0.05 were deemed DEGs and subjected to further analysis[20]. Furthermore, differentially expressed genes underwent KEGG functional analysis for additional characterization[21].
Gene clustering
-
Genes identified at the two stages underwent filtering with a coefficient of variation (CV) cutoff of > 0.4. The filtered genes were then subsequently grouped into distinct clusters utilizing the Mfuzz package with the fuzzy c-means algorithm[22] in R software version 3.6.1.
-
The transcriptome data were normalized using the normalized quantiles function within the R software package, and all DEGs between samples were selected for subsequent WGCNA[23]. The eigengenes of each module were computed, followed by cluster analysis of the modules. WGCNA was performed using the following parameters: minimum module size of 30, soft-threshold power of 13, and a merge cut height of 0.15. Eigengene values of modules associated with gallic acid, catechin, sennoside B, chrysophanol-8-O-glucoside, emodin-8-O-glucoside, aloe-emodin, rhein, emodin, chrysophanol, and physcion were calculated. Hub genes within significant modules were identified based on a coefficient > 0.6 and a p-value < 0.05. All genes and module eigengenes were subjected to expression level, Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses. The co-expression network of module eigengenes in the module of interest was visualized using Cytoscape (version 3.7.2)[24].
Co-expression network construction
-
Co-expression analysis was conducted using a Pearson correlation coefficient (PCC) threshold of ≥ 0.9 and a p-value < 0.01. Gene relationship pairs were determined based on miRNA targeting relationships, requiring a minimum of 5 miRNAs targeting each gene, a false discovery rate (FDR) of ≤ 0.01, and a p-value < 0.01. The relationship pairs for all DEGs were obtained and visualized using Cytoscape (version 3.7.2)[25].
GO and KEGG enrichment analysis
-
GO enrichment analysis of cluster genes and module genes was performed using the agriGO v2.0 database. Fisher's test and the multitest adjustment method of Yekutieli (FDR under dependency) were employed[26]. GO terms with a false detection rate (FDR) < 0.05 were deemed significantly enriched. The top 20 biological process terms were visualized using the REVIGO database (
http://revigo.irb.hr ) with default parameters to reduce the GO dimension. The KEGG enrichment analysis of DEGs was conducted using KOBAS software[27].RNA isolation and qRT-PCR verification
-
Total RNA was extracted from roots, rhizomes and leaves of R. officinale (six months, the seedlings were cultured in an incubator) using Trizol reagent (Invitrogen, USA), with three triplicates. The quality of total RNAs was detected by Nanodrop 2000 spectrophotometer and Agilent 2,100 Bio-analyzer. Quantitative real-time PCR (qRT-PCR) was performed on a Roche LightCycle 96 PCR System (Roche Applied Science, Mannheim, Germany) using a One Step SYBR PrimeScript PLUS RT-PCR Kit (TaKaRa Bio, Kyoto, Japan). Relative mRNA expression was analyzed with the comparative Ct method (2−ΔΔCᴛ) and normalized to β-actin as an internal reference gene. All primers were synthesized by Shenggong Bioengineering Co., Ltd. (Shanghai, China), and their sequences are listed in the Supplementary Table S2.
Statistical analysis
-
The data were presented as mean ± standard deviation (SD). Statistical differences among multiple groups were assessed using one-way analysis of variance (ANOVA) followed by Tukey's test.
-
In this study, the levels of ten bioactive components was evaluated across different years of root and rhizome specimens of R. officinale. The analysed compounds included gallic acid, catechin, sennoside B, chrysophanol-8-O-glucoside, emodin-8-O-glucoside, aloe-emodin, rhein, emodin, chrysophanic acid, and emodin monomethyl ether (Fig. 1a). Specifically, in root samples, seven bioactive components, including catechin, sennoside B, chrysophanol-8-O-glucoside, emodin-8-O-glucoside, aloe-emodin, emodin, and emodin monomethyl ether, exhibited higher concentrations in 4-year-old specimens compared to those aged 2 and 3 years (Fig. 1b). In rhizome samples, six bioactive components exhibited levels in 4-year-old specimens relative to those aged 2 and 3 years, including catechin, sennoside B, chrysophanol-8-O-glucoside, emodin-8-O-glucoside, aloe-emodin, and emodin monomethyl ether (Fig. 1c). These findings indicate the accumulation of six common bioactive components over time (Fig. 2b, c). Additionally, higher levels of chrysophanic acid was observed in 3-year-old specimens compared to those aged 2 and 4 years in both root and rhizome samples (Fig. 2b, c). Collectively, the results obtained suggest that 4-year-old R. officinale root and rhizome specimens exhibit elevated levels of bioactive components.
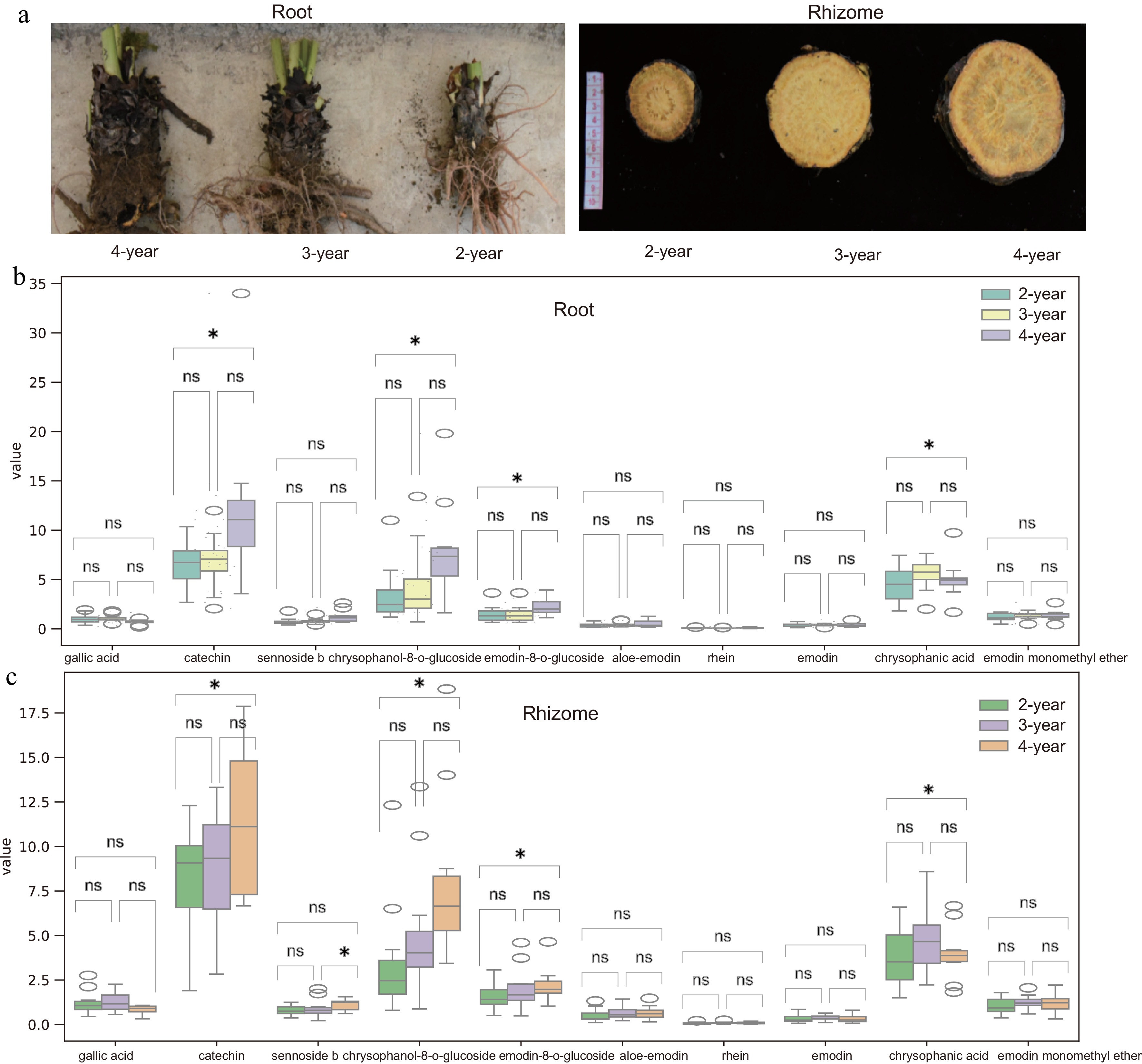
Figure 1.
Morphological and bioactive component changes in R. officinale roots and rhizomes at different ages. (a) Right to left: 2-, 3-, and 4-year-old roots; Left to right: 2-, 3-, and 4-year-old rhizomes. (b), (c) Bioactive compound content (dry weight) at different ages. Data represent mean ± SD (n = 3 biological replicates). Statistical significance was assessed by one-way ANOVA (* p < 0.05, ** p < 0.01).
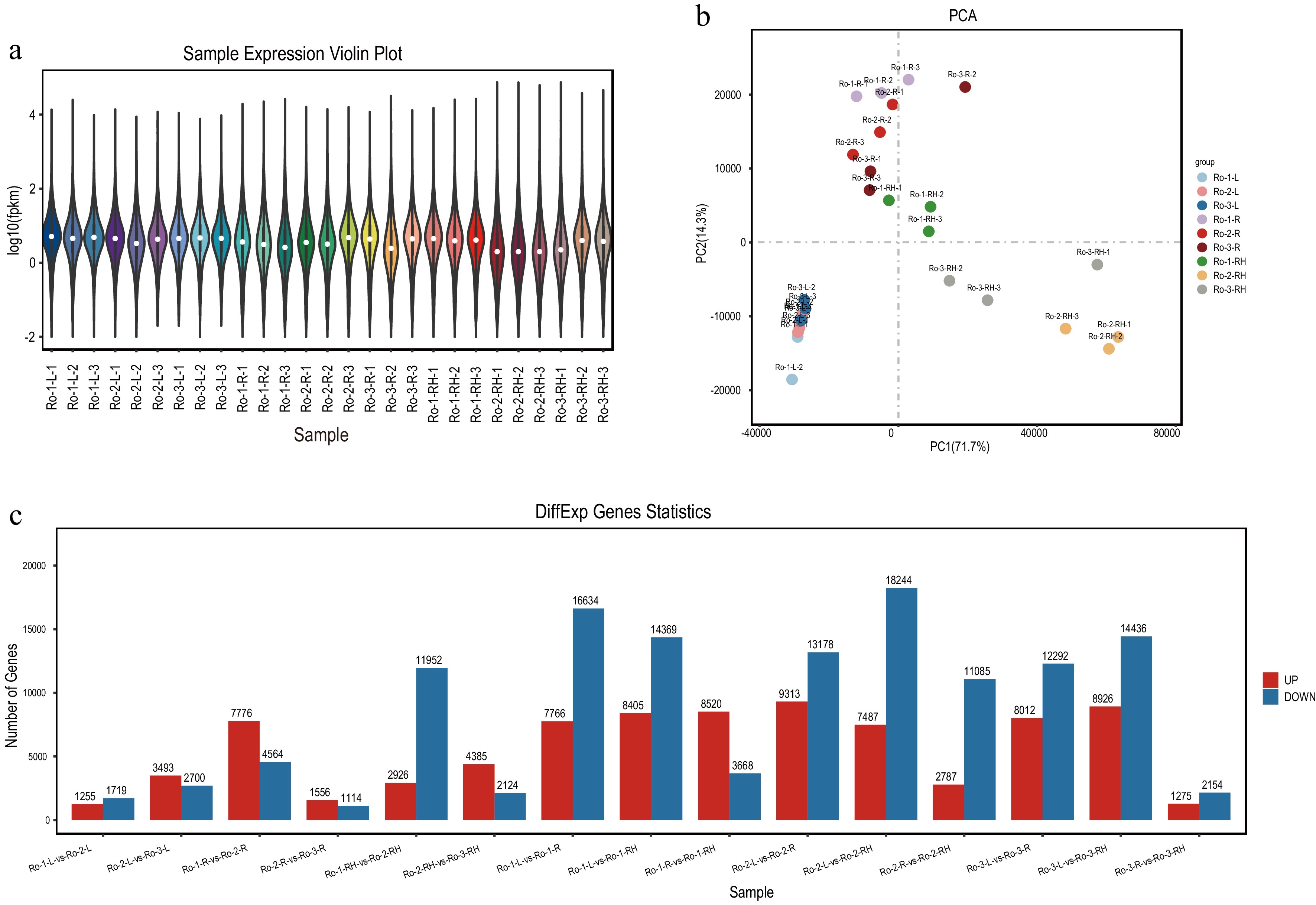
Figure 2.
The expression profiling of unigenes in roots, rhizomes and leaves of R. officinale at different ages. (a) Violin plot of unigenes with mean expression level in leaves, roots, and rhizomes. (b) The PCA analysis of unigenes based on the expression level in leaves, roots, rhizomes. (c) The statistics of DEGs among three groups of root, rhizome and leaf, respectively.
Identification and analysis of differentially expressed genes (DEGs)
-
RNA-seq analysis revealed consistent unigene expression patterns across roots, rhizomes, and leaves of Rheum officinale across different ages, as evidenced by the average Fragments Per Kilobase of transcript per Million mapped reads (FPKM) box plot and Principal Component Analysis (PCA) (Fig. 2a). PCA further illustrated distinct sample separations (Fig. 2b). Following differential expression gene (DEG) screening method, considerable numbers of upregulated and downregulated DEGs were identified: 1,255 up and 1,719 down DEGs between RO-1-L and RO-2-L; 3,493 up and 2,700 down DEGs between RO-2-L and RO-3-L; 7,776 up and 4,564 down DEGs between RO-1-R and RO-2-R; 1,556 up and 1,114 down DEGs between RO-2-R and RO-3-R; 2,926 up and 11,952 down DEGs between RO-1-RH and RO-2-RH; 4,385 up and 2,124 down DEGs between RO-2-RH and RO-3-RH; 7,766 up and 16,634 down DEGs between RO-1-L and RO-1-RH; 8,405 up and 14,369 down DEGs between RO-1-L and RO-1-RH; 8,520 up and 3,668 down DEGs between RO-1-R and RO-1-RH; 9,313 up and 13,178 down DEGs between RO-2-L and RO-2-R; 7,487 up and 18,244 down DEGs between RO-2-L and RO-2-RH; 2,787 up and 11,085 down DEGs between RO-2-R and RO-2-RH; 8,012 up and 12,292 down DEGs between RO-3-L and RO-3-R; 8,926 up and 14,436 down DEGs between RO-3-L and RO-3-RH; 1,275 up and 2,154 down DEGs between RO-3-R and RO-3-RH (Fig. 2c). These findings underscore the substantial presence of DEGs within each group.
Identification of DEGs in root of three ages using comparative transcriptome data
-
Based on the established criteria of |log2 fold change (FC)| > 1 and False Discovery Rate (FDR) < 0.05, a total of 3,525 upregulated and 6,043 downregulated DEGs were identified in the comparison between RO-1-R and RO-2-R in the roots. Similarly, in the roots, 3,525 upregulated and 6,043 downregulated DEGs were observed in the comparison between RO-2-R and RO-3-R, as well as between RO-1-R and RO-3-R (Fig. 3a−c). A total of 460 DEGs were detected across the three developmental stages of the roots (Fig. 3d; Supplementary Table S3). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed enrichment in a diverse array of functional pathways, including phenylpropanoid biosynthesis, biosynthesis of secondary metabolites, and anthocyanin biosynthesis, all of which are pertinent to anthraquinone biosynthesis (Fig. 3d). Among these, 23 DEGs (14 upregulated and 9 downregulated) were consistently differentially expressed across all three stages (RO-1-R, RO-2-R, and RO-3-R), encompassing CYP450 genes (CYP82G1, CYP71A26, CYPRO1, CYP736A117, CYP82C4, CYP72A15, CYP78A5, CYP82G, CYP73A4), WRKY genes (WRKY32), UGT73C5, and VIL1, VHA-a3, UPL1, UPL2, WAT1, and XTH15.
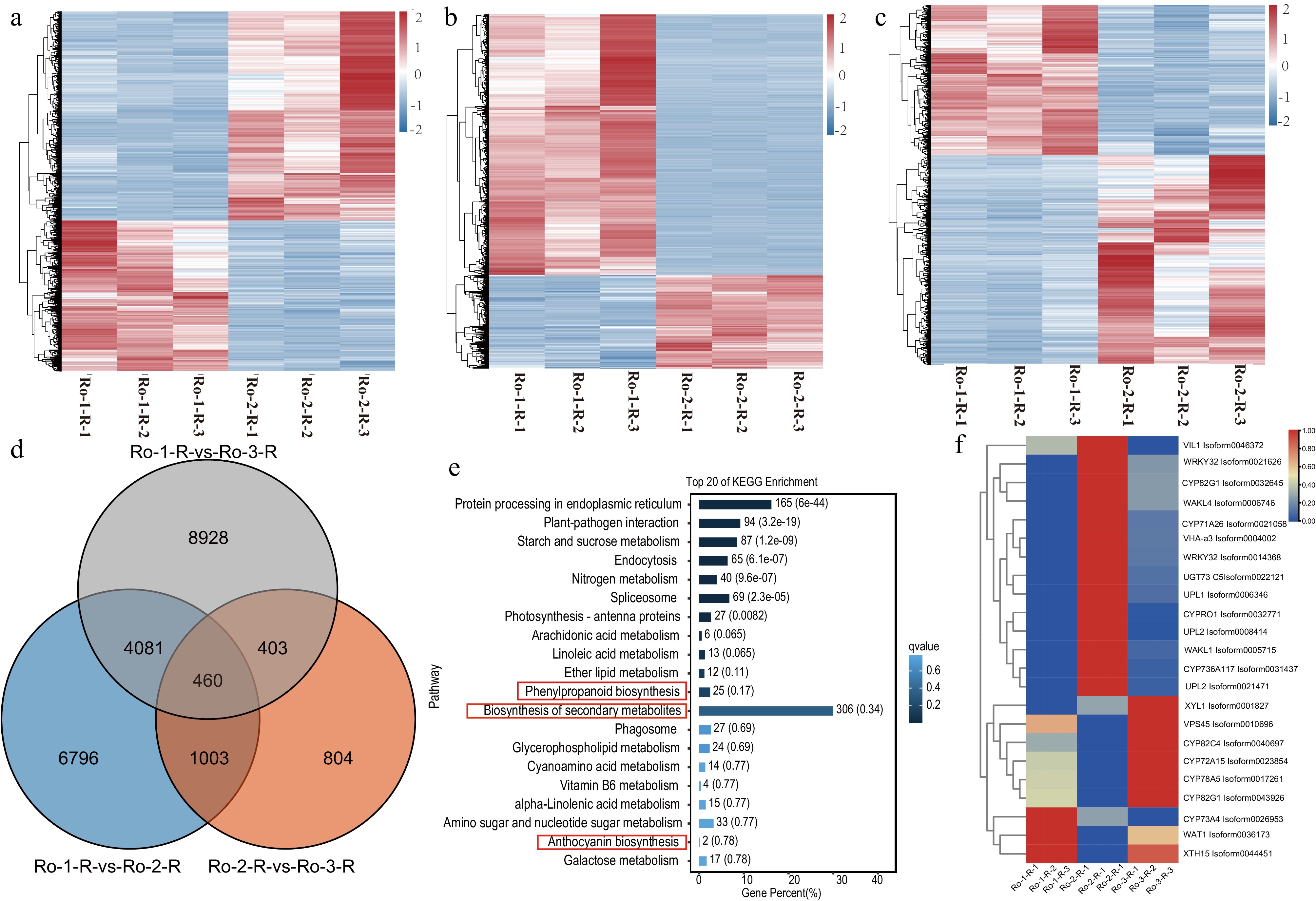
Figure 3.
The comparative transcriptomic analysis of R. officinale during three ages of roots. (a)−(c) The heatmap of DEGs during three ages of roots of R. officinale. (d) The Venn plot of DEGs at three ages of R. officinale. (e) The KEGG enrichment analysis of DEGs at three ages. (e) The heatmap of significantly differential unigenes during three stages of roots of R. officinale. Ro-1-R: 2021-9; Ro-2-R: 2021-9; Ro-3-R: 2022-9.
Identification of DEGs in rhizome of three ages using comparative transcriptome data
-
The comparative transcriptomic analysis of rhizome tissues revealed significant DEGs in various comparisons: 2,926 upregulated and 11,952 downregulated DEGs between RO-1-RH and RO-2-RH, 4,385 upregulated and 2,124 downregulated DEGs between RO-2-RH and RO-3-RH, and 2,786 upregulated and 6,052 downregulated DEGs between RO-1-RH and RO-3-RH (Fig. 4a−c). A total of 1,224 DEGs were identified across the three developmental stages (Fig. 4d). KEGG analysis unveiled enrichment in diverse functional pathways, notably including biosynthesis of secondary metabolites, flavonoid biosynthesis, and phenylpropanoid biosynthesis, all of which are pertinent to anthraquinone biosynthesis (Fig. 4d; Supplementary Table S4). Additionally, 14 DEGs (10 upregulated and four downregulated) were consistently differentially expressed across all three stages (RO-1-RH, RO-2-RH, and RO-3-RH), encompassing CYP450 genes (CYP75B137, CYP82G1, CYP82C4, CYP81Q32, CYP95, CYP76T24, CYP81Q32), WRKY genes (WRKY4, WRKY44, WRKY40, WRKY46), and UGT71A15 (Fig. 4e, f).
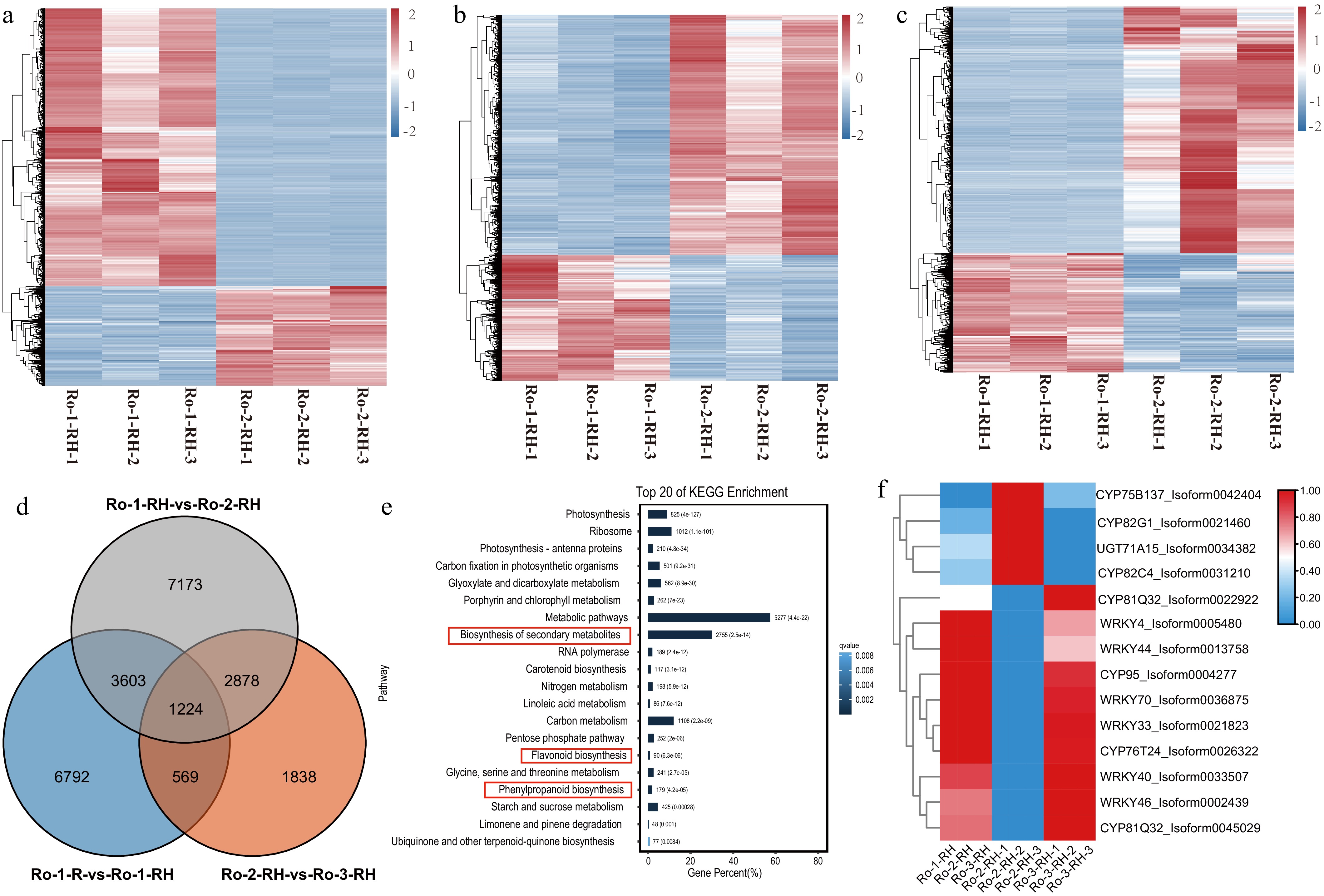
Figure 4.
The comparative transcriptome at three ages of rhizomes of R. officinale. (a)−(c) The heatmap of DEGs during three ages of rhizomes of R. officinale. (d) The Venn plot of DEGs during three ages of R. officinale. (e) The KEGG enrichment analysis of DEGs between CK and MeJA treatment. (f) The heatmap of significantly differential unigenes during three stages of rhizomes of R. officinale. RO-1-RH: 2020-9-1; RO-2-RH: 2021-9-1; RO-3-RH: 2022-9-1.
Transcriptome profiles of different ages of root and rhizome
-
To explore the functional dynamics during root development, the fuzzy c-means clustering algorithm was employed to cluster the expression patterns of all 13,964 genes with a high CV
(> 0.4) into nine co-expression clusters (Fig. 5a). Subsequently, GO enrichment analysis was performed, followed by dimensionality reduction to categorize these genes into functional groups within the clusters (Fig. 5b). In roots, GO enrichment analysis unveiled that cluster 10 comprised 1,826 genes associated with metabolic processes, DNA-binding transcription factor (TF) activity, and defense responses. KEGG enrichment analysis further revealed the involvement of these DEGs in biosynthesis of secondary metabolites, flavonoid biosynthesis, and phenylpropanoid biosynthesis, all pertinent to anthraquinone biosynthesis (Fig. 5c). Notably, cluster 10 (comprising 1,826 genes) was predominantly clustered into a number of gene families, primarily encompassing WRKY, CYP450, and Heat Shock Proteins (HSP) (Fig. 5d).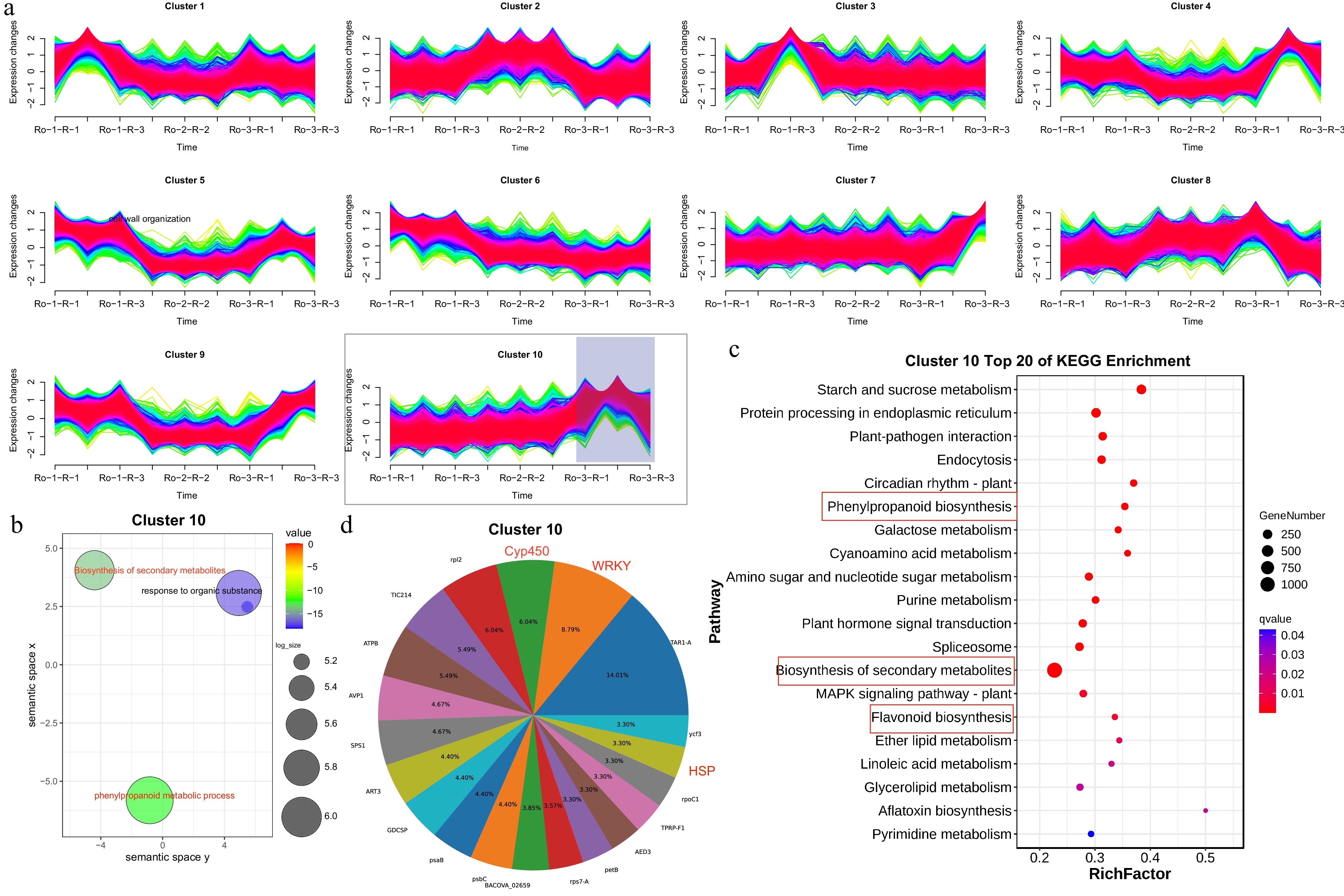
Figure 5.
Spatiotemporal gene expression pattern of R. officinale roots and functional enrichment analysis. (a) Cluster 1 to 10 showing DEGs in RO-1-R, RO-1-R, and RO-1-R. (b) GO enrichment pathway analysis of DEGs from cluster 10. (c) KEGG enrichment pathway analysis of DEGs from cluster 10. (d) The gene family cluster analysis of cluster 10.
During the developmental stages of the rhizome, the expression patterns of all 16,764 genes with a high CV (> 0.4) were organized into ten distinct expression clusters (Fig. 6a). Notably, cluster 9 comprised 3,301 genes associated with metabolic processes, TF activity, and defense responses, as determined by GO enrichment analysis (Fig. 6b). Additionally, KEGG enrichment analysis revealed that these DEGs were implicated in phenylpropanoid biosynthesis, biosynthesis of secondary metabolites, and flavonoid biosynthesis, suggesting their potential role in regulating anthraquinone biosynthesis (Fig. 6c). Cluster 9, consisting of 3,301 genes, was predominantly clustered into numerous gene families, with the top three gene families being WRKY, CYP450, and UGT (Fig. 6d).
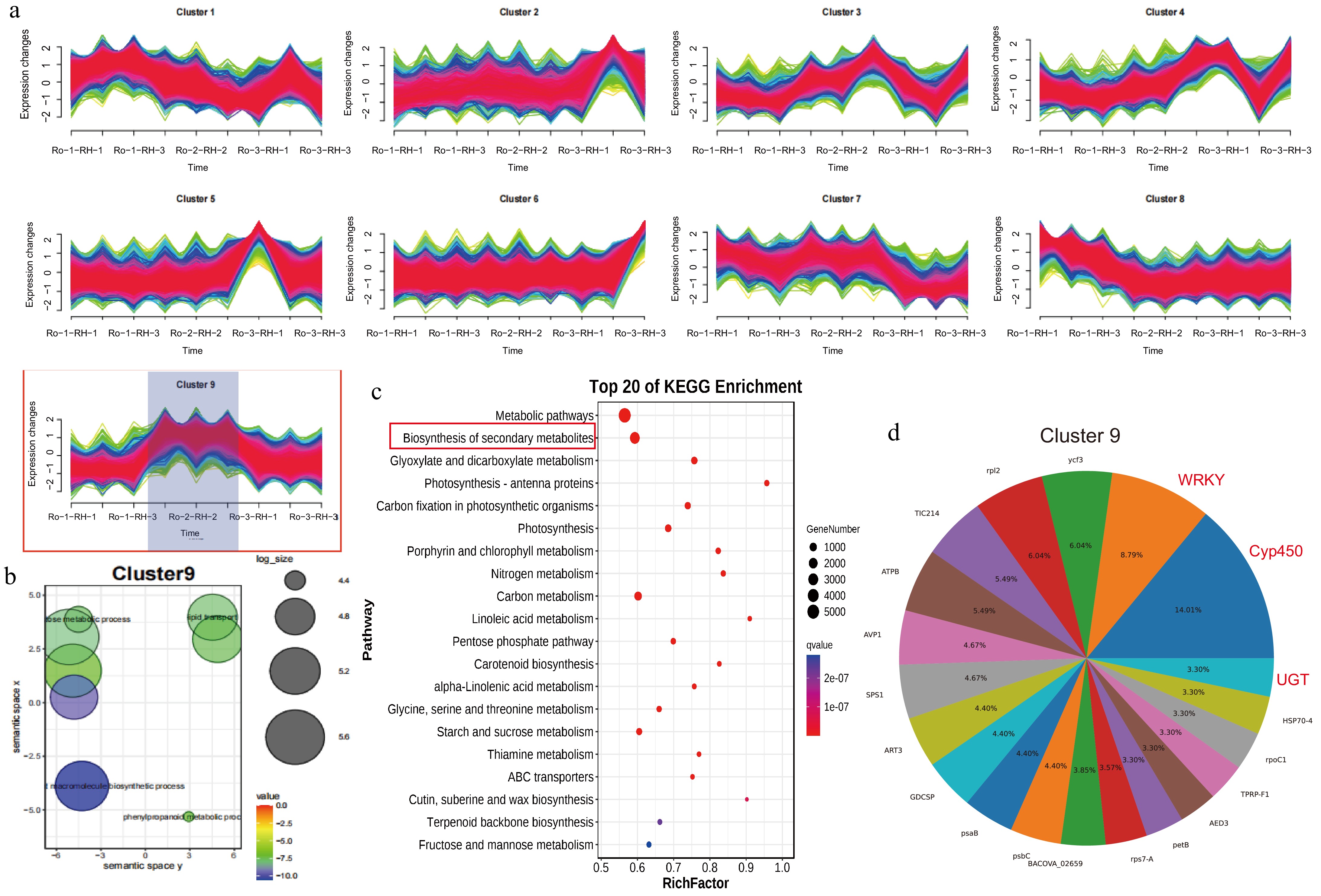
Figure 6.
Spatiotemporal gene expression patterns of R. officinale rhizomes and functional enrichment analysis. (a) Cluster 1 to 9 showing DEGs in RO-1-RH, RO-1-RH, and RO-1-RH. (b) GO enrichment pathway analysis of DEGs from cluster 9. (c) KEGG enrichment pathway analysis of DEGs from cluster 10. (d) The gene family cluster analysis of cluster 9.
Gene co-expression network
-
To unravel the gene regulatory networks associated with the biosynthesis of bioactive compounds in R. officinale roots and rhizomes, co-expression analysis and network construction were performed on 3,734 and 1,578 DEGs in root and rhizome, respectively. A soft-threshold power of 8 was utilized to define the adjacency matrix based on the criterion of approximate scale-free topology (Fig. 7a). Modules were identified using a minimum module size of 30, with the deepSplit sensitivity set to 2, and a cut height for merging modules of 0.2, indicating that modules with eigengenes correlated above 0.8 would be merged (Fig. 7d). The resulting network was constructed from the filtered probes, identifying 21 modules in the root and 6 modules in the rhizome (Fig. 7b−e).
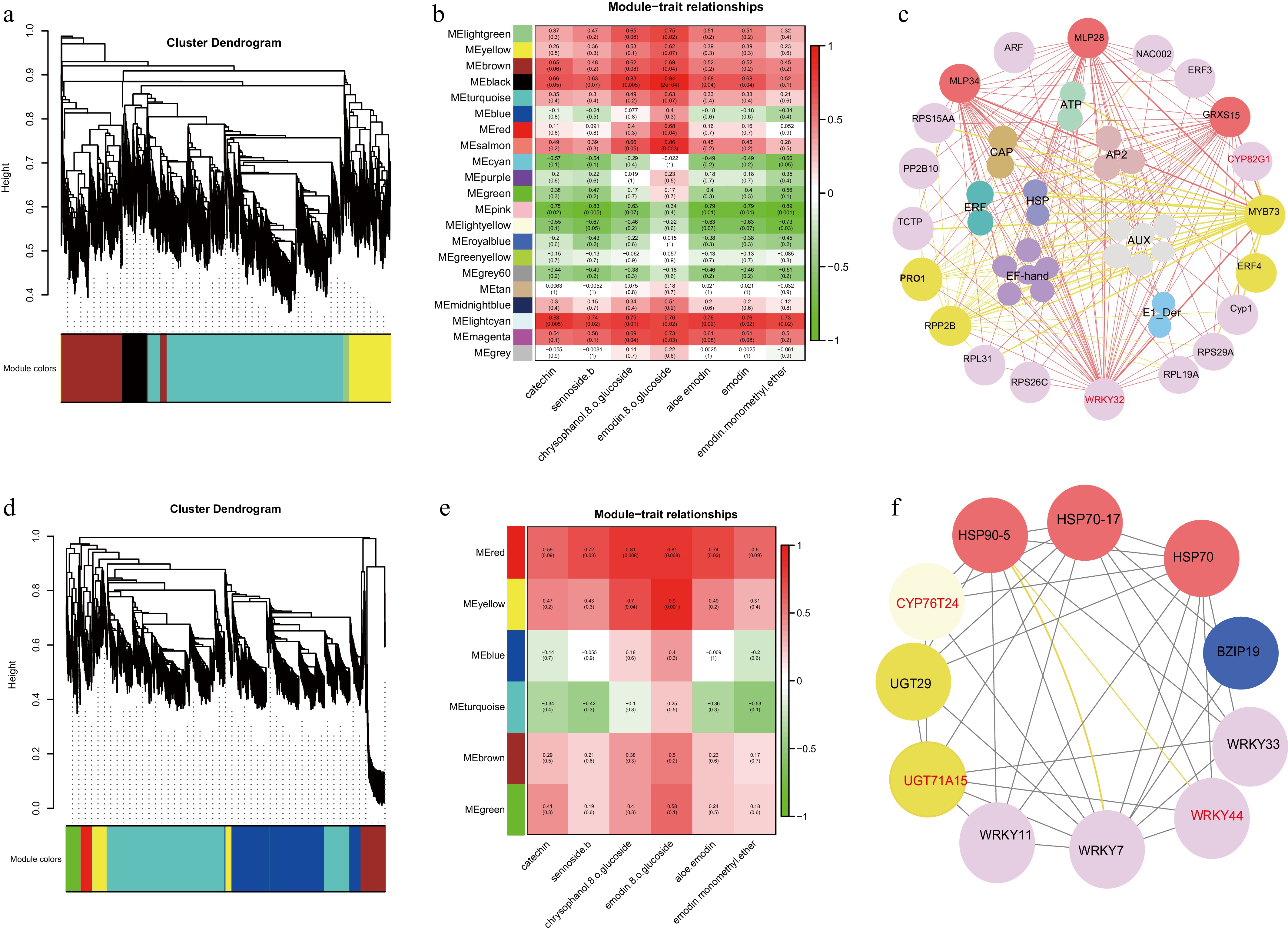
Figure 7.
WGCNA analysis was carried out in the roots and rhizomes of R. officinale. (a), (d) Network topology for different soft-thresholding powers. Numbers in the plots indicate the corresponding soft thresholding powers. The approximate scale-free topology can be attained at the soft-thresholding power of 8. (b), (e) Heatmap plot of the adjacencies of modules. Red represents high adjacency (positive correlation) and blue represents low adjacency (negative correlation). (c), (f) Transcriptome co-expression network diagram.
To elucidate the interactions among gene models, a network heat map of co-expression modules was constructed for the root transcriptome. Notably, seven modules exhibited high correlation with several bioactive components such as catechin, sennoside B, chrysophanol-8-O-glucoside, emodin-8-O-glucoside, aloe-emodin, emodin, and physcion, comprising the light green, yellow, brown, black, turquoise, light blue, and magenta gene modules (Fig. 7b). Subsequently, 20 hub genes were identified within each of these modules, elucidating their pivotal roles in root development and metabolite biosynthesis. Further analysis revealed that genes, such as WRKY32 and CYP82G1 located within the black module, exhibited strong connectivity with other hub genes, reinforcing their importance as key regulators in the biosynthesis of pharmacopoeia indicators (Fig. 7c).
In the rhizome tissues, DEGs were categorized into six distinct modules (Fig. 7e). Among these, four modules exhibited significant correlations with key bioactive compounds, including catechin, sennoside B, chrysophanol-8-O-glucoside, emodin-8-O-glucoside, aloe-emodin, and emodin monomethyl ether. These compounds were associated with the red, yellow, brown, and green gene modules (Fig. 7e). Subsequently, 11 hub genes were identified across the light green, yellow, brown, black, turquoise, light cyan, and magenta gene modules, emphasizing their crucial roles in regulating rhizome development and metabolite biosynthesis. Further network analysis unveiled the strong connectivity of CYP76T24, UGT71A15, and WRKY44 with other hub genes, underscoring their significance as key regulators in the biosynthesis of bioactive compounds (Fig. 7f).
qRT-PCR validation of RNA-seq data
-
To ensure the precision and reproducibility of the transcriptome analysis, five genes were selected for expression analysis in the leaves, rhizomes and roots of R. officinale. The results showed that CYP76T24 was highly expressed in the rhizomes and roots compared to leaves. CYP82G1 was highly expressed in the roots and rhizomes. UGT71A15 was highly expressed in the rhizomes. WRKY32 was highly expressed in roots. WRKY44 was highly expressed in the rhizomes (Fig. 8). These results indicate that these five genes are promising candidates involved in the biosynthesis of anthraquinone in R. officinale.
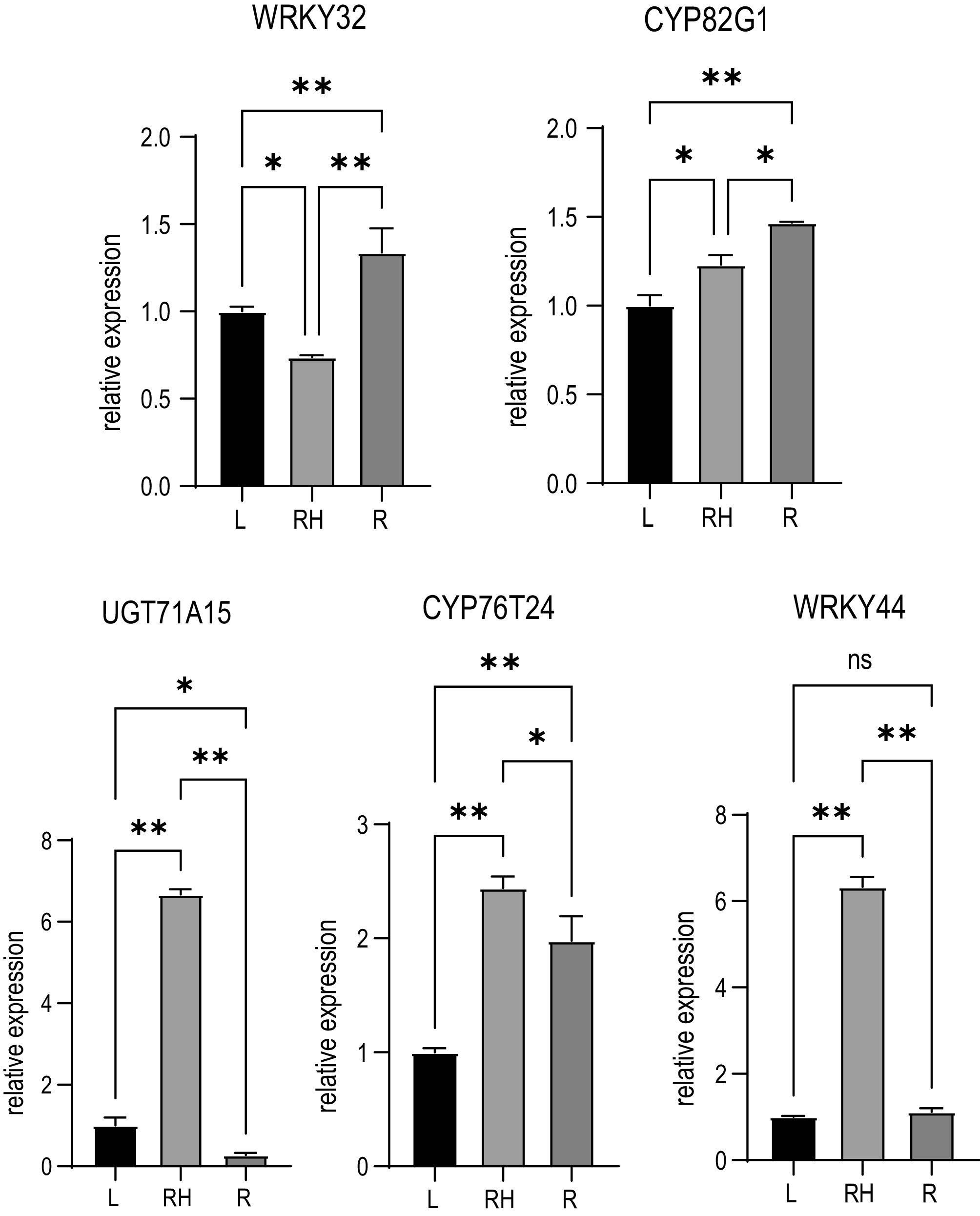
Figure 8.
qRT-PCR validation of gene expression for five candidate genes. Data represent mean ± SD (n = 3 biological replicates). Statistical significance was determined using Student's t-test (* p < 0.05, ** p < 0.01).
-
The roots and rhizomes of R. officinale are rich in diverse chemical constituents, with anthraquinones being the major bioactive components that exhibit diverse pharmacological effects[6−7,9]. The increasing demand for healthcare products derived from the dried roots and rhizomes of R. officinale underscores the necessity of developing appropriate strategies to optimize the yield and quality of medicinal materials with multifaceted biological activities[16,28]. To clarify the compositional changes and metabolic accumulation of hypoglycemic metabolites as influenced by harvest time, this study investigated the alterations in ten bioactive compounds in the roots and rhizomes of R. officinale across different years. Remarkably, seven and six bioactive components exhibited higher accumulation levels in 4-year-old specimens than in 2- and 3-year-old samples, respectively (Fig. 1)[12,15,29]. These findings highlights the accumulation of six common bioactive components over time, suggesting that the 4-year-old root and rhizome exhibited elevated levels of bioactive components.
The developmental procession and maturation of R. officinale root and rhizome exhibit strong correlations with metabolites accumulation dynamics, directly influencing medicinal yield and pharmacologically relevant quality parameters[30,31]. However, the regulatory network through which key genes contribute to the high content of bioactive components in the roots and rhizomes remains poorly elucidated. To bridge this knowledge gap, a multi-tissue transcriptomic strategy (leaf, root, rhizome) was implemented to establish high-resolution RNA-seq datasets, enabling systematic dissection of age-dependent metabolic regulation in root and rhizome (Fig. 2). A comprehensive transcriptomic analysis of 15 groups was performed to identify DEGs across various developmental stages (two, three, and four years of roots and rhizomes) (Fig. 2). A total of 460 DEGs were identified during three stages of root development, predominantly enriched in anthraquinone-related pathway including secondary metabolites biosynthesis, flavonoid metabolism, and phenylpropanoids production. Critical conserved DEGs—such as CYP450 genes (CYP82G1, CYP71A26, CYPRO1, CYP736A117, CYP82C4, CYP72A15, CYP78A5, CYP82G, CYP73A4), the transcription factor WRKY32, and the glycosyltransferase UGT73C5—demonstrated progressive expression elevation across developmental stages, implicating their functional centrality in root-specific phytochemical biosynthesis (Fig. 3). Parallel rhizome profiling revealed 1,224 temporally regulated DEGs, with analogous pathways enrichment patterns. Key regulators included CYP450 family members (CYP75B137, CYP82G1, CYP82C4, CYP81Q32, CYP95, CYP76T24, CYP81Q32), WRKY transcription factors (WRKY4, WRKY44, WRKY40, WRKY46, and UGT71A15, collectively orchestrating rhizome-specific metabolite accumulation[12,30].
Although transcriptional profiles of specific internodes have been generated, the transcriptional dynamics across different ages of roots and rhizomes in R. officinale remain largely unexplored[12,15,30]. This study presents the first comprehensive RNA-seq-based transcriptome analysis investigating the developmental processes of roots and rhizomes in R. officinale. 13,964 genes exhibiting a high CV were identified and subsequently classified into nine clusters for the root and ten clusters for the rhizome based on their expression patterns (Figs 5, 6). Within these co-expression clusters, the DEGs within cluster 10 (root) and cluster 9 (rhizome) were further scrutinized. These genes, implicated in the biosynthesis of secondary metabolites in R. officinale, were validated through GO and KEGG enrichment analyses. Moreover, these genes were categorized into different gene families, with the predominant types being CYP450, WRKY, and UGT, suggesting their involvement in the biosynthesis of secondary metabolites (Figs 5, 6). This extensive collection of gene clusters serves as a valuable and reliable resource for the improving R. officinale, substantially augmenting our understanding of the genetic mechanisms governing root and rhizome development in this species.
With the continual advancements in sequencing technology and the expanding repository of transcriptome data, WGCNA has emerged as a powerful tool for the rapid and efficient identification of genes or TFs associated with specific traits[31−33]. In our study, a comprehensive analysis was conducted, encompassing a total of 3,356 and 1,754 DEGs in the roots and rhizomes, respectively. This analysis entailed module division, correlation analysis between modules and traits, enrichment analysis, and transcription factor analysis. In the root, WGCNA identified the CYP19-3 and MYB73 genes, which exhibit high correlations with the accumulation of seven common bioactive components. These genes regulate the biosynthesis of catechin, sennoside B, chrysophanol-8-O-glucoside, emodin-8-O-glucoside, aloe-emodin, emodin, and emodin monomethyl ether, influencing the production of related secondary metabolites (Fig. 7). Similarly, in the rhizome, WGCNA revealed the regulatory roles of CYP76T24, UGT71A15, and WRKY44 in the biosynthesis of six common bioactive compounds, including catechin, sennoside B, chrysophanol-8-O-glucoside, emodin-8-O-glucoside, aloe-emodin, and emodin monomethyl ether (Fig. 7).
Transcription factors and modifying enzyme genes such as WRKY, CYP450, and UGT play pivotal roles in the biosynthesis of metabolites in medicinal plants[34−36]. CYP450s are extensively involved in the biosynthesis of secondary metabolites, encompassing isoprenoids, flavonoids, alkaloids, and phenolic compounds. Examples include CYP71AV9 and CYP71BL5 in Cynara cardunculus[34]. WRKY transcription factors exhibit notable correlations with the regulation of anthocyanin biosynthesis, as evidenced by MdWRKY11 in apples and PyWRKY26 in red-skinned pears[35]. Additionally, Pb-WRKY75 has been shown to promote PbMYB10b expression and activate anthocyanin biosynthetic genes DFR and UFGT in pears[36]. In our study, comparative transcriptome analysis revealed the involvement of CYP450, WRKY, and UGT73C5 in the accumulation of bioactive compounds in both roots and rhizomes (Figs 2−4). STEM analysis indicated that CYP450, WRKY, and UGT were the predominant types, underscoring their relevance to genes associated with the biosynthesis of secondary metabolites (Fig. 5, 6). Furthermore, WGCNA analysis identified WRKY32 and CYP82G1 in the root and CYP450, along with two UGT genes (UGT29 and UGT85A24), in the rhizome. These findings collectively highlighted the involvement of CYP450 and UGT genes in the biosynthesis of bioactive metabolites in R. officinale.
For R. officinale, as well as some other species in Rheum L., the roots and rhizomes are considered the primary and more effective medicinal parts compared to the leaves[37–38]. These underground structures contain higher concentrations of bioactive compounds, such as anthraquinones (e.g., emodin, rhein), tannins, and stilbenes, which are responsible for the plant's pharmacological properties, including laxative, anti-inflammatory, and antimicrobial effects[39]. In contrast, the leaves generally exhibit lower levels of these therapeutic compounds and may even contain toxic substances, such as oxalic acid. Thus, both traditional and modern pharmacopoeias prioritize the roots and rhizomes of R. officinale as the standard medicinal herb[40]. In the previous studies, comparative transcriptome analysis showed that the genes were highly expressed in the medicinal tissues represent its important function. For example, in Fritillaria unibracteata, comparative transcriptomics identified high expression levels in bulb tissues for genes such as ACAT, HMGR, MK, MVD and DXS, indicating their important role in the production of active steroidal alkaloids[41]. In addition, transcriptome analysis of these eight candidate genes showed higher expression levels in Artemisia argyi leaves compared to other tissues, suggesting their key roles in the biosynthesis of terpenoids[42]. In particular, a combined omics analysis of R. officinale revealed a significant amplification of genes related to anthraquinone biosynthesis, with many enzyme genes highly expressed in the roots. This may explain the significantly higher anthraquinone content observed in rhubarb roots compared to other tissues[43]. In this study, CYP76T24, CYP82G1, UGT71A15, WRKY32 and WRKY44 were found to be highly expressed in the roots and rhizomes of R. officinale. Among these, UGT71A15 and WRKY44 were specifically highly expressed in rhizomes, whereas CYP76T24 and CYP82G1 were highly expressed in both roots and rhizomes, and WRKY32 showed predominant expression in the roots (Fig. 8), suggesting that they may be key candidate genes for anthraquinone biosynthesis in R. officinale.
The heightened accumulation of bioactive compounds in 4-year-old R. officinale roots and rhizomes likely stems from a synergistic interplay of developmental programming, environmental adaptation, and stress-responsive mechanisms. Over time, the plant undergoes metabolic maturation, driven by age-dependent upregulation of key biosynthetic genes, such as CYP450s (e.g., CYP82G1, CYP76T24), WRKYFs (e.g., WRKY32, WRKY44), and UGTs (e.g., UGT73C5), which regulate anthraquinone and flavonoid biosynthesis. These genes exhibit sustained expression throughout secondary growth, enabling progressive metabolite biosynthesis and stabilization. Concurrently, prolonged environmental exposure—such as nutrient competition, seasonal stress, and pathogen pressure—may trigger adaptive resource allocation to underground organs, thereby amplifying precursor availability and defence-related metabolite production[44]. Stress-responsive pathways, mediated by WRKYs and CYP450s, further enhance anthraquinone synthesis as a survival strategy, linking metabolite accumulation to age-dependent stress memory. Modules identified through WGCNA (e.g., CYP19-3, UGT85A24) suggest coordinated regulation of metabolic clusters, integrating developmental timing (via phytohormones) and environmental cues. Thus, the 4-year peak reflects an evolved equilibrium where extended development, environmental resilience, and defence prioritization converge, positioning these genes as key factors for optimizing medicinal quality in R. officinale.
This study extends previous insights into anthraquinone biosynthesis in Rheum species by elucidating age-dependent regulatory mechanisms. Earlier studies identified tissue-specific expression of CHS genes (e.g., Chr1A.4008, Chr1B.3886) and non-CHS genes (e.g., Chr5B.4624) in R. officinale roots linked to anthraquinone production via the polyketide pathway[45]. Similarly, phylogenetic analysis in Polygonaceae highlighted PKS III genes (PL_741 PKSIII1, PL_11549 PKSIII5) as critical for polyketide biosynthesis, consistent with our findings on CYP450s and UGTs[46]. In R. tanguticum, tissue-specific expression of anthraquinone-related TFs (e.g., bHLH, MYB) further supports conserved regulatory networks[47]. However, our study uniquely identifies age-dependent dynamics in R. officinale: novel candidates like CYP76T24, WRKY44, and UGT85A24 show progressive upregulation in 4-year-old roots/rhizomes, correlating with anthraquinone accumulation. While previous studies emphasized CHS and UGT73 gene families in tissue specificity[45−47], our WGCNA analysis reveals co-expressed modules involving CYP450s, WRKYs, and UGTs that might drive metabolite biosynthesis over time. For instance, CYP19-3 and MYB73 emerged as hub genes in mature roots, highlighting temporal regulation previously unexplored. These findings bridge tissue-specific and developmental frameworks, providing new targets for optimizing harvest timing and metabolic engineering in Rheum species (Figs 6, 7 & 8).
-
In this study, a total of ten bioactive compounds were assessed in roots and rhizomes of R. officinale across different years. The results revealed the accumulation of six common bioactive compounds over time, including catechin, sennoside B, chrysophanol-8-O-glucoside, emodin-8-O-glucoside, aloe-emodin, and emodin monomethyl ether. Transcriptomic profilings revealed 23 consistent DEGs spanning all three developmental stages, predominantly from CYP450, WRKY, and UGT gene families, including UGT71A15 and UGT73C5. STEM analysis identified two significant expression patterns: root-specific cluster 10 (1,826 genes) and rhizome-specific cluster 9 (3,301 genes), both dominated by WRKY, CYP450, and UGT gene families. Weighted Gene Co-expression Network Analysis (WGCNA) further elucidated key regulatory relationships, with the black module in roots showing WRKY32 and CYP82G1 as central hubs with high connectivity scores, indicating their pivotal role in pharmacopoeia compound biosynthesis. Similarly, rhizome networks identified CYP76T24, UGT71A15, and WRKY44 as core regulatory nodes. These results systematically demonstrate the coordinated involvement of WRKY transcription factors, CYP450 enzymes, and UGT glycosyltransferases in secondary metabolite biosynthesis pathways. The integrated molecular data provided a framework for developing standardized cultivation strategies to improve medicinal production in R. officinale.
This research was supported by the Scientific Research Project of the National Natural Science Foundation of China (Grant Nos 82104334, 81973430), Shaanxi Institute of Basic Science (Chemistry and Biology) in 2023 (Grant No. 23JHQ025), Doctoral Scientific Startup Fund of Shaanxi University of Chinese Medicine, Youth Innovation Team Research Project of Shaanxi Provincial Education Department (Grant No. 24JP043), Shaanxi Provincial Department of Education Service Local Special Plan Project (Grant No. 24JC030), Natural Science Basic Research Program of Shaanxi (Grant No. 2024JC-YBQN-0837), and Discipline Innovation Team Project of Shaanxi University of Chinese Medicine (Grant No. 2019-QN01).
-
The authors confirm contribution to the paper as follows: conceptualization, data curation, writing - original draft: Li YM; HPLC and formal analysis: Wang XR; investigation: Li YM, Yang G; software: Yan TT, Hu XC; validation: Peng L; visualization: Gao J; methodology, project administration, writing - review & editing: Yan F, Zhang G. All authors have read and agreed to the published version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files. The raw reads were uploaded to the NCBI BioProject database under Accession No. PRJNA981643.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 HPLC detection method of ten bioactive compounds in R. officinale.
- Supplementary Table S2 The qRT-PCR primers used in this study.
- Supplementary Table S3 The differential gene list during three ages of root of R. officinale.
- Supplementary Table S4 The differential gene list during three ages of rhizome of R. officinale.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Li YM, Wang XR, Yang G, Yan TT, Hu XC, et al. 2025. Profiling the accumulation of ten bioactive compounds in Rheum officinale across different growth years using integrated HPLC and transcriptomic approaches. Medicinal Plant Biology 4: e022 doi: 10.48130/mpb-0025-0020 

Profiling the accumulation of ten bioactive compounds in Rheum officinale across different growth years using integrated HPLC and transcriptomic approaches
- Received: 13 February 2025
- Revised: 19 April 2025
- Accepted: 12 May 2025
- Published online: 26 June 2025
Abstract: Rheum officinale is a prominent traditional medicinal herb in the Chinese Pharmacopeia. Variations in the composition and accumulation of secondary metabolites in R. officinale have been observed to correlate with harvest periods. This study investigated the progressive accumulation of six bioactive compounds in the roots and rhizomes of R. officinale over three growth years. Comparative transcriptome analyses were performed across three developmental stages of both root and rhizome tissues. In the root tissues, 23 differentially expressed genes (DEGs) were observed, including CYP450, WRKY, UGT73C5, WAT1, and XTH15, which displayed significant differential expression patterns. Similarly, 14 DEGs in the rhizome tissues showed significant differential expression, encompassing CYP450, WRKY, and UGT71A15. Short Time-series Expression Miner (STEM) analysis revealed that cluster 10 (1,826 genes) in the root tissues was notably enriched in metabolic processes, predominantly involving WRKY, CYP450, and HSP gene families. In rhizome tissues, cluster 9 (3,301 genes) included numerous gene families, with WRKY, CYP450, and UGT as the top three. Additionally, Weighted Gene Co-expression Network Analysis (WGCNA) was used to construct networks for 3,734 and 1,578 DEGs in root and rhizome tissues, respectively. Hub genes such as WRKY32 and CYP82G1 exhibited robust connectivity with other genes, suggesting their pivotal roles in pharmacopoeia indicator biosynthesis in root tissues. Moreover, CYP76T24, UGT71A15, and WRKY44 were identified as crucial genes in pharmacopoeia indicator biosynthesis in rhizome tissues. Collectively, these comprehensive results underscore the involvement of CYP450 and UGT in the biosynthetic pathways of bioactive compounds in R. officinale.
-
Key words:
- Rheum officinale /
- Comparative transcriptome /
- STEM /
- WGCNA /
- Bioactive components