









-
Patchouli (Pogostemon cablin [Blanco] Benth.) is a medicinal and aromatic plant belonging to the genus Pogostemon of the Lamiaceae family. Its dried above-ground parts are used for medicinal purposes[1]. The Patchouli germplasm is a diverse resource cultivated primarily in the provinces of Guangdong, Guangxi, and Hainan (China)[2]. It has the functions of promoting the circulation of qi, harmonizing the middle burner, stopping vomiting, and dispelling summer heat[1]. It is used mainly to treat dampness obstruction syndrome and summer dampness syndrome, and has been regarded as an essential drug for treating summer dampness by successive generations of physicians. The essential oil extracted from patchouli contains a variety of active components, and is widely used in the medical, spice, and cosmetics industries[3−5]. Among the active ingredients, sesquiterpenoids are the main ones with medicinal effects, which include antibacterial[6,7], anti-inflammatory[8], anti-depressant[9], and mosquito repelling[10] effects. Among sesquiterpenoids, patchouli alcohol is an indicator component included in the Chinese Pharmacopoeia. It is widely used to evaluate the quality of broad-leaved patchouli-derived medicinal materials and broad-leaved patchouli essential oil. The 2020 edition of the Chinese Pharmacopoeia (Volume 1) stipulates that the content of patchouli alcohol in broad-leaved patchouli medicinal materials shall not be less than 0.10%, and that the content in broad-leafed patchouli essential oil shall not be less than 26%[1]. Currently, the global demand for patchouli is increasing annually; however, manually cultivated patchouli remains the primary source of patchouli oil, which limits its production. Additionally, in cultivation, the presence of intercropping obstacles severely affects the yield and quality of patchouli[11]; moreover, patchouli cuttings are the main materials cultivated, and this asexual reproduction method leads to slow growth, a high incidence of pests and diseases, and poor adaptability, thereby reducing the yield[12]. Although synthetic biology techniques enable cell factories to produce patchouli alcohol[13], how to increase the content of the active constituent of patchouli has attracted much attention.
To date, the biosynthetic pathway of sesquiterpenoids has been relatively fully elucidated. The synthesis process involves mainly the upstream mevalonate pathway (MVA), or the methylerythritol phosphate (MEP) pathway, as well as the downstream reactions catalyzed by farnesyl diphosphate synthase (FPPS), and sesquiterpenoid synthase (PTS)[14]. On the basis of the previously assembled whole genome of patchouli at the chromosomal level[15] and other related findings, advances have been made in identifying key enzyme-encoding genes involved in the sesquiterpenoid biosynthesis pathway. Studies have shown that patchoulol synthase (PatPTS) is a multifunctional terpenoid synthase in patchouli that plays a crucial role in regulating the biosynthesis of patchoulol, and can catalyze the conversion of farnesyl pyrophosphate (FPP) into patchoulol, and at least 13 other sesquiterpene products[16]. Additionally, in previous research, seven tandemly repeated PatPTS genes were found in patchouli, and may be the main factor contributing to the accumulation of patchoulol[15].
Many studies have revealed the crucial regulatory role of TFs in the terpene synthesis pathway. TFs influence the specific expression, temporal, and spatial expression patterns, and efficiency of enzyme-encoding genes[17]. This influence often leads to changes in the transcriptional activity of specific terpene biosynthesis genes, thereby affecting the content of terpene compounds. TFs are common genetic engineering tools. Relevant transcription factors mainly belong to families such as the APETALA2/ethylene responsive factor (AP2/ERF), WRKY, basic leucine zipper (bZIP), and basic helix-loop-helix (bHLH)[18,19]. Although some studies have revealed the regulatory effects of a few transcription factors, related research still has limitations, and a comprehensive study of the regulatory relationships between all the transcription factors and the sesquiterpenoids biosynthesis pathway in patchouli, at the genomic level, is lacking.
Gene co-expression networks are constructed on the basis of the degree of correlation between genes, and the correlations between all pairs of genes can be obtained, and a threshold can be set for filtering[20]. Weighted gene co-expression network analysis (WGCNA) is used to re-evaluate the correlations between gene pairs through exponential weighting of correlation coefficients to cover the information that was previously filtered out because of the low threshold[21]. To date, WGCNA, as a systematic biological analysis method, has been widely applied in studies on plant growth and development cycles, disease resistance and stress tolerance mechanisms, secondary metabolism regulation, etc.[22]. Therefore, a gene co-expression network for patchouli was constructed.
Over the past century, methodologies for investigating the biosynthesis of secondary metabolites have evolved considerably, transitioning from classical biochemical techniques to integrated approaches incorporating molecular biology and multi-omics technologies[23]. In this study, transcriptome analysis was conducted using samples of patchouli collected at different stages of growth and development, after stimulation with exogenous hormones and under adverse stress. On the basis of the relative expression levels of all the genes in the patchouli genome, a gene expression network for the entire genome of patchouli was constructed. Correlations among the expression of differentially expressed module genes, and the content of sesquiterpenoids in patchouli were analyzed. The co-expression relationships between genes encoding enzymes and transcription factors related to the differentially expressed genes were identified to construct the gene regulation network for the sesquiterpenoid biosynthesis pathway in patchouli. The regulatory relationships between transcription factors and the sesquiterpenoids biosynthesis pathway were analyzed to reveal a possible model of the biosynthesis of sesquiterpenoids in patchouli. This study provides new ideas for improving the active ingredients of patchouli, and lays a theoretical foundation for increasing the synthesis of active ingredients in medicinal plants, through the strategy of regulating their production via TFs, and cultivating high-quality varieties with high-quality active ingredients.
-
Patchouli plants were collected from Yaowang Mountain at the Guangzhou University of Chinese Medicine (23.03° N, 113.23° E) in Guangdong Province, China. After tissue culture, the patchouli plants are transferred to an artificial climate chamber (25 ± 2 °C). The plants were cultured in a growth chamber in the Research Center of the Institute of Medicinal Plant Physiology and Ecology, Guangzhou University of Chinese Medicine, Guangzhou, China.
Patchouli samples were gathered at different growth and developmental stages, specifically, multiple shoots were collected at 0, 15, 30, and 45 d, and leaf, stem, and root tissues were collected at 68 and 90 d. In addition, it also covered the young stem, old stem, young leaf, old leaf, and root for 2-, 4-, and 6-month-old patchouli plants (mature period). For hormone treatments, which was performed according to the method described by Wang et al., with improvements[24], 68-d-stage tissue cultures were sprayed with 300 μΜ methyl jasmonate (MeJA), salicylic acid (SA), abscisic acid (ABA), and 500 μM ethylene (ETH) in 0.5% (v/v) ethanol. For mock treatment, tissue cultures were sprayed with 0.5% ethanol. Leaf samples were washed and collected at different time points (0, 2, 4, 8, and 16 h), and stored at −80 °C. Results are shown in Supplementary Fig. S1. Compared with the negative control, the total sesquiterpene content of samples sprayed for 16 h with MeJA, 4 h with ABA, 4 and 8 h with MeJA + ABA showed higher. Then these samples were selected for RNA sequence (RNA-seq).
For salt treatment, 5-week-stage tissue, and 2-week-stage soil cultures were divided into Group A (foliar fertilizer solution), B (20 mM NaCl), C (40 mM NaCl), and D (80 mM NaCl). To prevent salt shock, the samples were added to the solution at the preset concentration twice within a 5-d period. After the final concentration was reached, 500 mL of the salt solution at the final concentration was poured onto the samples four times every 4 d, with six plants in each group, and three biological replicates. The samples were stored at −80 °C on the 9th and 17th d. This process was performed according to the method described by Yuan et al., with improvements[25]. Results are shown in Supplementary Fig. S2. Compared with the negative control, total sesquiterpene content of samples sprayed with 80 mM for 8 d, and 20 mM for 16 d were showed higher. Then these samples were selected for RNA-seq.
Gaschromatography-mass spectrometry (GC-MS) analysis
-
Adjustments were made on the basis of previously described methods[26]. GC-MS analysis was performed on a 7890A/5975C GC-MS unit (Agilent Technologies, Santa Clara, CA, USA) equipped with an HP-Intiowax polyethylene glycol column. The injector temperature was 250 °C. Helium was used as the carrier gas at a constant flow rate of 1.0 mL/min. For the patchouli alcohol determination in different accessions, 1 μL of sample was injected in the split mode (20:1) onto the column. The temperature gradient was started at 60 °C for 2 min, increased to 250 °C at a rate of 6 °C/min, and then held for 3 min. The mass spectrometry conditions were as follows: EI ion source, ion source temperature of 230 °C, the quadrupole temperature of 150 °C, the electron energy of 70 eV, an emission current of 34.6 μA, a multiplier voltage of 962 V, an interface temperature of 250 °C, and a mass range of 50–350 amu. The samples with higher content change compared with the control group were selected as RNA sequencing library building samples.
Construction of a gene co-expression regulatory network by WGCNA
RNA sequencing library construction and data analysis
-
Total RNA was extracted from samples using a HiPure Plant RNA Mini Kit (R4151) for RNA sequencing on the Illumina NovaSeq6000 platform (Illumina, USA) at Beijing Berry Genomics. The raw data for every samples were analyzed as previously described[27].
Soft threshold of WGCNA
-
The scale-free fit index was plotted using the function Sft
${\$} $ Gene co-expression network
-
To correct the correlation coefficients in the adjacency matrix, the function TOMsimilarity was used to transform the adjacency matrix into the topological overlap matrix (TOM). Aferwards, the hierarchical clustering method was used to divide the strongly correlated subnetworks according to the formula 1-TOM, after which the co-expressed gene modules were obtained. The main parameters were merge_height = 0.12, minmodulegene = 30.
Screening of key modules
-
Key enzyme genes were overlapped with module genes, and modules containing key enzyme-encoding genes were identified. Moreover, cocorrelation analysis was performed between the expression of the module genes and the relative content of sesquiterpenoid compounds in the samples of patchouli using the cor function in R language using RStudio software (v 1.2.5033), and candidate modules related to a high sesquiterpenoid content were identified as the basis for subsequent analysis.
Gene ontology annotation of key modules
-
The tools agriGO (v 2.0)[28] and Kobas (v 3.0)[29] were used to conduct GO annotation analysis of candidate module genes using the Arabidopsis annotation database.
Visualization of regulatory networks of sesquiterpenoid biosynthetic pathways
-
Using the function exportNetworkToCytoscape, regulatory relationships between genes in the candidate modules were derived. On the basis of the key enzyme-encoding genes and TFs members of the sesquiterpenoid pathway identified above, only the regulatory relationships between key enzyme-encoding genes and TFs were omitted. Afterward, the visualization tool Cytoscape (v.3.7.1)[30] was used to visualize the regulatory relationships between key enzyme genes of the sesquiterpenoid biosynthesis pathway and TFs, and a regulatory network of the sesquiterpenoid biosynthesis pathway was constructed.
Y1H assay
-
The prey construct was generated by cloning the full-length coding sequences of PatHDZIP116, PatSBP2, PatMYBR23, and PatMYB13 into the pGADT7 vector (pGADT7-TF), while the bait construct was generated by inserting the PatPTS9 promoter fragment into the pAbAi vector (pAbAi-PatPTS9pro). The obtained pAbAi-PatPTS9pro plasmid was digested with BbsI and subsequently introduced into the Y1HGold yeast strain to generate reporter strains. pGADT7-TF was transformed into the reporter strains, and the cells were then cultured in SD/-Leu medium with or without aureobasidin (AbA, 200 ng/mL) to verify the positive interactions. The yeast strains cotransformed with pGAD-p53 + p53-AbAi, and pGADT7 + pAbAi-PatPTS9pro were referred to as positive and negative controls, respectively. The sequences of the primers used are listed in Supplementary Table S1.
Dual-Luc
-
The PatPTS9 promoter was cloned and inserted into the pGreenII 0800-LUC vector (the primer sequences used are listed in Supplementary Table S1). Effector constructs for PatHDZIP116, PatSBP2, PatMYBR23, and PatMYB13 were generated by cloning the corresponding genes into the pGreenII 62-sk vector. The recombinant plasmids were subsequently transformed into Agrobacterium tumefaciens strain GV3101 (pSoup-p19; Weidi, Shanghai, China), which was subsequently infiltrated into Nicotiana benthamiana leaves. Equal volumes of the two bacterial suspensions were mixed at a 10:1 ratio and infiltrated into N. benthamiana leaves. Luciferase fluorescence was imaged at 40–48 h postinfiltration using a PlantView100 imaging system (BLT, China).
The fluorescence intensity was quantified using a luminometer; a bar graph was drawn based on the scanned values to show the intensity of fluorescence generated by the breakdown of the substrate by the luciferase enzyme and to determine the degree of interaction between the target proteins.
Transient overexpression
-
The CDSs of PatHDZIP116, PatSBP2, PatMYBR23, and PatMYB13 were cloned and inserted into the pJL TRBO vector at the PacI and NotI restriction sites (the primer sequences used are listed in Supplementary Table S1). The recombinant plasmids were subsequently transformed into Agrobacterium tumefaciens strain GV3101 (pSoup-p19). Successfully transformed single colonies were cultured in 50 mL of LB medium supplemented with 50 μg/mL kanamycin, and 25 μg/mL rifampicin at 28 °C with shaking at 225 rpm for approximately 24 h until the OD600 exceeded 0.6. The bacterial culture was centrifuged at 3,600 × g for 6 min, and washed once with MMA resuspension buffer (10 mM MES, pH 5.7; 10 mM MgCl2; 100 μM acetosyringone). The pellet was resuspended in fresh MMA buffer, and the solution was adjusted to an OD600 of 1.0. The cells were incubated in induction medium for 2–24 h at room temperature before infiltration into the abaxial surface of patchouli leaves using a 1-mL syringe with no needle. After infiltration, the plants were incubated at 25 °C for 72–96 h, after which the leaves were collected for patchoulol determination and total RNA extraction.
EMSA
-
EMSAs were performed to examine whether PatHDZIP116 and PatSBP2 could bind to the promoters of PatPTS9. To do this, two complementary 60 bp oligonucleotides containing the binding cis-elements were separately synthesized and labeled with biotin using the EMSA Probe Biotin Labeling Kit (Beyotime, cat GS008). The DNA gel mobility shift assay was performed using an EMSA kit (Beyotime, cat GS009) following the manufacturer's protocol. All assays were performed in three replicates. The primers used are listed in Supplementary Table S1.
Statistical analysis
-
GraphPad Prism 8 software was used for statistical analysis of the data using Student's t test and ANOVA. A p value < 0.05 was considered to indicate a statistically significant difference.
-
Patchouli (Pogostemon cablin [Blanco] Benth.) samples were collected at different growth cycle stages to ensure high variability in the patchouli alcohol content (Supplementary Tables S2 and S3). The stages at which the plant samples were collected encompassed the complete growth and development cycle (Fig. 1a). In addition to samples from the growing period, samples in which the patchouli alcohol content was significantly changed by hormone stress and salt stress were selected for the construction of the gene expression network. For hormone treatment, 2-week-stage samples were sprayed with MeJA for 16 h, ABA for 4 h, and MeJA + ABA for 4 and 8 h (Fig. 1b and Supplementary Fig. S1). For salt treatment, 5-week-stage samples were sprayed with 80 mM NaCl for 8 d or 20 mM NaCl for 16 d for the construction of the gene expression network (Supplementary Fig. S2).
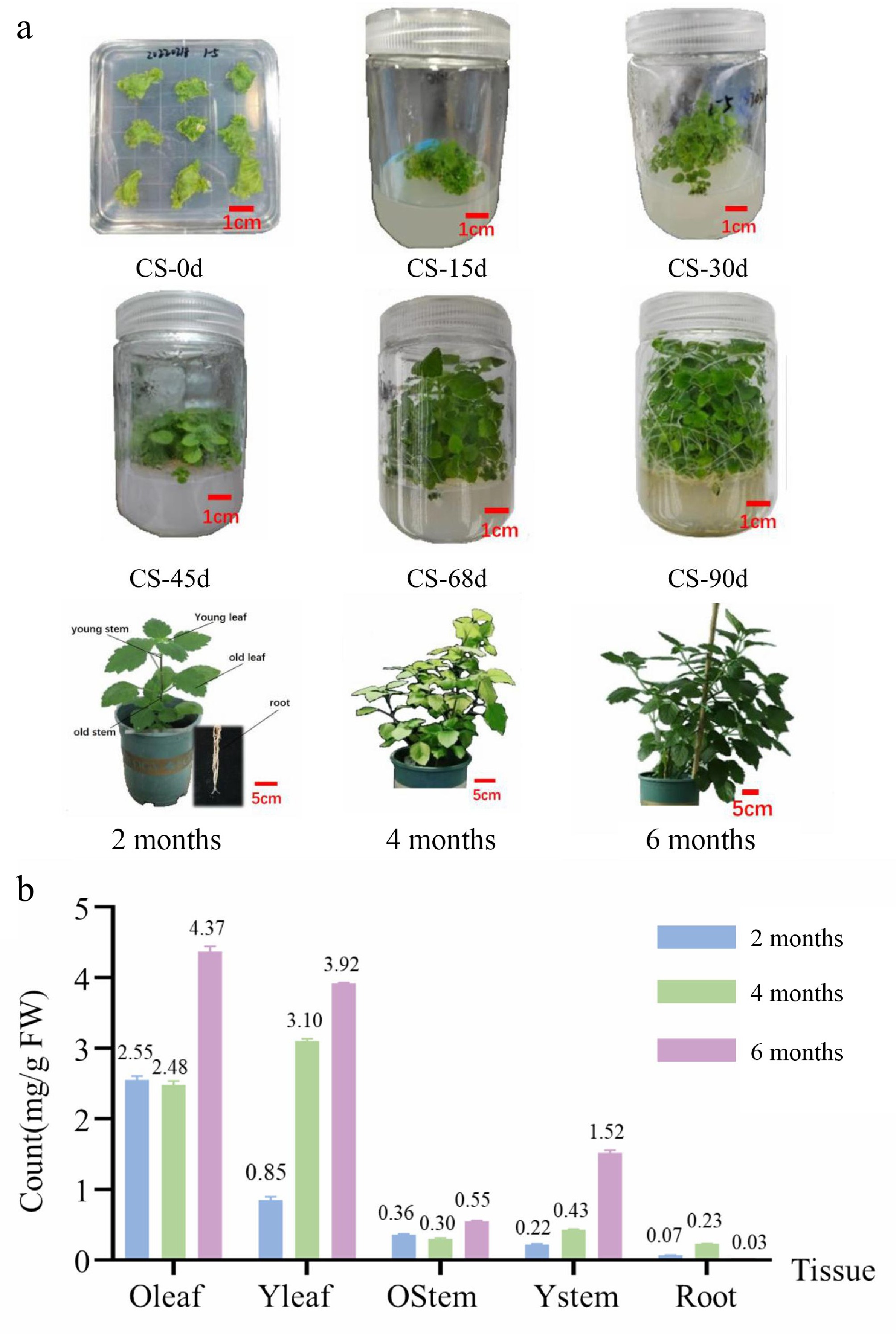
Figure 1.
Patchouli samples and total sesquiterpene compound content. (a) Samples from culture seeding (0, 15, 30, 45, 69, and 90 d), and soil growth (2, 4, and 6 months). (b) Total sesquiterpene compound content from different organization samples (2, 4, and 6 months). Total sesquiterpene compound content from culture seeding samples shown in Supplementary Tables S2 and S3.
Global gene expression profiling using RNA-seq was also carried out for the abovementioned plant samples. An average of more than 26 million high-quality reads (~6 Gb) was obtained for each sample, and the reads were aligned to the patchouli chromosome-level genome, with an average mapping rate of 90.81%. An average of 81,667 expressed genes was detected in each patchouli sample.
Identification of module genes whose expression was highly correlated with the sesquiterpenoid content and the terpenoid metabolic pathway
-
WGCNA was performed to identify candidate trait-linked modules on the basis of the expression profiles of 109,700 genes detected in the patchouli samples. The soft-thresholding power was calculated to be 9 (Supplementary Fig. S3), and a total of 159 modules containing 101,412 genes were identified (the salmon module to the lavender module) (Fig. 2a and Supplementary Table S4), while the remaining 8,288 genes were considered outliers and excluded from further analysis. Module-trait association studies were then performed, and 20 modules identified as potential modules involved in the terpenoids metabolic pathway for their significant correlation with sesquiterpenoid contents (Fig. 2b and Supplementary Table S5) (p ≤ 0.05). Besides, expression patterns of each gene in the selected modules were highly correlated with metabolite accumulation. Figure 2c shows the correlation analysis of module midnightblue genes and sesquiterpene contents of all patchouli samples.
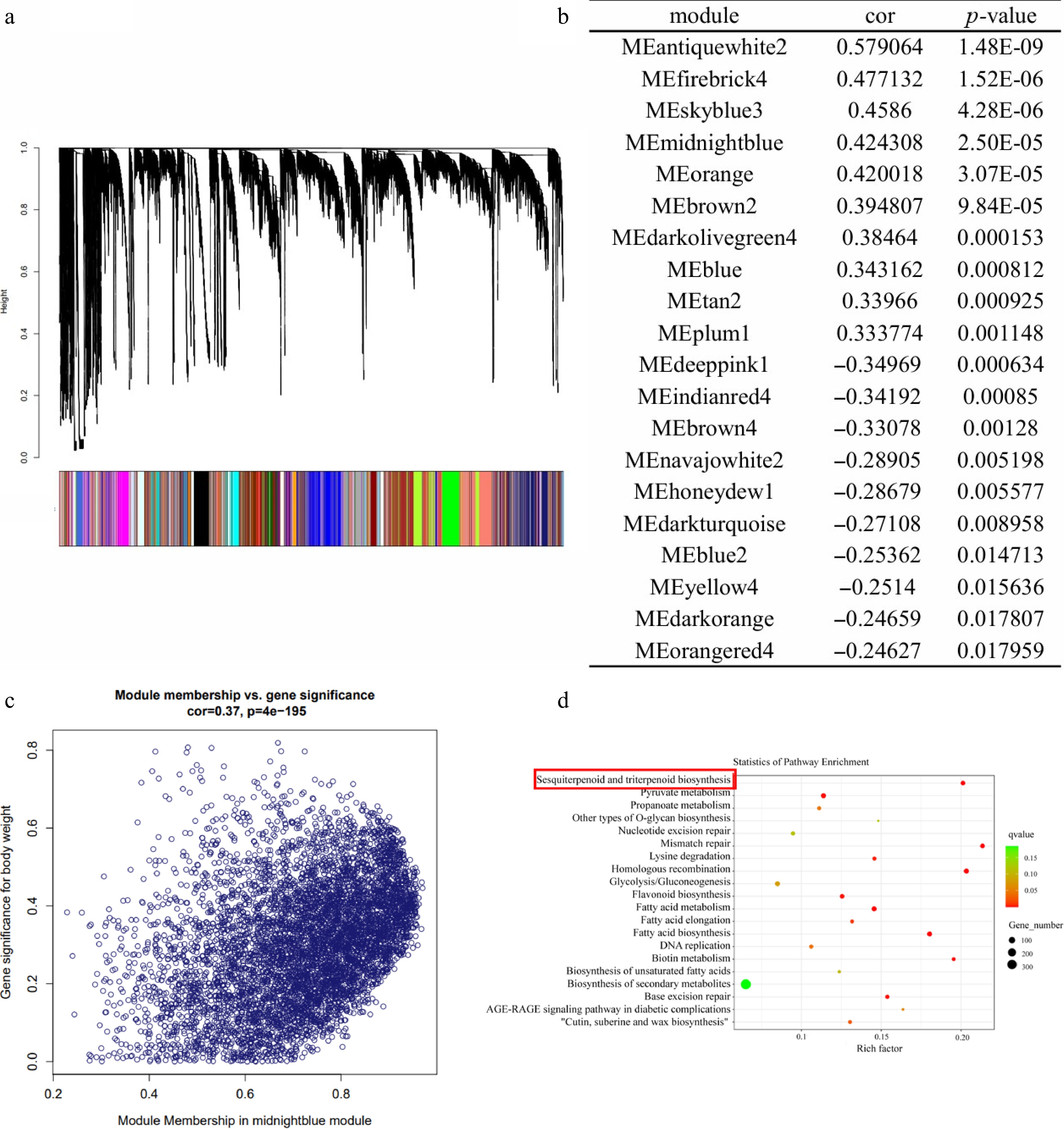
Figure 2.
Analysis of the co-expression network of patchouli genes. (a) Patchouli hierarchical clustering tree and its various modules. (b) Association analysis between modules and sesquiterpene content. (c) Correlation analysis of module midnightblue genes and sesquiterpene content of all patchouli samples. (d) GO annotation of Memidnightblue.
Using the patchouli genome database, key genes encoding enzymes involved in the terpene compound metabolic pathway were identified. Module genes were then overlapped with key enzyme-encoding genes, and 30 modules identified, comparing 63,768 key enzyme-encoding genes to identify potential modules. The numbers of genes in these 30 modules are shown in Supplementary Table S6. The three modules with the greatest numbers of key enzyme-encoding genes were the midnightblue module (28), blue module (14), and blue2 module (9).
Gene Ontology (GO) annotation of genes in the three terpenoid metabolic pathway-associated modules revealed many enriched processes putatively related to terpenoid metabolism (Fig. 2d and Supplementary Fig. S4). The enriched processes identified included sesquiterpenoid and triterpenoid biosynthesis (midnightblue module), and terpenoid backbone biosynthesis (blue2 module). Interestingly, in addition to revealing enrichment for the term 'Biosynthesis of secondary metabolites', GO analysis also revealed enrichment for 'valine, leucine, and isoleucine biosynthesis' (blue module), and 'steroid biosynthesis' (blue module 2).
Key enzyme-encoding genes and co-expression network of the terpenoid biosynthetic pathway
-
For the midnightblue module, the focus was on these key enzyme-encoding genes, and gene–trait association studies were performed using the 'corPvalueStudent' function in WGCNA, which revealed a total of 28 key terpenoid metabolism enzyme-encoding genes in the patchouli genome (Fig. 3a). The 28 key enzyme-encoding genes were components of the MEP pathway, and sesquiterpene (SesTPS) genes, which are the most crucial enzymes in the synthesis pathway, accounted for the greatest number of genes.
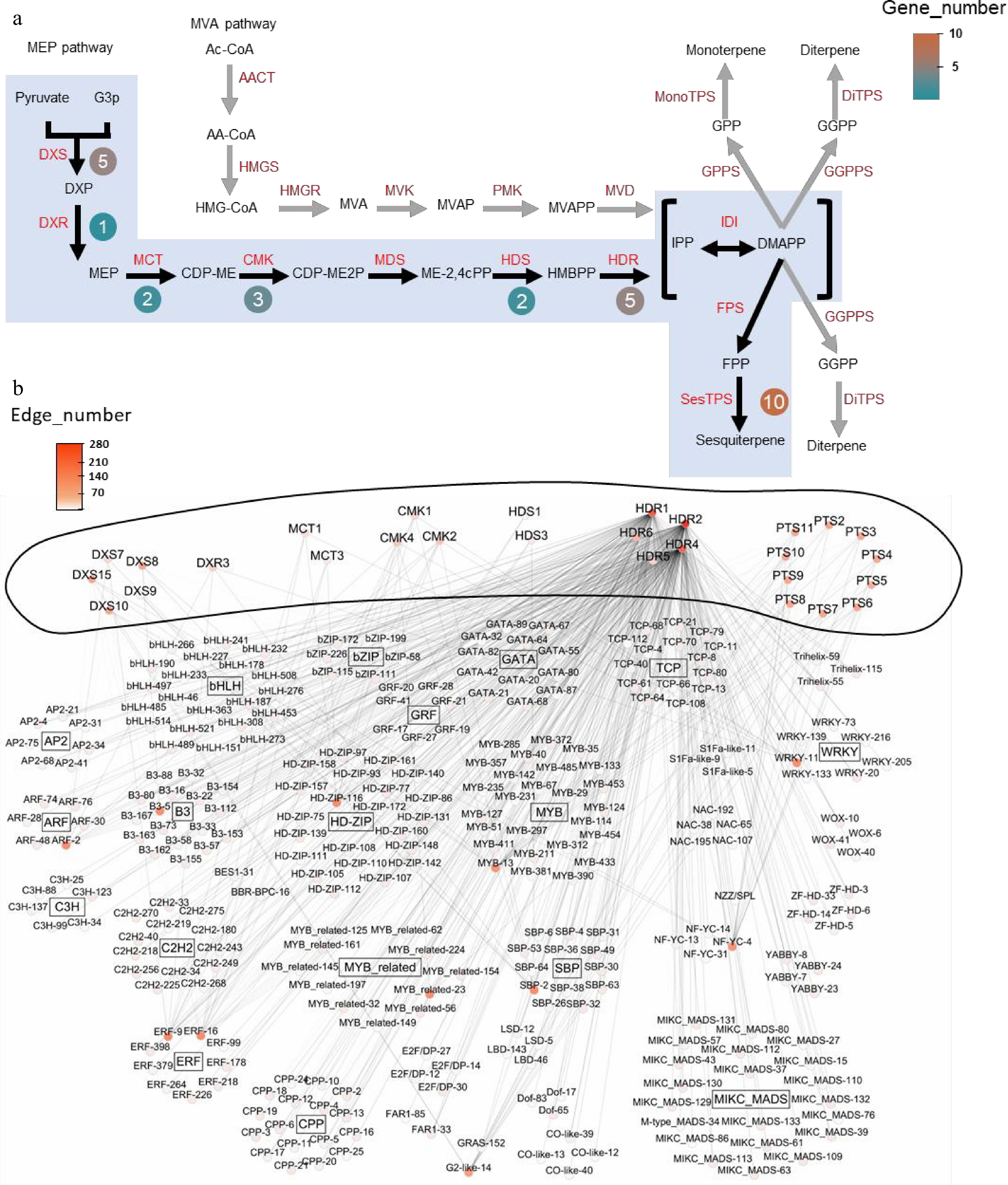
Figure 3.
Co-expression network and key enzyme genes from sesquiterpenoid metabolic pathway of patchouli. (a) Genes from sesquiterpenoid metabolic pathway in patchouli, adapted from Shen et al.[15]. Black arrows and blue shadows indicate key reaction steps. Genes in red indicate enzymes in the pathways. Round boxes next to the enzymes contain the numbers of core pathway genes. (b) Co-expression network of the sesquiterpenoid metabolic pathway. The outer ring of the network represents key enzyme genes implicated in sesquiterpenoid metabolic pathway, while inside are TFs. Nodes are labeled with gene names colored based on the edge number. TFs and pathway genes are connected via undirected edges. Internal connections among TFs and among pathway genes were not shown for clarity. Detailed IDs, names, and connection information for all genes in the network can be found in Supplementary Tables S7 and S8.
Moreover, a coexpression network of the genes encoding key enzymes involved in the terpenoid biosynthetic pathway was constructed, including transcription factors (TFs), and the 28 key enzyme-encoding genes, on the basis on the gene expression profiles of the patchouli samples at different growth stages, patchouli samples subjected to hormone treatment and patchouli samples subjected to salt treatment obtained by genome sequencing (Fig. 3b). In this network, GATA-20, GATA-32, GATA-80, and GATA-82 exhibited > 90% similarity with PatGT-1, which has been reported to be involved in terpenoid biosynthetic pathway regulation. In addition, the key enzyme-encoding genes were connected to many other TFs, collectively constituting a dense coexpression network (Supplementary Tables S7 and S8). Specifically, 11 TFs were found in the co-expression network with the most connections to sesquiterpene metabolism pathway enzymes, which were HD-ZIP-116, ERF-16, G2-like-14, ERF-9, MYB_related-23, B3-5, MYB-13, ARF-2, NF-YC-4, WRKY-11, and SBP-2.
PatPTSs are key enzymes in the patchouli sesquiterpene synthesis pathway. PatPTS2, PatPTS4, PatPTS7, PatPTS8, PatPTS9, PatPTS10, and PatPTS11 were included in this regulatory network (Fig. 3b and Supplementary Fig. S5). In addition, the binding sites in the PatPTS promoter sequences is shown in Supplementary Fig. S6; these binding sites may account for the potential regulatory relationships between PatPTSs and the abovementioned candidate TFs.
The aboveground parts of patchouli are used medicinally, and sesquiterpene metabolites accumulate in the aboveground parts. To identify genes that are related to the expression of metabolic pathway components in the regulatory network, the expression patterns of MEP, MVA, and downstream pathway components were evaluated, as shown in Fig. 4a−c. Genes in the MEP pathway were found to be relatively highly expressed in the leaves and stems. However, the expression patterns of the candidate TFs were diverse (Fig. 4d−g and Supplementary Fig. S7), and the expression patterns of HDZIP116, SBP2, MYBR23, and MYB13 were consistent with those of the MEP pathway components. The RNA-seq datasets were described in detail in our previous study[28].
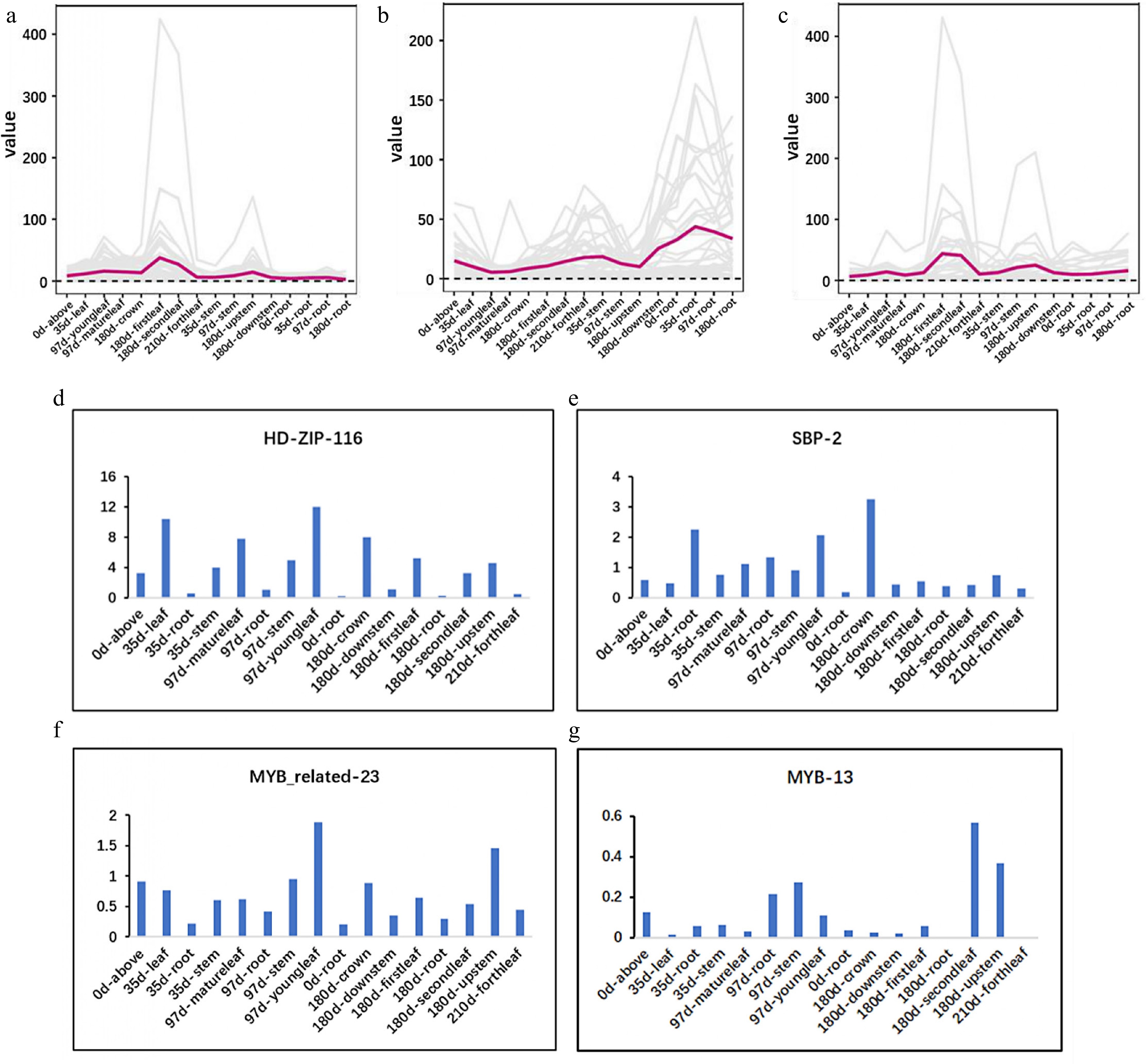
Figure 4.
Expression pattern of the candidate TFs is consistent with that of MEP pathway. (a)−(c) Expression of MEP, MVA, and downstream pathway. (d)−(g) Expression of HDZIP116, SBP2, MYBR23, and MYB13.
The homologs of other genes in the network have also been reported to regulate terpenoid biosynthetic pathway functions. In addition, the present network analysis revealed many unreported genes that strongly impact the function of terpenoid biosynthetic pathways in patchouli. These results highlight the complex regulation of the terpenoid biosynthetic pathway and indicate that many transcription factors are apparently involved in terpenoid biosynthetic pathway regulation.
Key TFs are potential regulators of the terpenoid biosynthetic pathway
-
To assess whether PatHDZIP116, PatSBP2, PatMYBR23, and PatMYB13 can bind to the PatPTS promoter, Y1H, and dual-LUC assays were performed using N. benthamiana. As shown in Fig. 5a, Y1H assays revealed that these four TFs interact with the promoter of PatPTS9, a key gene in the AD biosynthetic pathway. In addition, as shown in Fig. 5b, the activity of the PatPTS promoter was evaluated in the presence of the effectors PatHDZIP116, PatSBP2, PatMYBR23, and PatMYB13 in N. benthamiana leaf cells. As shown in Fig. 5c, the activity of the PatPTS promoter was suppressed by these four TFs. In summary, these four TFs suppress the expression of the PatPTS gene by directly binding to the PatPTS promoter.
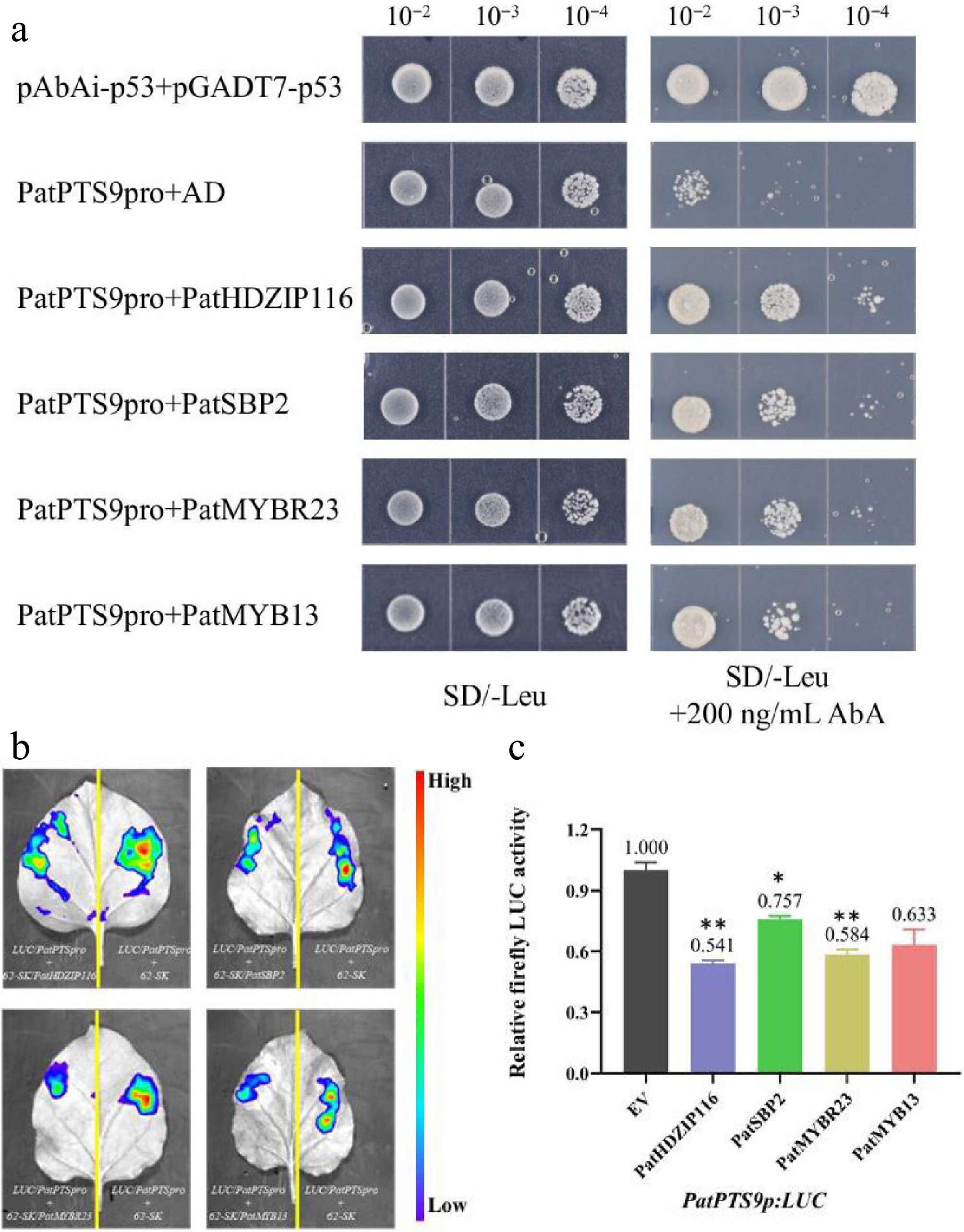
Figure 5.
PatHDZIP116, PatSBP2, PatMYBR23, and PatMYB13 repress the Sesquiterpene synthase gene PatPTS9. (a) Y1H assay confirming the interaction between the PatPTS9 promoter and four TFs. (b) Dual-LUC reporter imaging assay. N. benthamiana leaves were injected with the Agrobacterial GV3101-pSoup-p19 stains containing 62-sk + PatPTS9p:LUC and 62-sk:PatTF + PatPTS9p:LUC. Arrow position indicates the strongest fluorescence. (c) Relative firefly LUC activity assay in tobacco leaves. Error bars are shown with three biological replicates (Student's t-test: * p < 0.05, ** p < 0.01).
As for the other seven TFs, according to the binding element numbers of the 11 TFs, the other seven TFs were less likely to bind to PTS9 (Supplementary Fig. S5). Besides, expression of the others was low, which greatly reduced the success rate of Y1H assays, and might also significantly affect the efficiency and reliability of the Y1H assay.
Key TFs are involved in patchouli alcohol content accumulation
-
To investigate the role of PatHDZIP116, PatSBP2, PatMYBR23, and PatMYB13 in terpenoid biosynthesis, a transient overexpression assay involving these four TFs was conducted using patchouli leaves (Fig. 6a). After transient overexpression, total sesquiterpenoids and patchouli alcohol contents in different samples were detected by GC-MS, while the overexpression empty vector (EV-TO) was used as an experimental control. Results show (Fig. 6b) that the retention time of patchouli alcohol was 21.28 min, and the retention time of octadecane standard was 23.65 min. The contents of various compounds in the four experimental groups were lower than those in the control group.
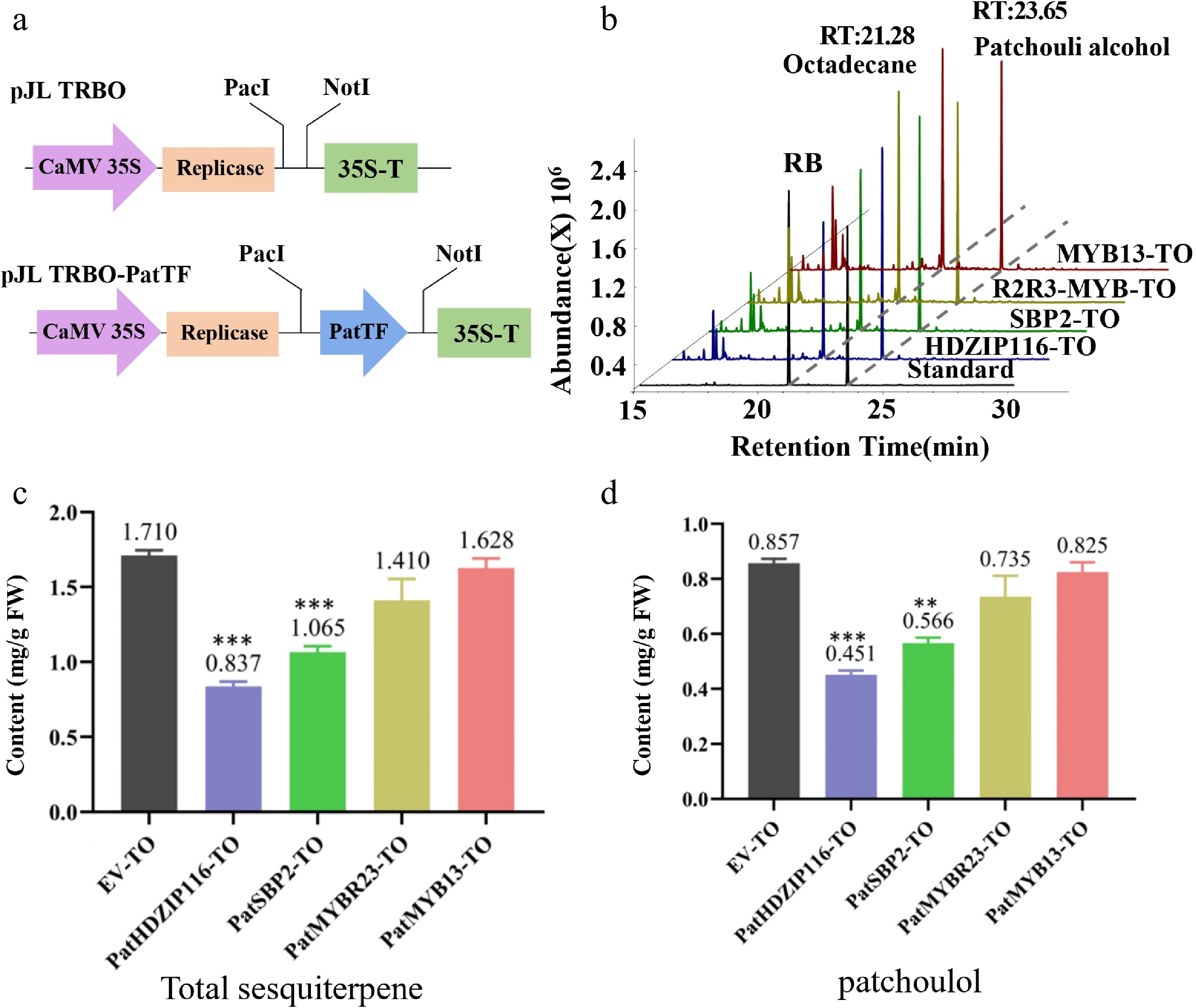
Figure 6.
Analysis of the transient overexpression (TO) of four TFs in patchouli. (a) Schematic diagram of pJL TRBO-PatTF vector construction. (b) The chromatogram of patchouli content detected by GC-MS. (c, d) Compared with EV-TO, the (c) total sesquiterpene and (d) patchouli alcohol content in leaves infiltrated with PatTF-TO was significantly reduced. Values are the mean ± SD of three biological replicates. Student's t-test: * p < 0.05, ** p < 0.01, *** p < 0.001. FW, fresh weight; TO, transient overexpression; EV, pJL-TRBO empty-vector.
As shown in Fig. 6c, d, the contents of patchouli alcohol and terpenoids in the PatHDZIP116-TO and PatSBP2-TO groups were significantly lower than those in the EV-TO group. The terpenoids are shown in Supplementary Table S9. In summary, the results demonstrated that PatHDZIP116 and PatSBP2 act as negative modulators of patchouli alcohol and terpenoid synthesis by inhibiting the expression of multiple genes in the patchouli alcohol and terpenoid biosynthetic pathway.
EMSA was performed further to determine whether PatHDZIP116 and PatSBP2 could directly bind to the PatPTS9 promoters in vitro. As shown in Supplementary Fig. S8, PatHDZIP116 and PatSBP2 bound to the PatPTS9 promoters.
Therefore, the following working model was proposed (Fig. 7): PatHDZIP116 and PatSBP2 mainly participated in patchouli alcohol biosynthesis by inhibiting the transcription of PatPTS9 and regulated MEP-pathway genes, forming a complex regulation mechanism. While PatMYBR23 and PatMYB13 had the ability to combine with the PTS promoter, no significant difference was found in the effect on patchouli alcohol content. Therefore, it was supposed that PatMYBR23 and PatMYB13, in addition to directly binding to the PatPTS9 promoter, could form protein complexes with other TFs for regulation.
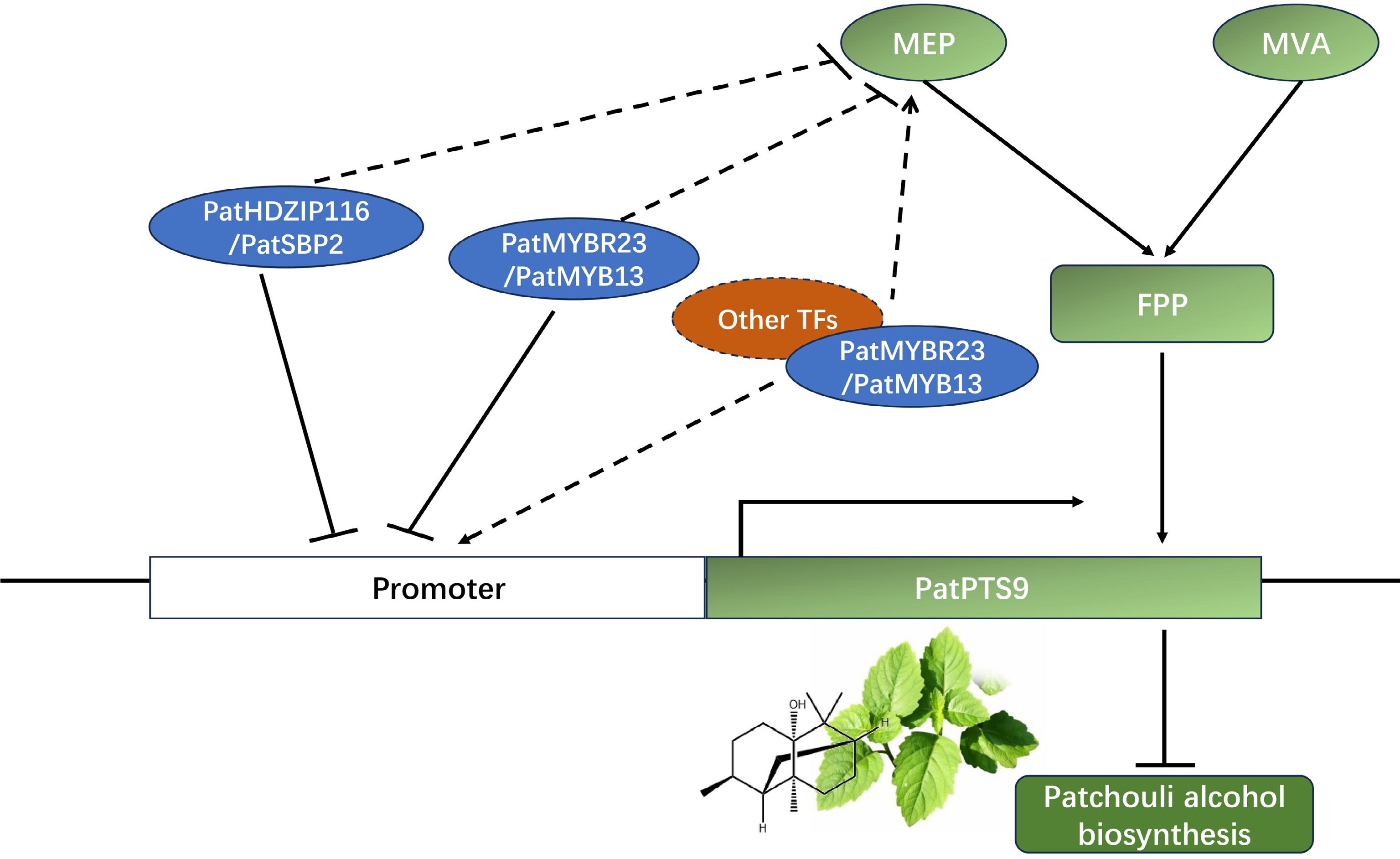
Figure 7.
Model describing the regulation function of PatHDZIP116, PatSBP2, PatMYBR23, and PatMYB13 in patchouli alcohol biosynthesis. PatHDZIP116 and PatSBP2 negatively regulated patchouli alcohol biosynthesis by inhibiting the expression of PatPTS9, while PatMYBR23 and PatMYB13 might had more complex regulation mechanism.
In summary, the present study has proved that PatHDZIP116 and PatSBP2 played a negative regulation role in patchouli alcohol biosynthesis, yet PatMYBR23 and PatMYB13 may had more complex regulation mechanism that require further investigation.
-
The present study was based on the transcriptome gene expression database and WGCNA to successfully construct a regulation network for the patchouli sesquiterpene metabolic pathway, which includes 286 TFs, and 28 pathway enzyme genes. According to the network, regulation relationships between potential TFs and pathway enzyme genes have been summarized. Through Y1H, dual-LUC, EMSA, and overexpression experiments, the regulation mechanisms were effectively verified as predicted by the network. In summary, this study not only systematically constructed and analyzed the regulation network of patchouli sesquiterpene metabolism, but also comprehensively explored all the TFs that might play regulatory roles in this pathway, and finally verified functions through various experiments.
The HD-zip TF family mainly depresses the accumulation of sesquiterpenoids in patchouli
-
As plants unique to the TF family, the HD-zip TF family proteins contain highly conserved HD and bZIP domains, which are involved in various stages of the plant life cycle, including growth, development, reproduction, differentiation, morphological formation, and responses to diseases and adverse conditions[31]. In plants, the first gene containing an HD domain was first discovered in maize, which could regulate leaf differentiation[32]. Subsequently, the gene was identified in various species such as Arabidopsis[33], tomato[34], maize[35], and wheat[36]. The HD-zip TF family was classified into four sub-families, according to structural characteristics, sequence conservation, physicochemical properties, and other features[37]. Among them, the HD-zip I sub-family mainly played a role in stress response, leaf development, and light signal transduction[38]. The HD-zip II sub-family was mainly involved in light response, photoprotection, and auxin signal transduction[39], while the HD-zip III sub-family regulated the initiation of lateral organs, embryonic development, leaf polarity, and pariental tissues[40], and the HD-zip IV sub-family plays a role in epidermal cell differentiation, hair root formation, anthocyanin accumulation, and root development process[41]. PatHDZIP116 in this study belongs to the IV sub-family.
PatbZIP44 was located in the cell nucleus in patchouli, which contained a zip domain. PatZIP44 could inhibit gene activity by combining the PatPTS gene promoter[42]. PatHDZIP116, which contained the same zip domain, could interact with the PatPTS promoter and depress the effects on the promoter activity, thereby reducing the accumulation of sesquiterpene compounds.
Regulatory functions of the SBP TF family are diversified in patchouli
-
SBP TFs could bind to the promoter of the key gene AQUAMOSA, named the Squamosa promoter binding protein (SBP)[43]. The family is also known as SPL TFs. The TF family participates in regulating the expression of downstream genes by binding to the GTAC motif of the downstream gene promoter. On the one hand, SBP TFs mainly regulate the development of plant leaves, fruit, and flowers, pollen formation of Arabidopsis, responses to GA signals, stress response, etc.[44]. There were 17 SPL TFs in Arabidopsis, and AtSPL4/5 may promote the transformation of floral meristem identity[45]. In the present study, PatSBP2, which was homologous with AtSBP4/5, has been confirmed to interact with the PatPTS9 promoter, and negatively regulate the accumulation of patchouli alcohol.
Furthermore, SBP TFs also participate in regulating secondary metabolic pathways of plants. For example, AtSPL9 competed with the flavonoid biosynthesis regulation gene TT8 for binding to PAP1, yet interfering with the formation of the MYB-bHLH-WD40 complex, affecting the expression of the key enzyme DFR for anthocyanin synthesis, and inhibiting the synthesis of anthocyanins[46]. In Artemesia annua, AaSPL9 could combine with the adenoid hair development-related AaHD1 promoter to activate expression of AaHD1, positively regulating the development of adenoid hairs, and thereby increasing the content of artemisinin[47]. Besides, miR156-targeted SPL TF could directly act on the sesquiterpene synthase PTS to regulate the content of patchouli alcohol[48]. Our previous research showed that PatSPL8/9 regulated the GA pathway by regulating the PatCPS promoter[49]. In the patchouli co-expression network, PatSPL9 regulated HDR1 and HDR2, which require further analysis.
MYB TFs are widely involved in the regulation of various pathways in plants
-
The MYB TF family is one of the largest in plants, they are involved in regulating secondary metabolic pathways, growth and development, and stress responses[50]. According to the MYB domain difference in quantity, the MYB TF family are divided into 4R, 3R, 2R, and the MYB-related sub-family. In the present study, PatMYBR23 belongs to the MYB-related, whereas MYB13 belongs to the 2R sub-family.
MYB in Arabidopsis is regulated in a variety of secondary metabolic pathways. For example, overexpressing AtMYB75 in Arabidopsis could positively regulate the accumulation of flavonoid metabolites[51], while overexpressing MYB3, MYB6, and MYBL2 reduced the anthocyanin content[52]. Besides, MYB TF regulated plant growth and development, like AtMYB21 and AtMYB24 which regulated the stamen development of Arabidopsis by combining with the JAZ protein[53].
In medicinal plants, there were many reports on regulating active components of MYB. AaMYB15 was the first R2R3-MYB negative regulator in Artemisia annua[54], while SmMYB11 in Salvia miltiorrhiza could positively regulate salvianolic acid by forming a protein complex with SmTTG1 and SmbHLH51[55]. Besides, overexpression of SmMYB1 significantly increased the accumulation of salvianolic acid and tanshinone in Salvia miltiorrhiza[56], while overexpression of SmMYB36 increased the accumulation of tanshinone, but decreased salvianolic acid[57]. Similarly, in patchouli, PatSWC4 could inhibit the activity of patchouli alcohol synthase by forming a protein complex with PatJAZ4[58].
Future research should further explore the interactions of the patchouli co-expression network. Despite the co-expression network, many TFs were found that have been shown to have regulatory roles for key sesquiterpene synthases. As is conjectured above, PatMYBR23/PatMYB13 might have more complex regulation mechanisms regarding about protein–protein. Finally, the present findings hold substantial potential value which contribute meaningfully to our understanding of secondary metabolism regulation in a non-model medicinal plants.
-
In this study, samples of various patchouli tissues were collected at different stages of growth and development, treated with exogenous hormones, and subjected to stress conditions, followed by transcriptome analysis. On the basis of relative expression data for all the genes in the patchouli genome, the first comprehensive gene expression network for patchouli was constructed. Moreover, the content of sesquiterpenoids in patchouli was measured, correlations between the expression of different module genes and sesquiterpenoid content were analyzed, and a regulatory network of the sesquiterpenoid biosynthesis pathways in patchouli was constructed. Systematic bioinformatics analysis of transcription factors was conducted to elucidate the regulatory relationships between transcription factors, and the sesquiterpenoid biosynthesis pathway. Through a yeast one-hybrid approach, the promoter sequence of the patchouli alcohol synthase gene PatPTS was selected as the site for gene regulation.
From the regulatory network of the sesquiterpenoid biosynthesis pathway in patchouli, a series of transcription factors were identified, and these transcription factors were isolated and cloned. Through the use of plant genetic engineering and metabolic analysis, the functions of transcription factors were studied to elucidate a possible model for the biosynthesis of sesquiterpenoids in patchouli, providing new ideas for increasing the content of medicinal active components of patchouli, and laying a theoretical foundation for the use of TF regulation strategies to increase the synthesis of bioactive components in medicinal plants and cultivate high-quality varieties with high-quality medicinal components.
This work was supported by the Guangdong Major Project of Basic Research (2023B0303000022), the National Natural Science Foundation of China (Grant Nos U22A20446, 32322007, 32400193), the Talent Support Project of Guangdong (2019TQ05N182), the China Postdoctoral Science Foundation (2024 M750662), the Fundamental Research Funds for the Central Public Welfare Research Institutes (ZZ16-ND-12, ZZ17-ND-12, ZZ16-YQ-048), and Jiangxi Provincial Key Laboratory for Sustainable Utilization of Traditional Chinese Medicine Resources (Platform ID: 2024SSY07081).
-
The authors confirm their contributions to the paper as follows: experimental design: Wang HB, Jin H, Zeng Y; execution of experiments: Yang C, Zeng Y, Wang C; data analysis: Zeng Y, Huang H, Chang N, Gao H; manuscript writing: Zeng Y; manuscript revision: Jin H, Wang HB, Zeng Y. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
-
The authors declare that they have no known competing financial interests or personal relationships that could have appeared toinfluence the work reported in this paper.
- Supplementary Table S1 List of primers.
- Supplementary Table S2 Total sesquiterpene compound content from patchouli culture seeding samples (0, 15, 30, 45, 68, and 90 d).
- Supplementary Table S3 Total sesquiterpene compound content from patchouli different samples (2, 4, and 6 months) and different organization.
- Supplementary Table S4 Construction of the patchouli gene co-expression module.
- Supplementary Table S5 The module was co-associated with sesquiterpene content.
- Supplementary Table S6 The module gene overlaps with the key enzyme gene of the terpene metabolic pathway.
- Supplementary Table S7 Detailed information of all the genes in the co-expression network of patchouli sesquiterpene regulation.
- Supplementary Table S8 The genes connections in the co-expression network of enzyme genes of terpenoid metabolic pathway.
- Supplementary Table S9 The content of sesquiterpenoids in each sample after overexpression of the candidate TFs genes.
- Supplementary Fig. S1 Patchouli samples with hormone-treated (A) and total sesquiterpene content of hormone-treated patchouli (B).
- Supplementary Fig. S2 Patchouli samples with salt-treated (A) and total sesquiterpene content of salt-treated patchouli (B).
- Supplementary Fig. S3 Confirmation of soft threshold (power).
- Supplementary Fig. S4 GO annotations of the module blue and blue2 genes.
- Supplementary Fig. S5 Eleven candidate TFs simultaneously regulate the PTS genes.
- Supplementary Fig. S6 The binding sites of the Pogostemon cablin PTS gene promoter sequence.
- Supplementary Fig. S7 Expression of candidate genes in different tissue sites.
- Supplementary Fig. S8 Electrophoretic mobility shift assays showing that PatHDZIP116 and PatSBP2 protein binds to PatPTS9 promoters.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zeng Y, Yang C, Wang C, Huang H, Chang N, et al. 2026. Global identification and functional validation of transcription factors regulating sesquiterpenoid metabolism in Pogostemon cablin. Medicinal Plant Biology 5: e008 doi: 10.48130/mpb-0026-0003 

Global identification and functional validation of transcription factors regulating sesquiterpenoid metabolism in Pogostemon cablin
- Received: 24 September 2025
- Revised: 12 December 2025
- Accepted: 09 January 2026
- Published online: 31 March 2026
Abstract: Patchouli is a medicinal plant belonging to the Pogostemon genus within the Lamiaceae family. It is composed mainly of sesquiterpenoid metabolites, and has various pharmacological effects. The mechanism underlying the transcriptional regulation of the patchouli alcohol has been reported, but the mechanism underlying the regulation of transcription factors (TFs) involved in the sesquiterpenoid metabolic pathway is still unknown. In this study, a regulatory network was constructed for the sesquiterpenoid metabolic pathway in patchouli by using a gene expression database and WGCNA. It was found that 286 TFs have potential regulatory relationships with 28 genes that encode enzymes in the sesquiterpenoid metabolic pathway. The regulatory mechanism of this network was successfully verified by yeast one-hybrid (Y1H), dual luciferase reporter assay (dual-LUC), transient overexpression, and electrophoretic mobility shift assay (EMSA). In conclusion, a regulation network was constructed for sesquiterpenoid metabolic pathways in Pogostemon cablin, and all the TFs that may have regulatory functions in sesquiterpenoid metabolic pathways were identified, and the regulatiory functions of transcription factors were successfully verified through bench experiments.