










-
Hydrogen energy is recognised as an important, long-term clean energy option for achieving carbon neutrality. Its use, however, is constrained by high flammability, a strong tendency for leakage, and embrittlement risks in distribution pipelines[1]. Storage and transportation remain challenging and expensive, restricting widespread implementation. Using ammonia (NH3) as a hydrogen carrier addresses these constraints: it contains ~17 wt% hydrogen, can be stored under near ambient conditions (~7.5 atm, 300 K), and is handled using the already established infrastructure[2]. Ammonia is transported and stored as the carrier and cracked on-site to supply hydrogen. The endothermic cracking of ammonia (2 NH3 → N2 + 3H2) provides a clean conversion pathway and a consistent hydrogen stream[3]. In addition, ammonia cracking can feed hydrogen to marine internal combustion engines for propulsion[4] and supply gaseous hydrogen to gas turbines for electricity generation[5]. Nevertheless, the cost and energy efficiency of current cracking systems remain limiting factors, underscoring the need for more effective catalysts[6].
The study of heterogeneous catalysts for ammonia cracking is extensive and encompasses both noble and base metals. The most extensively researched monometallic catalyst for ammonia decomposition is ruthenium (Ru)[6]. Its high intrinsic activity places Ru near the top of the volcano plot for optimal nitrogen binding. The dominant sites on Ru are the B5 ensembles, 3 Ru atoms on a (001) plane with two adjacent step atoms on a (101) plane, maximised on nanoparticles of ~2 nm[7]. This particle size is stabilised by an appropriate metal–support pairing, which maintains favourable kinetics[7]. Representative formulations, 4%Ru–5.4%Ba–7.9%Cs/Sibunit and 4%Ru–13.6%Cs/Sibunit, show high reactivity between 350 and 470 °C for both ammonia decomposition and reverse synthesis[8]. Beyond metal catalysts, compound-mediated routes using nitrogen-rich solids (imides, amides) can also decompose ammonia efficiently; however, their air instability and handling hazards limit their practical use[9]. Consequently, most development has focused on supported metal systems, where activity and durability are tuned through alloying, particle size control, surface basicity and engineered metal–support interactions[10].
On alumina, Ni/Al0.5Ce0.5Oₓ shows high activity with the lowest reported activation energy; Ce strengthens metal–support interactions, improves stability and promotes B5-like ensembles while facilitating removal of H2 from the surface[11]. Strong basicity donates electron density to adjust N binding at active sites and accelerate decomposition, resulting in a 25 wt% Ni on lanthanum hexaaluminate catalyst, achieving 85.75% NH3 conversion at 600 °C and 30,000 mL·gcat−1·h−1[12]. There is a general correlation: increased surface basicity is associated with more robust ammonia decomposition[13]. For instance, 30Fe/SiC-700 with a high basicity that was hydrothermally synthesised achieves a conversion rate of 90.16%[13]. A self-assembled Fe–Co/Al–CeZr catalyst achieves complete decomposition at 550 °C and 6,000 mL·g−1·h−1. The multi-shelled hollow MxOy nanosphere structure provides excellent stability and sintering resistance[14]. Strengthening the metal–support interaction is similarly effective: in Ru/Co–Al hydrotalcite, co-precipitation increases the density of oxygen vacancies density, lowers the cracking temperature and achieves 98.57% conversion at 500 °C[15].
Among the non-noble candidates, nickel is widely studied for ammonia decomposition. With appropriate supports and promoters, it can deliver high conversion[16−18]. The availability of Ni adsorption sites is increased, and dehydrogenation steps are facilitated by the higher density of acidic sites, resulting in ~72% conversion from a urine matrix with Ni/Ce-doped Al2O3[16]. Dopants with valences that differ from Al3+, such as Sr2+, Zr4+ and Ce4+, can significantly increase Ni activity on alumina[16]. Alkaline earth promoters like Ba, Sr, Ca and Mg increase the basicity and electron density around Ni on ceria. This facilitates the recombination and desorption of nitrogen, reducing hydrogen inhibition in the decomposition sequence[17]. Density functional theory supports these trends: Ni/BaZrO3 is predicted to have an overall barrier of ~0.90 eV, with Ni assisting N–H scission and supporting oxygen vacancies, further promoting the pathway[18]. Together, these findings emphasise the importance of catalyst platforms that maintain dispersion at elevated temperatures, regulate hydrogen inhibition and offer tunable basicity and vacancy populations in operating feeds[16−18].
These observations inevitably direct attention to catalyst platforms that maintain dispersion in hot H2 and NH3 conditions while simultaneously providing deliberate control over the basicity, reducibility and vacancy chemistry. ABO3 perovskites offer this combination. By using lower amounts of materials, they can release metal nanoparticles that stay intact and can be restored through redox reactions[19]. Through A site, B site and anion design, including oxynitrides such as hexagonal BaTiO3−xNy, they enable vacancy and basicity control that reduces the operating temperature and maintains activity in moist environments[20].
In this context, Co-based perovskites are attractive for NH3 cracking because of their ceramic stability, composition flexibility and cost, and because they can be programmed under working feeds to form anchored metal on basic oxides[19]. At the same time, anion-engineered perovskites, including oxynitrides, offer vacancy-rich supports that facilitate lower-temperature operation and enhanced moisture tolerance[20]. Prominent candidates for Co-based perovskites include LaNiO3 and LaCoO3[21]. La is an example of a rare-earth element that can reduce dependence on precious metals while maintaining high reactivity. The barrier for ammonia's dissociation is reduced when La is near Ni(111) according to surface calculations[22]. In direct comparisons, LaNiO3 frequently outperforms LaCoO3 because of the higher surface area and greater reducibility of Ni species[21]. Further performance optimisation can be achieved through adjusting the composition and alloying. For instance, the incorporation of Ce into LaCeNiO improves lower-temperature activity by increasing the surface area, enabling greater reducibility and guaranteeing that the basicity is appropriately set[21]. A perovskite approach can also conserve precious metals, as shown by the fact that alumina-supported LaFe0.9Ru0.1O3 achieves > 99% conversion at 500 °C while using less Ru than a traditional alumina-supported bimetallic Fe–Ru catalyst[23].
Within the perovskite family, activity trends become clear when exsolved metals and support cations are varied. A Co-based single-metal exsolution catalyst delivers 63.9% NH3 conversion with an H2 production rate of 21.4 mmol·gcat−1·min−1 at 30,000 mL·gcat−1·h−1 and 700 °C, outperforming Ni, Fe, NiFe, NiCo and CoFe variants on similar perovskite scaffolds[24]. For cobalt on alkaline earth cerate perovskites, the activity follows the trend 5Co–BaCeO > 5Co–SrCeO > 5Co–CaCeO > 5Co–MgCeO, underscoring the role of the support cation in basicity and metal–support interaction[25]. Oxynitride supports further shift performance, with Ni-loaded perovskite oxynitrides showing 2.5–18× the activity of Ni on oxides; lattice nitrogen and vacancy sites assist activation of ammonia rather than relying solely on the Ni surface[9].
In practice, the performance of mesostructured LaNiO3 is improved by the combination of textural templating and programmed exsolution. This is because the specific surface area is increased and the conversion rate reaches 89% at 550 °C and 30,000 mL·gcat−1·h−1 when the mesostructured LaNiO3 is prepared with an SBA-15 template, followed by the exsolution of finely dispersed Ni[26]. An A-site-deficient Sr0.9Ti0.8Ni0.1Co0.1O3−d exsolves 8–12 nm NiCo nanoparticles and outperforms Ni/SrTiO3 and Ni–Co/SrTiO3 benchmarks[27]. Perovskite concepts extend beyond purely thermal routes: titanate-based FexNi1−xTiO3 shows sensitivity to visible light for the conversion of aqueous ammonia under liquid plasma, giving an H2 production rate of ~140 L·g−1·h−1[28]. Self-combustion-derived La0.1Ce0.9NiO3 and La0.1Mg0.9NiO3 achieve 96% and 98% conversion, respectively, near 400 °C, with stable operation over 40 hours, attributed to their low Ni crystallite size and high basicity. This illustrates how perovskite catalysts can substitute for more expensive Ru systems in accessible temperature windows[29].
We investigated La(1−x)SrxCoO3 perovskite catalyst for NH3 decomposition. Sr, an alkaline earth element, was introduced at the A site to tune vacancy concentration, lattice reducibility and basicity. Prior studies on La(1−x)SrxCoO3 reported that increasing the Sr content increases the fraction of Co4+ and enhances the lattice's reducibility[30−32]. In our study, Sr was therefore used to adjust the reducibility and the vacancy population, which we probed through temperature-programmed measurements and related the observed activity to the density functional theory (DFT) analysis of vacancy-mediated steps. La(1−x)SrxCoO3 was prepared by a sol-gel route across a controlled series with x = 0, 0.2, 0.4, 0.6 or 0.8. Our approach involves the integration of temperature-programmed methods, steady state testing at a fixed gas hourly space velocity, and structural and surface characterisation to quantify performance. The correlations between the catalytic behaviour and the support characteristics, such as vacancy density, basicity and reducibility, were analysed. To quantify the impact of Sr-induced vacancy chemistry on the adsorption energies, dehydrogenation barriers and nitrogen association pathways, DFT mapped elementary steps on representative La(1−x)SrxCoO3 surfaces. This was achieved while maintaining the ABO3 framework as the structural platform.
-
La1−xSrxCoO3 perovskite at different compositions (x = 0, 0.2, 0.4, 0.6, 0.8) were prepared via the sol-gel technique for the decomposition of NH3. Analytical grade lanthanum nitrate hexahydrate (La(NO3)3•6H2O), strontium nitrate (Sr(NO3)2), cobalt nitrate hexahydrate (Co(NO3)2•6H2O) and citric acid were used for the synthesis of the perovskites. Each perovskite was prepared by dissolving a set amount of nitrate salt in 50 mL of deionised water before stirring in 0.961 g of citric acid to prepare a 0.1 mol·L−1 precursor solution. This mixture was heated to 80 °C while being stirred at 450 rpm for 2 h to form a viscous wet gel, which was then dried in an oven at 120 °C for 12 h. The dried gel was then calcined at 650 °C in a static air atmosphere for 3.5 h. To ensure the safe decomposition of citric acid, the calcination was initiated at 200 °C and held for 15 min before ramping to 650 °C at a heating rate of 10 °C·min−1. After cooling, the calcined perovskite was ground to fine powder form with a ceramic mortar.
Characterisation of the catalyst
-
X-ray diffraction (XRD) of the perovskite catalyst was conducted using a multifunctional powder X-ray diffractometer (Xpert Powder) from PANalytical. All possible diffraction directions of the lattice were obtained by scanning the sample within the 2θ angle range, while all the stages were identified through the use of the power diffraction file (PDF) reference database. The morphological structure and elemental composition of the perovskites were analysed via scanning electron microscopy (SEM) (Gemini 300 instrument, ZEISS) paired with energy-dispersive X-ray spectroscopy (EDS) using an INCAx-act detector (Oxford Instruments). Brunauer–Emmett–Teller (BET) analysis was performed to evaluate the specific surface area and pore distribution of the perovskites by using a gas adsorption instrument from Quadrasorb SI by Quantachrome Instruments. Carbon dioxide temperature-programmed desorption (CO2-TPD) was used to evaluate the basic strength and basic site distribution of the perovskites. The desorption of CO2 was detected by a gas chromatograph (HP5890) with a thermal conductivity detector (TCD), using the Autosorb-iQ-C chemisoption equipment from Quantachrome for testing. The reduction of the active components in the perovskites was investigated with the Autosorb-iQ-C chemisoption from Quantachrome Instruments via hydrogen temperature-programmed reduction (H2-TPR).
Ammonia decomposition
-
The perovskite reduction and ammonia decomposition were conducted in a 400-mm-long vertical quartz tube (inner diameter of 5 mm), whereby the perovskite was held at the centre of the tube with quartz wool. The amount of perovskite catalyst used was 0.08 g for all cases. A schematic diagram of the experimental setup is shown in Fig. 1. The H2, N2 and NH3 gas flows were independently controlled with mass flow controllers and connected to the reactor through a four-way connector. The quartz tube was first purged with 50 mL·min−1 of N2 while being heated to 550 °C at a heating rate of 10 °C·min−1. Then H2 was introduced into the quartz tube at 25 mL·min−1 while the N2 flow rate was reduced to the same flow rate (H2 : N2 = 1:1). The gas flow rate, with a space velocity of 37,500 mL·h−1·g−1, was maintained at 550 °C for 1 h. After the reduction was complete, the H2 supply was cut off and the perovskite catalyst was cooled to 300 °C in an N2 flow.
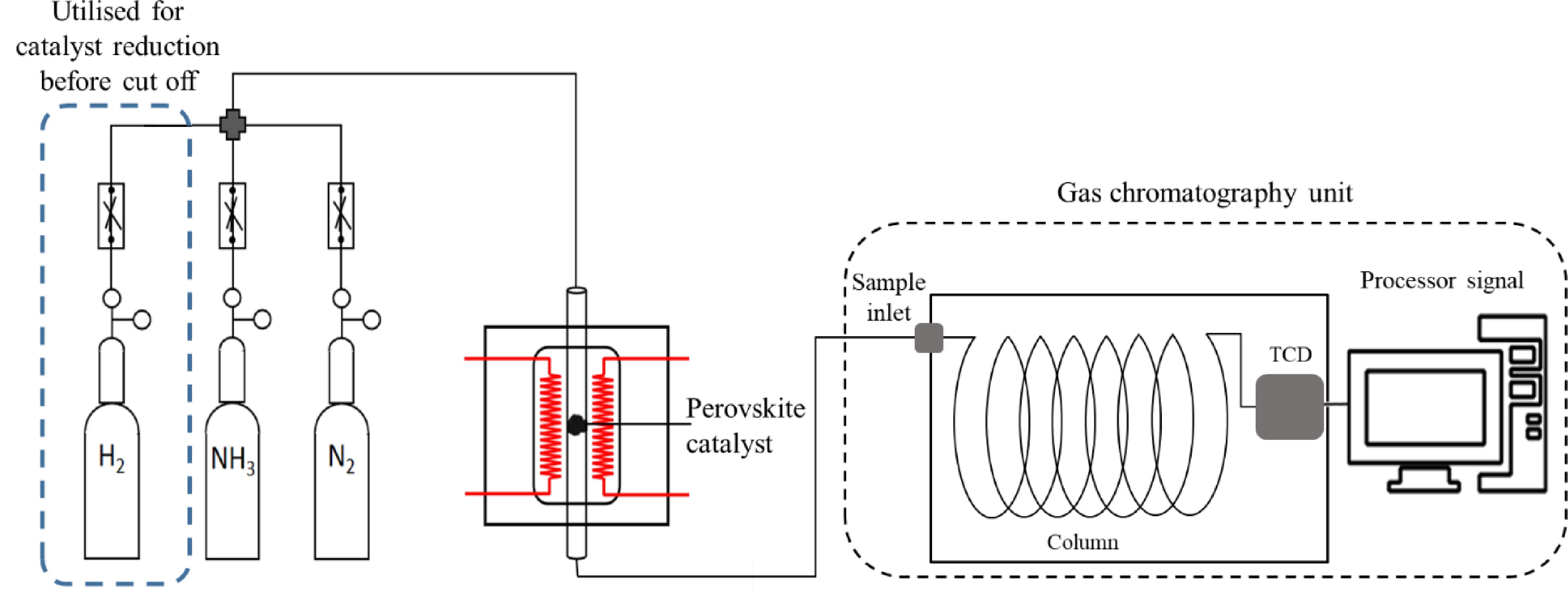
Figure 1.
Schematic of the experimental device for ammonia decomposition.
The decomposition of ammonia was evaluated at a temperature range of 300–650 °C. NH3 was first introduced into the quartz tube at a flow rate of 50 mL·min–1 while the N2 valve was turned off to allow 100% NH3 flow. The product gas was tested at every 50 °C increment by gas chromatography (GC) on a GC9720Plus (Fuli Instruments) equipped with a TCD to determine the hydrogen content. The GC column used in this study was a RubyBond Packed Column (Porapak Q 60-80 mesh ID.2 mm × 2 m). The hydrogen content was recorded once the value had stabilised for 10 min at the set temperature. Each point of measurement was repeated thrice to obtain an average value. The determined hydrogen content in the gas chromatograph was used to derive the ammonia conversion rate via Eq. (1).
$ {\mathrm{Conversion}\;\mathrm{of}\;\text{NH}}_{3}\left({\text{%}}\right)=\dfrac{{\mathrm{H}}_{2\text{out}}}{0.75}\times 100{\text{%}} $ (1) Density functional theory calculation
-
The DFT calculation was performed using the Vienna ab initio simulation package (VASP version 5.4.4)[33] to evaluate the mechanism of NH3 decomposition. The electron exchange–correlation interactions were treated via the generalised gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) functional[34−37]. To properly describe the strongly correlated 3d electrons of cobalt, the DFT + U approach was used, with an effective U parameter of 3.9 eV for Co 3d orbitals, which accurately reproduced the electronic structure of LaCoO3 perovskites[38]. The projector augmented wave (PAW) method was used to represent the interaction between valence electrons and ionic cores. The plane-wave energy cutoff was set to 500 eV, and structural optimisation was considered to have converged when the total energy changes fell below 1 × 10−4 eV and the forces on all the atoms were less than 0.03 eV·Å−1. For the La0.4Sr0.6CoO3 (001) surface, a 25 Å vacuum layer was constructed to eliminate periodic interactions. The Brillouin zone was sampled using a 3 × 3 × 1 Γ-centred k-point mesh for geometric optimisation and a 5 × 5 × 1 mesh for calculating the electronic structure. The adsorption energy (EAds) of NHx species was calculated as follows:
$ {E}_{Ads}={E}_{\left(NH{_{x}}/surface\right)}-\left[{E}_{surface}+{E}_{({{NH}_{x}})}\right] $ (2) where, E(NHₓ/surface) is the total energy of the adsorbed system, E(surface) is the clean surface energy and E(NHₓ) is the energy of the free adsorbate[39]. More negative EAds values indicate stronger adsorption. The reaction energy barriers (ΔEa) are determined as follows:
$ \Delta {E}_{a}=E\left(TS\right)-E\left(IS\right) $ (3) where, E(TS) and E(IS) are the transition state and initial state energies, respectively. Microkinetic analysis is performed using transition state theory (TST). The rate constant (k) for each elementary step is calculated as follows:
$ k=\left({k}_{B}T/h\right)exp\left(-\Delta {G}_{a}/{k}_{B}T\right) $ (4) where, kB is Boltzmann's constant, T is the temperature, h is Planck's constant and ΔGₐ is the Gibbs free energy of activation[40]. This approach allows an evaluation of temperature-dependent reaction rates and identification of the rate-limiting steps in the NH3 decomposition mechanism.
-
The XRD characterisation of La(1−x)SrxCoO3 (x = 0, 0.2, 0.4, 0.6, 0.8) across the diffraction angle range of 5°–90° is shown in Fig. 2. The LaCoO3 sample shows characteristic peaks at diffraction angles of 23.15°, 32.93°, 33.05°, 40.66°, 47.35° and 58.91°, consistent with the PDF standard card (PDF# 48-0123) for LaCoO3, indicating the high crystallinity of LaCoO3 synthesised via the sol-gel method. The XRD results of La(1−x)SrxCoO3 (x = 0.2, 0.4, 0.6, 0.8) showed that characteristic peaks appeared at diffraction angles of 23.145°, 32.97°, 33.044°, 47.373° and 58.835°, consistent with the PDF standard card (PDF# 48-0122) for La0.5Sr0.5CoO2.91. In addition, the Sr-doped samples also showed characteristic peaks at 25.11°, 25.79° and 36.56°, which is consistent with the PDF standard card (PDF# 99-0099) for SrCO3. Moreover, the doping of Sr2+ also results in various valence states of CoOx. The XRD results showed the presence of cobalt oxides in the form of CoO and Co3O4. As the Sr2+ doping increased, the content of the perovskite phase decreased, while the area of the SrCO3 and CoOx phases increased. Both the perovskite and CoOx phases contain the Co element, which is the active component for ammonia decomposition. On the other hand, the SrCO3 phase has no obvious catalytic effect. As an impurity phase, the content of SrCO3 gradually increases with the doping of Sr2+. Unlike the CoOx phase, the SrCO3 phase does not contain active components, and hence the generation of the SrCO3 phase could reduce the crystallinity of the perovskite structure.
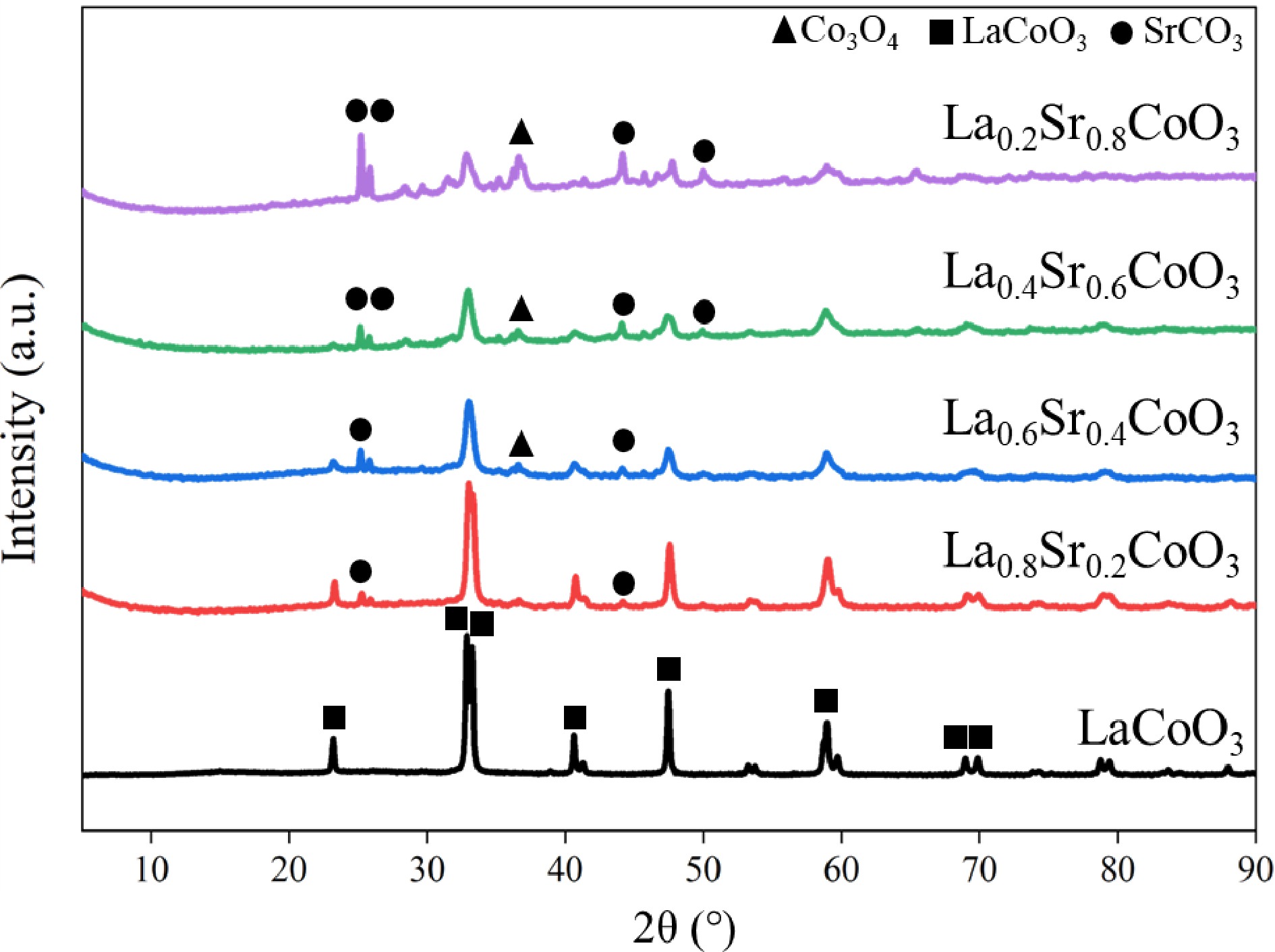
Figure 2.
X-ray diffraction pattern of La(1−x)SrxCoO3 perovskite.
The SEM and EDS images of La(1−x)SrxCoO3 (x = 0, 0.2, 0.4, 0.6, 0.8) perovskites at different magnifications are shown in Fig. 3. The ionic radius of Sr2+ (1.26 Å) is larger than that of La3+ (1.061 Å), and hence Sr2+ doping could potentially increase the oxidation state of cobalt (Co4+), leading to more oxygen vacancies. When the Sr2+ doping ratio increases to 0.8, the particle size of the perovskite becomes larger, which is presumed to be the SrCO3 phase. According to the EDS elemental mapping, as the Sr2+ doping ratio increases from 0.2 to 0.6, the elemental distribution of Sr starts to appear on the surface while the La gradually decreases, with the Co element being evenly mixed. However, for the La0.2Sr0.8CoO3, despite some patches of Sr detected on the surface, more O elements are evident, which could be caused by the migration of O atoms to the perovskite's surface. However, images of the individual elemental distribution on the perovskite's surface clearly show the presence of Sr in La0.2Sr0.8CoO3, as evident in Fig. 4q–t. Compared with the baseline LaCoO3 shown in Fig. 4a–d, no element of Sr is observed on the surface. As the doping of Sr increases, an increasing distribution of Sr can be observed, implying that the Sr elements are well dispersed on the catalyst's surface. A notable decrease in the La concentration is observed as the doping ratio of Sr2+ increases from 0.4 to 0.6, which is mainly attributed to the decrease in the mass fraction of the perovskite phase. The specific surface area (SSA) measured for the La0.4Sr0.6CoO3 is 38.05 m2·g−1, and the pore volume and average pore diameter are 0.063 cm3·g−1 and 3.32 nm, respectively, indicating the mesoporous structure of the catalyst. The SSA is slightly larger than the LaCoO3 catalyst reported in another study[41], which could range across 3.5–13.4 m2·g−1, depending on the calcination temperature.
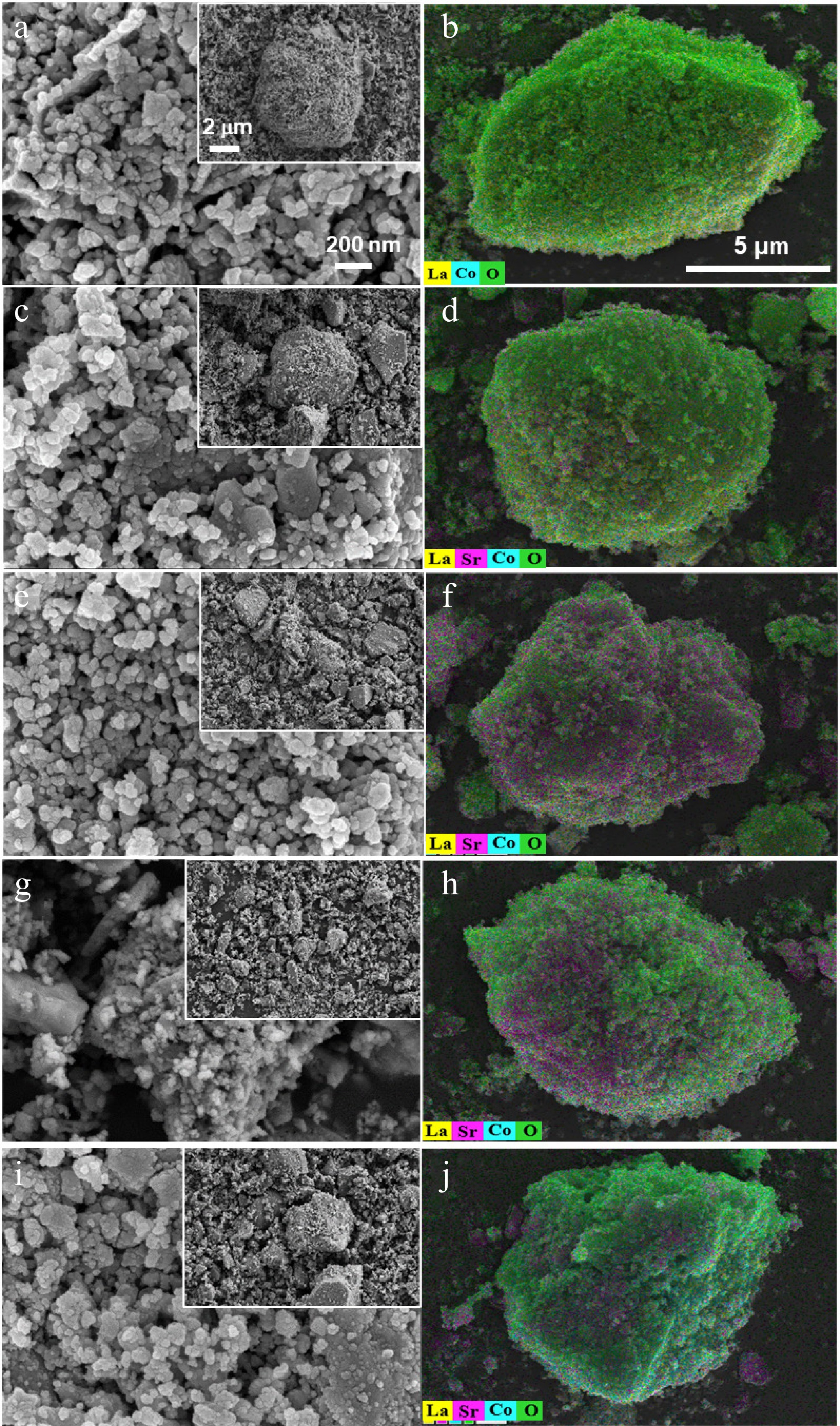
Figure 3.
SEM (first column) and EDS images (second column) of (a), (b) LaCoO3, (c), (d) La0.8Sr0.2CoO3, (e), (f) La0.6Sr0.4CoO3, (g), (h) La0.4Sr0.6CoO3 and (i), (j) La0.2Sr0.8CoO3.
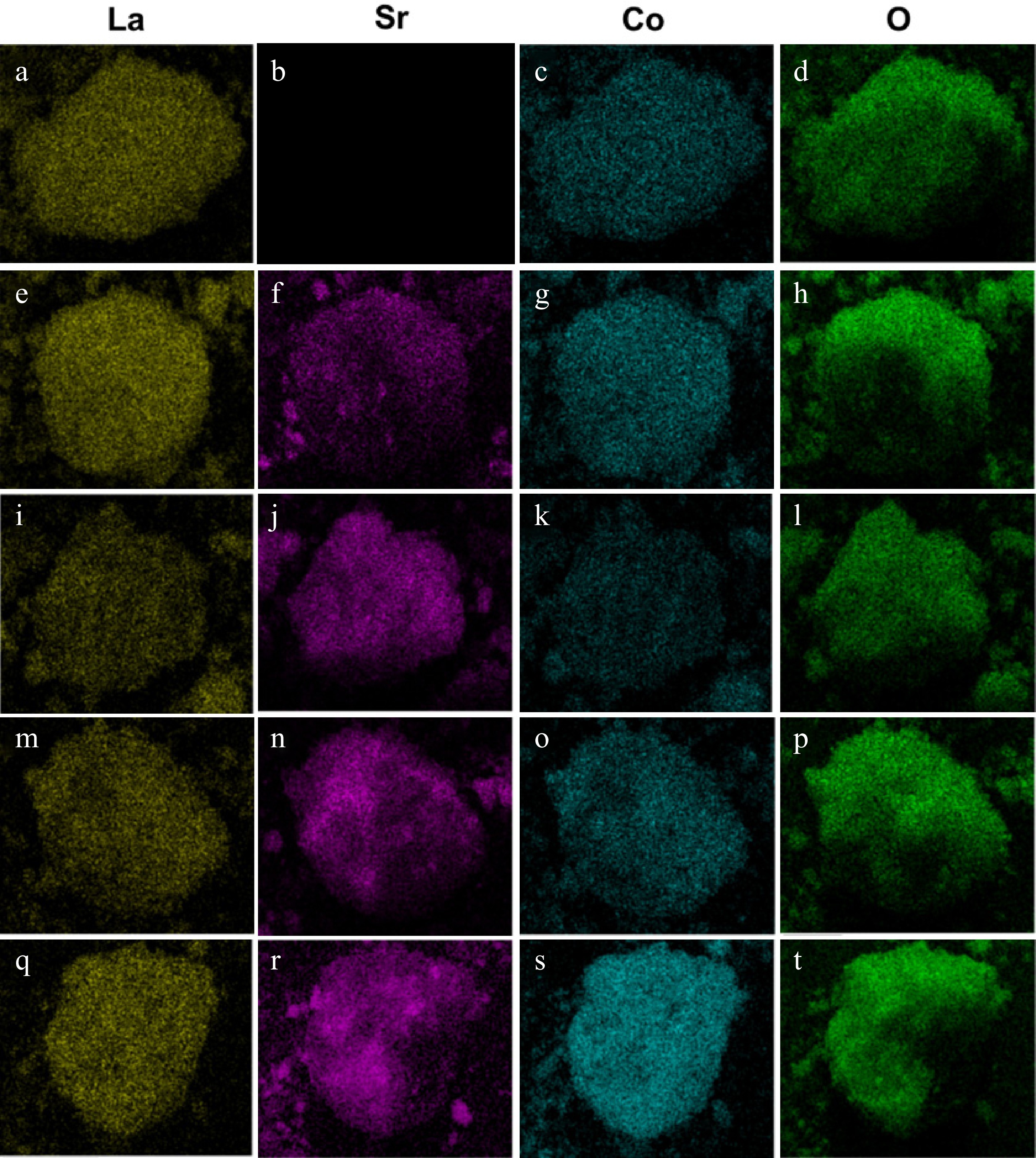
Figure 4.
Elemental distribution of La, Sr, Co and O for (a)–(d) LaCoO3, (e)–(h) La0.8Sr0.2CoO3, (i)–(l) La0.6Sr0.4CoO3, (m)–(p) La0.4Sr0.6CoO3 and (q)–(t) La0.2Sr0.8CoO3.
The H2-TPR curves of the La(1−x)SrxCoO3 catalysts are shown in Fig. 5a. For the LaCoO3 catalyst, there is an obvious TCD low-temperature peak (340 °C) and a TCD overlap peak in the temperature range of 400–750 °C, with the maximum peak appearing at 540 °C. The low-temperature peak is related to the reduction of unstable lattice oxygen in the LaCoO3 crystal, whereas the TCD overlap peak is the subsequent reduction of Co3+ → Co2+ and Co2+ → Co0[42], which can defined as follows:
$ {\text{LaCoO}}_{3}+\dfrac{1}{2}{\mathrm{H}}_{2}\rightarrow {\text{LaCoO}}_{2.5}+\dfrac{1}{2}{\mathrm{H}}_{2}\mathrm{O} $ (5) $ {\text{LaCoO}}_{2.5}+{\mathrm{H}}_{2}\rightarrow \dfrac{1}{2}{\mathrm{La}}_{2}{\mathrm{O}}_{3}+\mathrm{Co}+{\mathrm{H}}_{2}\mathrm{O} $ (6) 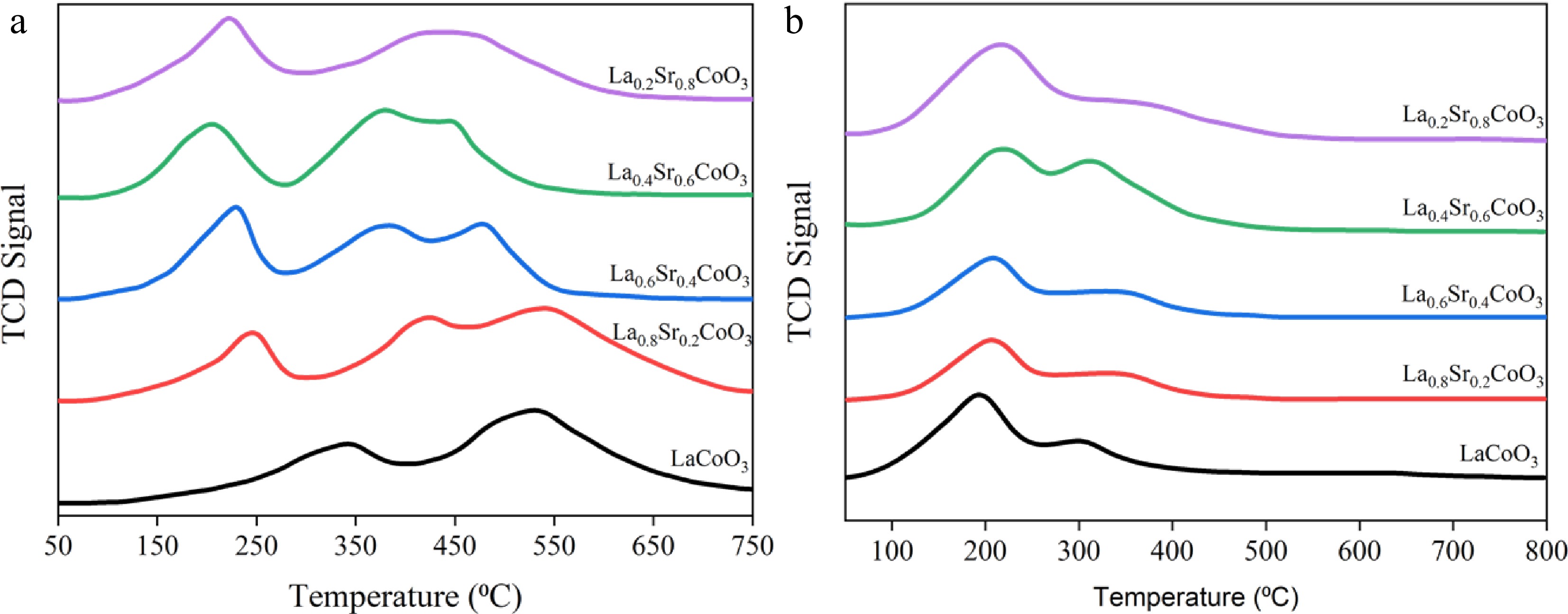
Figure 5.
(a) H2-TPR curve and (b) CO2-TPD image of La(1−x)SrxCoO3 perovskites.
From Fig. 5a, it can also be seen that doping with Sr2+ causes the TCD peaks to shift to a lower temperature, suggesting that doping promotes the Co reduction process in the perovskite. As the Sr doping increases from x = 0.6 to 0.8, the peaks shift to a higher temperature, indicating that beyond this point, excessive doping of Sr2+ reduces the perovskite's reducibility. Excessive Sr2+ doping leads to a significant production of the SrCO3 impurity phase which is difficult to reduce. In the TCD peak area, it is noticed that more Sr2+ doping increases the TCD peak area, leading to a higher consumption of H2 for reduction. At the temperature range of 50–200 °C, it is observed that doping with Sr2+ increased the consumption of hydrogen at low temperatures, which may be caused by Sr2+ causing oxygen to adsorb on the catalyst's near-surface region after the reduction treatment[43]. However, beyond x = 0.6, the reducibility of the La(1−x)SrxCoO3 catalyst reduces, thereby inhibiting the ammonia cracking activity.
The basicity of a catalyst plays a key role in the desorption of nitrogen atoms, which is the rate-determining step during ammonia cracking[41]. Alkaline earth elements are known to effectively improve the catalytic activity of perovskite catalysts by increasing the basicity of the catalysts[44]. The CO2-TPD method was used to measure the basicity of each perovskite, as shown in Fig. 5b. Each perovskite displays peaks in the low-temperature region (100–250 °C) and the medium-temperature region (250–500 °C), which represent weak and medium alkaline sites, respectively[45]. There is a slight shift to higher temperatures as Sr2+ doping increases from 0 to 0.6, indicating that Sr2+ doping enhances the alkalinity of the perovskite. For the low-temperature TCD signal, the peak area first decreases then increases as the Sr2+ doping increases from x = 0 to 0.6. There is a marked increase in the low-temperature peak area when Sr2+ doping is at x = 0.8, as a large amount of SrCO3 disrupts the perovskite's structure (as shown by the increase in the CoOx phase) and converts some of the medium basic sites into weak basic sites. This is also noticeable for the medium-temperature TCD peak areas (an initial increase followed by a decrease), where the number of medium-basic sites decreases at Sr2+ doping at x = 0.8.
These findings highlight the importance of an appropriate amount of Sr2+ doping to optimise the reducibility and increase the number of medium-basic sites, which are important properties that lead to an increase in ammonia decomposition activity. From the characterisation results, a Sr2+ doping rate of x = 0.6 shows the optimal reducibility with highest number of medium-basic sites present in the perovskite, suggesting the composition is most optimal for ammonia decomposition.
Catalytic ammonia decomposition performance
-
Ammonia decomposition is an endothermic reaction[46]. Full decomposition of NH3 can occur at relatively high temperatures, often requiring the catalyst to keep the reaction stable at temperatures beyond 600 °C[18]. In the present test, each La(1−x)SrxCoO3 perovskite was evaluated for its effect on ammonia decomposition from 300 to 650 °C, as shown in Fig. 6. At 600 °C, the order of catalytic activity for ammonia decomposition decreased as follows: La0.4Sr0.6CoO3 > La0.6Sr0.4CoO3 > La0.8Sr0.2CoO3 > LaCoO3 > La0.2Sr0.8CoO3. It is evident that Sr doping significantly increased the rate of ammonia decomposition up to the level of x = 0.6 before decreasing again at x = 0.8. La0.4Sr0.6CoO3 shows the highest catalytic activity with 91.92% conversion at 600 °C and nearly complete conversion at 650 °C. During the thermal decomposition of NH3, a series of reactions happen via the mechanism shown in Eqs (7)–(12):
$ \mathrm{N}{\mathrm{H}}_{3}\rightleftharpoons {\text{NH}}_{3,\,\text{ad}} $ (7) $ \mathrm{N}{\mathrm{H}}_{3,\,\text{ad}}\rightleftharpoons {\text{NH}}_{2,\,\text{ad}}+{\mathrm{H}}_{\text{ad}} $ (8) $ \mathrm{N}{\mathrm{H}}_{2,\,\text{ad}}+{\mathrm{H}}_{\text{ad}}\rightleftharpoons {\text{NH}}_{\text{ad}}+2{\mathrm{H}}_{\text{ad}}$ (9) $ \mathrm{N}{\mathrm{H}}_{3,\,\text{ad}}+2{\mathrm{H}}_{\text{ad}}\rightleftharpoons {\mathrm{N}}_{\text{ad}}+3{\mathrm{H}}_{\text{ad}} $ (10) $ 2{\mathrm{H}}_{\text{ad}}\rightleftharpoons {\mathrm{H}}_{2} $ (11) $2{\mathrm{N}}_{\text{ad}}\rightleftharpoons {\mathrm{N}}_{2} $ (12) 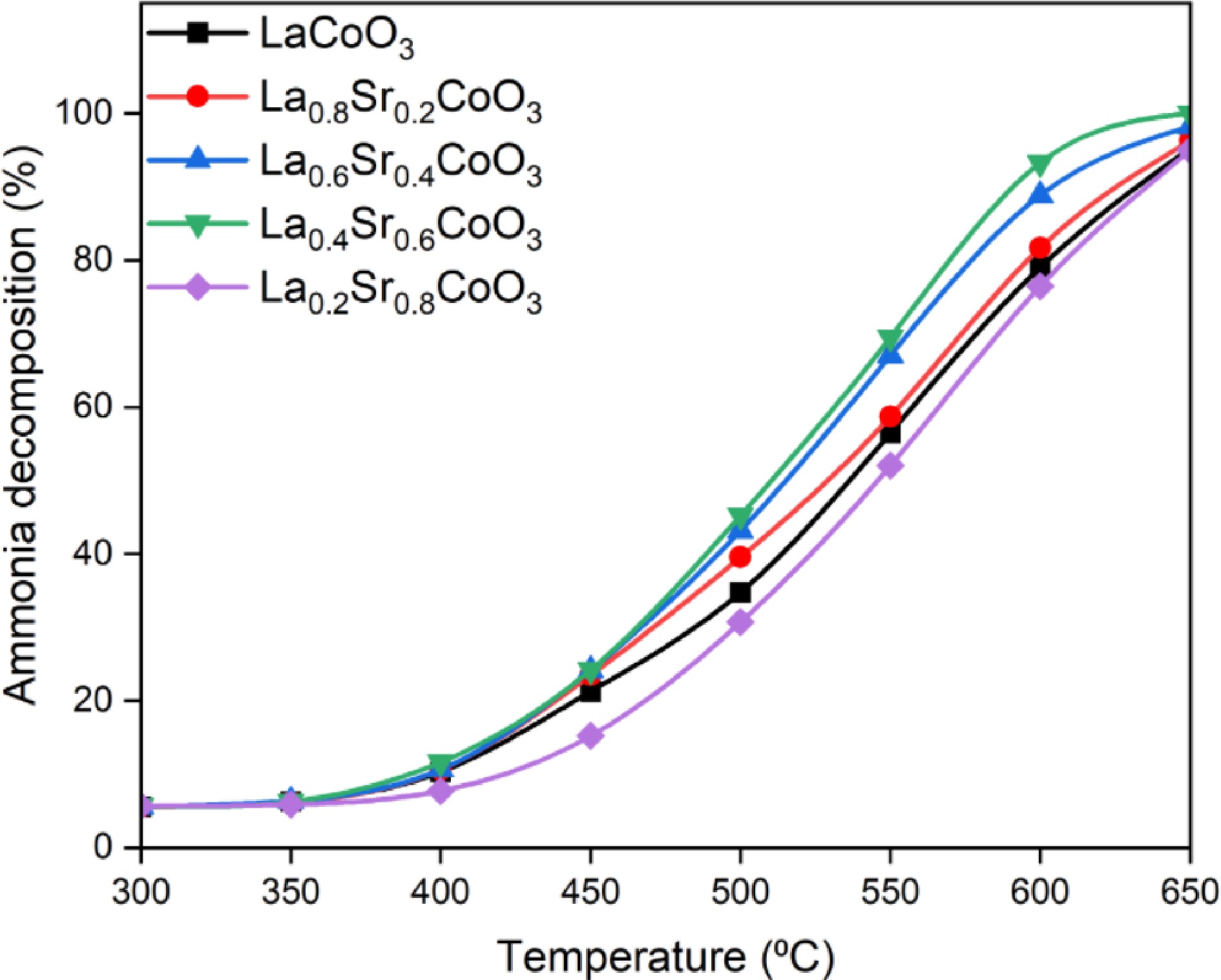
Figure 6.
Ammonia decomposition performance of La(1−x)SrxCoO3 perovskite between 300−650 °C.
The N recombination reaction (Eq. [12]) is the key determinant reaction that determines the rate of the reaction. If the binding energy of the N-N bond formation is not achieved, this will hinder the formation of N2, thereby slowing down the rate of ammonia decomposition. Ammonia decomposition mainly occurs at the basic sites on the perovskite's surface[47]. When the Sr2+ doping ratio is at 0.6, the TCD peak in the medium-temperature zone appears at the high-temperature zone with the largest area, indicating that La0.4Sr0.6CoO3 has higher basicity with more basic sites. The rate-determining step for ammonia decomposition is dependent on the type of catalyst and the operating conditions, which can be enhanced by selecting suitable supports, adjusting the appropriate temperature and gas composition and by optimising the catalyst's surface structure and active sites[48]. Figure 6 shows that the La0.4Sr0.6CoO3 catalyst achieved the highest conversion at 650 °C, which is attributable to the high basicity induced by the basic sites. This agrees with a previous study which concluded that a catalyst with higher surface basicity tends to possess stronger ability to decompose NH3[13]. In the case where Sr2+ doping is at 0.8, the alkalinity of the catalyst is significantly lower, which results in a lower ammonia conversion rate.
Test of the catalyst's stability and performance comparison
-
A continuous 12-h ammonia cracking test was conducted using the La0.4Sr0.6CoO3 catalyst to examine its stability performance. The experiment was performed at 550 °C at a constant flow rate and cracking temperature. The hydrogen content produced by thermal catalytic cracking was measured each hour to obtain the ammonia conversion rate. Figure 7 shows the result of the 12-h ammonia cracking conversion rate for the La0.4Sr0.6CoO3 catalyst. Throughout the test, the catalyst remained stable with a consistent conversion rate of 68%–70%. Despite the slight fluctuation, no significant deviation was observed, suggesting resistance to sintering and thermal deactivation during the test period. Note, however, that the test was conducted only for 12 h, whereas the stability of the catalyst for longer periods will require more in-depth tests to ensure its robustness and durability for industrial use.
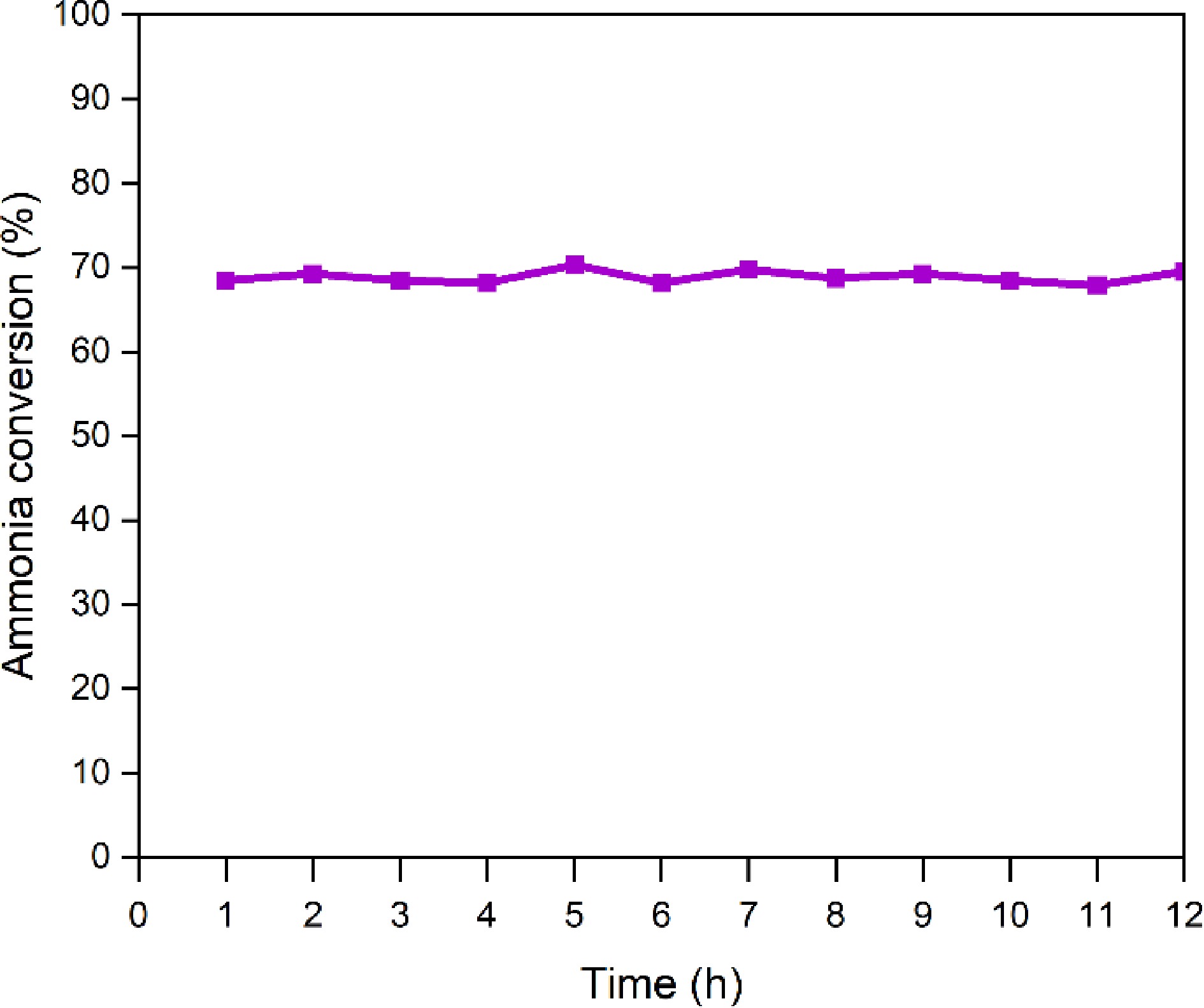
Figure 7.
Ammonia conversion rate of the La0.4Sr0.6CoO3 catalyst during the ammonia catalytic decomposition reaction.
Table 1 shows the performance of the newly developed perovskite-based catalyst (La0.4Sr0.6CoO3) compared with other thermal catalysts. Under reaction conditions of 500 °C and a gas hourly space velocity (GHSV) of 37,500 mL·h−1·g−1, the ammonia conversion rate of the present La0.4Sr0.6CoO3 catalyst is 43.2%, which is higher than the single-metal Co/γ-Al2O3 (21%) and Mo/γ-Al2O3 (22.4%) catalysts under a similar GHSV of 36,000 mL·h−1·g−1 at 500 °C[49]. However, for the γ-alumina supported Co–Mo catalyst, the ammonia conversion rate is 55% at the same temperature and GHSV, indicating the enhanced catalytic performance of the bimetallic catalyst[49]. Xie et al.[50] developed high-entropy alloy (HEA) catalysts comprising CoxMoyFe10Ni10Cu10 (x + y = 70) for ammonia decomposition, and reported that HEA-Co45Mo25 exhibited a conversion rate of 64.5% at 500 °C. The nitrogen-doped carbon nanotube (CNT)-supported noble metal (Ru) catalyst shows an ammonia conversion rate of 48% at 400 °C, albeit with a diluted mixture of NH3-He (1:2.4) and a GHSV of 6,000 mL·g−1·h−1[51]. Choudhary et al.[52] demonstrated that Ru/SiO2 can achieve an ammonia conversion rate of 64% at 500 °C. When Ni/SiO2 was doped with La, the performance was higher than that of the present La0.4Sr0.6CoO3 catalyst, but Ce doping resulted in a slightly lower ammonia conversion rate[53]. Strong catalytic performance was demonstrated by MnN-LiNH2 with a 75.2% conversion rate achieved at 465 °C, which is about twice as high as that of MnN-NaNH2 (32.0%) at the same temperature, indicating the strong promoting effect of the former alkali metal amides[54]. Nevertheless, the stability and the reactive performance of the present perovskite-based catalyst make it a prospective catalyst for thermal decomposition of ammonia.
Table 1. Performance of different catalysts in thermal catalytic decomposition of NH3.
Catalyst Metallic loading (wt %) GHSV (mL·g−1·h−1) Reaction temperature (°C) Ammonia conversion rate (%) Ref. Ru/N-CNT 7 6,000 400 48.0 [51] Ru/SiO2 10 30,000 500 64.0 [52] Co/γ−Al2O3 5 36,000 500 21.0 [49] Mo/ γ−Al2O3 5 36,000 500 22.4 [49] CoMo/ γ−Al2O3 5 36,000 500 55.0 [49] Fe5Co/CNTs 5 36,000 600 48.0 [55] *HEA-Co45Mo25 8.8 36,000 500 64.5 [50] Co7Mo3/MCM-41 5 36,000 500 51.8 [56] MnN-LiNH2 49.9 13,500 465 75.2 [54] MnN-NaNH2 46.7 13,500 465 32.0 [54] Ni-0.1La/SiO2 − 30,000 500 46.5 [53] Ni-0.1Ce/SiO2 − 30,000 500 40.2 [53] La0.4Sr0.6CoO3 − 37,500 500 43.2 This work *HEA, high-entropy alloy. DFT calculations
-
The high thermal stability of perovskite oxides makes them suitable for the catalytic decomposition of NH3 into N2 and H2[18]. Figure 8a summarizes the proposed NH3 decomposition pathway on the La(1−x)SrxCoO3 catalyst's surface. As shown in Fig. 8b, at a low Sr2+ substitution rate of x ≤ 0.6, the lattice contracts, consistent with the Co3+ → Co4+ oxidation that shortens the Co–O bond's length, whereas the b-axis remains nearly constant (~7.122 Å). Beyond x = 0.6, the volume expands, and the b-axis exhibits a maximum near x = 0.8, revealing a competing structural response such as octahedral tilting/orbital ordering. Overall, these trends reflect strong charge–lattice coupling, with a composition of x = ~0.6 providing a particularly favourable balance between structural distortion and the Co oxidation state. This balance correlates with improved NH3 cracking performance, potentially aided by enhanced redox flexibility and lattice-oxygen participation associated with changes in the Co3+/Co4+ ratio, which can accelerate surface reaction cycles.
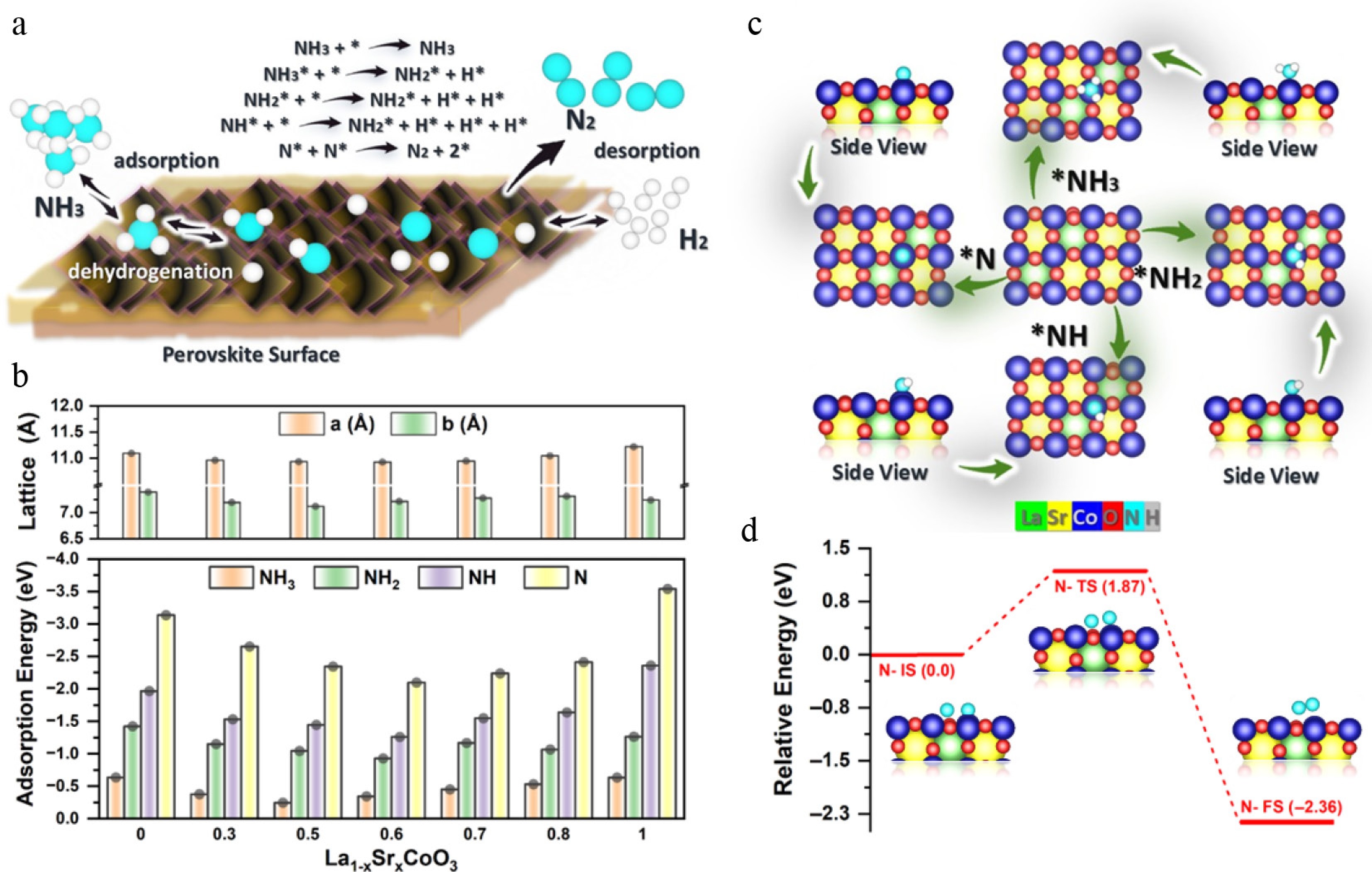
Figure 8.
(a) Pathway of the NH3 decomposition mechanism, with * denoting active sites. (b) Lattice contraction (x ≤ 0.6) to expansion (x > 0.6) and NHx adsorption energies (weakest at x = 0.6). Inset: NH3 on Co (1.82 Å). (c) Optimised NH3/NH2/NH/N structures. (d) N desorption profile: 1.87 eV barrier, exothermic (−2.36 eV) at x = 0.6.
The adsorption energies of NHx intermediates (NH3, NH2, NH* and N*) systematically weaken as the Sr content increases up to x = 0.6 (Fig. 8b, c). Specifically, the NH3* adsorption energy drops from –0.64 eV at x = 0 to –0.39 eV at x = 0.6, whereas N* binding weakens from –3.09 to –2.10 eV, representing an overall decrease of approximately 32%–35% in the adsorption strength. This pattern aligns with Co3+ → Co4+ oxidation, which reduces electron density at the active Co sites and consequently diminishes the binding strength of nitrogen-containing intermediates. Significantly, La0.4Sr0.6CoO3 reaches a balanced adsorption state, as shown in Fig. 8d, where NH2* (−0.92 eV) and NH* (−1.38 eV) are stabilised enough to facilitate the activation of NH3, yet not so strongly bound as to cause surface poisoning. The atomic structure for each model catalyst used in the DFT calculation is shown in the supplementary material (Supplementary Fig. S1).
As shown in Fig. 9a, decomposition of NH3 on La0.4Sr0.6CoO3 occurs through sequential dehydrogenation steps, NH3* → NH2* + H* → NH* + 2H* → N* + 3H*, with transition state barriers of 0.42, 1.10 and 1.54 eV, respectively (these energies are referenced to the preceding adsorbed state). The highest barrier is for the final N–H cleavage step (1.54 eV), which is lower than that reported for LaCoO3 (> 1.8 eV), consistent with the altered electronic structure at x = 0.6 (e.g., an increased Co4+ fraction). In addition to the monomeric route, Fig. 9b shows a cooperative dimer pathway in which two co-adsorbed NH3* species undergo stepwise dehydrogenation, (NH3* + NH3 → NH3 + NH2* + H*) → (NH3* + NH* + 2H*) → (NH3* + N* + 3H*), with transition state energies of 0.28, 1.34 and 1.30 eV, respectively. This dimer pathway presents barriers that are similar to or lower than those of the monomer route, as shown in Fig. 9b. It decreases the likelihood of strongly bound N* building up (final-state energy: ~0.82 eV), indicating a more favourable kinetic balance between activation of NH3 and N2 formation. Overall, the combined reaction profiles indicate that La0.4Sr0.6CoO3 is the most kinetically advantageous composition among those studied.
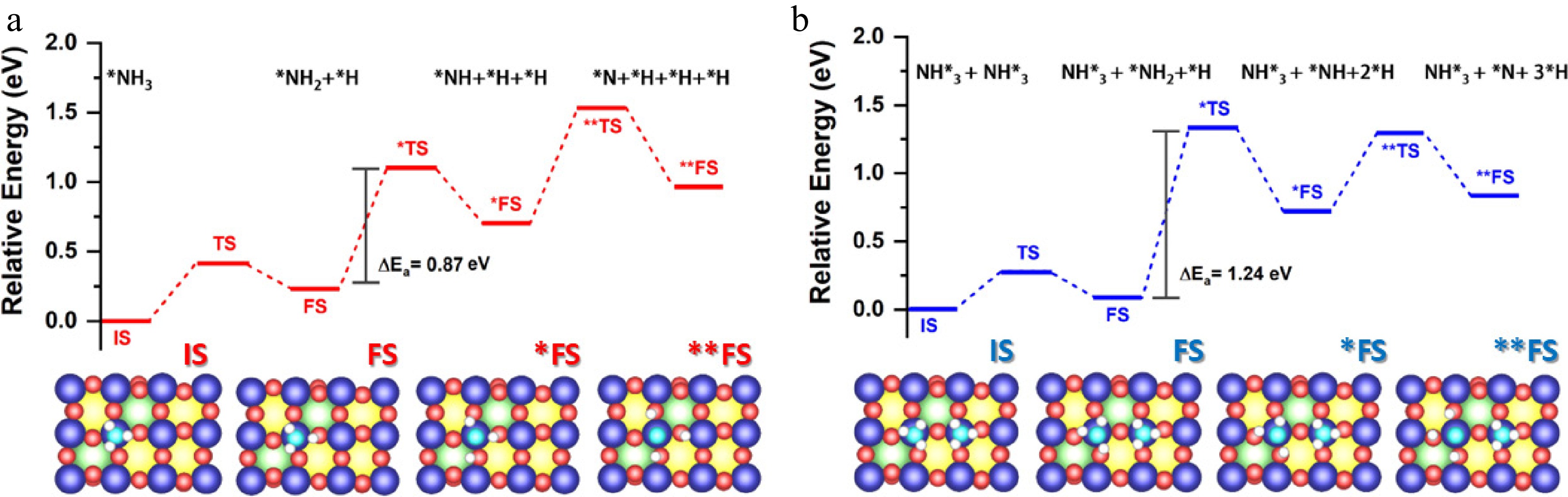
Figure 9.
(a) Mechanism of NH3 decomposition on La0.4Sr0.6CoO3 perovskite catalysts. Reaction pathway showing sequential dehydrogenation steps (NH3* → NH2* → NH* → N*) and N2 desorption, with energy distributions for key intermediates at different Sr doping levels (x = 0–1). (b) Optimised structures (top and side views) illustrating the dimerised decomposition mechanism at the active Co site, with bond lengths of 1.86 Å and relative energies indicated.
La0.4Sr0.6CoO3 outperforms both the Sr-deficient (x < 0.5) and Sr-rich (x > 0.7) analogues, which may be associated with an improved balance of surface adsorption strength, lattice distortion and redox behavior. However, its NH3 decomposition activity remains lower than that of precious metal catalysts and alkali metal amide systems, such as MnN-LiNH2[54] and Ru-KNH2/GNP[57], it should be noted that the present catalyst is expected to achieve even higher ammonia conversion rates at higher weights, hourly space velocities and ammonia concentrations. The La0.4Sr0.6CoO3 represents an optimally engineered perovskite catalyst for efficient NH3 cracking, achieving the crucial balance between NH3 activation and nitrogen desorption. Through systematic Sr doping, this composition overcomes the classic trade-off between activity and stability that plagues many transition metal catalysts. Future work should explore the interplay between Co4+/oxygen vacancy ratios and long-term stability under varying reaction conditions.
-
In the present work, a cobalt-based perovskite doped with Sr was developed for the decomposition of NH3 to produce hydrogen. Sr doping on the A-site of the perovskite surface enables a strong metal–support interaction that promote catalytic stability while maintaining the perovskite's structure. The results show that the decomposition rate of NH3 is enhanced with the use of the catalyst as the temperature increases, among which the La0.4Sr0.6CoO3 catalyst exhibited the highest catalytic activity at 650 °C with a decomposition rate of 98.89% with a NH3 flow rate of 37,500 mL·h−1·g−1. The high reactivity of La0.4Sr0.6CoO3 is attributable to the high basicity induced by the doping of Sr, which generated more basic sites. The result corroborates the DFT calculation, which showed that Sr doping at a rate of 0.6 exhibits a unique balance of structural, electronic and catalytic properties that optimises the NH3 cracking performance. This is attributed to the moderate adsorption energy is required for N2 desorption and the enhanced redox activity from Co4+ and oxygen vacancies. In general, the NH3 decomposition reaction pathway shows a sequential dehydrogenation step of NH3* → NH2* → NH* → N*, in which the La0.4Sr0.6CoO3 composition's optimal Co4+ concentration reduces the rate-limiting N-H cleavage barrier (1.54 eV) compared with LaCoO3 (> 1.8 eV). Moreover, the La0.4Sr0.6CoO3 perovskite also enables efficient NH3 cracking via a cooperative dimer mechanism. The lattice contraction of La0.4Sr0.6CoO3 perovskite further stabilises the intermediates, yielding lower barriers than monomeric pathways. Among the Sr-doped catalysts, the La0.4Sr0.6CoO3 strikes an ideal balance among adsorbate binding, lattice dynamics and redox activity, implying that the optimum Sr doping rate for LaCoO3 can enhance the reactivity for decomposing NH3. This work provides insight into the mechanism of perovskite-based catalysts for efficient hydrogen production via NH3 cracking.
The authors gratefully acknowledge the funding from the Scientific and Innovative Action Plan of Shanghai, the International Collaboration Research Fund by Science and Technology Commission of Shanghai Municipality (23160712400), the General Program of the National Natural Science Foundation of China (52476122), the Royal Society—National Natural Science Foundation of China (NSFC) International Exchanges Cost Share (W2521172) and the Shanghai Jiao Tong University (SJTU)-University of New South Wales (UNSW) Collaborative Research Program 2025.
-
The authors confirm their contributions to the paper as follows: conceptualization, funding acquisition, supervision: Chong CT; investigation, formal analysis: Zhang H, Goh BHH, Ahmed S; writing − review & editing: Chong CT, Zhang H, Goh BHH, Ahmed S, Jalili AR, Valera-Medina A, Xu H, Ajtai T. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
-
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
- Supplementary Fig. S1 The atomic structure for each model catalyst used in the DFT calculation.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Chong CT, Zhang H, Goh BHH, Ahmed S, Jalili AR, et al. 2026. Development of cobalt-based perovskite catalysts for hydrogen production via ammonia decomposition. Progress in Reaction Kinetics and Mechanism 51: e010 doi: 10.48130/prkm-0026-0005 

Development of cobalt-based perovskite catalysts for hydrogen production via ammonia decomposition
- Received: 03 December 2025
- Revised: 03 January 2026
- Accepted: 03 February 2026
- Published online: 07 May 2026
Abstract: The high cost associated with hydrogen storage and transportation pose a significant barrier to the widespread adoption of hydrogen energy. Utilising ammonia as a hydrogen carrier offers a promising solution but the development of an efficient catalyst is critical in reducing the costs of the process. In the present work, a new La(1−x)SrxCoO3 (x = 0, 0.2, 0.4, 0.6, 0.8) perovskite composite catalyst was synthesised via the sol-gel method for ammonia decomposition. The effect of the strontium (Sr) doping in the perovskite on ammonia's thermal decomposition was examined. The results show that Sr2+ doping can effectively enhance the catalytic activity of the catalyst, in which La0.4Sr0.6CoO3 catalysts exhibited the highest catalytic activity at 650 °C with a decomposition rate of 98.89% and a gas hourly space velocity of 37,500 mL·h−1·g−1. Density functional theory calculation reveals that the La0.4Sr0.6CoO3 exhibits a unique balance of structural, electronic and catalytic properties that optimises NH3 cracking performance with favourable N2 desorption characteristics. The enhanced redox activity from the optimal Co4+ concentration in La0.4Sr0.6CoO3 reduces the rate-limiting N-H cleavage barrier.