










-
The widespread contamination of aquatic systems by nitrate (NO3−), primarily originating from agricultural runoff and industrial effluents, presents severe risks to both environmental stability and human well-being[1,2]. Driven by ambient conditions, the electrocatalytic reduction of nitrate (NO3RR) serves as a dual-purpose solution, seamlessly integrating environmental remediation with the sustainable electrosynthesis of high-value ammonia (NH3)[3−6]. In stark contrast to the massive energy requirements of the traditional Haber–Bosch method and the sluggish reaction kinetics of the nitrogen reduction reaction (NRR), NO3RR leverages abundant aqueous nitrogen species and operates with highly favorable thermodynamics[7−9]. Nevertheless, deploying this technology for practical water purification is bottlenecked by the intricate multi-electron/proton transfer dynamics. This complexity not only produces a broad spectrum of nitrogenous intermediates, making high NH3 selectivity elusive, but also invites fierce competition from the parasitic hydrogen evolution reaction (HER), which aggressively consumes electrons and drastically diminishes overall yield[10−13]. Consequently, the central challenge in propelling NO3RR toward industrial-scale application lies in the rational design of electrocatalysts capable of specifically activating nitrate, directing the reaction pathway toward targeted NH3 synthesis, and robustly stifling the competing HER in aqueous media.
Both single-atom catalysts (SACs) and nanoparticle (NPs) architectures based on metal-nitrogen-carbon (M–N–C) matrices exhibit exceptional catalytic performance across a broad spectrum of electrochemical processes, including the reduction of oxygen, carbon dioxide, and nitrate, as well as hydrogen evolution[14−19]. The distinct appeal of SACs largely stems from their ultimate atomic efficiency and well-resolved geometric configurations, allowing for the accurate tailoring of the catalytic centers[20−25]. Nitrogen doping of the carbon skeleton increases the electron density of the support[26,27]. Concurrently, the strong synergistic interaction between the immobilized metal species and their adjacent nitrogen ligands modulates the local electronic microenvironment, systematically facilitating the binding and subsequent activation of reactant molecules[28−30]. Meanwhile, NPs provide abundant multi-site ensembles, metallic conductivity, and strong orbital hybridization that can accelerate multi-electron reactions and effectively activate chemical bonds through multi-point cooperative mechanisms[31−34]. Moreover, NPs allow higher metal loading and exhibit robust structural stability, ensuring durability in practical applications[35−37]. Despite these advantages, a direct mechanistic comparison between single-atom and nanoparticle active motifs for NO3RR remains insufficiently explored, leaving the optimal electronic and structural design principles unclear.
For certain metals with negligible spin polarization, such as ruthenium (Ru), SACs frequently demonstrate superior catalytic efficiency compared to their nanoparticulate forms across processes like hydrogen oxidation and oxygen evolution[38,39]. This suggests that in the absence of spin-related effects, the maximal atom utilization and well-defined coordination environment of SACs may provide greater advantages than the ensemble effects of NPs. In contrast, for redox reactions involving multiple electron transfer steps, the specific spin configuration of the active centers dictates both the reaction thermodynamics and the binding affinity of intermediates. An optimized spin configuration can facilitate orbital hybridization between metal d orbitals and reactant molecules, promote electron transfer, and enhance the redox capability of active sites.
Among transition metals, cobalt (Co) stands out as a highly viable option for electrocatalytic nitrate reduction, primarily because its inherently high-spin nature and adaptable 3d electron configuration grant it a pronounced binding affinity toward nitrogenous intermediate species. This elevated spin state is uniquely conducive to robust orbital overlap between the targeted reactant molecules and the Co d-band[40,41]. This promotes electron transfer, enhances adsorption, and improves the redox capability of active centers. Additionally, the tunable 3d orbitals enable strong electronic coupling with the carbon-nitrogen substrate[42,43]. This induces a rearrangement of surface charge, forming a localized electronic structure with high-spin character. Ultimately, this engineered configuration simultaneously amplifies intrinsic catalytic activity and streamlines the capture and activation of essential intermediates, thereby offering a favorable kinetic pathway for efficient NH3 synthesis. Structurally, Co-based catalysts can function as isolated single atoms in M–N–C SACs for precise catalysis, or utilize multi-site synergy in M–N–C NPs to activate complex reaction networks. The stable electronic interaction between Co and the carbon-nitrogen substrate also establishes a foundation for long-term catalyst stability. These properties together make Co an ideal choice for constructing high-performance NO3RR catalysts. Research to date has focused largely on tuning spin states in single atoms. Tuning cluster spin states, however, remains challenging due to thermodynamic constraints imposed by high surface energy.
In this study, we present a comprehensive comparative evaluation between cobalt nanoparticles (Co NPs) and their isolated single-atom counterparts (Co SACs) anchored on an N-doped carbon framework, both of which were derived through the pyrolysis of an identical ZIF-67 template. Moreover, we regulate the high-spin state of Co NPs. We demonstrate that Co NPs assemblies substantially outclass the single-atom centers in catalyzing the NO3RR, yielding striking improvements across NH3 production rates, Faradaic efficiency (FE), and target product selectivity. By integrating operando spectroscopy, electrochemical analysis, isotope labeling, and density functional theory (DFT) calculations, we uncover the underlying mechanism: the localized high-spin environment inherent to the Co NP ensembles fundamentally fortifies the binding affinity toward incoming nitrate species, enhanced orbital hybridization, accelerated direct hydrogenation pathways, and suppressed hydrogen evolution, thereby achieving exceptional NH3 yield and selectivity. This study provides fundamental insight into the structure–electronic–mechanistic relationships governing nitrate electroreduction and introduces high-spin cobalt nanoparticle ensembles as an advanced active-site architecture for sustainable NO3− degradation and NH3 electrosynthesis.
-
The fabrication of the Co SACs and Co NPs electrocatalysts utilized methanol, cobalt nitrate hexahydrate (Co(NO3)2·6H2O, 98%), 2-methylimidazole, N,N-dimethylformamide (DMF), zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 98%), and ultrapure water (H2O, 18.2 MΩ·cm). For the electrocatalytic evaluations, a 5 wt% Nafion dispersion (DuPont), anhydrous ethanol (CH3CH2OH, 99.7%), carbon paper (HCP030N), KOH (85%), and KNO3 (99%) were employed. Quantitative 1H NMR measurements of the generated ammonia relied on D2O, Maleic acid (98%), and isotopically labeled reagents, specifically 15N-KNO3 (> 99.9%) and 15N-NH4Cl (> 99.9%).
Synthesis of catalysts
-
Synthesis of Co SACs: initially, a metallic precursor solution was prepared by dissolving 0.546 g of Co(NO3)2·6H2O and 0.558 g of Zn(NO3)2·6H2O in 15 mL of methanol. This mixture was rapidly combined with a second methanolic solution containing 0.616 g of 2-methylimidazole (15 mL). Following 4 h of continuous ambient-temperature stirring, the resulting solid was isolated via centrifugation. The product was subjected to consecutive washing cycles with DMF and methanol, followed by overnight desiccation under vacuum at 75 °C. To yield the final Co SACs, the dried powder was pyrolyzed in a tube furnace under a continuous nitrogen flow, dwelling at 900 °C for 3 h with a heating ramp of 5 °C min−1.
Synthesis of Co NPs: the nanoparticle variant was synthesized following a similar protocol but with adjusted precursor ratios. Specifically, 0.546 g of the cobalt precursor and 1.674 g of the zinc precursor were dissolved in 40 mL of methanol, which was then swiftly mixed with 40 mL of a methanolic 2-methylimidazole solution (2.464 g). The reaction mixture was stirred for an extended duration of 12 h. All subsequent washing, drying, and carbonization procedures were identical to those described for the Co SACs.
Synthesis of NC substrate: a cobalt-free, nitrogen-doped carbon (NC) reference sample was fabricated utilizing the same methodology as the Co SACs, deliberately omitting the addition of the cobalt nitrate precursor.
Electrochemical measurements
-
Electrocatalytic evaluations were conducted using a standard CHI760E electrochemical workstation (Chenhua, Shanghai, China) connected to a two-compartment H-type cell, compartmentalized by a Nafion 117 proton-exchange membrane. The standard three-electrode configuration consisted of an Hg/HgO reference electrode, a Pt mesh counter electrode, and the prepared catalyst serving as the working electrode. The cathodic chamber was filled with a mixed electrolyte comprising 1 M KOH and 0.5 M KNO3, which was completely deaerated with argon for 30 min prior to testing. Comprehensive protocols regarding working electrode preparation, specific testing procedures and quantitative product analysis are detailed in Supplementary Text S1 of the Supporting Information.
Characterizations
-
Comprehensive descriptions of the instrumental techniques utilized to probe the morphological, structural, and chemical properties of the catalysts—including synchrotron X-ray absorption spectroscopy (XAS)—are provided in Supplementary Text S2. Furthermore, in-depth methodologies for the operando attenuated total reflectance-Fourier transform infrared (ATR-FTIR) measurements, differential electrochemical mass spectrometry (DEMS) analysis, and density functional theory (DFT) computations are elaborated in Supplementary Texts S3, S4, and S5, respectively.
-
According to the impregnation-pyrolysis method illustrated shown in Fig. 1a, M–N–C electrocatalysts (Co-SACs and Co-NPs) were synthesized. Initially, highly crystalline Zeolitic Imidazolate Framework-67 (ZIF-67) polyhedra were grown to serve as the structural foundation. During the subsequent 900 °C thermal treatment, the specific formation of either isolated atomic sites or metallic clusters was dictated simply by modulating the initial molar ratios of the precursor salts[44]. Morphological examination via scanning electron microscopy (SEM) and transmission electron microscopy (TEM) confirmed that the pristine ZIF-67 displayed a characteristic rhombic dodecahedral geometry (shown in Supplementary Fig. S1). After pyrolysis, Co SACs retained this rhombic dodecahedral framework structure, while Co NPs exhibited Co nanoparticles embedded within the dodecahedral framework (shown in Fig. 1b, c, and Supplementary Fig. S2). Elemental mapping through energy-dispersive X-ray spectroscopy (EDS) verified the homogeneous dispersion of Co, N, and C across both architectures, shown in Fig. 1d, e. To definitively confirm the atomic-level dispersion within the Co SACs, high-angle annular dark-field scanning TEM (HAADF-STEM) was utilized. As highlighted by the red circles in Supplementary Fig. S3, isolated bright dots corresponding to individual Co atoms are uniformly scattered across the N-doped substrate, ruling out the presence of nanoscale aggregates. Crystallographic analysis via X-ray diffraction (XRD) in Fig. 1f corroborated these structural variances. The Co NPs yielded prominent diffraction signals matching the (111), (200), and (220) facets of metallic cobalt, whereas the Co SACs presented a featureless profile devoid of crystalline metal peaks. Furthermore, quantitative Inductively coupled plasma optical emission spectrometry (ICP-OES) measurements determined the precise Co mass fractions to be 0.48 wt% for the Co SACs and 14.39 wt% for the Co NPs. These indicate that Co is highly dispersed in Co SACs and appears as nanoparticles in Co NPs. Raman spectra of samples displayed two typical defect-induced D-band (1,361 cm−1) and graphitic G-band (1,579 cm−1) as shown in Supplementary Fig. S4[45]. The lower ID/IG ratio of Co SACs (0.94) compared to that of Co NPs (1.06) indicates a higher degree of graphitization in Co SACs and increased defect sites in Co NPs. Additionally, the saturation magnetization intensity (SMI) reflects the magnetic strength of the catalysts shown in Fig. 1g. The Co NPs exhibited an SMI of 12.98 emu g−1, drastically surpassing the near-zero value of the Co SACs (0.09 emu g−1), though predictably trailing bulk pure Co nanoparticles (146 emu g−1) due to the substantial mass contribution of the non-magnetic carbon support[36,46]. This decrease is attributed to the presence of carbon, which reduces the SMI of Co NPs. These findings suggest that Co NPs supported on nitrogen-doped carbon exhibit a larger net magnetic moment, more unpaired electrons, and higher spin than Co SACs. Such characteristics are essential for enhancing the adsorption strength and reduction capability of the catalysts.
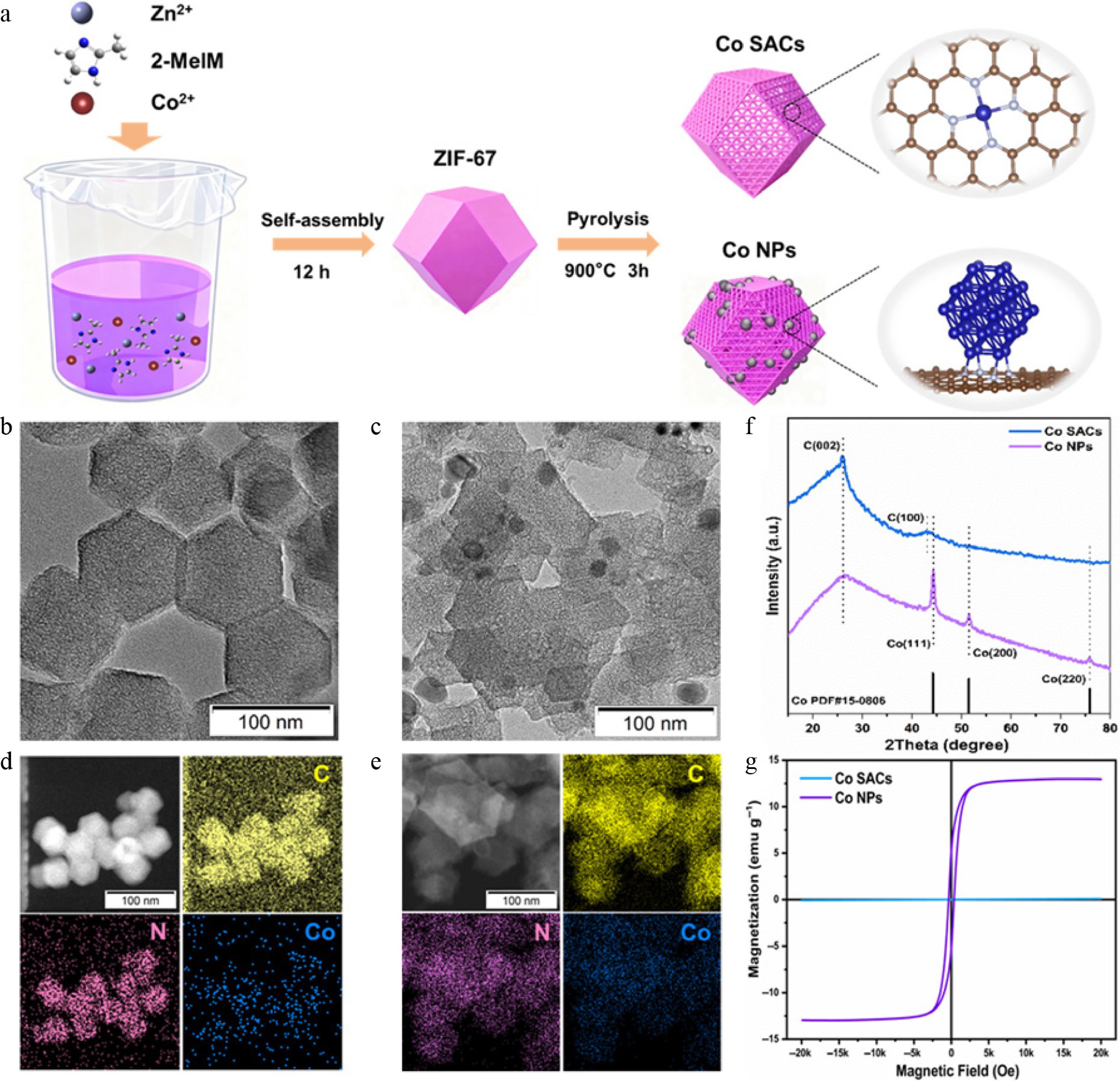
Figure 1.
(a) Schematic illustration detailing the synthetic pathways of ZIF-67, Co SACs, and Co NPs. (b), (d) TEM and EDS mapping images of Co SACs. (c), (e) TEM and EDS mapping images of Co NPs. (f) XRD patterns of Co SACs and Co NPs. (g) Magnetic-field-dependent magnetization for Co NPs at room temperature.
To gain deeper insights into the chemical states and local coordination environments of the catalysts, the synthesized materials were subjected to X-ray photoelectron spectroscopy (XPS) alongside X-ray absorption spectroscopy (XAS). As illustrated by the Co 2p XPS profiles shown in Fig. 2a, the Co centers within the Co SACs exist dominantly in a Co2+ oxidation state, evidenced by the primary Co 2p3/2 and Co 2p1/2 signals at 779.5 and 794.7 eV, accompanied by their respective shake-up satellites at 784.1 and 803.0 eV. Conversely, the Co NPs sample displayed a mixed-valence character containing both oxidized Co2+ features (located at 780.9 and 796.2 eV with satellites at 785.7 and 802.7 eV) and a distinct zero-valent metallic Co0 peak emerging at 778.5 eV[47]. The N 1s spectra, as shown in Fig. 2b, can be accurately deconvoluted into four distinct components: pyridinic N, pyrrolic/Co-N, graphitic N, and oxidized N. For Co SACs, these peaks are located at approximately 398.3, 399.6, and 400.8 eV, respectively. Notably, the corresponding N 1s peaks for Co NPs (398.6, 399.9, and 401.1 eV) exhibit a consistent positive shift of ~0.3 eV to higher binding energies[48]. The XPS results indicated that Co NPs exhibited positive shifts in binding energies compared to Co SACs. This indicates that the presence of active site groups in Co NPs reduces the electron cloud density of the entire N-doped carbon framework and Co atoms, resulting in a higher binding energy. For the Co SACs, the data strongly support the covalent anchoring of Co atoms to neighboring nitrogen sites, establishing a robust Co-Nx geometry.
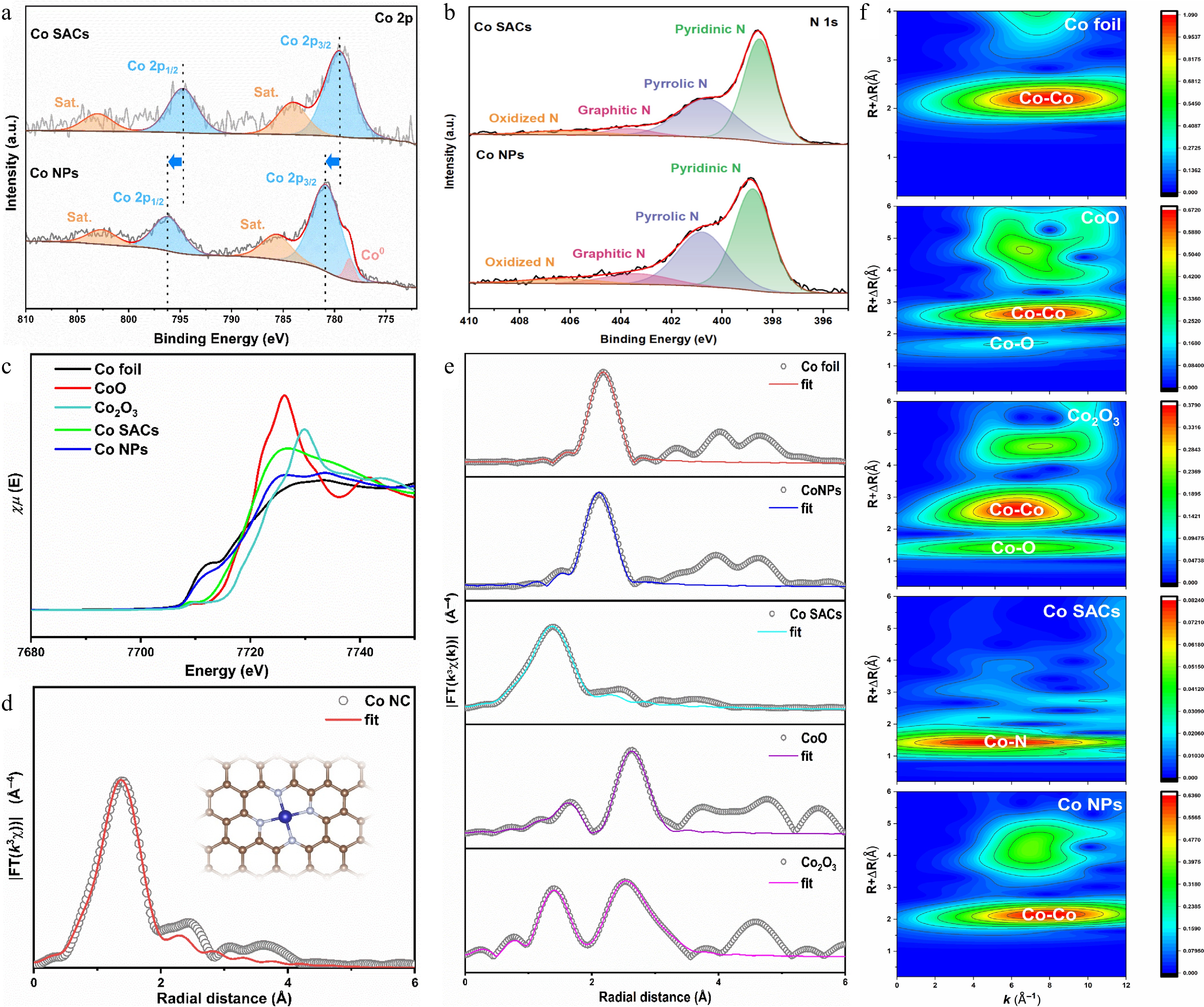
Figure 2.
(a) Co 2p XPS spectra of Co SACs and Co NPs. (b) N 1s XPS spectra of Co SACs and Co NPs. (c) XANES spectra at Co K-edge for Co foil, CoO, Co2O3, Co SACs, and Co NPs. (d) Coordination number fitting of FT-EXAFS for Co SACs. (e) FT-EXAFS spectra of Co foil, CoO, Co2O3, Co SACs, and Co NPs. (f) Wavelet transform (WT)-EXAFS spectra of Co foil, CoO, Co2O3, Co SACs, and Co NPs.
Synchrotron-based X-ray absorption spectroscopy, encompassing both the near-edge (XANES) and extended fine structure (EXAFS) regions at the Co K-edge, was utilized to probe the atomic-level coordination and intrinsic electronic states of the cobalt species[49]. The Co K-edge XANES spectra, as shown in Fig. 2c, demonstrate that the absorption edges of both catalysts lie between those of Co foil and CoO, indicating oxidation states of Co atoms intermediately between Co0 and Co2+. Notably, Co NPs exhibit closer resemblance to metallic Co0, whereas Co SACs show higher similarity to Co2+. Analysis of the Fourier-transformed EXAFS (FT-EXAFS) data shown in Fig. 2e highlights a dominant scattering signal at 1.38 Å for the Co SACs, which directly originates from the primary Co–N coordination sphere. Crucially, the complete absence of Co–Co metallic scattering paths—typically present in the bulk foil—verifies that the cobalt centers are exclusively dispersed as isolated atoms across the N-doped framework[50]. Quantitative modeling of these EXAFS oscillations shown in Fig. 2d and Supplementary Table S1 extracts an atomic coordination number of approximately 4, firmly establishing a classic Co–N4 geometric architecture[51]. In contrast, Co NPs display a Co–Co peak at 2.12 Å, consistent with that of Co foil, confirming the formation of cobalt nanoparticles with a metallic crystal structure, harmonizing with XRD and HAADF-STEM findings. To achieve enhanced spatial (R) and momentum (K) resolution, we further processed the Co K-edge EXAFS oscillations using wavelet transform (WT) algorithms shown in Fig. 2f, enabling a more detailed deconvolution of the structural configurations[52]. The intensity maximum at 5.3 Å−1 in Co SACs indicates the presence of Co–N or Co–O coordination. Significantly, no intensity maximum corresponding to Co–Co contributions (8.0 Å−1) was observed for Co SACs, ruling out any metal-derived crystalline structure. Conversely, the intensity maximum at 8.0 Å−1 in Co NPs matches perfectly with the Co foil, further validating the formation of metallic cobalt nanoparticles.
Electrocatalytic NO3RR performance
-
To evaluate the electrocatalytic reduction of nitrate, we performed standard assessments within a compartmentalized H-cell system. The tests utilized an Ar-saturated 1 M KOH electrolyte, both with and without the addition of 0.5 M KNO3 as the target reactant (Supplementary Fig. S5). As depicted by the linear sweep voltammetry (LSV) profiles shown in Fig. 3a, the introduction of nitrate triggers a surge in cathodic current density across all tested materials, confirming active NO3RR electrocatalysis. Co NPs showed the lowest onset potential (−0.1 V vs RHE) and the fastest current density decrease compared to Co SACs. Co NPs outputted an impressive 250 mA·cm−2 at −0.7 V vs RHE, which distinctly overshadows the 184 mA·cm−2 recorded for the Co SACs under identical conditions. This demonstrates the superior nitrate-reduction activity of Co NPs. Corroborating this kinetic advantage, the Nyquist plots derived from electrochemical impedance spectroscopy (EIS, Supplementary Fig. S6) reveal a drastically diminished charge-transfer resistance (Rct) for the Co NPs, indicating faster NO3RR kinetics for Co NPs.
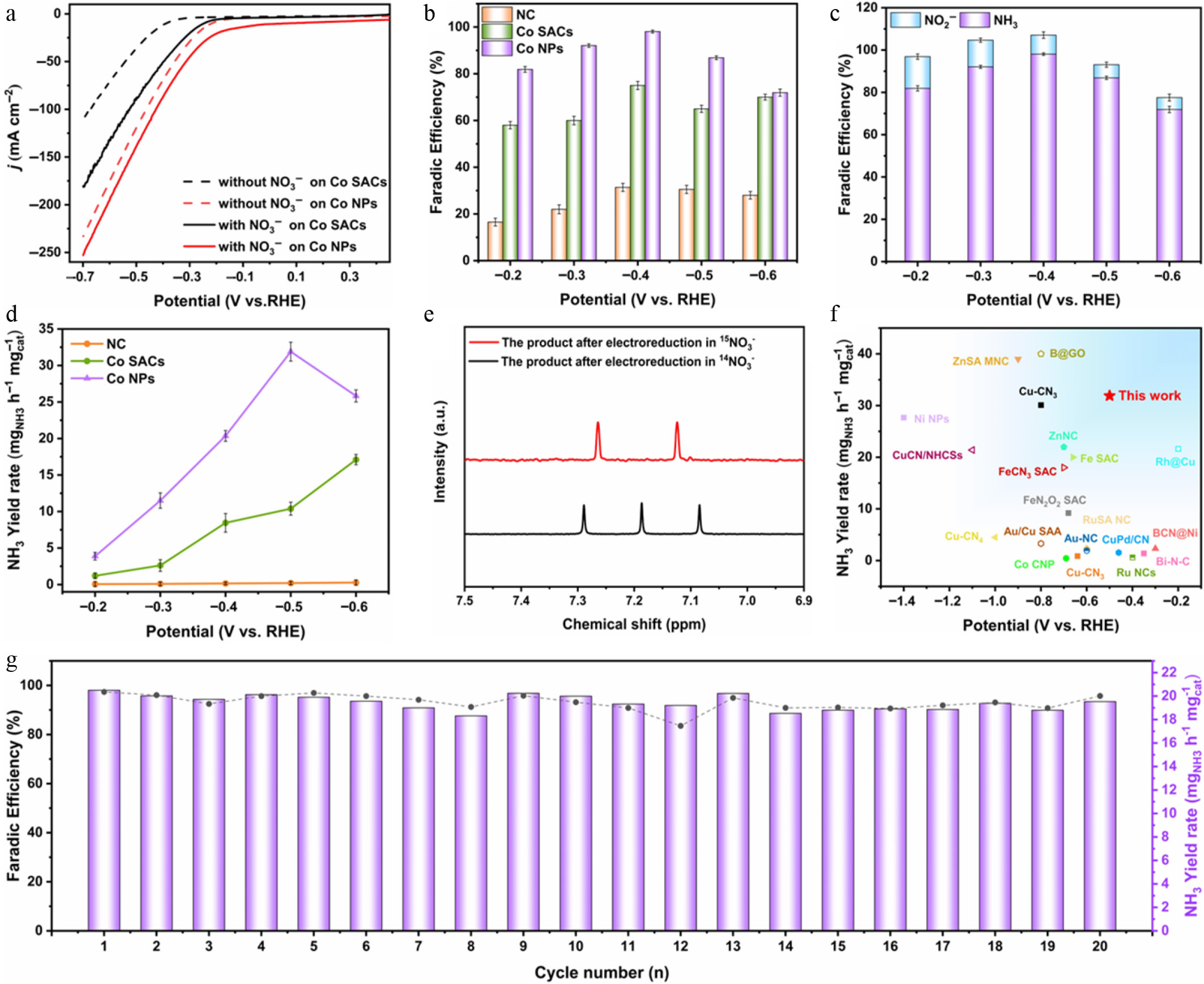
Figure 3.
(a) LSV of Co SACs and Co NPs in electrolyte with and without 0.5 M KNO3. (b) FE of NH3 for NC, Co SACs, and Co NPs. (c) FE of NH3 and NO2− for Co NPs. (d) NH3 yield rate for NC, Co SACs, and Co NPs. (e) 1H NMR spectra of the postreaction electrolyte using 0.5 M K15NO3 or K14NO3 as the precursor. (f) Comparison of the NO3RR performance of Co NPs in this work with state-of-the-art literature results. (g) Cyclic stability test of Co NPs over 20 consecutive cycles.
To further evaluate NO3RR performance, i-t curves were recorded between −0.2 and −0.6 V vs RHE for all samples (shown in Supplementary Figs S7–S9). The UV-visible spectrophotometric colorimetric method was used to quantify the amounts of NH3, NO2, and NO3 in the electrolyte after i-t tests, and calculate the FE of NH3 and NO2 (Supplementary Figs S10–S12). Co NPs exhibited higher NH3 FE than both NC and Co SACs at all potentials, as illustrated in Fig. 3b, achieving a maximum FE of 98.07% at −0.4 V vs RHE. Conversely, Co SACs exhibited inferior activity with an FE of 75% under identical conditions. The metal-free NC showed negligible NO3RR catalytic capability, with FE below 30%, indicating that the Co centers act as the indispensable active sites dictating the targeted catalytic transformation. More abundant Co active sites in Co NPs significantly enhanced NO3RR activity. Notably, applying voltages more negative than −0.4 V vs RHE triggered a noticeable decline in NH3 selectivity for the Co NPs, despite a continuous upward trend in overall current density. This suggests that competing HER and alternative nitrogenous byproducts like NO2− and N2 suppress NH3 yield. Therefore, the FE of the main byproduct NO2− was measured to assess selectivity. Figure 3c shows that NO2− was the dominant product for Co NPs at low potentials. With increasingly negative potentials, the FE of NO2− gradually decreased, while HER became the primary competing reaction affecting NH3 selectivity. Figure 3d presents the NH3 yield rates for NC, Co SACs, and Co NPs at different potentials. The NH3 yield rates were 0.269, 17.105, and 25.833 mgNH3 h−1 mgcat−1 for NC, Co SACs, and Co NPs when polarized at −0.6 V vs RHE. Co NPs achieved a maximum NH3 yield rate of 31.891 mgNH3 h−1 mgcat−1 at −0.5 V vs RHE, which is approximately 158 times and three times higher than that of NC, and Co SACs, respectively. To further unravel the intrinsic origin of this heightened electrocatalytic activity, cyclic voltammetry (CV) was deployed to extract the double-layer capacitance (Cdl) as a reliable proxy for the electrochemically active surface area (ECSA), as provided in Supplementary Figs S13, S14. The Cdl of Co NPs was as high as 92.20 mF·cm−2, which substantially eclipses the Co SACs at 48.82 mF·cm−2. The results collectively confirm the remarkable activity of Co NPs for NH3 synthesis via NO3RR. Co NPs also exhibited superior catalytic performance in an advanced oxidation process for peroxymonosulfate activation to degrade organic pollutants compared to Co SACs[36,53]. This further confirms that Co NPs possess unique structural advantages across multiple catalytic reactions. It is worth noting that while the significantly higher Co loading in Co NPs compared to Co SACs naturally contributes to the higher overall current density and absolute NH3 yield, the drastic leap in FE cannot be solely explained by the increased number of active sites. A mere increase in mass loading would proportionally amplify both NO3RR and the competing NO2RR and HER. Therefore, the near-unity selectivity of Co NPs intrinsically stems from an optimized electronic structure that fundamentally alters the active sites’ binding affinity, highly favoring nitrate reduction while suppressing NO2− and H2 evolution.
To conclusively trace the elemental origin of the synthesized ammonia during the electrocatalytic process, isotopic labeling experiments employing 15N were executed, with the resulting products analyzed via 1H nuclear magnetic resonance (NMR) spectroscopy, as presented in Fig. 3e. Utilizing 15NO3− as the reactant generated a distinct doublet signal corresponding to 15NH4+, while 14NO3− produced triplet peaks corresponding to 14NH4+[54]. This unambiguously demonstrates that nitrogen from the catalyst does not participate in the reaction, and NH3 is derived entirely from the targeted reduction of the initial nitrate. Figure 3f summarizes recent reported data on FE and yield for NH3 synthesis using non-noble metal, noble metal, and nanoparticle-based M–N–C catalysts. As detailed extensively in Supplementary Table S2, the concurrent realization of an ultrahigh FE and exceptional NH3 production rates places the present Co NP system well above most previously reported systems. The stability of Co NPs was evaluated via 20 consecutive cycles at −0.4 V vs RHE. Figure 3g shows that the NH3 yield rate remained stable at approximately 19.466 mgNH3 h−1 mgcat−1, with consistently maintained at ~93.03% of FE. This robust performance confirms the high activity and catalytic durability of Co NPs, demonstrating great potential for practical industrial application, and laying a solid experimental foundation for advancing NO3RR to NH3 synthesis technology from laboratory research toward industrialization.
Electrochemical in-situ spectroscopic analysis
-
To capture the dynamic evolution of transient surface species during electrocatalytic nitrate reduction, operando ATR-FTIR spectroscopy was deployed across both the Co NPs and Co SACs (Fig. 4a, b, and Supplementary Fig. S14). Figure 4a shows the ATR-FTIR spectra of Co NPs collected from open-circuit potential (OCP) to −0.7 V vs RHE. A gradual increase in the vibrational band intensity of *NO3 at 1,201–1,231 cm−1 indicates continuous nitrate consumption during electrolysis. Concurrently, the peak intensities for hydrogenated intermediates (*NH2 at ~1,400, 1,507, and 3,272 cm−1 in Supplementary Fig. S15; *NH at 1,615 cm−1), deoxygenated intermediates (*NO2 at ~1,281 cm−1; *NO at 1,573 cm−1), and the product (*NH3 at 1,428−1,442 cm−1) progressively increased[49,55,56]. These observations suggest that Co NPs effectively activate NO3−, facilitating the formation of various deoxygenated and hydrogenated intermediates, ultimately leading to NH3 formation. In contrast, the ATR-FTIR spectra for Co SACs in Fig. 4b showed much weaker signals for both hydrogenated and deoxygenated intermediates. Notably, a peak corresponding to *NH2OH at 1,156 cm−1 grew gradually in the Co SACs system, whereas it was barely detectable for Co NPs. These differences indicate that the abundant Co sites in Co NPs effectively stabilize key reactive intermediates, supplying ample hydrogenated species for subsequent *NH3 formation. For Co SACs, *NH3 may proceed via the *NH2OH intermediate, but the overall catalytic performance for NH3 synthesis remains comparatively weak. To quantitatively analyze intermediates, DEMS was utilized to detect intermediates and products during the Co NPs catalyzed NO3RR process, as shown in Fig. 4c. The mass-to-charge ratio (m/z) signals at 15 (NH), 16 (NH2), 17 (NH3), 28 (N2), 30 (NO), 31 (NOH), 33 (NH2OH), and 46 (NO2−) were monitored. Notably, strong signals for NH3 (1 × 10−11) and NH2 (9 × 10−12) indicate efficient hydrogenation steps on Co NPs. Weak signals for NH (4 × 10−13) and N2 (1 × 10−14), alongside faint signals for NO2− (2 × 10−14), NO (4 × 10−13), and NOH (5 × 10−14), suggest rapid conversion of these intermediates on Co NPs. Almost no signal was detected for NH2OH. The intensities of these peaks decreased significantly after multiple cycles, likely due to uneven electrolyte distribution caused by the lack of stirring. The results of ATR-FTIR and DEMS jointly demonstrate that Co NPs effectively adsorb and activate NO3−, converting it to NH3 primarily via a direct pathway involving deoxygenated intermediates (*NO2, *NO, *NOH) followed by hydrogenation to *NH and *NH2. In contrast, Co SACs exhibit weaker capability for NO3− adsorption and activation, proceeding through both direct and indirect pathways involving multiple intermediates (*NO2, *NO, *NH2OH), resulting in significantly lower catalytic performance compared to Co NPs.
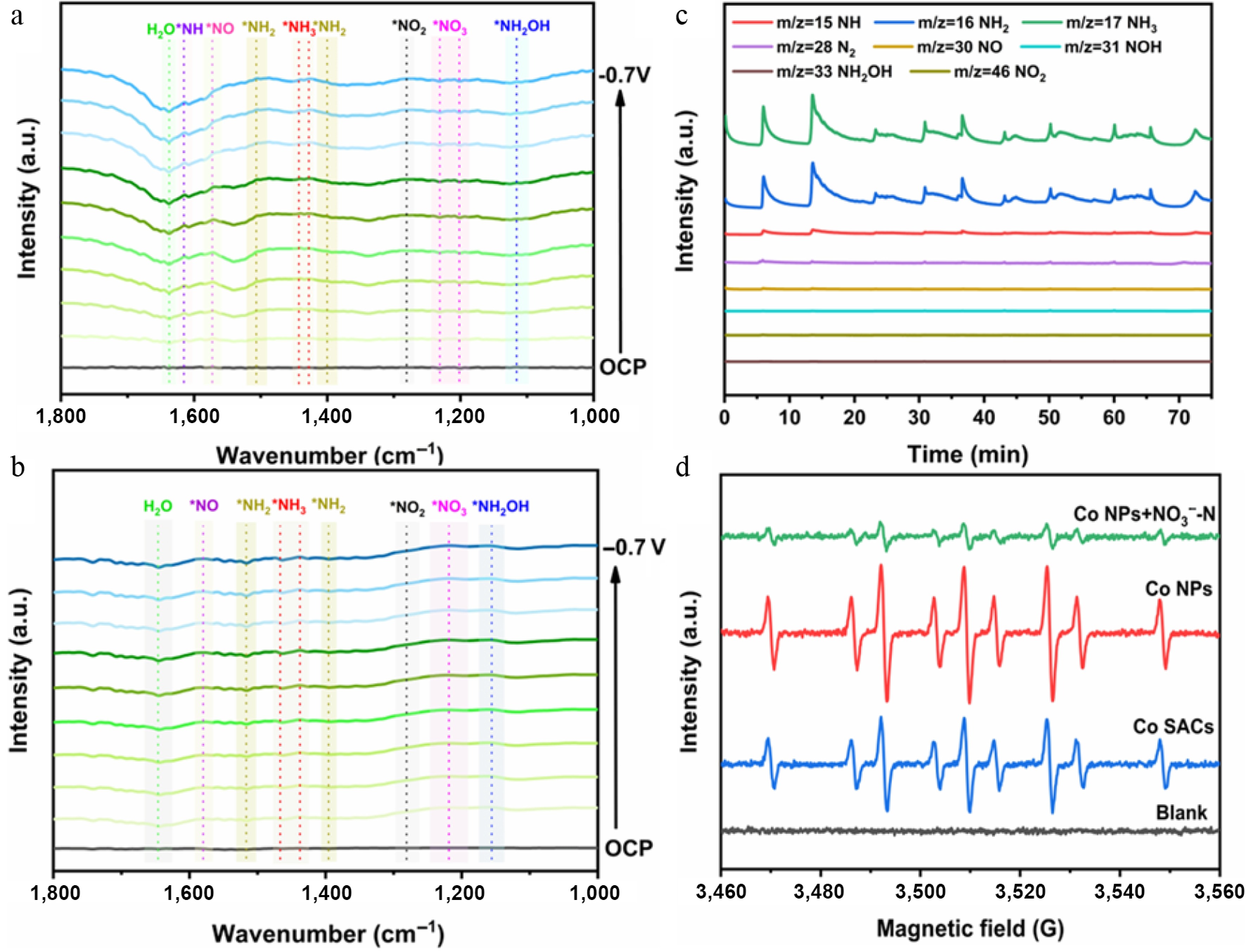
Figure 4.
Operando ATR-FTIR spectra of (a) Co NPs, and (b) Co SACs at various applied potentials. (c) DEMS analysis of Co NPs. (d) EPR spectra of blank control, NC, Co SACs, and Co NPs.
In addition, recent studies indicate that the electrocatalytic NO3RR can proceed via a direct electron-driven mechanism or a pathway heavily mediated by active hydrogen species (H*). Within aqueous electrolyte environments, the cathodic reduction of water yields highly reactive H* intermediates, which act as potent reducing agents capable of attacking and cleaving the stable N–O bonds of NO3−. To experimentally capture and validate the generation of these transient species, electron paramagnetic resonance (EPR) measurements were conducted utilizing 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as a radical scavenger to form the detectable DMPO–H* spin adduct. When evaluated in pure KOH electrolyte (without NO3−) shown in Fig. 4d, a highly resolved multi-line EPR spectrum—exhibiting the classic splitting pattern with an intensity ratio of 1:1:2:1:2:1:2:1:1:1—was clearly recorded, unambiguously proving the in-situ generation of H*. Co NPs exhibited the strongest DMPO–H* signal, followed by Co SACs, indicating the superior H* generation capability of Co NPs. Upon introducing 0.5 M KNO3, this prominent radical signal for the Co NPs underwent a drastic attenuation[57]. Such a sharp decline strongly implies that the freshly formed H* on the metallic surface is swiftly depleted to hydrogenate adjacent, chemisorbed nitrogenous intermediates during the active NO3RR. This dynamic consumption directly validates the indispensable function of water-derived active hydrogen in driving the catalytic conversion. These results demonstrate that Co NPs optimize reactant adsorption and generate abundant H* on their numerous active sites, thereby accelerating the protonation process and promoting efficient NH3 synthesis via NO3RR.
Theoretical study of NO3RR
-
DFT calculations further elucidate the reaction mechanism. Figure 5a illustrates the reaction pathways (direct and indirect routes) for ammonia synthesis via NO3RR on Co NPs and Co SACs. Initially, NO3− adsorbs on Co sites (via end-on and side-on configurations) and undergoes hydrogenation and deoxygenation to form the *HNO2 intermediate. Further hydrogenation and deoxygenation generate *NO. Subsequently, the distinct adsorption structures of *NO (N-end or O-end) bifurcate the reaction into direct and indirect pathways for NH3 formation, with each step involving at most one (H+ + e−) pair. To accurately unravel the NO3RR mechanism, we calculated the detailed reaction free energy changes (ΔG) for four distinct pathways on Co NPs and Co SACs (Supplementary Figs S16, S17). The results indicate that Co NPs favor the direct pathway (Path 1) for NH3 synthesis, while Co SACs predominantly follow the indirect pathway (Path 4), consistent with the ATR-FTIR and DEMS observations. Figure 5b compares the reaction free energy profiles of Co NPs and Co SACs during NO3RR. First, Co NPs exhibit stronger NO3− adsorption than Co SACs. The theoretical limiting potential (UL) (see Supplementary Tables S3 and S4 for detailed values) was calculated to evaluate the catalytic activity. Co NPs show a UL of −0.43 V in Path 1, while Co SACs exhibit a UL of −0.72 V in Path 4. This indicates the potential-determining step (PDS) on Co NPs has a lower energy barrier, which accounts for the superior NO3RR performance and provides compelling evidence for the significantly enhanced NH3 synthesis activity on Co NPs. Furthermore, we calculated the *H adsorption energy (ΔG*H) on Co NPs and Co SACs (shown in Supplementary Fig. S18), a critical parameter for the competing HER. The ΔG*H value on Co NPs (0.81 eV) is significantly higher than that on Co SACs (0.11 eV), indicating that Co NPs suppress H2 formation and thereby achieve higher FENH3, consistent with experimental observations.
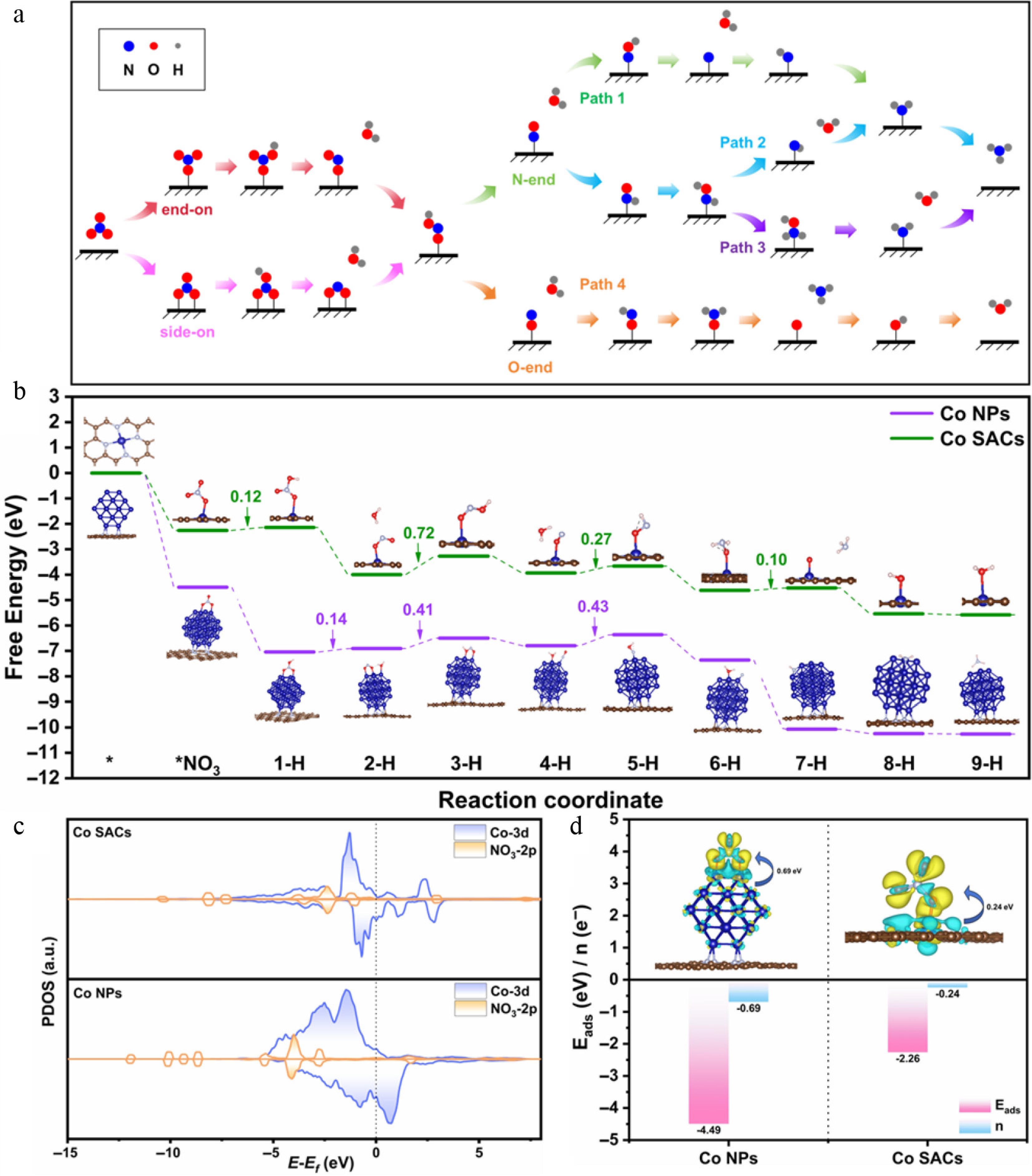
Figure 5.
(a) Reaction schematic for NO3RR. (b) Reaction energy barrier diagrams of Co NPs and Co SACs. (c) PDOS of Co NPs and Co SACs after adsorption of *NO3. (d) The ΔG*NO3 and charge transfer number of Co NPs and Co SACs.
These DFT models evaluate the thermodynamic and electronic properties strictly at the single active-motif level, which effectively isolates the inherent catalytic activity from the macroscopic metal mass loading. These results explicitly confirm that the superior NO3RR performance of Co NPs is fundamentally driven by intrinsic electronic effects rather than simply a higher quantity of active sites. The origin of catalytic activity was investigated in depth from the perspectives of electronic structure and charge transfer. Total density of states (TDOS) analysis shows that both Co SACs and Co NPs exhibit continuous electron occupation near the Fermi energy (Ef) (shown in Supplementary Fig. S19). Notably, the electron density near Ef is significantly higher in Co NPs than in Co SACs, indicating superior charge transport capability in Co NPs. Projected density of states (PDOS) analysis, as shown in Fig. 5c, further reveals that the Co 3d orbitals of Co SACs exhibit a relatively symmetric density of states, consistent with low-spin Co atoms lacking unpaired electrons. In contrast, Co NPs exhibit asymmetric and spin-polarized electronic states near the Ef, originating from unpaired electrons on Co atoms within the nanoparticle clusters. This indicates a higher spin state. To elucidate the role of the nitrogen-doped carbon substrate, we further calculated the TDOS for pristine Co nanoparticles without substrate interaction (Supplementary Fig. S20). While these unsupported Co NPs also exhibit spin-polarized features, their electron density near the Ef is markedly lower than that of the nitrogen-doped carbon-supported Co NPs. This comparison demonstrates that the nitrogen-doped carbon substrate plays a crucial role in amplifying the high-spin states of Co NPs by donating additional electron density, thereby elevating the near-Ef electronic states and enhancing the overall reactivity. This is consistent with the SMI results. The regulation of the spin state of Co NPs by NC carriers is essentially governed by a profound metal-substrate synergistic effect, which is dominated by interfacial electron transfer and orbital redistribution. Driven by the interfacial work function difference, the N-doped carbon substrate donates additional electron density to the metallic Co clusters. These injected electrons selectively populate the Co 3d orbitals at the asymmetrical metal-substrate interface. Governed by intra-atomic exchange interactions (Hund’s rule), this electronic influx undergoes spatial redistribution, which maximizes the number of unpaired electrons. Consequently, the nitrogen-doped carbon substrate essentially acts as an electronic promoter that amplifies the high-spin states of Co NPs. Furthermore, a substantial overlap exists between the Co-3d and NO3-2p orbitals in Co NPs, demonstrating stronger hybridization with reactant orbitals and a significant enhancement of the interacting electronic states. The unique electronic structure of Co NPs—characterized by near-Fermi electronic states and a high-spin state—directly leads to their stronger *NO3 adsorption capability (−4.49 vs −2.26 eV) and greater electron transfer number (0.69 vs 0.24 e−) as shown in Fig. 5d. The enhanced orbital hybridization and charge transfer ability optimize the binding energy with reaction intermediates, effectively lowering the energy barrier of the PDS and thus improving the overall catalytic performance. This is consistent with the unique activity advantages of Co NPs reported previously. Recent studies also indicate that the Co nanoparticle structure can promote efficient charge transfer, thereby significantly enhancing performance in various catalytic reactions[36,53,58,59]. Therefore, this study reveals at the electronic level that the abundant active sites on the Co NP surface enables stronger electronic interaction and orbital hybridization with *NO3. This weakens the N–O bond, promotes nitrate activation, and lays the foundation for subsequent hydrogenation and deoxygenation steps, providing solid theoretical support for the experimentally observed outstanding performance.
-
In conclusion, we demonstrate that N-doped carbon-supported Co NPs constitute a highly efficient and selective electrocatalyst for NO3RR, significantly outperforming Co SACs. The Co NPs achieve an outstanding FENH3 of up to 98.1% and a high NH3 yield rate of 31.9 mgNH3 h−1 mgcat−1, together with excellent operational stability. Operando ATR-FTIR and DEMS measurements reveal that Co NPs favor a direct nitrate reduction pathway involving rapid deoxygenation and hydrogenation intermediates, whereas Co single-atom sites predominantly follow an indirect mechanism. DFT calculations identify the electronic origins of this performance enhancement, showing that Co NPs possess high-spin electronic states, increased near-Fermi-level charge density, stronger Co-NO3− orbital hybridization, and weaker hydrogen adsorption, which collectively lower reaction energy barriers and suppress competing HER. These findings establish high-spin Co NPs ensembles as a superior catalytic motif for multielectron nitrate reduction, highlighting the importance of spin polarization, metallic conductivity, and multi-site orbital coupling in complex electrochemical transformations. More broadly, this work offers a generalizable strategy for designing advanced electrocatalysts that couple wastewater denitrification with sustainable ammonia production, advancing the development of next-generation nitrogen-cycle technologies.
-
The authors confirm their contributions to the paper as follows: writing − original draft, validation, software, project administration, formal analysis, data curation, conceptualization: Xu D; validation, formal analysis: Yan B; software, investigation: Liu T; software: Wu H; supervision, funding acquisition: Wang J; supervision: Chen G; writing − review and editing, funding acquisition: Cheng Z. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed in the current study are available from the corresponding author on reasonable request.
-
The authors gratefully acknowledge the National Natural Science Foundation of China (52322005, 52400179, and 52176197), the Young Scientific and Technological Talents (Level One) in Tianjin (QN20230114), the Tianjin Science and Technology Committee (24JCJQJC00140), and the China Postdoctoral Science Foundation (GZC20241199, 2024T010TJ, and 2024M762352).
-
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
-
accompanies this paper online at: https://doi.org/10.48130/prkm-0026-0013.
- Supplementary Table S1 EXAFS fitting parameters at the Co K-edge for various samples (Ѕ02 = 0.78).
- Supplementary Table S2 Comparison of the NO3RR activity and NH3 yield performance among nitrogen-doped carbon-supported Co NPs and the recently reported M-N-C-based electrocatalyst.
- Supplementary Table S3 The Gibbs free energy change of each reaction step of path 1 for electrocatalytic NO3RR to NH3 by Co NPs.
- Supplementary Table S4 The Gibbs free energy change of each reaction step of path 4 for electrocatalytic NO3RR to NH3 by Co SACs.
- Supplementary Text S1 Electrochemical measurements.
- Supplementary Text S2 Characterization.
- Supplementary Text S3 Operando ATR-FTIR characterization.
- Supplementary Text S4 DEMS measurement.
- Supplementary Text S5 Theoretical calculation methods.
- Supplementary Fig. S1 TEM image of ZIF-67.
- Supplementary Fig. S2 SEM image of Co NPs.
- Supplementary Fig. S3 HAADF-STEM image of Co SACs.
- Supplementary Fig. S4 The Raman shift of Co NPs and Co SACs.
- Supplementary Fig. S5 Photograph of the H-cell setup.
- Supplementary Fig. S6 Nyquist plots for EIS charge transfer resistance tests of Co SACs and Co NPs.
- Supplementary Fig. S7 The i-t curve of Co NPs electrocatalyzing NO3RR for 2 h at −0.2 to −0.6 V vs. RHE.
- Supplementary Fig. S8 The i-t curve of Co SACs electrocatalyzing NO3RR for 2 h at −0.2 to −0.6 V vs. RHE.
- Supplementary Fig. S9 The i-t curve of NC electrocatalyzing NO3RR for 2 h at −0.2 to −0.6 V vs. RHE.
- Supplementary Fig. S10 Concentration-absorbance UV-visible calibration curves of NH3-N.
- Supplementary Fig. S11 Concentration-absorbance UV-visible calibration curves of NO3-N.
- Supplementary Fig. S12 Concentration-absorbance UV-visible calibration curves of NO2-N.
- Supplementary Fig. S13 Cyclic voltammograms of Co SACs and Co NPs.
- Supplementary Fig. S14 ECSA and Cdl of Co SACs and Co NPs.
- Supplementary Fig. S15 Operando ATR-FTIR spectra of Co NPs and Co SACs.
- Supplementary Fig. S16 Reaction barrier diagrams of the four reaction pathways of Co NPs.
- Supplementary Fig. S17 Reaction barrier diagrams of the four reaction pathways of Co SACs.
- Supplementary Fig. S18 The adsorption energy of Co NPs and Co SACs for *H and *NO3.
- Supplementary Fig. S19 TDOS profiles of Co SACs and Co NPs.
- Supplementary Fig. S20 TDOS profiles of Co NPs without nitrogen-doped carbon substrate.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Xu D, Yan B, Liu T, Wu H, Wang J, et al. 2026. High-spin cobalt nanoparticles on nitrogen-doped carbon for superior electrocatalytic nitrate reduction to ammonia from water. Progress in Reaction Kinetics and Mechanism 51: e015 doi: 10.48130/prkm-0026-0013 

High-spin cobalt nanoparticles on nitrogen-doped carbon for superior electrocatalytic nitrate reduction to ammonia from water
- Received: 24 February 2026
- Revised: 17 March 2026
- Accepted: 10 April 2026
- Published online: 30 May 2026
Abstract: The electrochemical reduction of nitrate (NO3RR) presents an environmentally viable strategy for simultaneously mitigating nitrogen-based water pollution and synthesizing green ammonia (NH3). Nevertheless, the overall efficacy of this process is severely hindered by the sluggish kinetics inherent to its multi-electron transfer steps, coupled with intense competition from the hydrogen evolution reaction (HER). Metal-based single-atom catalysts (SACs) with low-spin effects, such as Cu and Ru, exhibit great performance in the NO3RR. However, this study demonstrates that nitrogen-doped carbon–supported cobalt nanoparticles (Co NPs) significantly outperform Co SACs in NO3RR. Under alkaline conditions, the Co NPs architecture achieved a remarkable Faradaic efficiency of 98.1%, alongside an exceptional NH3 yield rate of 31.9 mgNH3 h−1 mgcat−1. In-situ electrochemical spectroscopy reveals that Co NPs promote a direct nitrate-to-ammonia pathway involving rapid deoxygenation and hydrogenation intermediates, whereas Co SACs favor indirect routes with sluggish *NH2OH formation. Density functional theory calculations uncover that Co NPs exhibit high-spin electronic states, elevated near-Fermi-level electron density, and stronger Co-3d/NO3−-2p orbital hybridization, leading to enhanced nitrate adsorption, lower potential-determining energy barriers, and weakened hydrogen adsorption that suppresses HER. Notably, these high-spin states are further enhanced by the nitrogen-doped carbon substrate, which increases the near-Fermi-level electron density and boosts catalytic activity. These controllable high-spin structures and the electronic synergistic effect enable accelerated nitrate activation, efficient proton–electron transfer, and improved ammonia selectivity. This work establishes high-spin cobalt nanoparticle ensembles as a superior active-site motif for NO3RR, and provides a fundamental design principle for next-generation electrocatalysts that integrate wastewater denitrification with sustainable ammonia synthesis.