










-
Organic silicon compounds, known for their chemical stability and thermodynamic properties are integral to nanomaterial synthesis, chemical vapor deposition (CVD), and flame retardants[1−6]. Among these, octamethylcyclotetrasiloxane (D4, [CH3]8Si4O4]), with the structure unit (-[CH3]2SiO-), is prevalent. As a key precursor, D4 forms silica nanoparticles or films via high-temperature pyrolysis and oxidation. Its pyrolysis kinetics are crucial for material synthesis efficiency and product characteristics[7]. Additionally, D4 is frequently found in landfill syngas[8−10], yet its use in combustion systems is hindered by the uncertain pyrolysis behavior of silicon compounds, especially D4. The pyrolysis of D4 is intricate, involving numerous intermediates and pathways, leading to an incomplete understanding of its kinetics. Thus, investigating the high-temperature pyrolysis kinetics of D4 is essential for optimizing industrial processes and informing combustion models of complex silicon-based systems.
Recent studies on the pyrolysis kinetics of D4 reveal significant limitations, particularly in temperature and pressure ranges. Davidson & Thompson[11] investigated D4 decomposition in a static reactor at low temperatures (762–842 K), proposing a mechanism for D3 formation via dimethyl siloxane monomer (D1) elimination, with an activation energy of 301 kJ/mol. However, this research did not address kinetic behavior at higher temperatures (> 1300 K), crucial for nanomaterial synthesis[12]. Sanogo & Zachariah[7] extended the temperature range (1,058–1,197 K) using a fast flow reactor, identifying a secondary pathway for D5 decomposition into D3 and D2, yet the mechanism did not fully elucidate methane (CH4) formation. Additionally, intermediate detection remains challenging, with ongoing debate over the reaction pathway. Khabashesku et al.[13] identified D3, SiO, and small hydrocarbons in D4 pyrolysis products via matrix-isolated infrared spectroscopy, but failed to directly observe the key intermediate D1, suggesting its rapid decomposition into SiO and free radicals. Almond et al.[14], using G3-level quantum chemical calculations, suggested that D1 decomposes into CH3 and CH3SiO via Si-C bond cleavage, initiating a chain reaction that produces CH4 and C2H2. However, the stability and decomposition pathway of D1 at high temperatures remained disputed, particularly the direct formation of CH4, which lacks full validation. Additionally, the mechanism model has its limitations. Sela et al.[12] developed a D4/D3 sub-mechanism comprising 19 reactions between 1,160 and 1,600 K, integrating shock tube experiments with high repetition rate time-of-flight mass spectrometry (HRR-TOF-MS). They observed a significantly higher decomposition rate for D4 compared to D3. Nonetheless, the model underestimates CH4 experimental values, indicating potentially overlooked rapid channels. Furthermore, discrepancies in predictions for products like C2H2, where experimental values exceed simulations, highlight the need for refinement in the secondary reaction pathway.
This study investigates the high-temperature pyrolysis kinetics of D4 using molecular dynamics simulations and gas chromatography, extending its reaction kinetics beyond 1,500 K. It proposes new reaction channels to elucidate the CH4 generation pathway, enhancing the understanding of CH4 formation observed by experiments and simulations. By integrating experimental and theoretical approaches, the study unveils the formation and transformation pathways of intermediates, such as the open chain structure I-Si4O4C8H24, and clarifies the contribution of some important reactions to pyrolysis kinetics and product distribution. This research aims to elucidate the multi-scale kinetic behavior of D4 during high-temperature pyrolysis, providing a more precise theoretical foundation for the synthesis and combustion simulation of silicon-based materials.
-
Figure 1 shows the schematic diagram of the experimental setup and product detection of D4 pyrolysis. The experiments were conducted in a flow tube reactor (about 980 cm3). Argon (Ar) served as the carrier gas to introduce D4 into the reactor. Before each pyrolysis experiment, the reactor was purged with Ar to remove air, with the flow rate of 200 mL/min during heating to the desired temperature. Once enough D4 was introduced, the reactor's front and rear valves were closed to allow complete pyrolysis of D4. After pyrolysis, the valves were reopened, Ar was reintroduced, and pyrolysis products were collected at the quartz nozzle. Throughout this process, the reactor maintained a constant temperature, while liquid D4 was heated to its boiling point in an oil bath and transported into the reactor by Ar. Experiments were conducted at 803 K and 843 K, with products collected and analyzed using gas chromatography. The other conditions included reactor pressure (3−6 MPa), carrier gas flow rate (200 mL/min), D4 vapor feed rate (76 g/min), residence time (60 s), and D4 initial concentration (2.65 × 10−4 mol/mL). In addition, reproducibility tests showed that the uncertainties of measured temperature, pressure, and flow rate were ± 1 °C, ± 0.3 MPa, and ± 2.5 mL/min, respectively.
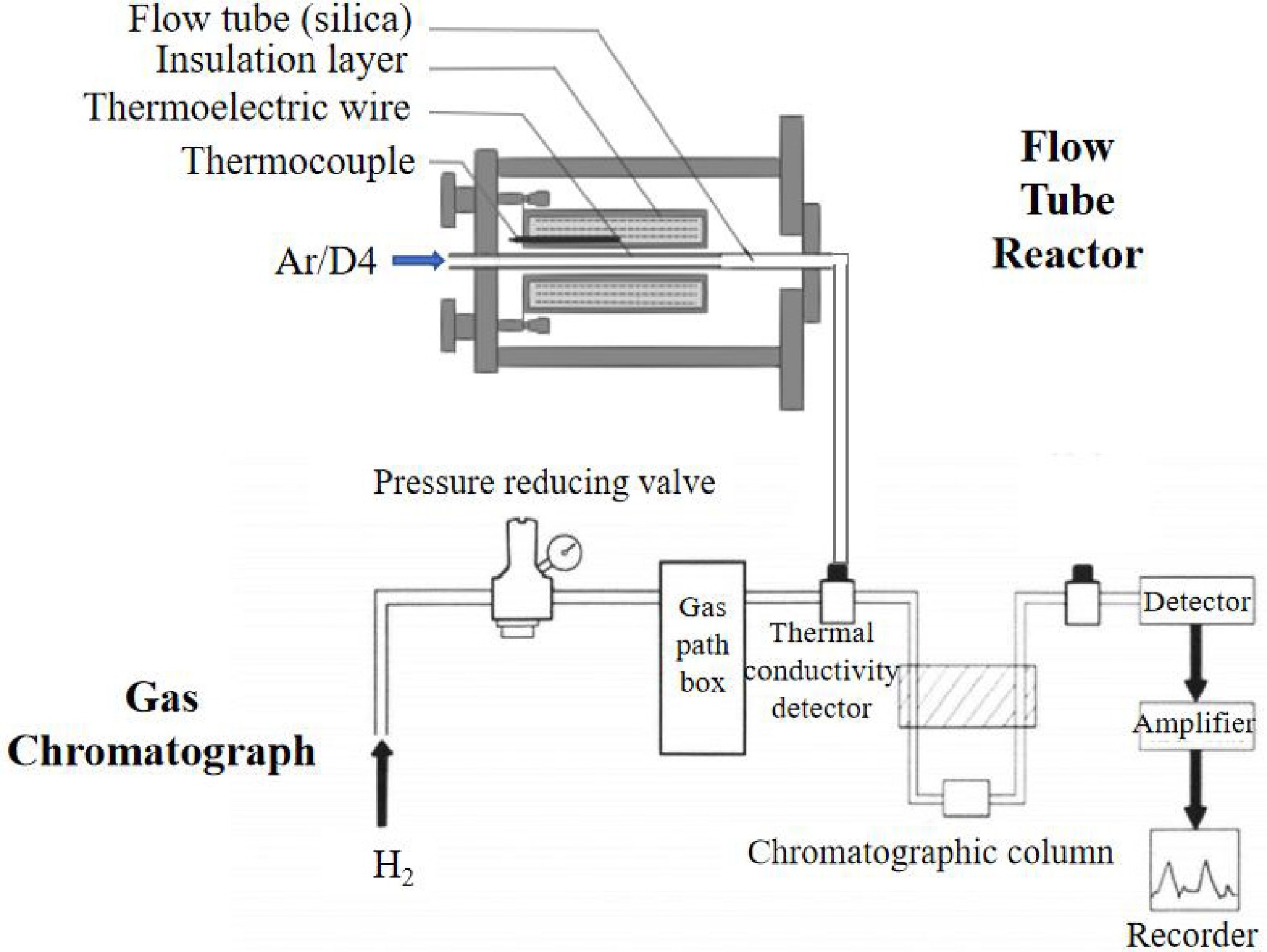
Figure 1.
Experimental setup and product detection of D4 pyrolysis.
In the gas chromatograph, pyrolysis products were introduced into the thermal conductivity detector and propelled through the column by a carrier gas, typically hydrogen or helium. Within the chromatographic column, sample components interacted with a stationary phase, such as a silica gel, undergoing adsorption, desorption, and dissolution. Variations in boiling point, polarity, and adsorption properties resulted in different movement speeds, facilitating separation. The separated components entered the detector, which recorded their retention time and peak area. A data processing system then analyzed these metrics to determine component content. Retention time, the duration for a component to reach peak height, correlates with boiling point; higher boiling points result in slower peak times and longer retention. This experiment employed a hydrogen flame ionization detector (FID), with hydrogen supplied by a generator. An external standard method was used for integration, employing the standard gas to create a reference sample.
ReaxFF molecular dynamics simulations
-
Traditional empirical force fields necessitate predefined atomic connectivity for molecular dynamics simulations and cannot model bond formation or breaking in chemical reactions. In contrast, the ReaxFF force field utilizes 'bond order' based on interatomic distances to simulate complex reactions without preset pathways[15]. This bond order allows ReaxFF to determine interaction parameters, including atomic parameters, bond stretching, angle bending, dihedral twisting, conjugation, coordination corrections, and hydrogen bonding. The electronegativity equalization method (EEM) dynamically updates system charges, enhancing data accuracy. Compared to other key-level methods, ReaxFF offers superior accuracy and reactivity. In ReaxFF, molecular energy is expressed via bond order unless a bond interaction occurs, i.e.,
$ \begin{matrix}{E}_{system}={E}_{bond}+{E}_{over}+{E}_{under}+{E}_{lp}+{E}_{val}+{E}_{tor}+{E}_{conj}+{E}_{Coulomb}+{E}_{vdWaals}\\ \end{matrix} $ (1) where, Esystem refers to the total energy of the system, and Ebond represents the bond energy. Eover and Eunder represent the energy correction terms for over and under coordination, respectively. Elp represents the energy term for lone pair electrons, and Eval represents the energy term for bond angles. Etor represents the energy term for dihedral angles, EvdWaals represents the energy term for van der Waals forces, and ECoulomb represents the energy term for Coulomb forces. The specific expressions for the above energy terms are found in the references[16].
ReaxFF molecular dynamics simulations were performed using the Amsterdam Modeling Suite (AMS) software with a reactive force field from PDMS decomposition[17], previously validated in other organosilicon systems[18,19]. The molecular structure of D4, sourced from the NIST Chemistry WebBook, is depicted in Fig. 2. The size of simulation box was 68 Å × 68 Å × 68 Å, containing fifty D4 molecules at a gas density of 0.07866 g/mL. The simulations employed Canonical Ensemble (NVT) with temperatures ranging from 1,500 to 2,600 K to explore the high-temperature pyrolysis mechanism of D4. Under the NVT ensemble, with a fixed density, the pressure increases with temperature, and the highest pressure can reach about 80 bar. The total simulation duration spanned 100 to 250 ps, with a time step of 0.1 fs, and a bond order cutoff of 0.3 was applied. To improve statistical reliability, it was noted that we had performed three independent runs at each temperature with different initial configurations, and averaged the results to minimize stochastic fluctuations. Additionally, we mainly focused on early-stage kinetics (≤ 200 ps), where D4 decomposition and primary product formation dominate.
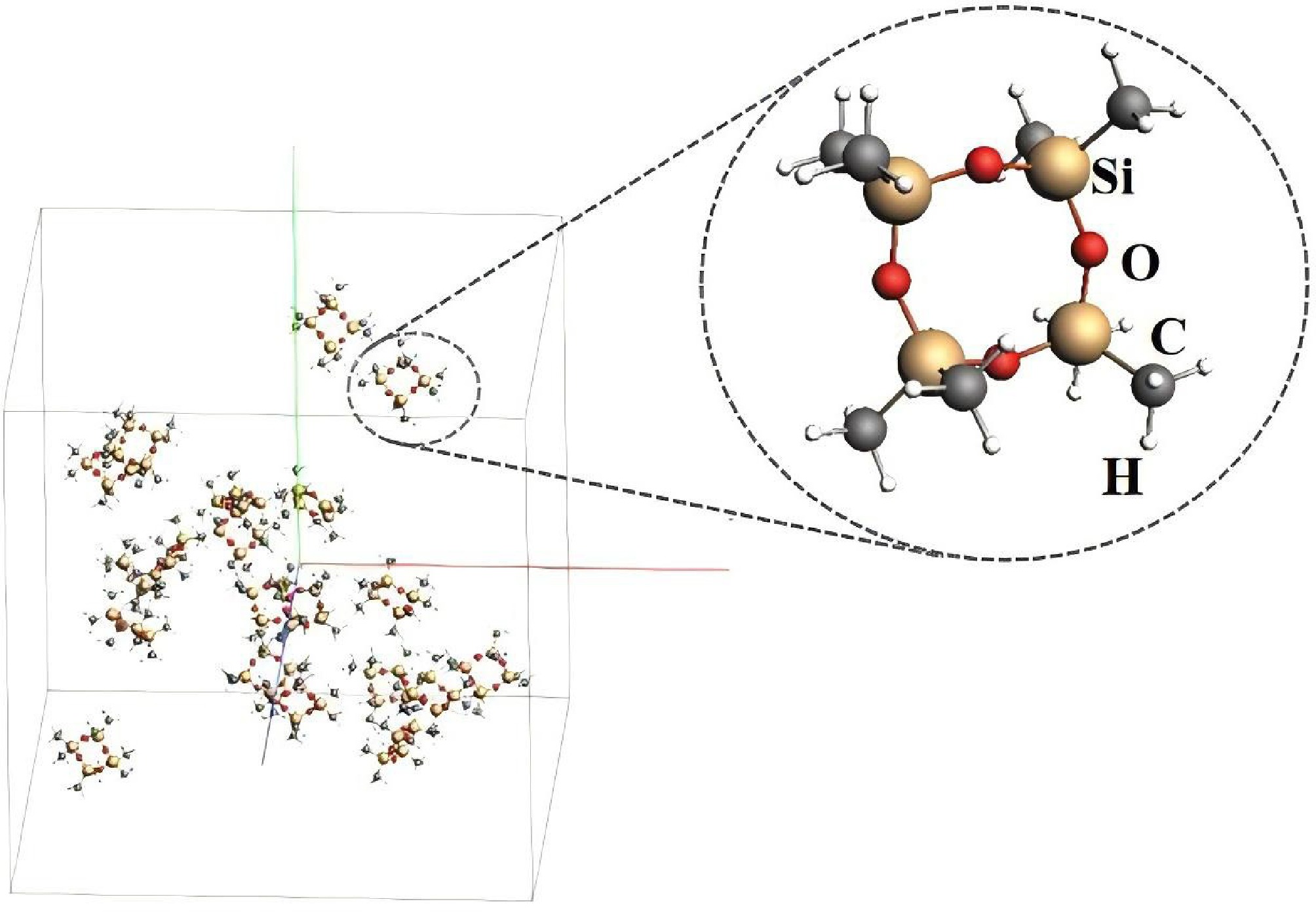
Figure 2.
The initial geometric configuration of D4 pyrolysis for ReaxFF MD simulations.
Validation of the reactive force field
-
To verify the accuracy of the force field used in ReaxFF MD simulations, density functional theory (DFT) calculations were conducted with Gaussian 09 software, and the results from DFT calculations were compared with ReaxFF simulations. The B3LYP method with a 6-31G(d) basis set was employed in the DFT calculations. Figure 3 shows the potential energy surface scans at different molecular structures (different bond lengths and angles). Good agreement is obtained between ReaxFF simulations and DFT calculations in the relative energies of different Si-C bond lengths and Si-O bond lengths, and the relatively small errors are presented in most scanning points of Si-O-Si angles and Si-O-Si-O dihedral angles, which indicates that the reactive force field used in the ReaxFF simulations is acceptable.
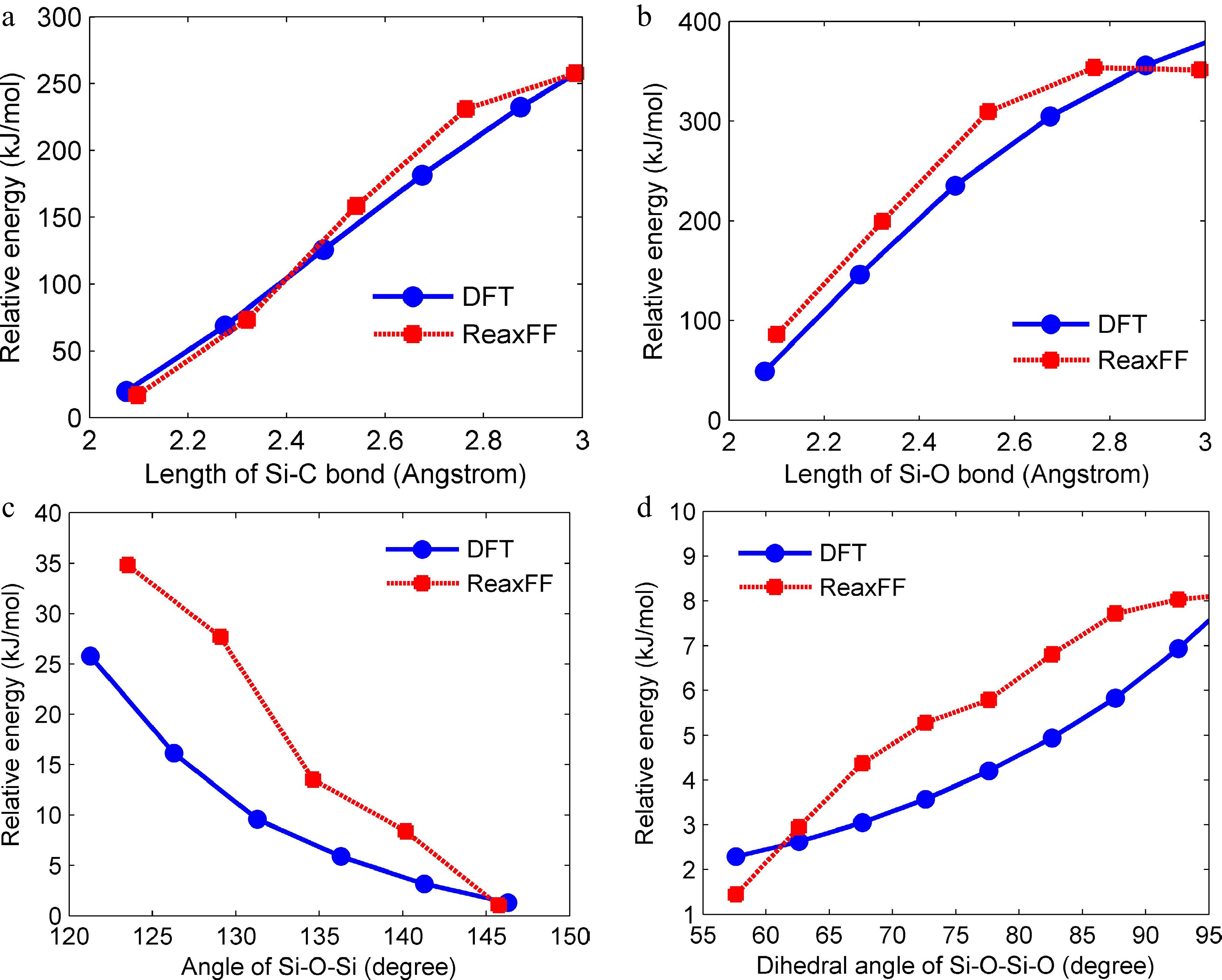
Figure 3.
Potential energy surface scans by ReaxFF simulations and DFT calculations with different bond lengths of (a) Si-C bonds, (b) Si-O bonds, and angles of (c) Si-O-Si bonds, and (d) Si-O-Si-O bonds. The relative energy is obtained by subtracting the initial molecular structure energy.
-
In this section, D4 pyrolysis experiments were conducted at 803 K and 843 K in a flow tube reactor. The hydrocarbon products were analyzed in detail by gas chromatography (GC). Each experiment was repeated three times with identical conditions, and the reproducibility tests indicated that the uncertainties of the measured mole fractions of D4 pyrolysis products was less than ± 0.01%.
Figure 4 shows the chromatographic spectral lines of hydrocarbons produced after D4 pyrolysis at 803 K and 843 K, where the corresponding product names are presented in Table 1. The detailed chromatographic data are shown in Tables 2 and 3. The data clearly reveals that pyrolysis temperature plays a significant role in regulating product distribution, especially the key effect on methane (CH4) formation. At 803 K, the main products detected are C2-C4 hydrocarbons such as ethane (C2H6), ethylene (C2H4), propane (C3H8), propylene (C3H6), butane (C4H10), and butene (C4H8). These products indicate that at this temperature, the initial cleavage of D4 molecules mainly involves the cleavage of Si-O-Si bonds and the migration, recombination, or elimination of methyl (-CH3) groups, forming free radicals or fragments containing 1 to 2 carbon atoms, which in turn generate the observed C2-C4 alkanes and olefins by recombination, disproportionation, or hydrogen extraction reactions. When the temperature was raised to 843 K, the spectrum of products changed significantly. In addition to the C2-C4 products described above, methane (CH4) becomes an extremely prominent product (its prominent peak is clearly visible in Fig. 4). This contrast strongly suggests that methane formation is a process with high activation energy in the pyrolysis of D4, and is extremely temperature-sensitive. The temperature of 803 K may be below the effective initiation temperature for this critical path, or its reaction rate may be too low to produce detectable amounts of methane at this temperature. A temperature difference of only 40 K from 803 K (no detection), to 843 K (mass production), results in a significant change in methane from nothing, strongly suggesting the existence of a specific temperature window or activation energy barrier. Below this threshold (e.g., 803 K), the reaction path leading to C2-C4 products dominate; above this threshold (e.g., 843 K), new pathways leading to methane formation are opened or accelerated.
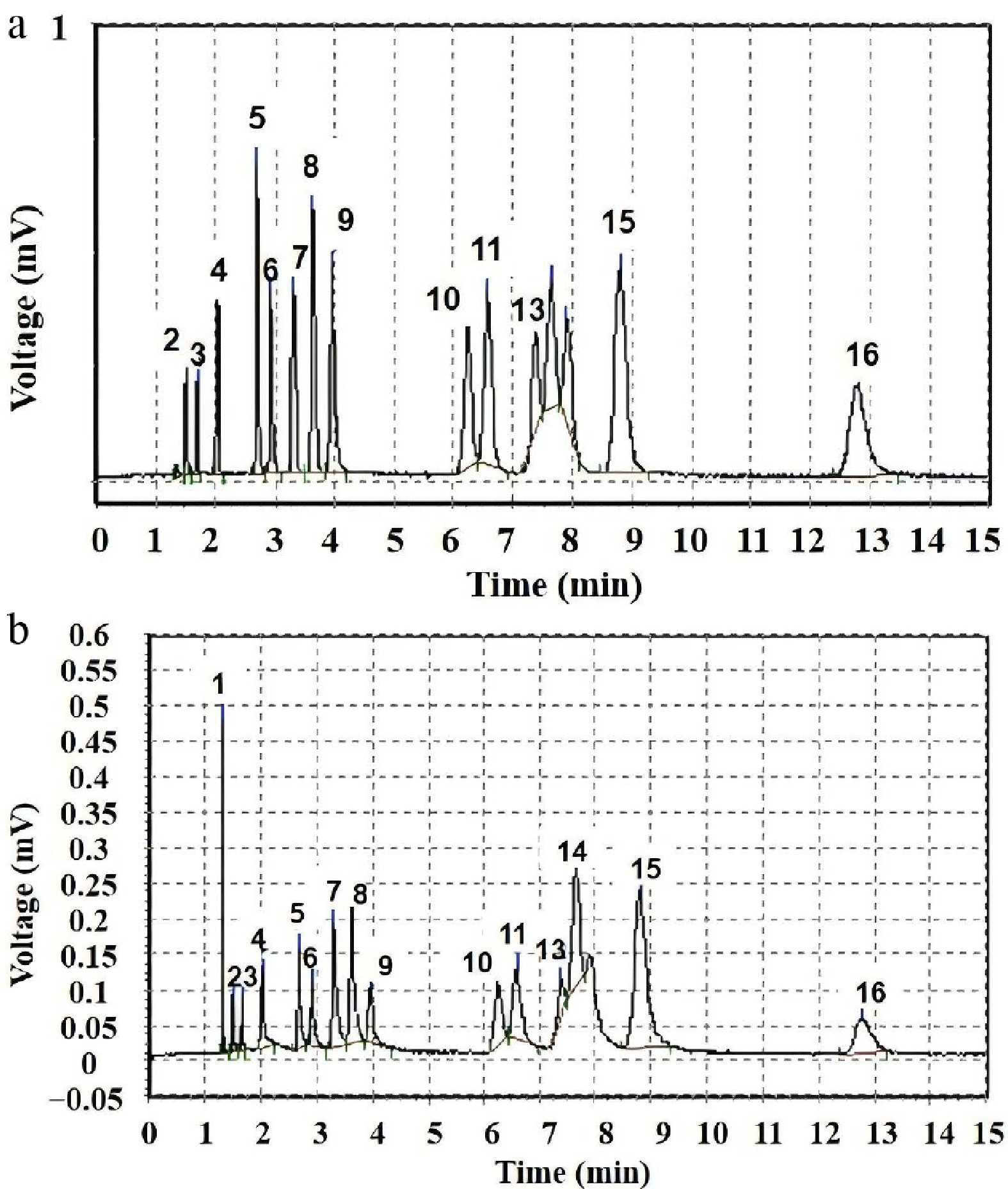
Figure 4.
The detected results of hydrocarbons from D4 pyrolysis by gas chromatography, where (a) and (b) correspond to the temperatures of 803, and 843 K, respectively.
Table 1. Component names represented by peak number in gas chromatograms.
Peak No. Component name Peak No. Component name 1 Methane 9 Propadiene 2 Ethane 10 1-Butene 3 Ethylene 11 trans-2-Butene 4 Propane 12 Isobutylene 5 Cyclopropane 13 iso-Pentane 6 Propylene 14 cis-2-Butene 7 Isobutane 15 n-Pentane 8 n-Butane 16 1,3-Butadiene Table 2. Chromatographic data of pyrolysis products at 803 K.
Peak
no.Retention time
(min)Peak height
(μV)Peak area
(μV·s)Mole fraction
(%)1 / / / / 2 1.515 237.000 414.300 0.0131 3 1.682 195.500 380.200 0.0134 4 2.032 398.458 922.800 0.0215 5 2.698 703.759 2,155.450 0.0434 6 2.932 408.000 1,415.500 0.0337 7 3.315 400.000 1,644.500 0.0369 8 3.640 579.756 2,698.850 0.0447 9 3.965 450.385 2,339.200 0.0531 10 6.240 308.933 2,575.000 0.0497 11 6.582 373.814 3,285.500 0.0599 12 7.382 213.667 1,584.700 0.0395 13 7.640 278.303 2,327.700 0.0449 14 7.907 227.150 2,038.000 0.0393 15 8.807 450.358 6,256.600 0.0678 16 12.798 193.846 3,925.900 0.0646 Note: Argon is the primary species in the gas mixture. Table 3. Chromatographic data of pyrolysis products at 843 K.
Peak
no.Retention time
(min)Peak height
(μV)Peak area
(μV·s)Mole fraction
(%)1 1.323 453.000 738.900 0.0763 2 1.507 78.167 170.900 0.0054 3 1.673 77.600 167.750 0.0059 4 2.023 111.909 388.800 0.0090 5 2.690 144.276 532.450 0.0107 6 2.923 90.135 389.500 0.0093 7 3.315 177.308 980.700 0.0161 8 3.640 200.472 1,165.600 0.0193 9 3.965 75.037 467.900 0.0106 10 6.257 85.795 780.750 0.0151 11 6.598 100.742 1,009.600 0.0184 13 7.390 50.371 365.200 0.0091 14 7.657 157.459 1,475.000 0.0502 15 8.798 220.842 3,427.950 0.0372 16 12.790 48.941 978.600 0.0161 The possible methane formation pathways include: (1) direct dehydrogenation/cleavage of methyl radicals. Higher temperatures lead to higher energy collisions, which may prompt the methyl radical (·CH3) produced by the cleavage of D4 to directly abstract hydrogen atoms from other molecules (e.g., another methyl group, a silomethyl group, or a larger hydrocarbon radical) to form CH4. For example: ·CH3 + R-H -> CH4 + R· (R-H may be another methylsilyl group, intermediate molecule, or product molecule); (2) deep cleavage of the silyl group. D4 cleavage initially produces Si-CH3 bonds containing intermediates (e.g. silicon radical R3Si· or silylene R2Si:). At elevated temperatures, the Si-CH3 bond on these intermediates may undergo homolysis to produce methyl radicals (·CH3), which can then rapidly participate in the reaction to form CH4 (e.g., hydrogen extraction reaction above, or a combination with other methyl radicals to form ethane; but ethane is already present at 803 K, indicating that methyl radical formation has occurred at 803 K, but not enough, or different routes make CH4 difficult to form). The more critical high temperature pathway may be the direct cleavage of Si-C bonds accompanied by H transfer: Si-CH3 → Si· + ·CH3 (this step may be slower at lower temperatures), or more complex intramolecular rearrangement leading to direct removal of CH4, and high temperature provides sufficient energy for Si-C bond cleavage; (3) secondary reactions are enhanced. The initial C2-C4 olefins (such as ethylene and propylene) are less stable at high temperatures, and further cracking, dehydrogenation, or radical attack reactions may occur. These secondary reactions may also contribute to CH4. For example, pyrolysis or radical-induced cracking of ethylene can produce methyl radicals and methylene groups, which in turn produce methane. Higher free radical concentrations at 843 K may also promote such secondary reactions.
From 803 K (dominated by C2-C4), to 843 K (dominated by C2-C4 + CH4), the transition of product distribution modes, and the reconstruction of D4 pyrolysis paths with temperature are clearly demonstrated. The presence of methane marks a significant increase in the depth of pyrolysis, involving higher energy demand bond breakage (e.g., Si-C bond homolysis, methyl deep dehydrogenation) or more intense secondary reactions. In addition, the extreme sensitivity of methane formation to temperature indicates that the reaction path has a high apparent activation energy. This experimental phenomenon provides a key constraint condition for the subsequent construction of a detailed D4 pyrolysis model, which must include elementary reaction steps that become important only at high temperatures, and accurately describe CH4 formation (e.g., specific Si-C bond cleavage, rapid hydrogen extraction of methyl radicals, etc.). The main microscopic mechanism leading to CH4 formation and silicon-containing species detection can be observed by ReaxFF MD simulations.
A single molecule pyrolysis by ReaxFF MD simulations
-
To understand the chemical bond breaking of the D4 molecule, a single-molecule pyrolysis was investigated using the ReaxFF molecular dynamics simulation method, and the pyrolysis pathway was analyzed. The total simulation duration was 200 ps using the NVT ensemble, and the time step was 0.2 fs. A pyrolysis temperature of 2,000 K was employed for accelerating the occurrence of chemical reactions.
The pyrolysis pathway of a single D4 molecule is shown in Fig. 5. The initial step of pyrolysis is the cleavage of the Si–C bond to remove the methyl group, as in reaction R1. This is because the bond energy of the Si-C bond (about 377 kJ/mol) is weaker than that of other chemical bonds (such as the Si-O bond at 540 kJ/mol)[7], so this bond breaks first when pyrolysis occurs. Subsequently, a ring- opening reaction of the molecule takes place, that is, the Si-O bond breaks, causing the molecule to transform into a ring- opening structure, as in reaction R2. Thereafter, another methyl group is removed and one of the methyl groups is transferred; the removed methyl group is located on the Si atom, attached to the O atom on the O side of the just broken Si-O bond, as shown in reaction R3. The transferred methyl group is located on one of the Si atoms of the unbroken Si-O bond, and is transferred to the Si atom of the just broken Si-O bond, as shown in reaction R4. Further, the molecule undergoes a dehydrogenation reaction to lose a hydrogen atom (R5), in which the C-H bond is broken because the bond energy of the C-H bond is greater, relative to the Si-C bond and the Si-O bond. The methyl group (methylene CH2) that loses a hydrogen atom is also removed, as in reaction R6. A series of demethylation reactions (R7 to R10) then occur, leaving only one methyl group on the final molecule. CH3O4Si4 molecules are then formed through ring-opening and H-transfer reactions. The CH3O4Si4 molecule then decomposes into HO4Si3 and CH2Si. Through ReaxFF molecular dynamics simulation, it is found that these two substances are the final products of single-molecule pyrolysis of D4 at 2,000K and cannot be decomposed again. It can be seen from the energy evolution of Fig. 6 that when pyrolysis proceeds to reaction R13, the overall energy of molecules maintains relative equilibrium. To decompose these two products further, it is necessary to increase the pyrolysis temperature. The study of a single molecule pyrolysis can not only clarify the reactions caused by chemical bond breakage, but also help to explain the pyrolysis reaction path of actual D4 precursor.
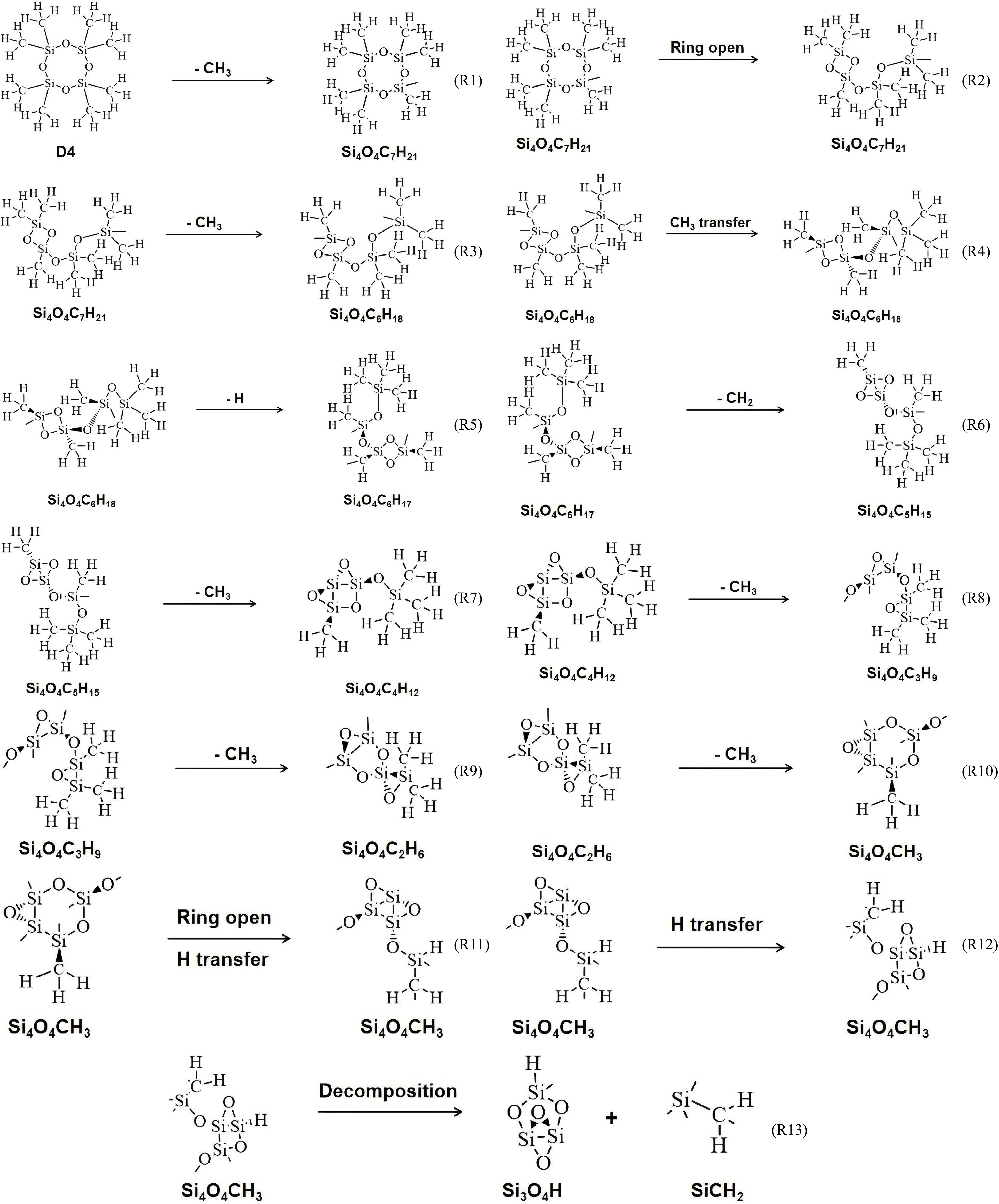
Figure 5.
Pyrolysis pathways of a single D4 molecule at 2,000 K.
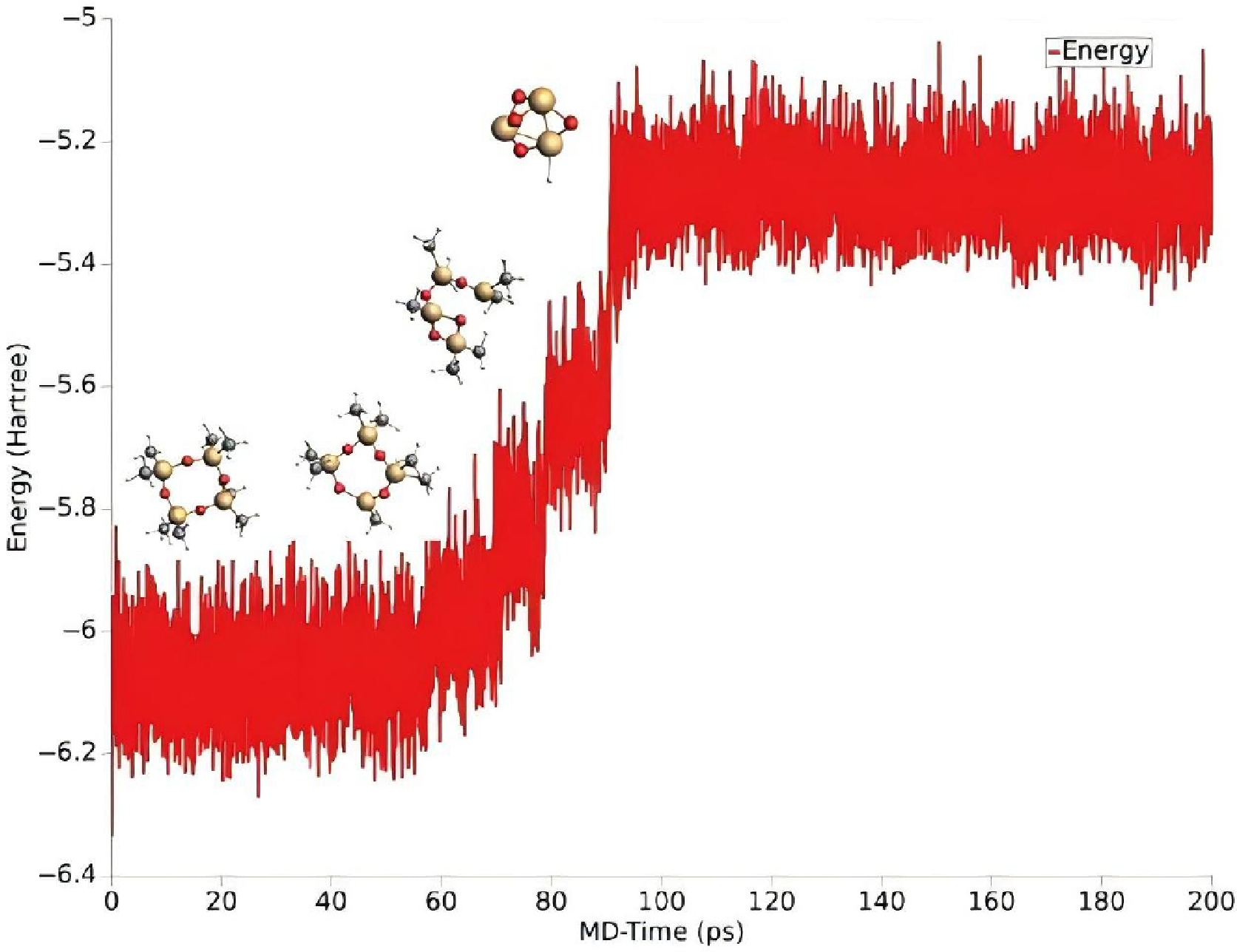
Figure 6.
Energy evolution during a single D4 molecule pyrolysis at 2,000 K.
Pyrolysis kinetics of D4 by ReaxFF MD simulations
-
In this section, the pyrolysis kinetics of D4 was investigated under the condition of 50 molecules, and gas density of 0.07866 g/ml using the ReaxFF molecular dynamics simulations at a temperature range of 1,500−2,600 K. The simulation time is set to 150−250 ps and the time step is 0.1 fs.
Figure 7 shows the variation of D4 molecular number with time at different temperatures. Results showed that the decomposition rate of D4 increased significantly with the increase in temperature. At 1,500 K, only about 25% of D4 decomposes within 250 ps, while at 2,600 K, the decomposition is basically completed within 150 ps, which accords with the basic law of high temperature promoting reactions. By fitting the D4 concentration decay curve, it was found that the D4 pyrolysis followed a first-order reaction kinetics model[3], i.e.,
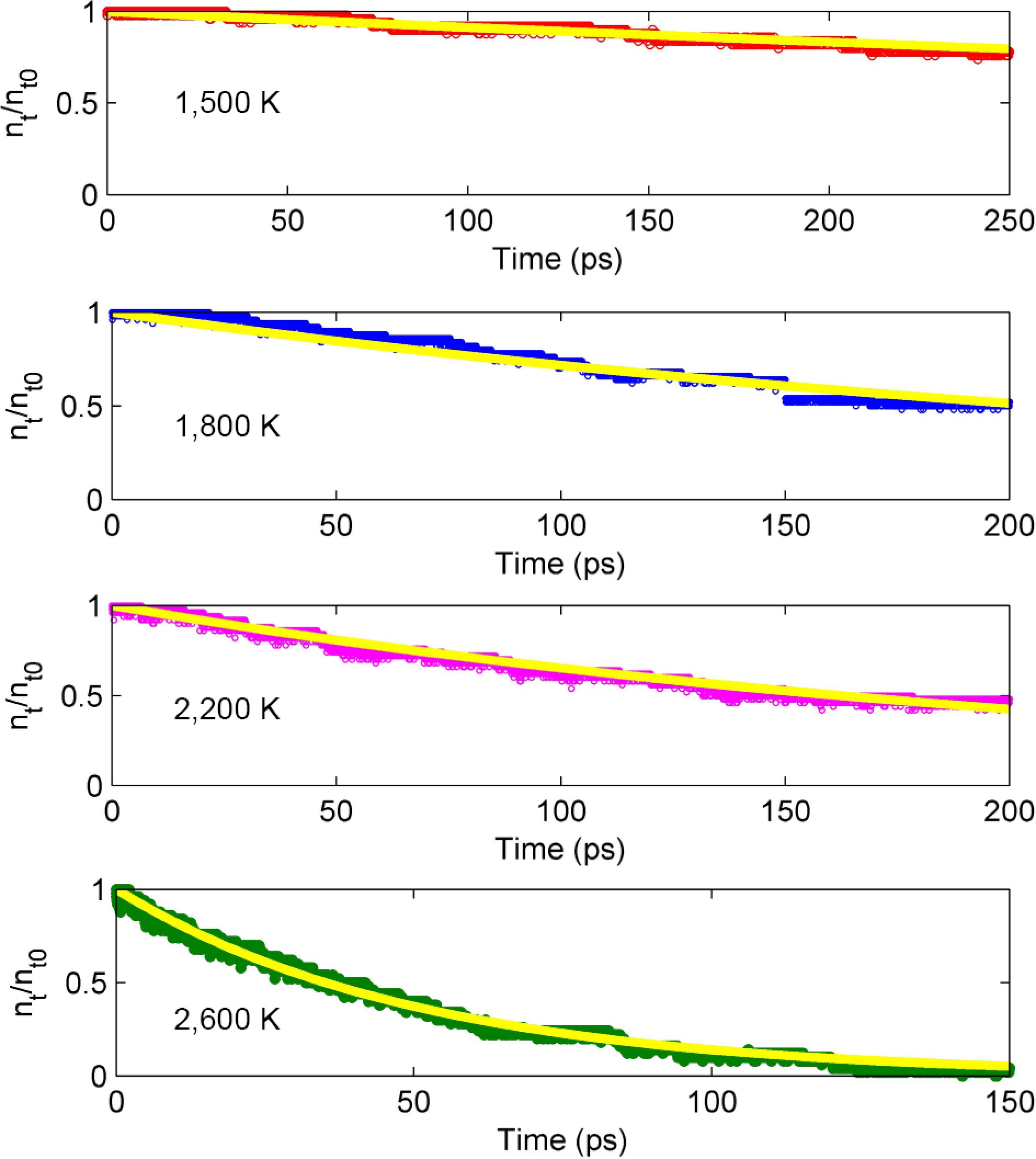
Figure 7.
Evolution of D4 molecular number at the temperature of 1,500−2,600 K. The solid line represents data fitting.
$ \frac{\text{d}n}{\text{d}t}=\text-{k}_{\text{dec}}n $ (2) where, kdec is the rate constant for D4 decomposition, and n is the number of D4 molecules. By integrating Eq. (2), the evolution of the number of D4 molecules with time is obtained as follows:
$ {n}_{\text{t}}/{n}_{\text{0}}=\text{exp[-}{k}_{\text{dec}}(t-{t}_{\text{0}})] $ (3) where, nt and n0 are the number of D4 molecules at the time of t and t0, respectively. t0 refers to the time when D4 begins to decompose. These results are consistent with the experimental results in fast flow reactors[7], which indicates that single-molecule decomposition is the dominant pathway for D4 pyrolysis.
Based on the first-order kinetic model, the reaction rate constant kdec values at various temperatures were calculated (Fig. 8a). The results show that there is a significant linear relationship between In(kdec) and 1/T (R2 > 0.9), which verifies that the pyrolysis reaction follows the Arrhenius equation: kdec= A·exp(-Ea/R/T). In order to investigate the effects of reaction conditions, the kdec of this study was analyzed jointly with the experimental data from Sanogo et al.[7] (1,058−1,197 K, ~18 mbar), and Sela et al.[12] (1,160−1,600 K, ~2.0 bar) (Fig. 8b). In these middle-low-temperature and low-pressure experiments, the obtained reaction rate constants are much smaller than the high-temperature and high-pressure simulated values in this work, and at some of the same temperature points (such as 1,500 K), the deviation is over 50%. This indicates that the increase in temperature and pressure can greatly promote the thermal decomposition of D4.
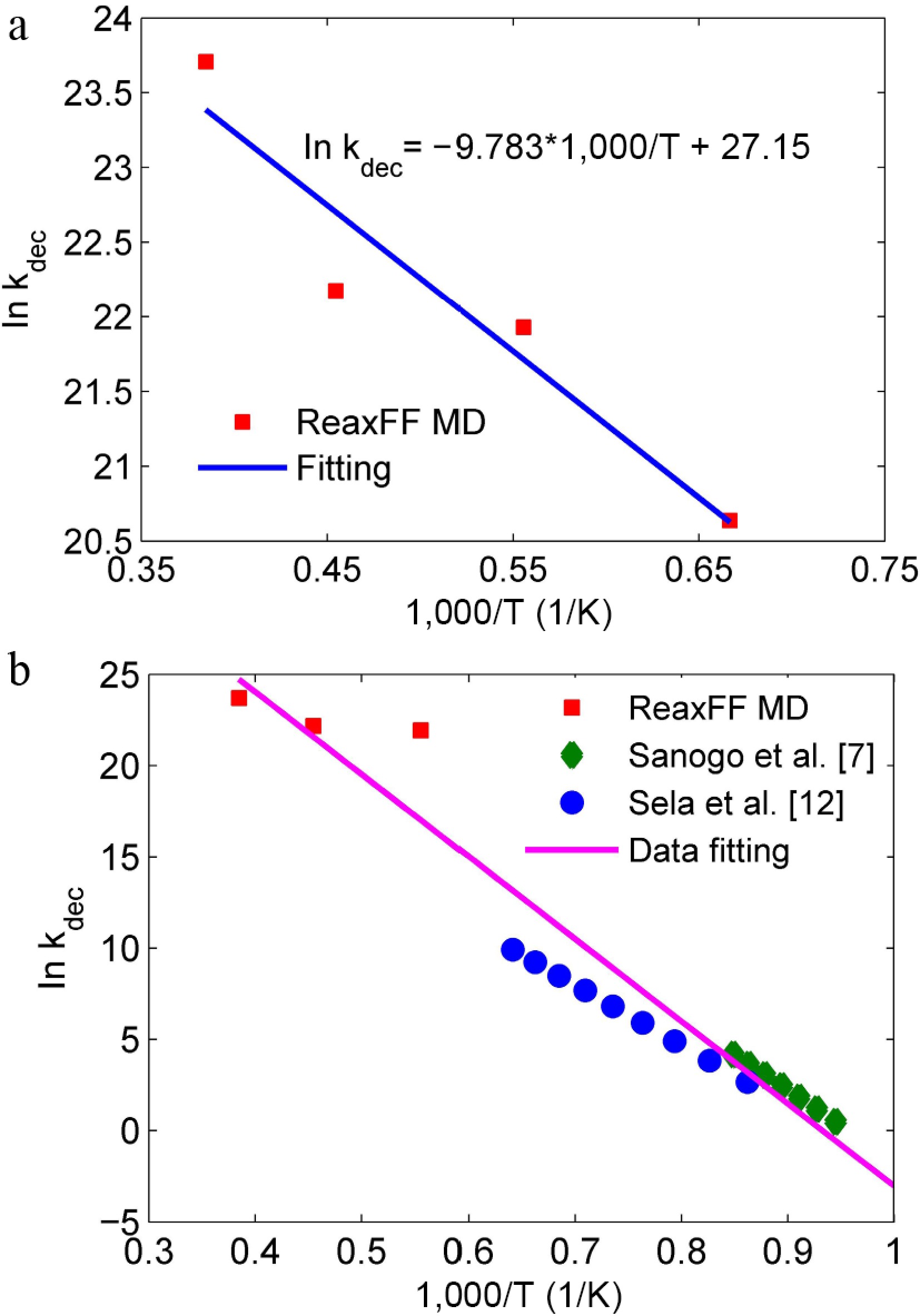
Figure 8.
Reaction rate constants of D4 pyrolysis at the temperatures of (a) 1,500−2,600 K by ReaxFF MD simulations, and (b) 1,058−2,600 K from the references.
By fitting the reaction rate constants obtained by ReaxFF MD simulations at 1,500~2,600 K, the apparent activation energy Ea of D4 decomposition is found to be 81.3 ± 4.2 kJ/mol, which is significantly lower than that of Davidson et al.'s[11] low temperature static reactor experiment (301 kJ/mol), Sanogo & Zachariah's fast flow reactor experiment (about 320 kJ/mol), and Sela et al.'s shock tube experiment (273.2 kJ/mol). The difference is due to the high temperature (1,500−2,600 K) and the high pressure (~80 bar) employed in this study, which is quite different from the experimental conditions (< 1,500 K, < 2.0 bar) in the references[7,12]. This may reveal the contribution of new reaction channels at high temperature and high pressure. Figure 9 shows the main paths of D4 initial thermal decomposition by reaction trajectory analysis of ReaxFF MD simulations. It is found that there are totally five reaction paths for D4 initial decomposition, where the paths ① to ④ had been reported in the past[12]. A new reaction channel ⑤ (i.e., Si-C bond homolysis) is found in this work by ReaxFF MD simulations. Also, at the current conditions of high temperature and high pressure, the reaction paths ③ and ④ that radicals participatein are significantly enhanced due to the existence of a great amount of free radicals (·CH3 and ·H) in the reactive system. The total percentage of these two reaction paths reaches 70%. While the activation energies of these two reaction paths are relatively low, 76 kJ/mol, and 25.6 kJ/mol[12], respectively. Consequently, the fitted activation energy of 81.3 kJ/mol is obtained and essentially characterizes the effective apparent activation energy of the initial thermal decomposition of D4.
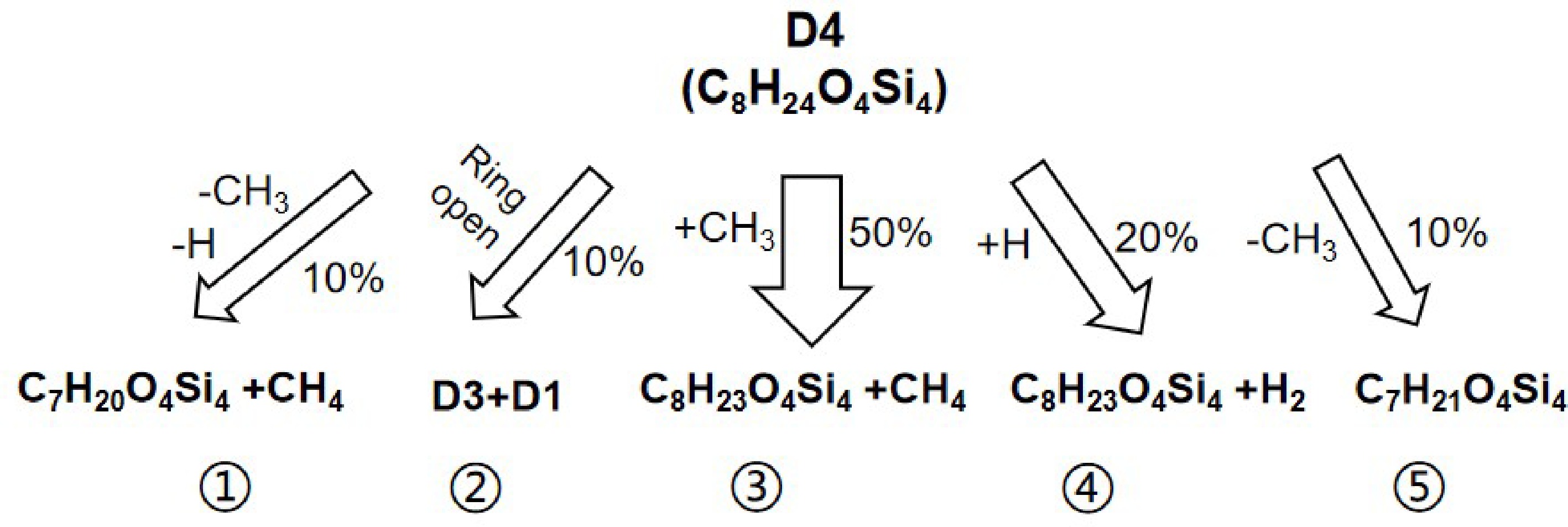
Figure 9.
Initial thermal decomposition paths of D4.
In addition, the effective pre-exponential factor can be obtained by intercept calculation of the fitting line, and gives InA = (27.15 ± 1.35) (unit of A: s−1). A high A value indicates that the pyrolysis of D4 involves complex transition state structure, which is consistent with the multi-step reaction characteristics of D4 thermal decomposition.
Combining the single-molecule simulation results, the microscopic mechanism of temperature effects on reaction pathways is elucidated as follows: (1) Initial bond cleavage selectivity. Above 1,500 K, Si-C bond cleavage (bond energy 377 kJ/mol) remains the primary initiation step (R1), but the rate of ring-opening reactions (Si-O cleavage) significantly increases. High temperatures enhance molecular internal energy transfer, promote the release of strain energy in rigid-ring structures, and accelerate ring-opening reactions, forming linear siloxane intermediates (e.g., -Si4O4C8H24). (2) Enhanced free radical chain reactions. The concentration of methyl radicals (·CH3) sharply rises at high temperatures (R1/R3/R6 in Fig. 5), leading to two acceleration effects: direct hydrogen extraction (·CH3 + R-H → CH4 + R·, where R is silicon or hydrocarbon), explaining the sudden increase in CH4 at 843 K; and secondary cracking catalysis, where free radicals attack intermediates (e.g., open-chain siloxanes), inducing Si-O/Si-C bond homolysis and forming an autocatalytic cycle. (3) Competition for methane generation channels. At temperatures exceeding 2,000 K, Si-C bond homolysis (R1) coexists with intramolecular rearrangements (e.g., methyl migration of R4), generating H and CH2, which contribute to methane formation via CH2 + H2 → CH4, aligning with the C1/C2 product ratio changes in GC results.
Mechanism of CH4 generation
-
The formation of methane (CH4) during the pyrolysis of D4 is a critical process that exhibits strong temperature dependence, as revealed by both experimental and molecular dynamics (MD) simulation results. This section elucidates the mechanistic pathways for CH4 generation, integrating insights from flow tube experiments (803–843 K) and ReaxFF MD simulations (1,500–2,600 K), and highlights the key reaction steps and their kinetic implications.
Based on experimental findings, it was found that CH4 was undetectable at 803 K, while C2-C4 hydrocarbons (e.g., ethane, ethylene) dominated the product spectrum. At 843 K, CH4 production increased dramatically, indicating a high activation energy threshold (> 803 K) for its formation. This suggests that CH4 generation is initiated by temperature-sensitive bond cleavages (e.g., Si–C homolysis) or radical-driven pathways. In addition, from ReaxFF MD simulation results, CH3 and CH4 formation became significant above 1,500 K, with their yields increasing exponentially with temperature (Fig. 10).
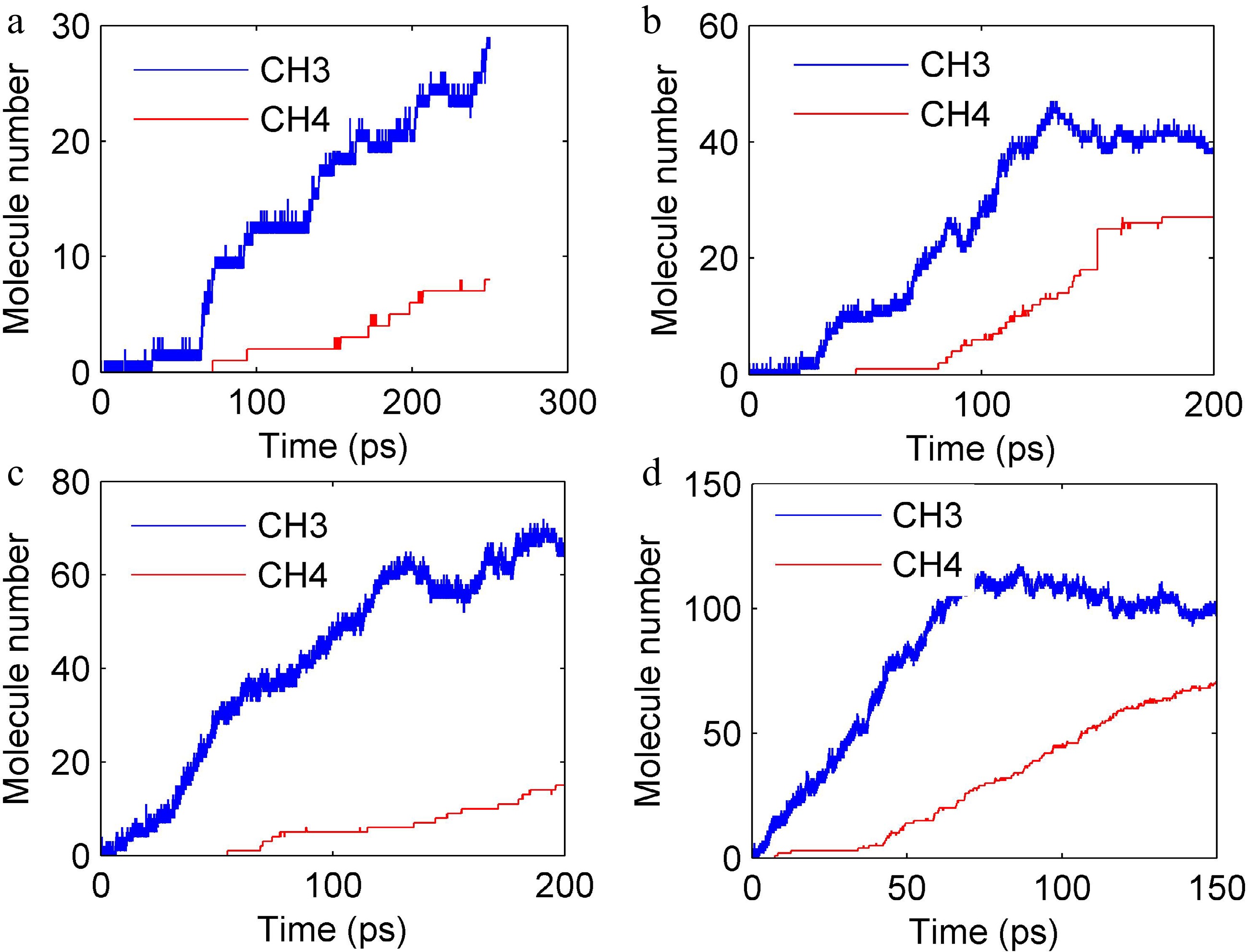
Figure 10.
Evolution of CH4 and CH3 molecules number at different temperatures. (a) 1,500 K, (b) 1,800 K, (c) 2,200 K, and (d) 2,600 K.
Also, by reaction event counting of CH4 formation across the trajectories of MD simulation at 2,000 K, 11 reactions are identified involved in CH4 generation during the D4 pyrolysis process. Of these, ten reactions result in CH4 production via CH3 hydrogen abstraction, comprising about 91% of the total. The frequency of CH4 production events is 19, with 18 instances attributed to CH3 hydrogen abstraction, making up approximately 94.7% (details about these reactions can be found in the Supplementary File 1). Therefore, the simulations identified methyl radicals (·CH3) as the primary precursor for CH4, consistent with the experimental hypothesis of radical-mediated pathways. Based on the combined evidence, the following mechanistic routes are proposed:
(1) Si-C bond homolysis and methyl radical release represent the primary reaction pathway: Si-CH3 → Si· + ·CH3. The relatively weak Si-C bond (377 kJ/mol) preferentially dissociates at elevated temperatures, liberating ·CH3 radicals. This cleavage is rate-limiting at temperatures such as 803 K, but becomes relatively fast above 843 K. ReaxFF MD simulations validate that Si-C bond cleavage (R1 in Fig. 5) is the principal initiation step, with ·CH3 concentrations showing a strong correlation with CH4 yields (Fig. 10).
(2) Hydrogen abstraction by methyl radicals occurs as follows: ·CH3 + R-H → CH4 + R· (where R is a Si- or hydrocarbon fragment). This chain-propagation step has a low activation barrier, accounting for the sharp increase in CH4 observed at 843 K in experiments. The reaction is driven by high ·CH3 concentrations at elevated temperatures. Both experimental and simulation data show CH4 formation coinciding with ·CH3 accumulation (Fig. 10). ReaxFF MD simulations also indicate that ·CH3 abstracts hydrogen from intermediates such as open-chain siloxanes (e.g., I-Si4O4C8H24).
Intramolecular rearrangement and secondary cracking involve methyl migration and dehydrogenation, exemplified by reactions like R4 in Fig. 5, which generates CH2 and H. These intermediates recombine to form CH4 (CH2 + H2 → CH4). Secondary cracking of C2-C4 hydrocarbons (e.g., C2H6 → ·CH3 + ·CH3) provides additional ·CH3 for hydrogen abstraction. Above 2,000 K, these pathways prevail in simulations, consistent with experimental findings of extensive pyrolysis at 843 K.
At lower temperatures (803 K), CH4 is absent due to insufficient energy for Si-C bond homolysis and limited ·CH3 availability, with C2-C4 products forming via Si-O cleavage and methyl recombination. In contrast, at higher temperatures (≥ 843 K), Si-C cleavage accelerates, releasing ·CH3, and radical chain reactions and intramolecular rearrangements enhance CH4 production. The experimental detection of CH4 at 843 K corroborates MD-predicted ·CH3-mediated pathways. Simulations accurately reproduced the Arrhenius parameters (Ea = 81.3 kJ/mol, ln A = 27.15), confirming the predominance of radical-driven mechanisms at elevated temperatures.
In summary, CH4 generation during D4 pyrolysis is governed by two temperature-dependent regimes: (1) Below 803 K, CH4 formation is kinetically inhibited due to high Si-C cleavage barriers. (2) Above 843 K, Si-C homolysis and radical chain reactions prevail, with ·CH3 hydrogen abstraction as the main CH4 source. These findings refine kinetic models by integrating high-temperature pathways and highlight the significance of ·CH3 dynamics in predicting CH4 yields for industrial applications, such as silicon-based material synthesis and waste gas combustion.
-
This study elucidates the high-temperature pyrolysis kinetics of D4 through a combined experimental and ReaxFF MD simulation approach. Flow tube experiments identified a temperature-dependent transition in product distribution, with CH4 formation becoming significant only above 843 K, underscoring the role of Si-C bond cleavage and radical-driven pathways. ReaxFF simulations revealed first-order decomposition kinetics (1,500–2,600 K) with an apparent activation energy of 81.3 kJ/mol, markedly lower than low-temperature literature values, due to the enhancement of the D4 decomposition channel that radicals participate at high temperatures. The simulations further validated ·CH3 as the key intermediate for CH4 production, aligning with experimental observations. These findings resolve longstanding discrepancies in D4 pyrolysis mechanisms and provide a robust kinetic framework for industrial applications at high temperatures, such as silicon nanomaterial synthesis and landfill gas combustion modeling. Future work should extend experimental validation to higher temperatures and explore the role of pressure effects on pathway selectivity.
The authors express their gratitude to the support from Soochow Municipal laboratory for low carbon technologies and industries.
-
The authors confirm contribution to the paper as follows: study conception and design, draft manuscript preparation: Wang N, Huang Y; data collection: Wang N, Sun J, Huang Y; analysis and interpretation of results: Wang N, Sun J, Huang Y, Shen G. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and analyzed during the current study are available from the corresponding author on reasonable request.
-
The authors declare that they have no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/prkm-0026-0008.
- Supplementary File 1 Supporting materials to this study.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang N, Sun J, Huang Y, Shen G. 2026. Exploring pyrolysis kinetics of octamethylcyclotetrasiloxane by experiments and ReaxFF molecular dynamics simulations. Progress in Reaction Kinetics and Mechanism 51: e016 doi: 10.48130/prkm-0026-0008 

Exploring pyrolysis kinetics of octamethylcyclotetrasiloxane by experiments and ReaxFF molecular dynamics simulations
- Received: 22 July 2025
- Revised: 24 January 2026
- Accepted: 04 March 2026
- Published online: 08 June 2026
Abstract: The pyrolysis kinetics of octamethylcyclotetrasiloxane (D4) at high temperatures, crucial for silicon-based material synthesis and waste-to-energy applications are not fully understood. This work carries out this research by combining flow tube experiments (803–843 K), with ReaxFF molecular dynamics (MD) simulations (1,500–2,600 K). Experimentally, a significant increase in methane (CH4) production at 843 K—compared to its absence at 803 K—suggests a high activation energy barrier for CH4 formation, which is overcome via Si-C bond cleavage, and methyl radical (·CH3) chain reactions. ReaxFF MD simulations indicate that D4 decomposition follows first-order kinetics at high temperatures, with an apparent activation energy of 81.3 ± 4.2 kJ/mol, much lower than the 273.2–320 kJ/mol reported at lower temperatures. This difference arises from the enhancement of the D4 decomposition channel in which radicals participate in at high temperatures. Both experiments and simulations confirm that hydrogen abstraction by ·CH3 radicals (·CH3 + R-H → CH4 + R·) is the primary route for CH4 production. The developed multi-scale kinetic model offers crucial insights for optimizing silicon-based material synthesis and simulating silicon-containing gas combustion.
-
Key words:
- Octamethylcyclotetrasiloxane /
- Pyrolysis /
- Reaction kinetics /
- ReaxFF /
- Molecular dynamics simulation