






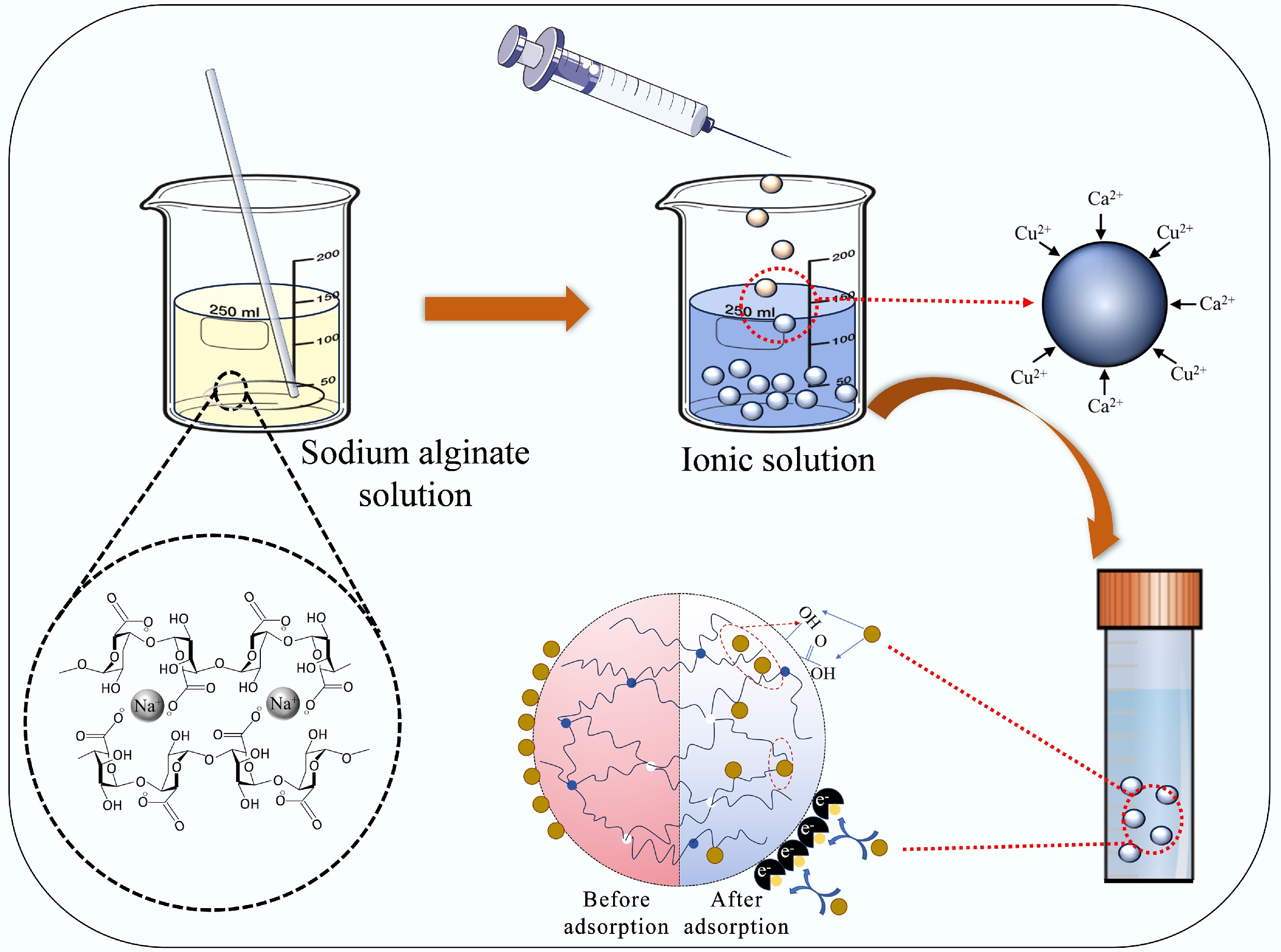



-
Amid accelerating urban-industrial megatrends, global energy demand is surging. As nuclear energy is low-cost, highly efficient, and clean, it is increasingly utilized worldwide[1]. Uranium is a primary fuel for nuclear power. Nevertheless, uranium is a highly-radioactive element that can apparently affect the natural environment and human health if not properly managed[2]. Hence, the sequestration of uranium from uranium-bearing effluents is imperative. Many techniques, such as biological treatment, precipitation, chemical reduction, ion-exchange, and adsorption, have been engineered for the selective capture and subsequent recovery of U(VI)[3−6]. Among them, adsorption has rapidly risen to prominence because of its exceptional uptake efficacy, low energy consumption, and simple operation. Adsorbents constitute the pivotal determinant of the adsorption process. Hence, engineering a high-performance adsorbent remains a formidable bottleneck[7]. A variety of sorbent materials—including clay[8], mesoporous silicon[9], carbon nanotubes[10], graphene oxide[11], ion-exchange resins[12], and polysaccharides[13]—has been engineered for uranium sequestration. Among these adsorbents, polysaccharides have been extensively applied for U(VI) sequestration due to their natural abundance, environmental compatibility, and oxygen-rich functional groups that provide intrinsic affinity for U(VI). Sodium alginate (SA), an inexpensive, biodegradable, nontoxic linear polysaccharide rich in hydroxyl and carboxylic groups[14], possesses a chain architecture built from mannuronic acid (M) and guluronic acid (G) blocks, and is extracted from brown seaweeds[15]. The carboxylic groups of this polysaccharide can attract multivalent metal cations via an ion-exchange mechanism. Carboxylic groups can be used for the ionotropic gelation of sodium alginate for preparing hydrogel beads[16]. So far, sodium alginate has been used as a selective adsorbent for U(VI)[17]. In particular, research on sodium alginate gel beads has received extensive attention. For example, Zhang et al.[18] entrapped Ti3C2Tx within sodium alginate, yielding Ti3C2Tx/SA gel beads through ionic cross-linking. They identified the Ti3C2Tx/SA-30% formulation as optimal, delivering a saturation uptake of 92.17 mg/g. Li et al.[19] prepared an SA-GO composite hydrogel for uranium sequestration by encapsulating graphene oxide into sodium alginate, reporting a saturation uptake of 149.76 mg/g. Yi et al.[15] crafted sodium alginate (SA)/polyvinyl alcohol (PVA)/graphene oxide (GO) hydrogel microspheres via a facile route, and reported that SPG hydrogel microspheres achieved a maximum adsorption capacity of 403.78 mg/g for UO22+.
Divalent metal ions, such as Sr2+, Ba2+, Ca2+, Sr2+, and Ni2+, can interact with the functional groups of alginates, acting as a linchpin for regulating the characteristics of hydrogels. The Ca2+ ions are most commonly employed to produce alginate gels, but the tenuous skeletal connectivity and feeble mechanical resilience of calcium alginate gels limit their utility for uranium-containing wastewater[20]. Alginate's divalent-cation avidity is reported to diminish along the sequence: Cu > Cd > Ba > Sr > Ca > Co > Ni[21], and thus the copper alginate gel molecular chains exhibit superior structural stability compared with calcium alginate[22]. Copper alginate gels have attracted attention across diverse fields by nature of their expansive interfacial area and reactive loci for sequestering a broad spectrum of contaminants, however, their application as U(VI) adsorbents is constrained by the challenge of achieving both high structural stability and superior adsorption performance simultaneously[23].
In this work, sodium-alginate-based composite gel microspheres (SA-x: SA-Ca, SA-Cu, and SA-CC) were prepared by a drop-injection method. The SA-CC formulation demonstrated particularly exceptional U(VI) sequestration performance, achieving outstanding adsorption efficacy that substantially exceeds that of other reported sodium alginate-based uranium adsorbents, while maintaining robust reusability. The synthesized SA-x gels were characterized. Meanwhile, the properties of adsorbents and the influence exerted by adsorbent dose, solution pH, and coexisting ions on the sequestration process were investigated. Moreover, the adsorption mechanism of U(VI) on the adsorbents was unraveled using SEM, FTIR, and XPS. These findings provide a mechanistic foundation for scaling up SA-based microspheres in industrial wastewater treatment, particularly highlighting Cu2+ cross-linking as a strategy to enhance structural stability.
-
Uranium nitrate (UO2[NO3]2·6H2O) and chlorphosphonazo-III (C22H16Cl2N4O14P2S2) were purchased from Shanghai Aladdin Bio-Chem Technology Co., Ltd, Shanghai, China. Sodium alginate (C6H7NaO6), sodium chloride (NaCl), copper chloride (CuCl2), hydrochloric acid (HCl), sodium hydroxide (NaOH), and calcium chloride (CaCl2) were bought from Chengdu Chron Chemical Reagent Co., Ltd, Chengdu, China.
Synthesis of samples
-
SA-based composite gel microspheres were synthesized via a simple method building on antecedent reports (Fig. 1). In brief, 3.0 g of sodium alginate was homogeneously dispersed in 97.0 mL of deionized water and magnetically stirred for 20.0 min to form a uniform solution. Next, the solution was added to a calcium chloride solution (0.04 mol/L) using a 10.0 mL syringe equipped with a 0.5 mm inner-diameter needle from a dropping height of 0.3 m to produce SA-Ca beads. The concentration of the cross-linking solution was selected based on established protocols to ensure adequate gelation. This process yielded spherical SA-Ca beads with an average diameter of 2.0 mm (Supplementary Fig. S1). Finally, the SA-Ca beads were washed with distilled water five times, and then stored in a bottle with DI water. Through the above method, the corresponding SA-Cu and SA-CC gel microspheres were obtained by changing the types of metal ions in the gel microspheres to a copper chloride solution (0.04 mol/L) and a calcium chloride/copper chloride solution (0.02/0.02 mol/L), respectively[12].
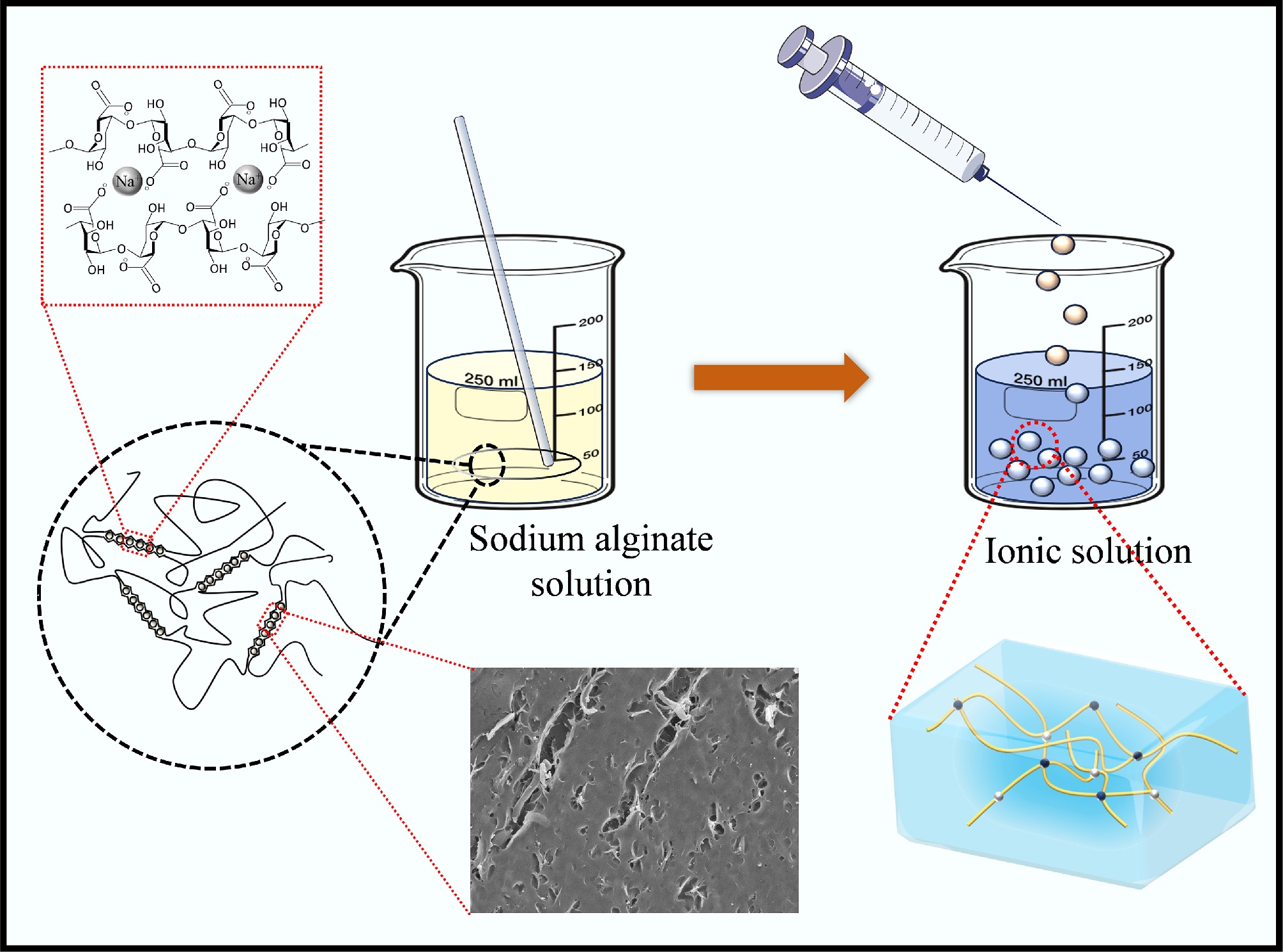
Figure 1.
The production process of sodium alginate composite gel microspheres.
Characterization
-
The functional groups on their surface, as well as the morphology and structure, thermal stability, and surface chemical composition of the adsorbents were analyzed and characterized using a scanning electron microscope (SEM), Fourier transform infrared spectroscopy (FTIR), X-ray diffractometer (XRD), and X-ray photoelectron spectroscopy (XPS).
Batch adsorption experiments
-
An array of batch sorption experiments were executed to evaluate the effects of adsorbent dose, reaction time, solution pH, and coexisting ions on U(VI) removal by SA-Ca, SA-Cu, and SA-CC. These experiments were conducted in 20.0 mL borosilicate vials. In brief, 1 mg of adsorbent was added to 10.0 mL of uranium solution (10–100 mg/L) and stirred at a constant temperature for a predetermined period. The solution was filtered, and the U(VI) concentration was determined using chlorphosphonazo-III. The removal efficiency (E, %) and adsorption capacity (Qe, mg/g) were calculated using Eqs (1) and (2), respectively.
$ E\; (\text{%})=\dfrac{C_0-C\mathrm{_e}}{C_0}\times100\text{%} $ (1) $ Q\mathrm{_e}=\dfrac{V\left(C_0-C_{\mathrm{e}}\right)}{m} $ (2) where, C0 (mg/L) and Ce (mg/L) are the initial concentrations, and the concentration measured at the equilibrium time, respectively. m (mg) is the weight of adsorbent, and V (L) is the aqueous solution volume.
Desorption and recyclability
-
To explore the reusability of SA-CC, adsorption-desorption cycles were carried out. First, 1 mg of adsorbent was placed in 10.0 mL of 0.10 mol/L HCl and allowed to react for 12.0 h at room temperature. After sedimentation, the supernatant was collected to determine the concentration of U(VI). The adsorbent was washed with DI water five times, and a newly prepared U(VI) solution was used for the next adsorption experiment.
-
FTIR was employed to analyze the functional groups and chemical bonds of the SA-x. In Fig. 2a, the intense peaks near 3,435 and 1,650 cm−1 are attributed to O–H stretching modes. The wide band centered near 2,911 cm−1 corresponds to the C–H stretching modes, the peak at 1,081 cm−1 is ascribed to the C=O stretching vibration, and the peak near 1,380 cm−1 is attributed to the symmetric stretching vibration of the carboxylate group (–COO−). Differences in the band intensities among different gel spheres indicated that the affinity of different ions for carboxyl groups on the single chain of alginate varied; SA-CC and SA-Cu exhibit stronger bands than SA-Ca, suggesting superior U(VI) adsorption properties[24]. To further reveal the microstructures of SA-x, XRD patterns were obtained for SA-Cu, SA-Ca and SA-CC. As displayed in Fig. 2b, the XRD pattern of SA-x presents a weak and broad diffraction peak at 20.19°, indicating a rather amorphous structure[25]. Comparison of the patterns revealed that the three adsorbents possess similar crystal structures, implying that the reaction with calcium and copper ions did not alter the alginate structure. XPS was further deployed to elucidate the chemical compositions of SA-x. Notably, the survey spectrum of SA-CC (Fig. 2c) exhibits four characteristic peaks at binding energies of 932.1, 532.8, 343.5, and 285.6 eV, which are assigned to Cu2p, O1s, Ca2p, and C1s, respectively[26], confirming the successful incorporation of Ca and Cu ions into SA-CC.
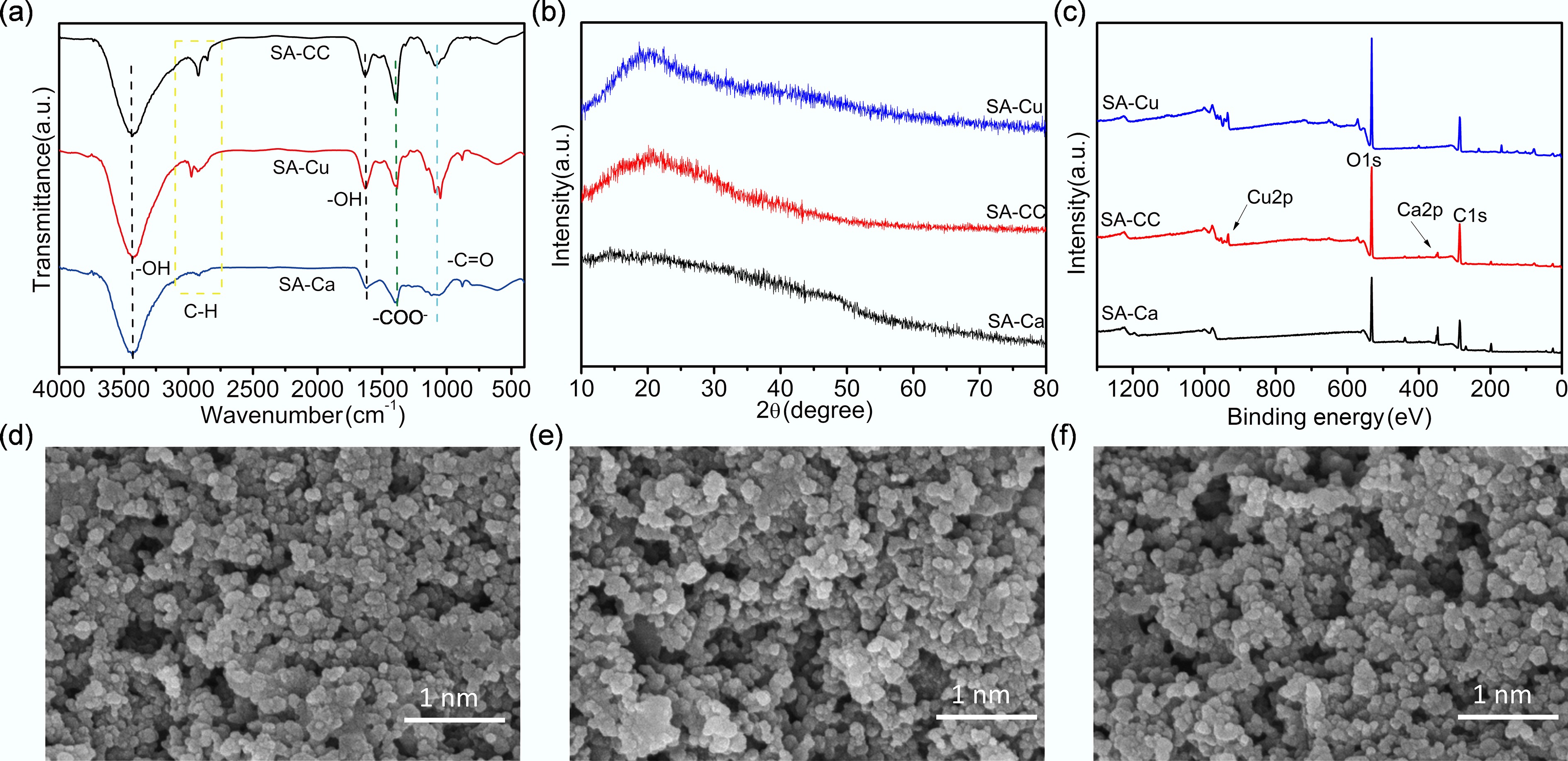
Figure 2.
(a) FTIR spectra, (b) XRD patterns, and (c) XPS spectra of SA-Ca, SA-CC, and SA-Cu, SEM images of (d) SA-Ca, (e) SA-Cu, and (f) SA-CC.
SEM was employed to characterize the morphology of SA-x. In Fig. 2d–f, the SA-x composite microspheres were spherical with smooth surfaces, whose dimensional profile serves as a critical determinant for microsphere deployment viability[27]. A diminished microsphere diameter amplified specific surface area, which in turn intensifies U(VI) adsorption capacity[28]. The SA-x microspheres exhibit uniform size and good dispersion, indicating that full contact between adsorbate and adsorbent is ensured by the large surface area[29]. As displayed in Fig. 2f, partial aggregation of SA-Cu occurred because the high concentration of copper ions exhibited a stronger affinity for sodium alginate.
Adsorption
Adsorption effect of pH and adsorbent dose
-
Solution pH governs the distribution of uranyl species and the surface-charge state of the sorbent, rendering it a critical parameter. With trace additions of HCl or NaOH, the pH was swiftly adjusted within the range of 2.0–6.0. As depicted in Fig. 3a, the amount of U(VI) adsorbed by SA-Cu, SA-Ca, and SA-CC increased sharply from pH 2.0 to 4.5. At pH 4.5, SA-CC achieved its peak U(VI) removal efficiency of 93.4%. Subsequently, a declining trajectory was observed from pH 4.5 to 6.0. The experiments were limited to pH ≤ 6.0 because, at higher pH values, UO2+ ions undergo significant hydrolysis and precipitation as polynuclear species or insoluble hydroxides, which would obscure the evaluation of true adsorption capacity by introducing a competing precipitation mechanism. At low pH values, U(VI) existed predominantly as UO22+, which competed with H+ for binding sites on SA-CC, resulting in reduced U(VI) uptake.
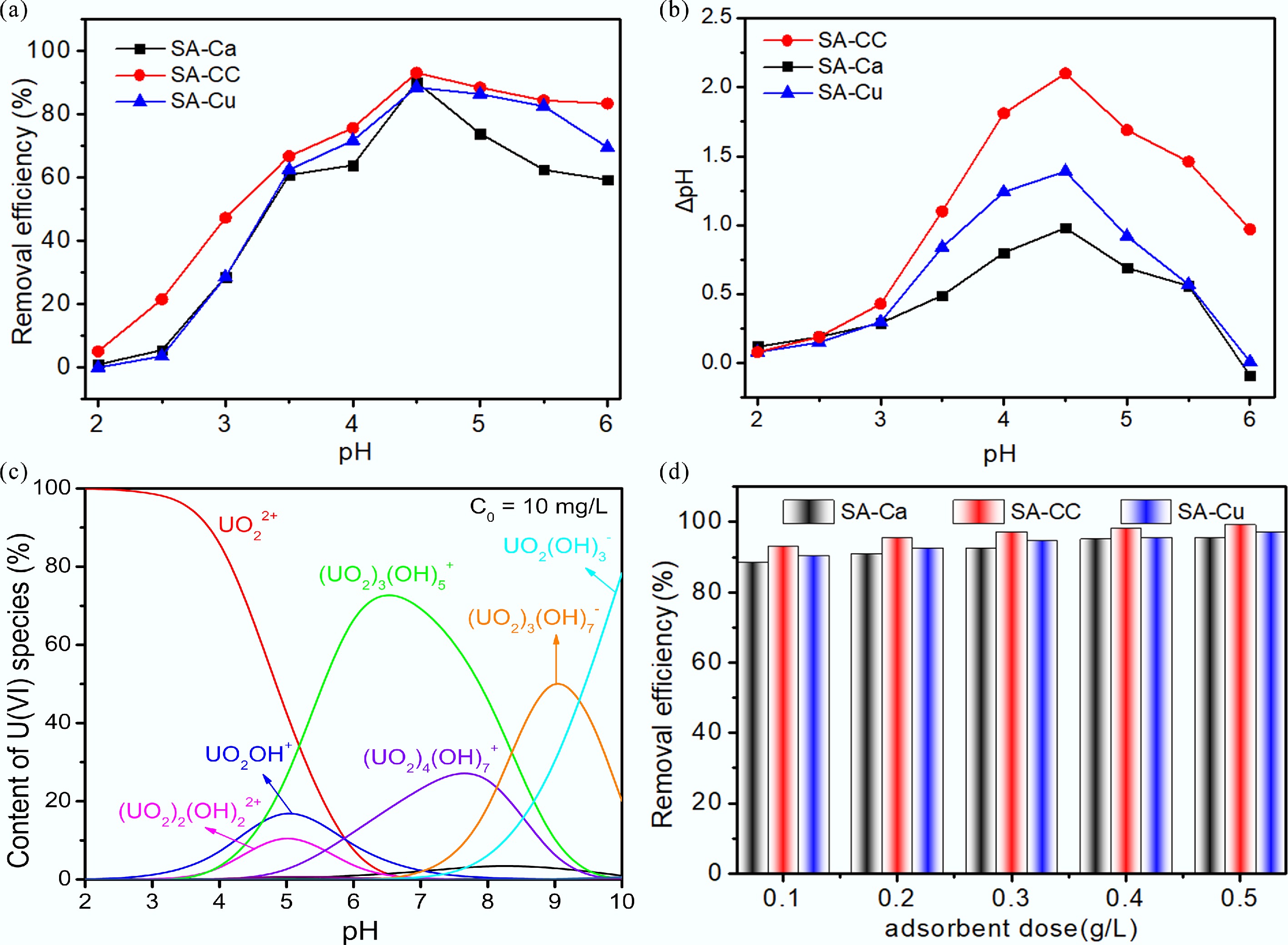
Figure 3.
(a) Effect of pH on U(VI) removal; (b) Zero charge of samples at various pH values (ΔpH = pHfinal – pHinitial); (c) Speciation of U(VI) at pH (2–10); (d) Effect of adsorbent dose on U(VI) removal.
When the solution pH reached 4.5, the H+ concentration decreased gradually, allowing a fraction of U(VI) to coordinate with the oxo-rich domains of SA-CC, yielding surface complexes. Figure 3b displays the surface potential of SA-Ca, SA-Cu, and SA-CC at various pH values, suggesting that SA-Ca, SA-Cu, and SA-CC were negatively charged when pH < pHpzc. Using Visual MINTEQ 3.1, the U(VI) speciation across the tested pH range was simulated (Fig. 3c). The U(VI) species experienced a charge transition from positive to negative at pH > 4.5; the abundance of cationic moieties (i.e., [UO2]3[OH]5+) diminished in solution, while the proportion of anionic species (i.e., [UO2]3[OH]7-) increased accordingly. In addition, the electrostatic repulsion between U(VI) and SA-CC intensified as pH increased, resulting in diminished U(VI) adsorption onto SA-CC.
Adsorbent dosage was also a crucial factor affecting adsorption efficiency, as it determined the equilibrium between adsorbents and U(VI) in the system. The effect of adsorbent dosage was investigated[30]. In this experiment, 1, 2, 3, 4, and 5 mg of SA-Cu, SA-Ca, and SA-CC were individually employed. Contact parameters were fixed at pH 4.5, with 10.0 mg/L U(VI) for 10.0 h. Figure 3d shows that the removal efficiencies of SA-Cu, SA-Ca, and SA-CC rapidly increase to 97.1%, 95.6%, and 99.2%, respectively. The uptake efficiency of SA-Cu, SA-Ca, and SA-CC for U(VI) also increased as the adsorbent dosage increased when the doses were less than 3 mg. The progressive increase in uptake efficiency upon elevating the adsorbent dosage is ascribed to greater abundance of accessible binding active sites[31]. Nevertheless, when the doses of SA-Cu, SA-Ca, and SA-CC exceeded 3.0 mg, almost all the U(VI) was immobilized by the active sites on the adsorbents, generating a significant solid accumulation effect[19], which led to consistently high U(VI) removal efficiencies for SA-Cu, SA-Ca, and SA-CC. The influence of initial U(VI) concentration on adsorption was also investigated under the optimal pH condition (Supplementary Fig. S2). The SA-CC adsorbent maintained a relatively high removal efficiency ( > 90%) even at an initial concentration of 100 mg/L, demonstrating its superior adsorption capacity and site availability compared to SA-Ca and SA-Cu.
Adsorption kinetics
-
Figure 4a displays the time-resolved U(VI) uptake profiles of SA-Cu, SA-Ca, and SA-CC. An initial surge in sorption efficiency occurred within the first 4.0 h, followed by a gradual approach to equilibrium beyond 11.0 h. To ensure that the adsorption equilibrium of U(VI) was achieved with SA-Cu, SA-Ca, and SA-CC, a contact time of 15.0 h was selected for subsequent experiments. At the beginning of the reaction, the adsorbent surface exhibited a high abundance of active adsorption sites, resulting in high adsorption efficiency. As contact time progressed, the population of active adsorption sites on the SA-CC surface gradually diminished; a concomitant decline in adsorption efficiency ensued.
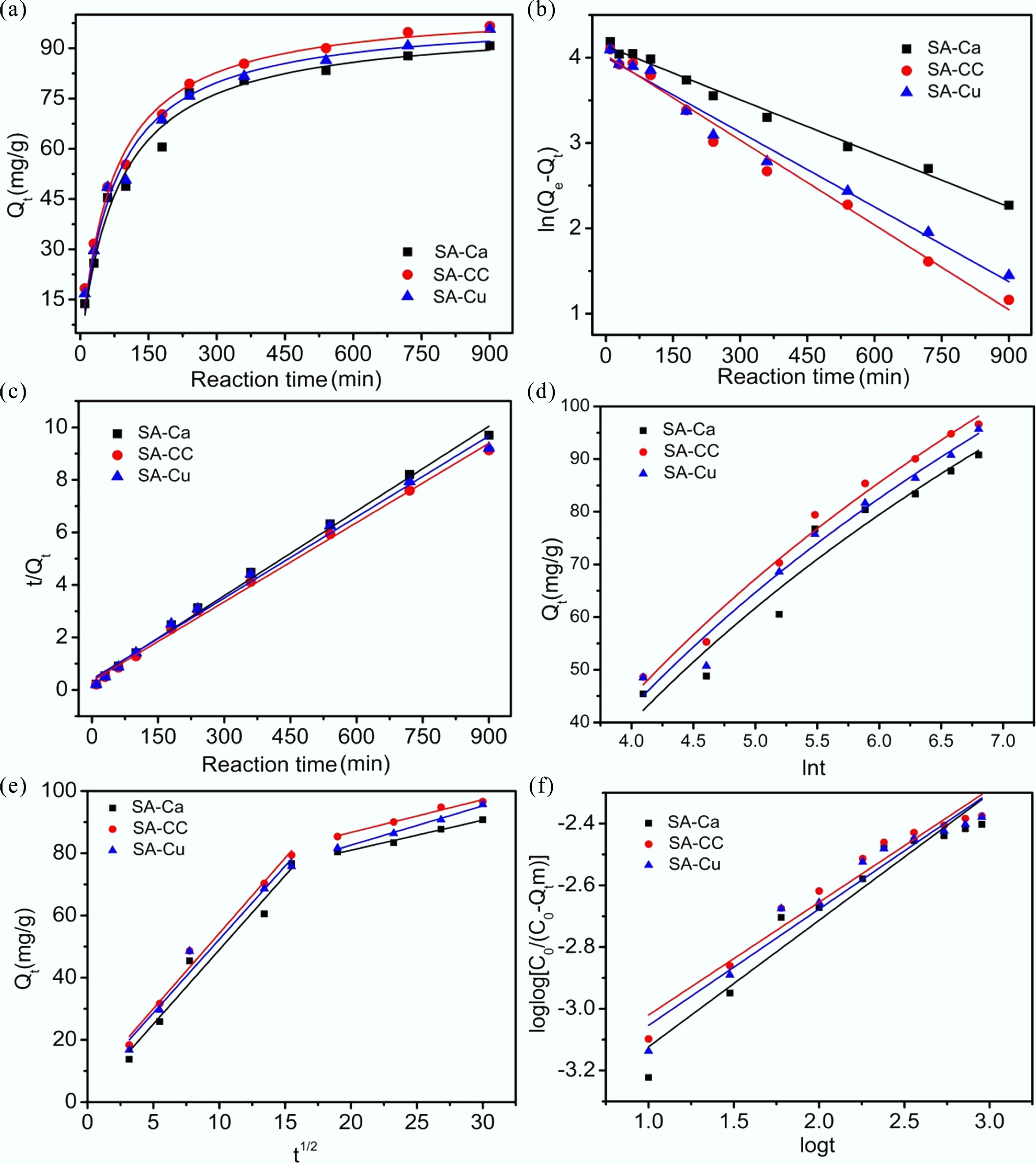
Figure 4.
(a) Effect of contact time on U(VI) removal; (b) Pseudo-first-order kinetic models; (c) Pseudo-second-order kinetic model; (d) Elovich kinetic model; (e) Intraparticle diffusion kinetic model; (f) Bangham kinetic model of U(VI) removal on SA-based materials.
In this work, to gain deeper mechanistic insight into the adsorption behavior of uranium on SA-Cu, SA-Ca, and SA-CC, different kinetic models (pseudo-second-order, pseudo-first-order, intraparticle diffusion kinetic, Elovich, and Bangham model) were employed to fit the adsorption experimental data (see Supplementary Information). The kinetic fitting curves are displayed in Fig. 4b–f, while the corresponding parameters are tabulated in Supplementary Tables S1 and S2, and Supplementary Figs S3 and S4. Surpassing those of all rival kinetic models, the correlation coefficients of the pseudo-second-order model (R2 = 0.98) were the highest, indicating that U(VI) adsorption on the gel microspheres followed pseudo-second-order kinetics. These findings demonstrate that the adsorption process of U(VI) on SA-Cu, SA-Ca, and SA-CC was primarily a chemical adsorption process.
Adsorption isotherms
-
Adsorption isotherms provide insights into the behavior of U(VI) at the phase interface of adsorbents. Therefore, Freundlich, Temkin, Langmuir, Dubinin-Radushkevich, and Redlich-Peterson isotherm models were employed to fit the experimental data and to describe the process of U(VI) adsorption onto SA-Cu, SA-Ca, and SA-CC. Figure 5 displays the Langmuir and Freundlich isotherms in both their linear and nonlinear forms. The Langmuir model exhibited the highest correlation coefficient (0.99), outperforming those of the Freundlich (0.98) and all other models. Figure 5a reveals that, after an initial rapid ascent, the adsorption capacity rose more gradually before finally plateauing. These results suggested that U(VI) adsorption by the gel microspheres proceeded via monolayer coverage under the investigated concentrations, accompanied by negligible lateral interactions among the adsorbed species. It is evident from Fig. 5 that SA-Cu, SA-Ca, and SA-CC exhibit pronounced adsorption capacity toward U(VI). The absence of a distinct Henry's law region within the studied concentration range indicates that non-specific physisorption contributes negligibly to the overall U(VI) uptake, confirming the dominance of specific chemisorption mechanisms. Among them, SA-CC achieved the saturation adsorption ceiling of 394.85 mg/g, surpassing all previously documented gel-based microsphere sorbents (Table 1). The lower intensity of the –OH peak for SA-Ca compared to SA-CC implies that calcium ions were not sufficiently cross-linked with SA, resulting in fewer oxygen-containing functional groups for U(VI) adsorption. As the high copper-ion concentration exhibited a stronger affinity for sodium alginate and caused particle agglomeration in SA-Cu, the adsorption capacity of SA-Cu toward U(VI) was diminished.
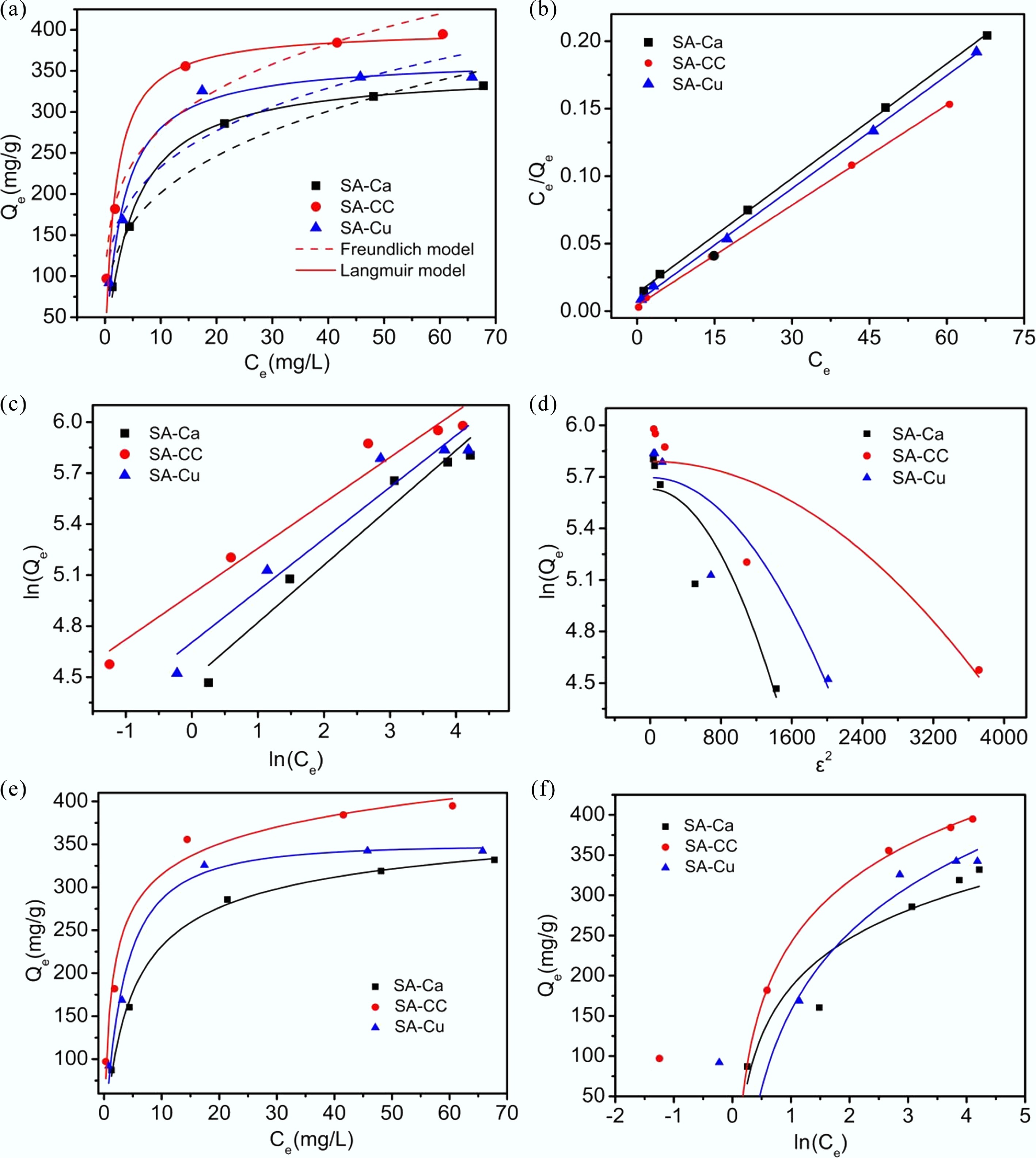
Figure 5.
Adsorption isotherms of U(VI) on SA-based materials at 298.15 K. (a) Nonlinear Freundlich and Langmuir isotherms; (b) Linear Langmuir isotherm; (c) Linear Freundlich isotherm; (d) D-R isotherm; (e) R-P isotherm; (f) Temkin isotherm.
Table 1. Comparison of the maximum adsorption capacity of uranium (VI) on various adsorbents
Effect of coexisting ions
-
The adsorption capacity of U(VI) on adsorbents in a complex environment can be influenced by coexisting ions. All adsorption experiments were conducted at a controlled temperature of 298.15 K. The impacts of various coexisting anions and cations on U(VI) adsorption by SA-Cu, SA-Ca, and SA-CC are illustrated in Fig. 6a, b; each coexisting ion was present at 10 mg/L. Figure 6a shows that the order of the influence on sorption efficiency among coexisting cations is Ca2+ < Mg2+ < Zn2+ < Na+ < K+, indicating that Na+ and K+ have little effect on the U(VI) uptake, with adsorption capacities remaining above 90%, whereas Ca2+ exerts the strongest effect. This is because divalent cations are more readily captured by negatively charged adsorbents than monovalent cations[29]. The influence of competing ions on U(VI) sequestration by SA-Cu, SA-Ca, and SA-CC was investigated using NaCl and CaCl2 at different concentrations (1, 10, and 100 mg/L). As evident from Fig. 6c, the U(VI) adsorption efficiency decreased slightly as the Na+ concentration increased, which is due to intensified competition between Na+ and UO22+ for surface coordination sites as ionic strength increases. As Ca2+ concentration increased, U(VI) adsorption efficiency decreased markedly (Fig. 6d); at a Ca2+ concentration of 100 mg/L, only 50.1% U(VI) removal was achieved with SA-CC. Ca2+ had a more pronounced inhibitory effect on the U(VI) sequestration than Na+, further confirming that the adsorbents preferentially bind high-valence ions[32].
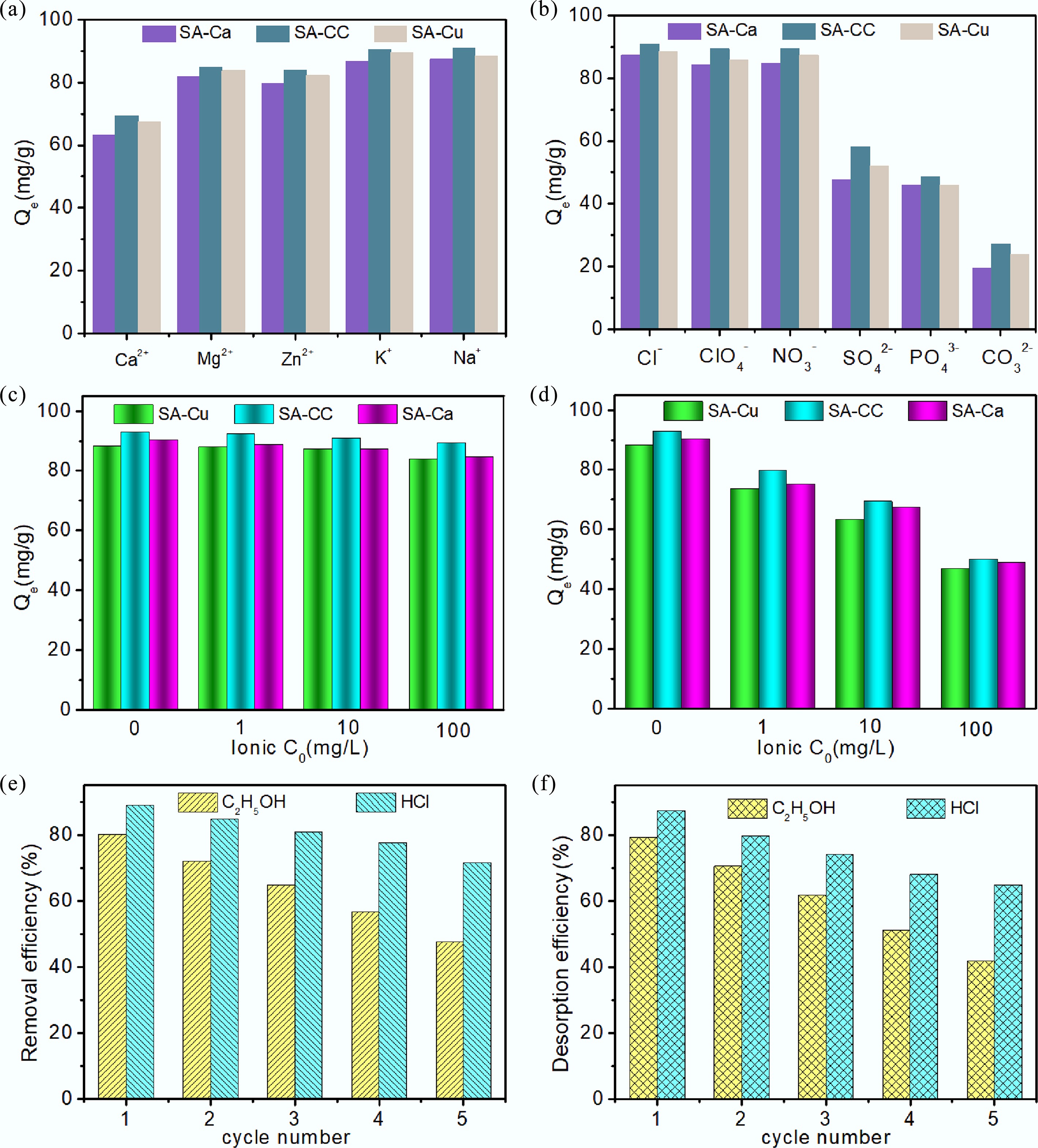
Figure 6.
Influence of (a) cations, and (b) anions on the removal of uranium (VI), influence of ionic strength of (c) NaCl, and (d) CaCl2 on the removal of uranium (VI), (e) removal efficiency, and (f) desorption efficiency of U(VI) onto SA-CC in five cycles with CH3OH and HCl.
The order of influence of coexisting anions on U(VI) adsorption efficiency is depicted in Fig. 6b. The impact decreases in the sequence: CO32– < PO43– < SO42– < NO3– < ClO4– < Cl–, indicating that Cl–, NO3– and ClO4– ions exert relatively weak effects on U(VI) removal, whereas PO43– and CO32– react with UO22+ to form stable complexes such as UO2CO3, UO2PO4–, UO2(CO3)34– and UO2(CO3)22–. Occupancy of the active adsorption sites by these complexes leads to diminished adsorption performance. Additionally, under the same background ion conditions, SA-CC maintains the highest adsorption efficiency among the three adsorbents.
Desorption and reusability
-
Adsorption-desorption cycles were conducted to evaluate the reusability of SA-CC. In this experiment, 0.1 mol/L HCl and C2H5OH (adjusted to pH = 1) were used as elution solvents. Figure 6e, f illustrates how the cycle number influences both removal and desorption efficiencies across five sequential adsorption-desorption cycles[33]. It can be seen from Fig. 6e that HCl exhibited the best desorption effect for uranium. No obvious decline in adsorption efficiency towards U(VI) was observed during the first three cycles, indicating that the proposed absorbents show a good reusability. A decline in uptake performance was noted after three cycles, attributable to incomplete removal of residual U(VI) from SA-CC. Nevertheless, the adsorbents retained 70% of their initial adsorption capacity after five cycles[3]. The results confirm that SA-CC is a high-performance, economically viable sorbent for U(VI) removal in practical applications. The enhanced adsorption capacity and reusability of SA-CC can also be attributed to its improved structural stability. Macroscopic observation (Supplementary Fig. S5) revealed that SA-CC and SA-Cu microspheres maintained integrity with minimal swelling during the shaking process, whereas SA-Ca microspheres underwent significant swelling and were susceptible to fragmentation. This observation confirms that Cu2+ cross-linking effectively enhances the mechanical strength of the alginate matrix, which is crucial for long-term practical application.
Mechanism
-
SEM imaging was employed to visualize the SA-CC morphology both prior to, and following U(VI) adsorption. Figure 7a shows that the initial adsorbent, with a rough and uneven surface, and a porous structure, provided SA-CC with an expansive specific surface area, thereby enhancing its adsorption performance. Following U(VI) uptake, obvious differences in surface morphology were observed; the adsorbent became smooth and compact. Compared with Fig. 7a, b reveals surface-adsorbed attachments on the absorbent after U(VI) adsorption, confirming the surface-immobilized uranium species on SA-CC hydrogel microspheres.
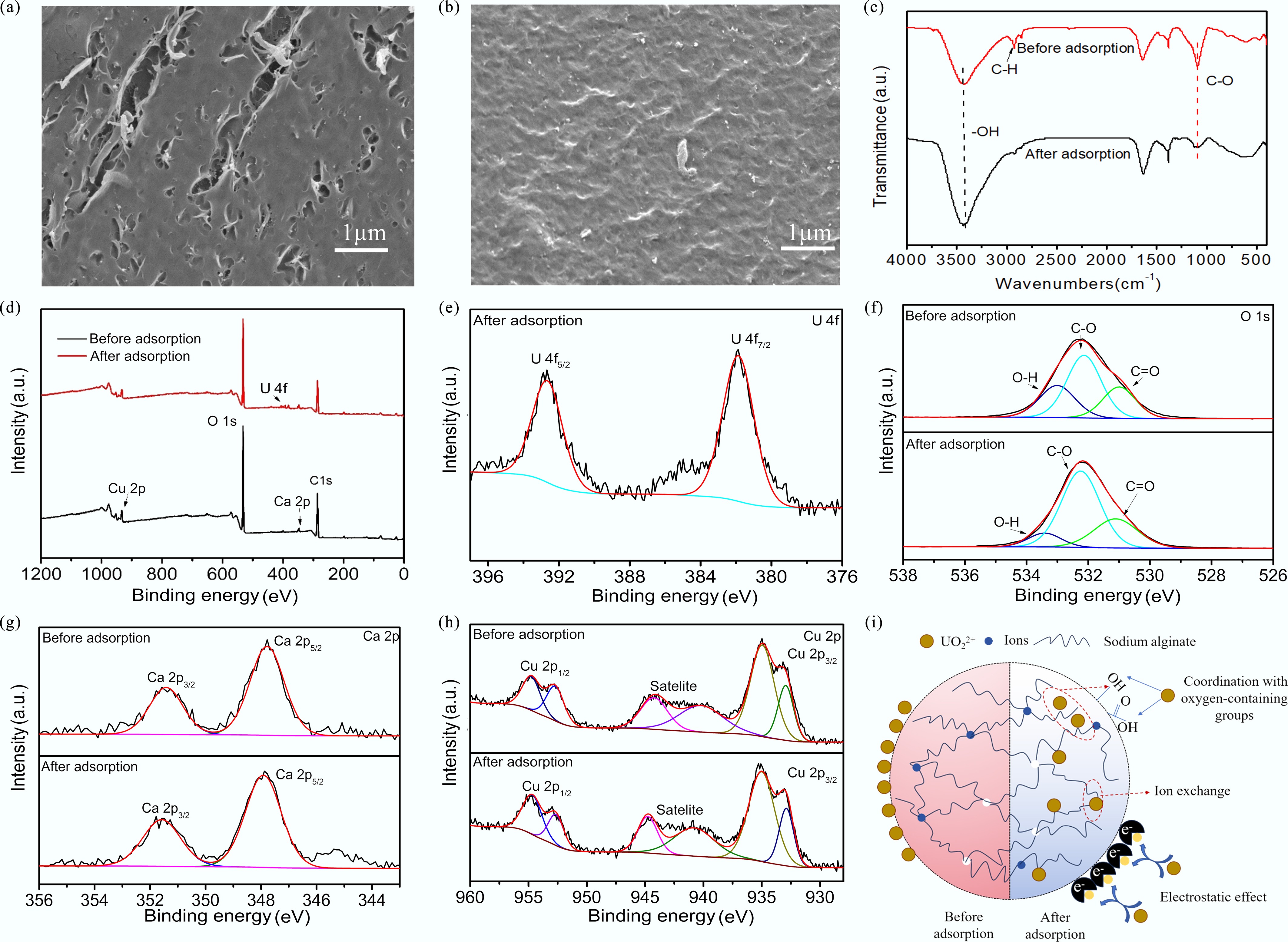
Figure 7.
(a) SEM images of SA-CC before adsorption; (b) SEM images of SA-CC after adsorption; (c) FTIR spectra of SA-CC before and after adsorption; (d) XPS spectra of SA-CC before and after adsorption; High-resolution XPS spectra of (e) U 4f, (f) O 1s, (g) Cu 2p, and (h) Ca 2p of SA-CC before and after U(VI) adsorption; (i) Possible mechanism for the interaction between SA-CC and U(VI).
Depicted in Fig. 7c, the intensities of C–H and C–O bonds are weakened after U(VI) adsorption on SA-CC[34]. The post-adsorption FT-IR spectra of SA-CC show only minor shifts; the peaks at 3,435 and 1,650 cm−1 are slightly shifted[35]. This indicates that O–H groups associated with these bands interacted with U(VI), reducing the vibrational energy requirement, and causing the corresponding absorption bands[36].
The elemental composition of SA-CC was determined using XPS. As shown in Fig. 7d, the full spectrum indicates the presence of Ca2p, O1s, C1s, and Cu2p peaks before adsorption, while a U4f peak appears and the intensities of the other peaks decrease after adsorption. After adsorption, the peaks of U4f7/2 and U4f5/2 appeared at 381.84 and 392.66 eV, respectively (Fig. 7e), confirming that uranium was successfully immobilized in SA-CC[37]. To elucidate the binding mode between U(VI) and SA-CC, O1s, Ca2p, and Cu2p were examined. As shown in Fig. 7f, the O1s spectrum is composed of three peaks at 531.7, 532.8, and 533.7 eV, corresponding to C–O, C=O, and O–H, respectively[38]. After uranium adsorption, the O–H peak shifts to 534.1 eV, and its relative content decreased by 16.3%, indicating that the U(VI) adsorption involves complexation between –OH and U(VI)[39]. From Fig. 7g, h, the Ca2p3/2 and Ca2p1/2 peaks are located at 347.9 and 341.6 eV, while those centered at 953.6 and 933.9 eV are assigned to Cu2p1/2 and Cu2p3/2[40,41]. It was evident that the binding energies of Ca2p and Cu2p shifted to higher values by 0.2 and 0.5 eV, respectively, demonstrating that Ca2+ and Cu2+ participate in U(VI) adsorption through ion-exchange with UO22+[42].
Based on the above analyses, three possible mechanisms account for the U(VI) uptake by SA-CC (Fig. 7i): (1) Electrostatic attraction between the negatively charged SA-CC surface and the positively charged U(VI) ions[43]; (2) Complexation of U(VI) with –OH and –COOH groups on the SA-CC surface; (3) Ion exchange where U(VI) ions occupy Ca and Cu sites on the surface of SA-CC. The increased U(VI) adsorption capacity of SA-CC, compared to that of SA-Cu and SA-Ca, could primarily be attributed to the higher abundance of –OH groups present in SA-CC.
-
In this study, the microscopic morphology of sodium-alginate-based composite gel microspheres was observed using SEM. The results indicated that SA-CC had a porous structure and was further characterized by XPS and FTIR. The findings revealed that the SA-CC's surface was densely populated with oxygen-containing functional groups and that Ca/Cu was uniformly introduced into SA-CC. Experimental datasets were regressed against both the isotherm and kinetic models, suggesting that the pseudo-second-order model and the Langmuir model provided the best depiction of SA-CC’s U(VI) uptake profile, thereby evidencing that the adsorption of U(VI) by SA-CC was a homogeneous monolayer chemisorption process. SA-CC exhibited the highest adsorption efficiency, delivering an uptake of 394.85 mg/g. Additionally, SA-CC demonstrated good recyclability and maintained a robust adsorption efficiency after five cycles. These findings indicate that sodium-alginate-based composite gel microspheres offered exceptional promise for field-scale remediation of uranium-laden effluents.
-
It accompanies this paper at: https://doi.org/10.48130/scm-0026-0005.
-
Not applicable.
-
The authors confirm their contributions to the paper as follows: Yuzhi Xie, Sai Yang, and Huanhuan Dong conceived the concept and orchestrated the experimental blueprint. Yuzhi Xie and Sai Yang excuted catalyst preparation and full analytical profiling. Lechang Xu, Jie Kang, and Xudong Wu performed the adsorption experiments. Xue Jiang, Wenli Zhou, Jin Zhang, Xi Wu, and Jian Zhang performed the SEM, TEM, XPS, and FTIR characterization. Yuzhi Xie and Sai Yang jointly drafted the manuscript. All authors analyzed the findings, provided editorial input, reviewed the results, and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
-
This work was supported by NSFC (No. U23A20105 and U2267224), Sichuan Natural Science Foundation Innovation Research Group Project (No. 2024NSFTD0012), Sichuan Provincial Natural Science Foundation Project (No. 2025ZNSFSC0949), Innovation Development Fund of China Seawater Uranium Extraction Technology Innovation Alliance (No. CNNC-HSTY-2024-016), and the Research Fund of SWUST for PhD (No. 23zx7110).
-
The authors declare no competing interests.
-
# Authors contributed equally: Yuzhi Xie, Sai Yang, Huanhuan Dong
Full list of author information is available at the end of the article. - The supplementary files can be downloaded from here.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Xie Y, Yang S, Dong H, Wu X, Xu L, et al. 2026. High-capacity and selective adsorption of uranium by porous sodium alginate composite gel microspheres. Sustainable Carbon Materials 2: e016 doi: 10.48130/scm-0026-0005 

High-capacity and selective adsorption of uranium by porous sodium alginate composite gel microspheres
- Received: 20 November 2025
- Revised: 02 January 2026
- Accepted: 21 January 2026
- Published online: 13 March 2026
Abstract: Efficient adsorption and recycling of uranium resources are pivotal to the enduring advancement of the atomic-energy sector. Three different sodium-alginate-based composite gel microspheres (SA-Cu, SA-Ca, and SA-CC) were successfully prepared using the drop-injection method with copper chloride, calcium chloride, and a mixed solution of both, respectively. The results showed that the metal-modified sodium alginate gel microspheres exhibited a porous structure with plentiful oxo-type surface functionalities. The U(VI) adsorption performance of the samples was evaluated across a matrix of controlled variables. The results revealed that U(VI) uptake by the gel microspheres reached equilibrium within 900 min, with SA-CC delivering a robust U(VI) scavenging performance (394.85 mg/g). Kinetic profiling aligned with the pseudo-second-order framework, while equilibrium data conformed to the Langmuir formalism, collectively evidencing single-layer, site-uniform chemical sequestration of U(VI) on the gel microspheres. In addition, most of the co-existing anions and metal cations exerted negligible perturbation on the U(VI) sequestration of SA-CC. SA-CC exhibited good recyclability with a robust U(VI) adsorption efficiency after five cycles. It was hypothesized that the high adsorption efficiency was closely related to electrostatic attraction, ion exchange, and coordination of U(VI) on SA-CC. These results suggested that sodium-alginate-based composite gel microspheres could be a robust adsorbent with potential application in U(VI) removal.
-
Key words:
- Sodium alginate /
- Gel microspheres /
- Uranium(VI) /
- Adsorption