









-
Glycoprotein 130 (gp130), also known as IL-6β or CD130, is a widely expressed transmembrane receptor that serves as the principal signal transducer for the IL-6 cytokine family[1]. Studies have shown that gp130 triggers the release of inflammatory factors after binding to IL-6, thereby sustaining chronic inflammation[2], and is a precursor to immune evasion and tumorigenesis[3]. The gp130 protein has been an important therapeutic target due to its significant functions in both inflammatory and oncogenic processes. In various tumor tissues, gp130 is continuously activated to form the IL-6/IL-6R/gp130 complex with signal transduction function. Multiple downstream signaling pathways are then activated to promote tumor progression. Targeting and inhibiting gp130 signaling may not only block various tumorigenic signals mediated by the gp130 protein for oncogenic therapy, thereby enhancing anti-tumor efficacy, but also effectively alleviate immunosuppression caused by IL-6/IL-6R-targeted therapies. Therefore, targeting gp130 represents a promising strategy for developing anti-tumor therapies with superior therapeutic efficacy.
The IL-6/gp130 signaling axis promotes tumor progression through the trans-signaling pathway[4]. IL-6 binds to soluble sIL-6Rα to form a binary complex, which then combines with the D2 and D3 domains of membrane-bound gp130 to form a ternary complex. This complex undergoes homodimerization through the D1 domain of gp130 to form a hexameric complex with signal function[5]. The complex then activates the downstream JAK2/STAT3 pathway, inducing STAT3 phosphorylation and nuclear translocation. This process ultimately regulates the transcription of target genes and tumorigenesis.
Crystal structure analysis from the IL-6/IL-6R/gp130 hexamer (PDB: 1P9M) highlights the gp130 D1 domain as a critical interface, which interacts with the Trp157 residue and the hydrophobic Leu57 residue on IL-6[6]. These interactions are essential for the homodimerization process that drives signal transduction. Based on this, we aimed to design small molecule inhibitors targeting the gp130 D1 domain to block the interaction between IL-6 and gp130, and consequently exhibit antitumor activity.
Current research on gp130 inhibitors remains limited, and only a few small-molecule inhibitors have been reported. While Madindoline-A was identified as the first natural product inhibitor targeting gp130, its clinical transformation has been limited by a low binding affinity (KD = 288 μmol·L−1) and a challenging synthesis[7]. LMT-28, which exhibits improved affinity (KD = 7.4 μmol·L−1), has been investigated primarily in arthritis, with its potential antitumor effects remaining unexplored[8,9]. SC144, an orally active gp130 inhibitor, exerts antitumor effects by inducing gp130 phosphorylation (Ser782) and downregulating glycosylation, though its gp130 binding affinity remains uncharacterized[10,11]. GP130-IN-1 (Compound 49) arrests cells in G0/G1 phase via targeting gp130, demonstrating potent activity against triple-negative breast cancer (IC50 = 9.82 μmol·L−1, SI > 16), but the affinity is also absent[12]. Bazedoxifene has also been reported as a novel gp130 inhibitor (KD = 182 μmol·L−1) by suppressing the gp130/STAT3 pathway[13,14]. Our previous work revealed that the Bazedoxifene analog 10a has demonstrated activity against non-small cell lung cancer (IC50 = 6.16 μmol·L−1, KD = 3.8 μmol·L−1) by downregulating the gp130/STAT3 pathway[15]. Despite these compounds having potential antitumor activity, none of them have entered clinical studies due to their limitations. Therefore, the development of novel gp130 inhibitors remains critical for oncology drug discovery.
In the context of data-centric drug discovery, the scarcity of GP130-targeted compounds remains a major challenge to directly construct reliable activity prediction models. In contrast, STAT3, as a key downstream effector in the gp130 signaling pathway has been extensively studied in preclinical research and has accumulated many inhibitors with diverse chemical structures. Given the tight functional correlation between gp130 and STAT3 on the IL-6/gp130/JAK/STAT signaling axis, this data availability provides a theoretical basis for using the STAT3 inhibitor dataset as the information source task within the transfer learning framework. Transfer learning methods can then be used to provide an important data basis for the discovery of gp130-targeted compounds.
In this study, we employed a two-step drug design strategy to identify novel small-molecule inhibitors targeting gp130. Firstly, a computational framework based on transfer learning was used to screen the candidate compounds in the natural product library, and a priority ranking assisted by this model was established. Based on this, evodiamine was selected as a representative scaffold for further exploration. Secondly, considering the structural and biosynthetic similarities between evodiamine and rutaecarpine, a series of derivatives based on both scaffolds were synthesized and systematically evaluated. Structural optimization was performed based on their antitumor activity at the cellular level, ultimately selecting the candidate compound 8a. In vitro and in vivo experiments demonstrated that 8a directly targets gp130, suppresses the IL-6/gp130/STAT3 signaling pathway and induces apoptosis via the mitochondrial pathway, thereby exhibiting significant antitumor activity.
-
To construct the activity prediction model, we collected data on IL-6-mediated STAT3 inhibitors and non-inhibitors from the STAT3In dataset[16]. All compounds were derived from the AID 862 bioassay in the PubChem Bioassay database (
https://pubchem.ncbi.nlm.nih.gov/bioassay/862 ), which was conducted through high-throughput screening based on a luciferase reporter system in STAT1-deficient U3A human fibrosarcoma cell lines.In this primary screen, all tested compounds were experimentally evaluated under identical assay conditions and explicitly annotated as active or inactive according to assay-defined inhibition thresholds. From 194,698 tested compounds, 1,565 inhibitors (label = 1) and 1,671 non-inhibitors (label = 0) with available three-dimensional structural information as provided in PubChem were retained, as such records typically reflect more complete and well-defined molecular annotations. This criterion also provided a consistent filtering rule applied to both active and inactive compounds, and helped balance the dataset size, given the large number of inactive entries in the original screen. Following the criteria from prior studies, the curated dataset was randomly divided into training and validation sets at a 4 : 1 ratio.
GP130 inhibitor dataset
-
To implement transfer learning and adapt the STAT3-based model to the gp130 target, we compiled a small literature-curated dataset comprising 45 molecules with experimentally reported gp130-related activity from published studies[6,8,10,15,17−21]. Molecules with an IC50 value below 20 μmol·L−1 were classified as inhibitors, resulting in 37 inhibitors and eight non-inhibitors. The molecular structures (SMILES) and corresponding binary activity labels used for model fine-tuning are provided in Supplementary Material 1.
Candidate compound library
-
We began the virtual screening using the commercially available natural product library from Selleck Chemicals (SelleckChem Natural Products Database,
www.selleckchem.com/screening/natural-product-library.html ), known for its structural diversity and relevance to drug discovery. To ensure data quality, we first cleaned the compound collection using RDKit. Any entry with an unreadable SMILES string was removed, and duplicate structures were filtered out by comparing canonical SMILES. After this processing, we obtained a final set of 2,560 unique and valid compounds for subsequent analysis.STAT3 activity prediction model
-
We built an activity prediction model for STAT3 as a basis for later transfer to gp130. Following common practice, we split the STAT3 dataset randomly into training and validation sets in a 4 : 1 ratio. To find the best model setup, we used Bayesian optimization (via the HyperOpt package) to optimize key hyperparameters, including hidden dimensions, network depth, dropout rate, and the number of feed-forward layers. The search space and the final hyperparameter values are summarized in Supplementary Material 2 (Table S1). The model with the strongest validation performance was selected for subsequent transfer learning.
Transfer learning and model fine-tuning
-
The pre-trained STAT3 model was then fine-tuned on our gp130 inhibitor dataset. To avoid overfitting and enhance model adaptability, we kept the parameters of the D-MPNN encoder fixed and only updated the classification layer. This allowed us to keep the molecular features learned from the STAT3 dataset, while using fewer trainable parameters. During model development, 40 molecules were used for fine-tuning, while five molecules were set aside for qualitative evaluations. The fine-tuned model was later applied to virtual screening and compound prioritization.
Model evaluation strategy
-
To assess the model performance under data-scarce conditions, we compared two versions of the model: a baseline version trained from scratch on the limited gp130 dataset, and a transfer-learning version that started with knowledge from a pre-trained STAT3 encoder. For both training strategies, we applied the same repeated five-fold cross-validation with identical data partitions, model architecture and hyperparameter settings. Model performance was quantified using the area under the receiver operating characteristic curve (AUROC), the area under the precision-recall curve (AUPRC), and the Recall@3 rate as ranking-oriented metrics. The results were presented as mean ± SD across all runs and folds.
ADMET-based pre-screening
-
To screen out compounds with poor drug-like properties, we conducted an in silico ADMET prescreening of the 2,560 compounds in the candidate library using ADMETlab 3.0[22]. We used a set of key properties to filter the library, and the specific thresholds can be found in Supplementary Material 2 (Table S2). Any compound that failed to meet all the criteria was removed.
Compound scoring and ranking
-
We then scored the remaining compounds with our fine-tuned gp130 activity prediction model. The model assigned each compound a prediction score, which estimates its relative likelihood of inhibiting gp130. Based on these scores, we ranked all compounds and selected the top 20% for further study.
Final selection using the MCE-18 metric
-
To further explore novel structures in chemical space, we used the MCE-18 metric to assess the chemical complexity. MCE-18, or Medicinal Chemistry Evolution 2018, is a well-established measure that can effectively score molecules by novelty in terms of their cumulative sp3 complexity. It can highlight compounds with more unique and elaborate scaffolds. Among the top candidates, the compound with the highest MCE-18 score was chosen as our lead hit for follow-up studies.
Molecular docking studies
-
To evaluate the binding mode of 8a, we performed molecular docking using the Schrödinger software suite. The crystal structure of the human gp130 D1 domain (PDB ID: 1P9M) was retrieved from the Protein Data Bank. The protein structure was prepared by adding missing atoms, assigning bond orders, and adjusting protonation states to pH 7.0 using the Protein Preparation Wizard and the Epik module. Energy minimization was performed using the OPLS4 force field with default parameters.
The 3D structure of 8a was generated and energetically minimized with LigPrep, setting ionization states to reflect physiological pH. A docking grid was generated and centered on residue Asn92. We used Glide in Extra Precision (XP) mode to dock the ligand flexibly into the binding site. The resulting poses were ranked by GlideScore, and the top-scoring pose was chosen for further analysis.
The protein-ligand interactions in the top pose were visualized and analyzed using PyMOL and Maestro. We inspected hydrogen bonds, hydrophobic contacts, and overall shape complementarity to evaluate the binding mode.
General information for chemistry
-
All chemicals were obtained from commercial suppliers and used without further purification. The reaction progress was monitored via analytical thin layer chromatography (TLC) on precoated silica gel GF254 plates (Qingdao Haiyang Chemical Plant, Qingdao, China), and the spots were detected under UV light (254 nm). The melting point was measured on an XT-4 micromelting point instrument. IR (KBr-disc) spectra were recorded with a Bruker Tensor 27 spectrometer.
1H NMR and 13C NMR spectra were measured on a Bruker ACF-500 spectrometer at 25 °C and referenced to TMS. Chemical shifts are reported using the residual solvent line as an internal standard. Splitting patterns were designed as s, singlet; r.s, broad single; d, doublet; t, triplet; and m, multiplet.
The purities of all the compounds used for biological evaluation were confirmed to be greater than 95% through analytical HPLC performed with an Agilent 1100 HPLC System. Mass spectra were obtained on an MS Agilent 1100 Series LC/MSD Trap mass spectrometer (ESI-MS) and a Mariner ESI-TOF spectrometer (HRESI-MS), respectively. The 1H NMR spectra, 13C NMR spectra, purities, and mass spectra of the compounds could be found in the supporting information.
Cell lines and cell culture
-
All cell lines were purchased from the Cell Bank of the Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). Human colonic carcinoma HT-29 cells, human colonic carcinoma HCT116 cells, human liver cancer L-02 cells, human embryonic kidney 293T cells, human normal lung epithelial Beas-2B cells, and human normal breast MCF-10A cells were cultured in Dulbecco modified Eagle medium (DMEM, SenBeiJia Biological Technology Co., Ltd., Nanjing, China) supplemented with 10% (V/V) fetal bovine serum (FBS, Lanzhou Rongye Biotechnology Co., Ltd., lanzhou, Gansu, China), 50 µg·mL−1 penicillin (SenBeiJia Biological Technology Co., Ltd., Nanjing, China) and 50 µg·mL−1 streptomycin (SenBeiJia Biological Technology Co., Ltd., Nanjing, China). Human colonic carcinoma SW480 cells and human normal colonic epithelial HIEC-6 cells were cultured at Roswell Park Memorial Park Institute-1640 (RPMI-1640, SenBeiJia Biological Technology Co., Ltd., Nanjing, China) medium supplemented with 10% (V/V) FBS. All the cell lines were incubated at 37 °C in a humidified atmosphere containing 5% CO2.
Cell viability assays
-
The cell viability was measured via the CCK-8 method. The cells (HT-29, HCT116, SW480, HIEC-6, MCF-10A, L-02, 293T, and Beas-2B) were seeded into 96-well plates at a density of 4,000 cells/well and allowed to attach overnight. The tested compounds were first dissolved in dimethyl sulfoxide (DMSO, SenBeiJia Biological Technology Co., Ltd., Nanjing, China) and serially diluted with the cell growth medium. Subsequently, the cells were treated with serial concentrations of the compounds for 48 h. The final working concentration of DMSO was maintained below 0.1% (V/V). The CCK-8 reagent was prepared with the corresponding medium as a 10% mixed solution, and 100 µL of the mixture per well was incubated with the treated cells at 37 °C in a 5% CO2 incubator for 2–4 h in the dark. The absorbance of each well was read at 450 nm via a multifunctional plate reader (Biotech Synergy H1, Agilent, USA). Cytotoxic activity was expressed as the IC50 value. All the experiments were performed three times.
Isothermal titration calorimetry
-
The affinity of compound 8a for the gp130 protein was measured via a Malvern Panalytical MicroCal PEAQ-ITC (UK). A 4 mM stock solution of compound 8a was prepared in DMSO and diluted to 200 μmol·L−1 in protein dilution buffer (137 mmol·L−1 NaCl, 2.7 mmol·L−1 KCl, 10 mmol·L−1 NaH2PO4, 2 mmol·L−1 KH2PO4, pH 7.4), with a final DMSO concentration of 5% (V/V). A 25 μM solution of gp130 protein (300 μL) was placed in the sample cell, and 200 μmol·L−1 of compound 8a (100 μL) was titrated into the cell in 20 injections with a 75 s interval. The data was analyzed via MicroCal PEAQ-ITC analysis software to calculate the KD value. Control measurements were performed by titrating the compound into the buffer, buffer into the protein solution at the same concentration, and buffer into the buffer to account for the dilution heat and background noise.
Drug affinity responsive target stability (DARTS) assay
-
The DARTS assay was used to detect the binding of compound 8a to the gp130 target. Human colonic carcinoma HT-29 cells were lysed via an NP-40 (Beyotime, Nanjing, China) lysis buffer supplemented with protease and phosphatase inhibitors. The lysates were then incubated with increasing concentrations of compound 8a (10–1,000 μmol·L−1) or the DMSO control at room temperature for 1 h. Proteolysis was followed by the addition of protease pronase at a ratio of 1 : 1,000 (pronase : lysates) and incubated for 30 min at room temperature. To stop proteolysis, 5 × sodium dodecyl sulfate (SDS) sample loading buffer was added at a 1 : 4 ratio to each sample, which was subsequently boiled at 100 °C. The resulting protein samples were separated via 8% sodium dodecyl sulfate (10%) polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed via western blot as described below.
Co-immunoprecipitation (Co-IP) assay
-
Co-IP was performed in HEK293 cells that had been co-transfected with plasmids expressing Flag-gp130 and Myc-IL-6R. After 24 h, the transfected HEK293 cells were treated with compound 8a (12 μmol·L−1) for 4 h and then stimulated with recombinant human IL-6 (25 ng·mL−1) for 30 min. The transfected cells were collected and lysed in cold RIPA lysis buffer supplemented with protease and phosphatase inhibitors. Precleared HEK293 cell lysates were then subjected to immunoprecipitation with anti-Flag M2 (Sigma) and protein A/G PLUS-Agarose (Santa Cruz) at 4 °C. After washing with whole-cell extraction buffer, the beads were boiled in 1 × SDS-PAGE loading buffer. Myc-tagged and Flag-tagged proteins were immunoprecipitated and detected with anti-Myc (CST) and anti-Flag (Sigma) antibodies, respectively.
Western blot
-
Protein levels were determined by Western blot. MDA-MB-231 cells were incubated with increasing concentrations of compound 8a (3–12 μmol·L−1) for 48 h. After trypsinization, the cells were treated with 1 × RIPA lysis buffer (50 mmol·L−1 Tris-HCl, pH 7.4; 150 mmol·L−1 NaCl; 0.25% deoxycholic acid; 1% NP-40; 1 mM EDTA, and the protease inhibitor PMSF, Amresco, Solon, USA) to extract total protein. An aliquot of protein from the total cell lysates (40–80 µg per lane) was separated via SDS-PAGE (Bio-Rad Laboratories, Hercules, CA), wet-transferred to PDVF membranes (Bio-Rad Laboratories, Hercules, CA), and blotted with primary antibodies specific for cleaved caspase-9, caspase-9, cleaved caspase-3, caspase-3, PARP, cleaved PARP, gp130, STAT3, p-JAK2, JAK2, p-STAT3 (Y705), Bcl-2, cyclin D1 and GAPDH, which were probed with secondary isotype-specific antibodies tagged with horseradish peroxidase (Cell Signaling Technology). The bound immunocomplexes were detected via a ChemiDOC™ XRS + system (Bio-Rad Laboratories, Hercules, CA, USA).
Electrophoretic mobility shift assay
-
In the experiment, appropriate HT-29 cells were inoculated into 6-well plates and cultured until the cell confluence reached 60%–70%. The cells were treated with compound 8a (3, 6, or 12 µmol·L−1) or DMSO at gradient concentrations in complete medium for 48 h, the cells were collected, and nuclear protein was extracted for concentration determination. Consistent with the above methods, probe labeling, and protein and DNA binding were performed according to the instructions of the electrophoretic mobility shift assay (EMSA)/Gel-Shift Kit (Binetian, China)[23]. The oligonucleotide and protein complexes were separated via 8% SDS-PAGE. After electrophoresis, the oligonucleotide and protein complexes were dried on a dry gel, and autoradiography was subsequently performed. A 32P-labeled oligonucleotide hSIE probe was used to bind STAT3 (5′-AGCTTCATTTCCCGTAAATCCCTA-3′)[24].
Cellular thermal shift assay
-
HT-29 cells were seeded in 10 cm culture dishes and cultured until they reached 70%−80% confluence. The cells were treated with compound 8a or DMSO for 1 h and then collected. The cells were resuspended in PBS (containing 1 × protease inhibitor) and divided into 11 aliquots, each with a volume of 28 μL. A gradient temperature (40−60 °C) was set in the PCR instrument within 8 min, and then rapidly cooled. The samples were subjected to three freeze-thaw cycles in liquid nitrogen, followed by centrifugation at 12,000 r·min−1 for 10 min at 4 °C to collect the supernatant. The abundance of gp130, JAK2, and STAT3 proteins was detected by Western blotting, and the Tm values were analyzed using Image J and fitted with GraphPad Prism software.
gp130 knockdown
-
The gp130-targeted siRNA (General Biol, Anhui, China) was transfected into HT-29 cells by Lipofectamine 2000 (Beyotime, Shanghai, China). After 48 h, cells were collected and spread in 96-well plates at a rate of 5,000 cells per well. The plates were treated with gradient concentrations of compound 8a for 48 h, and the cell viability was tested by the CCK-8 method. At the same time, cells were collected and confirmed to be successfully knocked down through Western blot experiments. The target sequences of siRNA were as follows: siRNA-NC: 5′-UUCUCCGAACGUGUCACGUTT-3′, siRNA-1: 5′-GUGCAUCGCACCUAUUUAATT-3′, siRNA-2: 5′-GAGUGCUGUUCUGCUUUAATT-3′.
Cell apoptosis analysis
-
Apoptosis was determined by flow cytometry in accordance with the manufacturer's instructions. An Annexin V-FITC/PI double-staining assay was used to analyze apoptosis, in which 3.0 × 105 HT-29 cells were incubated with increasing concentrations of compound 8a for 48 h. Then, the cells were harvested, washed, and stained with 5 µL of Annexin V-FITC and 10 µL of propidium iodide at room temperature in the dark for 15 min. The cells were then analyzed via flow cytometry (488 nm excitation and 525 nm emission filters) via a BD Accuri C6 flow cytometer (Becton & Dickinson Company, Franklin Lakes, NJ, USA).
Colony formation assay
-
The anti-proliferative activity of compound 8a was measured via colony formation assays. HT-29 cells were plated in 6-well culture plates (1,000 cells per well) and treated with 8a at different concentrations for 48 h. Then, the culture medium was discarded, and the cells were sub-cultured in fresh medium. The cells were allowed to grow for 10‒14 d until colonies were visible. The cells were fixed with 4% paraformaldehyde. The cells in the plates were then stained with crystal violet solution (Sigma Aldrich, St. Louis, MO, USA), and the resulting colonies were photographed manually.
The metabolic stability in rat liver microsomes assay
-
Compound 8a (or evodiamine) was prepared into a 1 mg·mL−1 solution using DMSO. After thoroughly mixing rat liver microsomes (RLM, 59.23 mg·mL−1) with 50 μL of 4.066 mg·mL−1 MgCl2, 50 μL of PBS (pH 7.0), and 10 μL of compound 8a (or evodiamine), the mixture was incubated at 37 °C for 5 min, and then 33.3 mg·mL−1 NADPH was added to initiate the reaction. The reaction was terminated using cold glibenclamide acetonitrile solution (100 μg·mL−1) at time points of 0, 5, 10, 30, 60, and 120 min. Supernatant was collected following centrifuging at 12,000 r·min−1 for 10 min, and HPLC chromatography (Agilent, USA) used to detect the peak area. The remaining percentage of each compound over time was quantified, and the in vitro half-life (t1/2) and intrinsic clearance (CLint) were calculated. The t1/2 in vivo used the equation: t1/2 = ln2/slope. And the CLint used the equation: CLint = 0.639/t1/2 · μL (incubation)/mg (microsomes).
Animal experiments
-
Nude mice were injected in the right flank area with 5 × 106 human colonic carcinoma HT-29 cells in 100 μL of PBS. After the tumor volume reached approximately 70 mm3, the mice were randomly assigned to four groups (5 mice/group). Nude mice were intravenously administered compound 8a (10 or 20 mg·kg−1), bazedoxifene (20 mg·kg−1) or vehicle daily and monitored every 3 d. Drugs were dissolved in 60% physiological saline with 40% polyethylene glycol for in vivo administration. Tumor growth was determined by tumor volume, which was calculated according to the 8a equation V = 0.52 × a2 × b (where a is the smallest superficial diameter and b is the largest superficial diameter). After 21 d of treatment, all the mice were killed, and the tumors were segregated, weighed, and stored at −80 °C for later use.
Statistical analysis
-
The experimental results were processed via GraphPad and one-way analysis of variance. All the experimental groups consisted of three repeated experiments, and the results are noted in the form of mean ± SD (** P < 0.01; *** P < 0.001).
-
We applied a transfer learning model for compound prioritization (Fig. 1a), which was implemented using the Chemprop framework. This model has a directed message passing neural network (D-MPNN) encoder and a feedforward neural network (FFN) classifier[25,26]. Based on the functional relationship between gp130 and STAT3 signaling, we defined the pre-training task as predicting the inhibitory activity of compounds against STAT3. The model first learned the molecular representations from the larger STAT3 dataset, then fine-tuned itself with the smaller gp130 dataset. During fine-tuning, the parameters from the encoder layers were frozen to retain the general chemical information that had already been learned, while the classification layer was updated using gp130-related data. This design allowed the model to prioritize the compounds that have a higher possibility to inhibit gp130 with the limited training data.
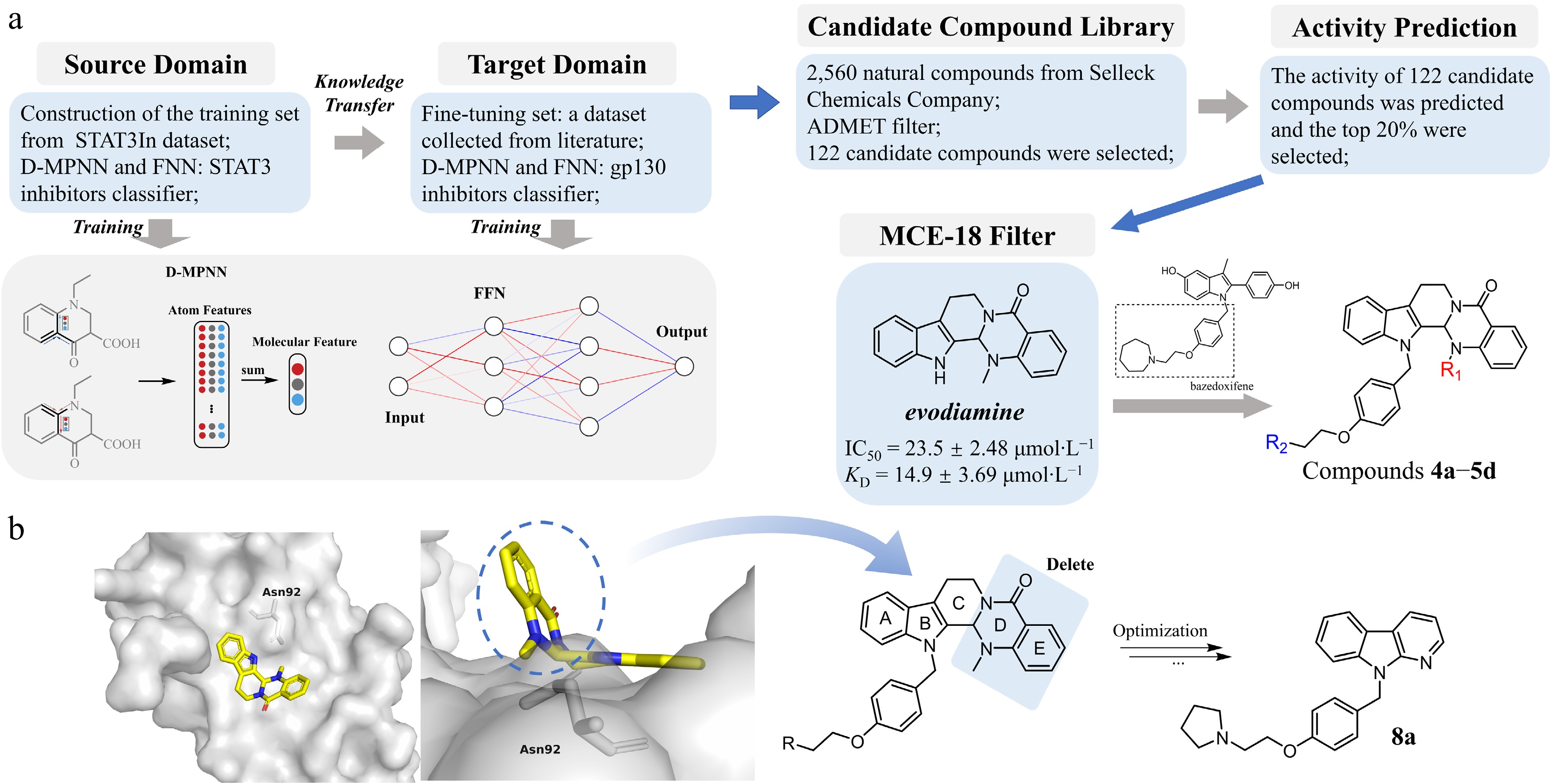
Figure 1.
Design strategy and structure optimization of novel gp130-targeting compounds. (a) Preliminary identification of lead compounds. A transfer learning strategy was adopted to construct a gp130 inhibitor classification model integrating a Directed Message Passing Neural Network (D-MPNN) and a Feedforward Neural Network (FNN). After pretraining on the known STAT3 inhibitor (source Domain), the pretrained model was fine-tuned using the gp130-related compounds (Target Domain) to create the final gp130 inhibitor classifier. This classifier was then used to screen a library of 2,560 natural compounds. After ADMET screening, 122 candidate drugs with relatively high activity scores were obtained. These candidate drugs were further evaluated through the MCE-18 index, and ultimately evodiamine was selected as the primary hit. It shows weak anti-tumor activity and binding affinity with gp130 against colorectal cancer HT-29 cells (with the IC50 of 23.5 ± 2.48 µmol·L−1, and the KD of 14.9 ± 3.69 µmol·L−1). A hybridization strategy combining the evodiamine scaffold with the linker moiety from bazedoxifene led to the synthesis of derivative series 4a−5d. (b) Structural optimization based on anti-tumor activity at the cellular level. Left panel (front and top view): Molecular docking of evodiamine with the gp130 D1 domain reveals its binding pose and key interactions, providing a structural basis for modification. Based on the docking analysis (right panel) and supported by in vitro antitumor activity data, iterative structural modifications were performed on the evodiamine-bazedoxifene hybrid scaffold. This rational design process ultimately yielded the optimized candidate compound 8a.
Evaluations of the transfer-learning algorithm
-
To examine the contribution of the transfer-learning algorithm, we compared two approaches: a baseline model that was trained only on the limited gp130 dataset, and a transfer-learning model that was discussed above. We ran both models 20 times using the five-fold cross-validation and acquired 100 independent evaluation results. The evaluations were conducted under identical conditions, including data partitions, model architecture, and hyperparameters. The results are shown in Supplementary Material 2 (Table S3).
Across all the evaluation metrics, the transfer-learning model demonstrated better performance. It achieved higher average scores in AUROC and AUPRC, meaning it was better at distinguishing active from inactive compounds, even when the data was imbalanced. It also had a better Recall@3 rate, which shows it was more effective at placing active compounds near the top of rankings than the baseline model. Importantly, the results of the transfer-learning model varied less across all runs, indicating it was more stable and reliable when trained on small amounts of data.
Overall, these findings confirm that when the training data for the main task is insufficient, applying the transfer learning algorithm based on the knowledge from a related database may enhance the reliability of the final model.
Qualitative model consistency on a held-out gp130 set
-
During model development, we set aside a small number of gp130-related compounds as a quick sanity check. After fine-tuning the model on the training set (40 compounds), we ran the model on the five remaining compounds. The predictions fully matched their reported activity labels. While five compounds are too few for statistical validation, this check helped confirm that the model behaved reasonably and did not produce any obvious errors on the test structures. Along with the previous cross-validation results, this check supports the suitability of the model for prioritizing compounds in a practical virtual screening workflow.
ADMET-based filtering
-
Before applying the activity prediction model, we conducted an in silico ADMET prescreening to prioritize compounds with favorable drug-like properties. The initial compound library consisted of 2,560 compounds from the natural product subset of the Selleck Chem Bioactive Compound Library, all of which were standardized and duplicates were removed.
We evaluated each compound using the ADMETLab 3.0 platform, which provides machine learning-based predictions for drug-related properties. Based on a series of property thresholds (Supplementary Material 2: Table S2), only 122 compounds passed this initial screen, representing approximately 4.8% of the original library. Although this step greatly reduced the number of candidates, such a low pass rate is not unusual for natural products, which often fall outside typical drug-like chemical space. The goal of this screening step was to prioritize compounds with potentially favorable pharmacokinetic properties for subsequent activity evaluations.
Virtual screening and compound selection
-
We ran the fine-tuned model on the 122 compounds that passed the ADMET prescreening. The model produced a score for each compound, which is an estimation for its relative likelihood of activity. We then ranked all compounds based on these scores and focused on the top-ranking candidates for closer inspection.
The complete ranked list of all 122 compounds, together with their predicted scores and annotations, is provided in Supplementary Material 3. Among the top candidates, evodiamine stood out for a notably higher MCE-18 score, suggesting it had greater structural complexity and potential novelty compared to other high-ranking compounds. This external screening step also served as a generalization check, confirming it could produce meaningful predictions even for compounds that were structurally different from those in the training set.
Given its strong performance across both ADMET filtering and model scoring, evodiamine was chosen for follow-up experimental testing.
Drug design based on cell-level antitumor activity
-
Evodiamine and rutaecarpine are structurally similar alkaloids extracted from the traditional Chinese medicine Evodia rutaecarpa. Studies have reported that they have antitumor activity against colorectal cancer[27,28]. Our previous research found that the linker of bazedoxifene has a positive effect on targeting the gp130 protein[15]. Therefore, we combined the linker of bazedoxifene with evodiamine and rutecarpine to design compounds 4a−5d. However, cell-based assays showed that compounds 4a−5d exhibited weak anti-tumor activity against HT-29 cells in vitro (Table 1). Further analysis through molecular docking (Fig. 1b) and affinity assays (Supplementary Material 2: Fig. S1a) revealed that the non-planar structure of evodiamine leads to the twisting of the D and E rings, which prevents effective interaction with the Trp157 site. Consequently, we attempted to remove the D and E rings and optimized the structure of the compounds based on their in vitro antitumor activity. Ultimately, candidate compound 8a was obtained, and further studies were conducted.
Table 1. Antiproliferative activity of compounds 4a−9d in HT-29 cells (48 h).

Compd. R1 R2 IC50a (μmol·L−1) Compd. R1 R2 IC50a (μmol·L−1) 4a 

17.01 ± 0.89 7a 

9.39 ± 0.71 4b 

10.58 ± 1.06 7b 

9.67 ± 0.17 4c 

32.88 ± 1.55 7c 

> 60 4d 

16.72 ± 0.88 7d 

14.37 ± 1.21 5a 

15.15 ± 1.29 8a 

4.57 ± 0.16 5b 

15.70 ± 1.24 8b 

6.57 ± 0.91 5c 

13.68 ± 0.42 8c 

28.35 ± 1.47 5d 

17.73 ± 1.31 8d 

6.58 ± 1.21 6a 

6.18 ± 0.83 8e 

> 60 6b 

5.77 ± 0.79 8f 

> 60 6c 

17.57 ± 0.63 8g 

> 60 6d 

7.55 ± 0.92 BFb / / 10.03 ± 1.02 a IC50: 50% inhibitory concentration (mean ± SD of three experiments). b Bazedoxifene, used as positive control. Chemistry
-
As depicted in Fig. 2, the target compounds were synthesized via two distinct pathways, depending on the substituent R. For the synthesis of analogues bearing aliphatic heterocycles, nucleophilic substitution of chlorides 1a−1d with 4-(hydroxymethyl) phenol was performed under phase-transfer conditions (NaOH, TBAB) to yield alcohols 2a−2d. These intermediates were converted to the corresponding benzyl chlorides 3a−3d using thionyl chloride, followed by N-alkylation with scaffolds 4−8 to afford the final compounds 4a−8d. Conversely, for the aromatic series, alcohols 1e−1g were coupled with 4-hydroxybenzaldehyde via a Mitsunobu reaction, followed by in situ reduction with NaBH4 to provide benzyl alcohols 2e−2g. Subsequent chlorination with SOCl2 yielded intermediates 3e−3g, which underwent nucleophilic substitution with 9H-pyrido[2,3-b] indole to produce compounds 8e−8g. The final compounds were confirmed by IR, HRMS (ESI), 1H NMR, and 13C NMR.
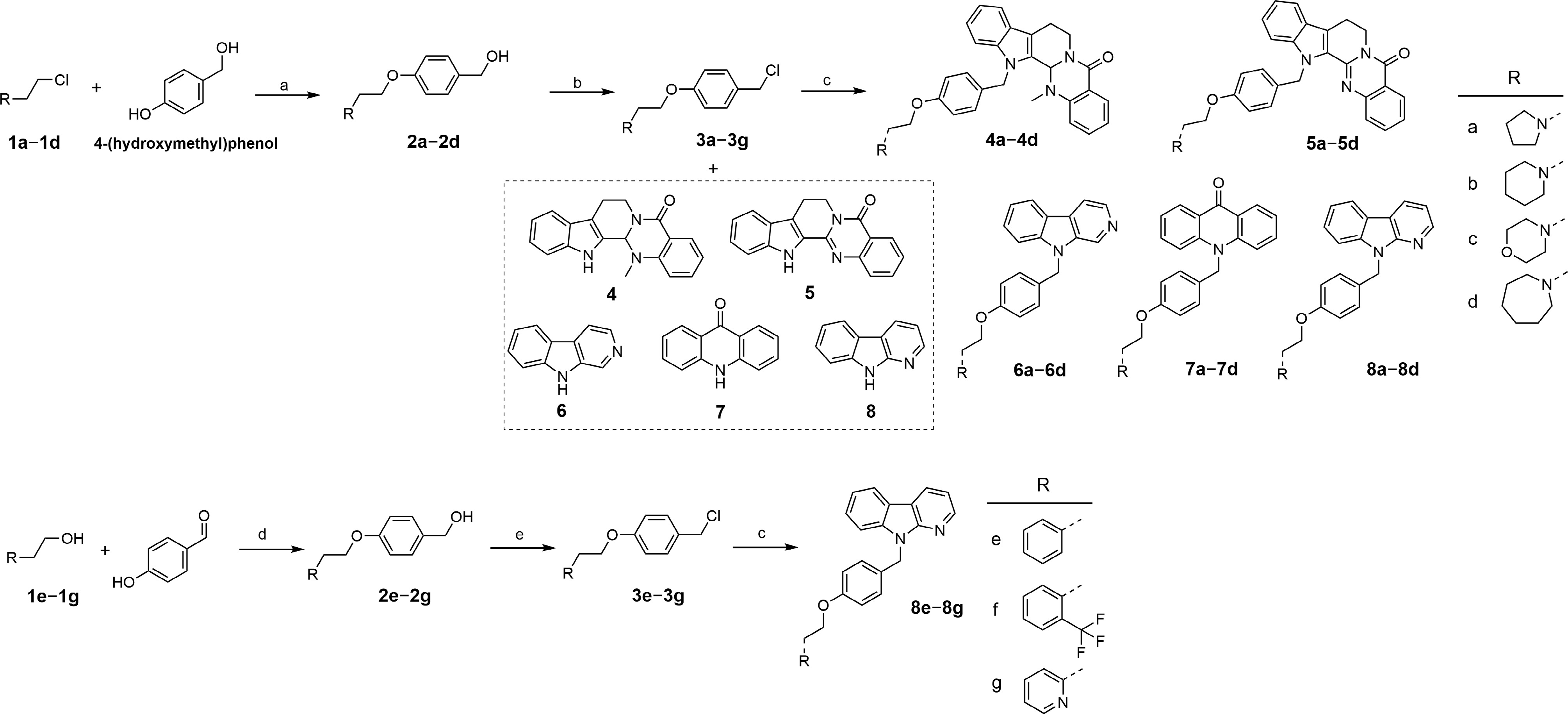
Figure 2.
The synthesis of targeted compounds. (a) NaOH, TBAB, H2O/toluene, 85 °C, 2 h; (b) SOCl2, THF, 0 °C to r.t., overnight; (c) NaH, anhydrous DMF, 0 °C to r.t., overnight; (d) (1) PPh3, DIAD, 4-hydroxybenzaldehyde, THF, 0 °C to r.t., 2 h; (2) NaBH4, MeOH, r.t., 2 h; (e) SOCl2, anhydrous CH2Cl2, 0 °C to r.t., overnight.
Antitumor activity on HT-29 cells
-
The study assessed the proliferation inhibitory activity of the synthesized compounds on HT-29 cells by the CCK-8 assay (Table 1). Preliminary screening showed that compounds 4a−5d exhibited weak anti-tumor activity. In contrast, compounds 6a−6d and 8d had superior inhibitory effects compared to 7a−7d. This difference may stem from the different parallel arrangements of the pyridine ring and the indole ring, leading to variations in the exposure of nitrogen atoms, which affects the hydrophobic interactions of the compounds with Gly95 and nearby amino acids on the gp130 protein. In terms of side chain modifications, aliphatic structures (a−d) usually exhibited better antitumor activity than aromatic ring structures (e−f), while compounds containing a morpholine ring showed generally poorer activity. This might be due to the lone pair electrons on the morpholine ring interfering with the interaction between the compound and hydrophobic amino acid residues such as Val93, Cys6, and Phe36 on gp130. Additionally, compounds with similar activity on HT-29 cells (6a, 6b, 6d, 8a, 8b, and 8d) were further evaluated for their cell inhibitory activity in other colorectal cancer cell lines (Supplementary Material 2: Table S4) and normal cell lines (Supplementary Material 2: Table S5). The comprehensive data showed that compound 8a exhibited significant antitumor activity and good selectivity. Therefore, compound 8a was prioritized as a candidate for further research.
Compound 8a directly binds to the gp130 protein
-
Compound 8a demonstrated superior anti-tumor activity compared to bazedoxifene, evodiamine, and rutecarpine (Fig. 3a). To explore the interaction between 8a and gp130, we used Isothermal Calorimetric Titration (ITC) to test the binding affinity of compound 8a with gp130. The dissociation constant (KD) was calculated to be 2.17 μmol·L−1 by the MicroCal PEAQ-ITC analysis software, demonstrating that 8a can directly target the gp130 protein and exert its antitumor effects (Fig. 3b). Compared to evodiamine (KD = 14.9 μmol·L−1), rutecarpine (KD = 24.6 μmol·L−1), and bazedoxifene (KD = 38.4 μmol·L−1), 8a (KD = 2.17 μmol·L−1) exhibited significantly higher affinity for gp130 (Supplementary Material 2: Fig. S1). In addition, the results showed that the binding of 8a to gp130 was an endothermic process (Fig. 3b).
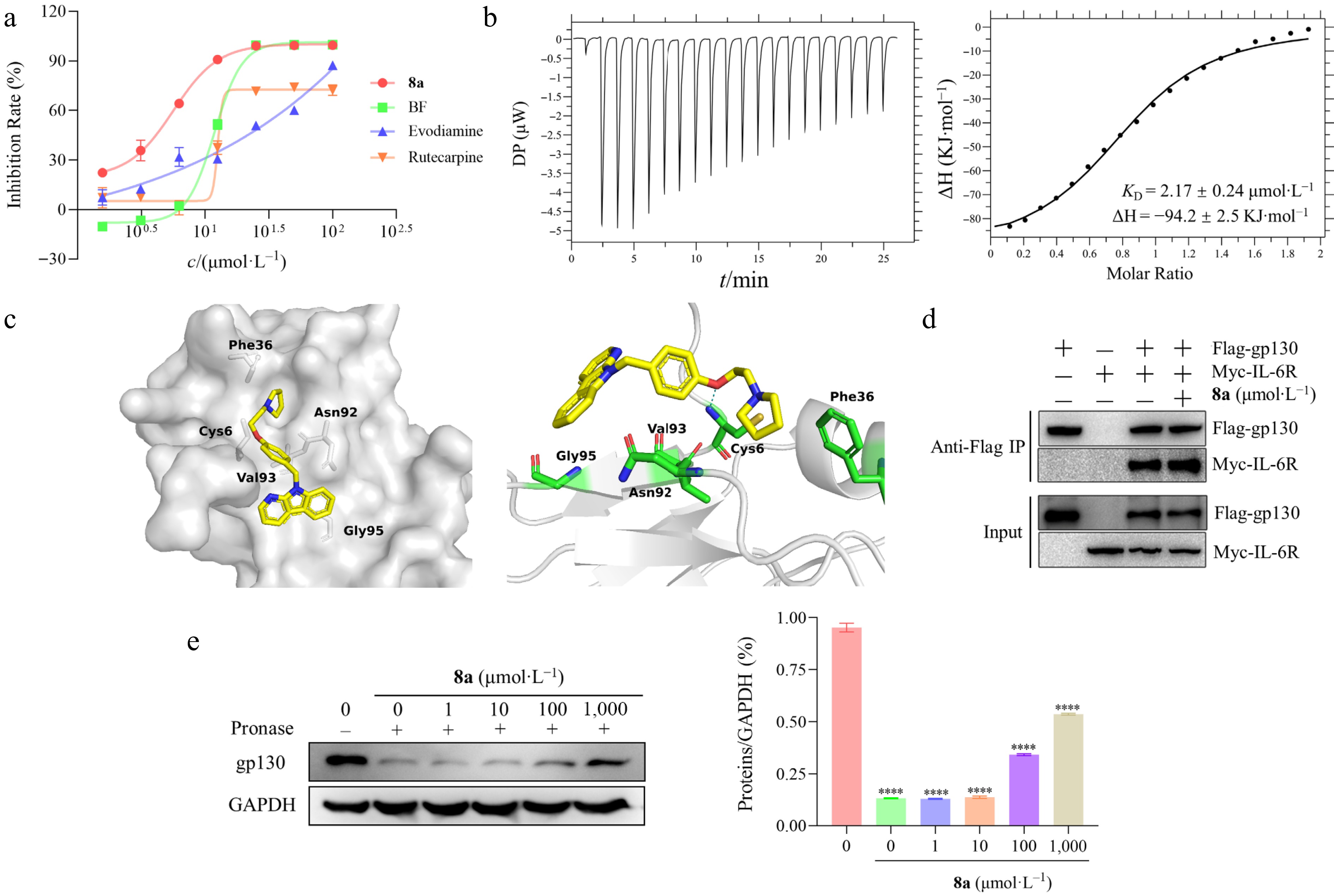
Figure 3.
Study on the interaction between 8a and gp130 protein. (a) The antiproliferation activities of evodiamine (IC50 = 23.50 ± 2.48 μmol·L−1), rutaecarpine (IC50 = 20.41 ± 2.35 μmol·L−1), bazedoxifene (IC50 = 10.03 ± 1.02 μmol·L−1), and 8a (IC50 = 4.57 ± 0.16 μmol·L−1) against HT-29 cells in vitro. (b) Thermodynamic characterization of the interaction between 8a and the gp130 protein. The direct binding affinity of compound 8a to gp130 protein was determined by isothermal titration calorimetry (ITC) at 25 °C, and the binding isotherms were fitted by the unit point model of the PEAQ-ITC software. The left figure shows the thermodynamic curve of the binding of 8a to the gp130 protein, and the titration curve indicates that the binding of 8a to the gp130 protein is an exothermic process. The right figure shows that the equilibrium dissociation constant (KD) is 2.17µmol·L−1 by integrating with isotherms. (c) Molecular docking predicts the 8a binding mode. Molecular docking simulation of compound 8a (yellow rod) within the D1 domain of gp130 (gray surface). This diagram includes a general view (left) and a detailed view (right), that the specific hydrogen bond interactions formed between 8a and the key amino acid residues of gp130 (the blue dotted line). (d) The compound 8a treated the HEK293 cells that were co-transfected with flag labeled gp130, and myc labeled IL-6Rα, and then collected cell lysates or co-immunoprecipitation assay analysis. The results revealed that 8a did not disrupt IL-6/IL-6R/gp130 complex formation. (e) The binding of compounds to target proteins protect proteins from protease hydrolysis. HT-29 cells were treated with 8a, and the lysates were collected for DARTS assay to analyze the relative abundance of the gp130 protein. The left figure shows the protein abundance by Western blot, and the right figure shows the quantitative analysis of protein abundance.
To further explore the binding mode between 8a and the gp130 protein, we employed molecular docking technology to predict the binding effect (Fig. 3c). The results showed the linker of 8a forms hydrogen bonds with Cys6. The tetrahydropyrrole ring interacts with Phe36 through van der Waals forces. The indole nucleus has a hydrophobic interaction with Gly95.
Studies demonstrated that the D1-3 domains of the gp130 protein are involved in signal transduction, while the D2 and D3 domains are related to the formation of IL-6/IL-6R/gp130 trimers, and the D1 domain is involved in the dimerization of the trimer complex[29]. Based on the above protein binding model, we further explored whether compound 8a interferes with the interaction between IL-6R and gp130 through co-immunoprecipitation (Co-IP) experiments. The results showed that treatment with 8a did not disrupt the binding between IL-6R and gp130 (Fig. 3d). This finding indicates that 8a is not targeting on the D2 or D3 domain of gp130 and therefore does not affect the formation of the IL-6/IL-6R/gp130 ternary complex. Considering the critical role of the D1 domain previously identified, we speculate that 8a may inhibit the homodimerization of the formed IL-6/IL-6R/gp130 ternary complex by targeting the D1 domain of gp130, and blocking its downstream oncogenic signaling to exert an anti-tumor effect.
In addition, we verified the targeting effect of 8a on gp130 by Drug Affinity Responsive Target Stability (DARTS) technology. The experiment revealed that 8a could increase the protein abundance of gp130 in a concentration-dependent manner (Fig. 3e). This result confirmed that the binding of 8a enhances the protein stability of gp130, thereby providing varying degrees of resistance against proteolytic digestion.
Compound 8a downregulated the JAK2/STAT3 signaling pathway
-
JAK2 and STAT3 are important downstream signaling targets of gp130 and play significant roles in tumor progression. To determine whether compound 8a acts its antitumor effects through the gp130/JAK2/STAT3 signaling axis, we first employed the cellular thermal shift assay (CETSA) to evaluate its binding to key proteins within this pathway. The results indicated that 8a substantially increased the thermal stability of gp130. In contrast, it only marginally stabilized JAK2 and had no discernible stabilizing effect on STAT3 (Fig. 4a–d). It suggests that gp130 is the primary direct molecular target of 8a within this pathway, and that the compound interferes with downstream signaling by specifically engaging this receptor.
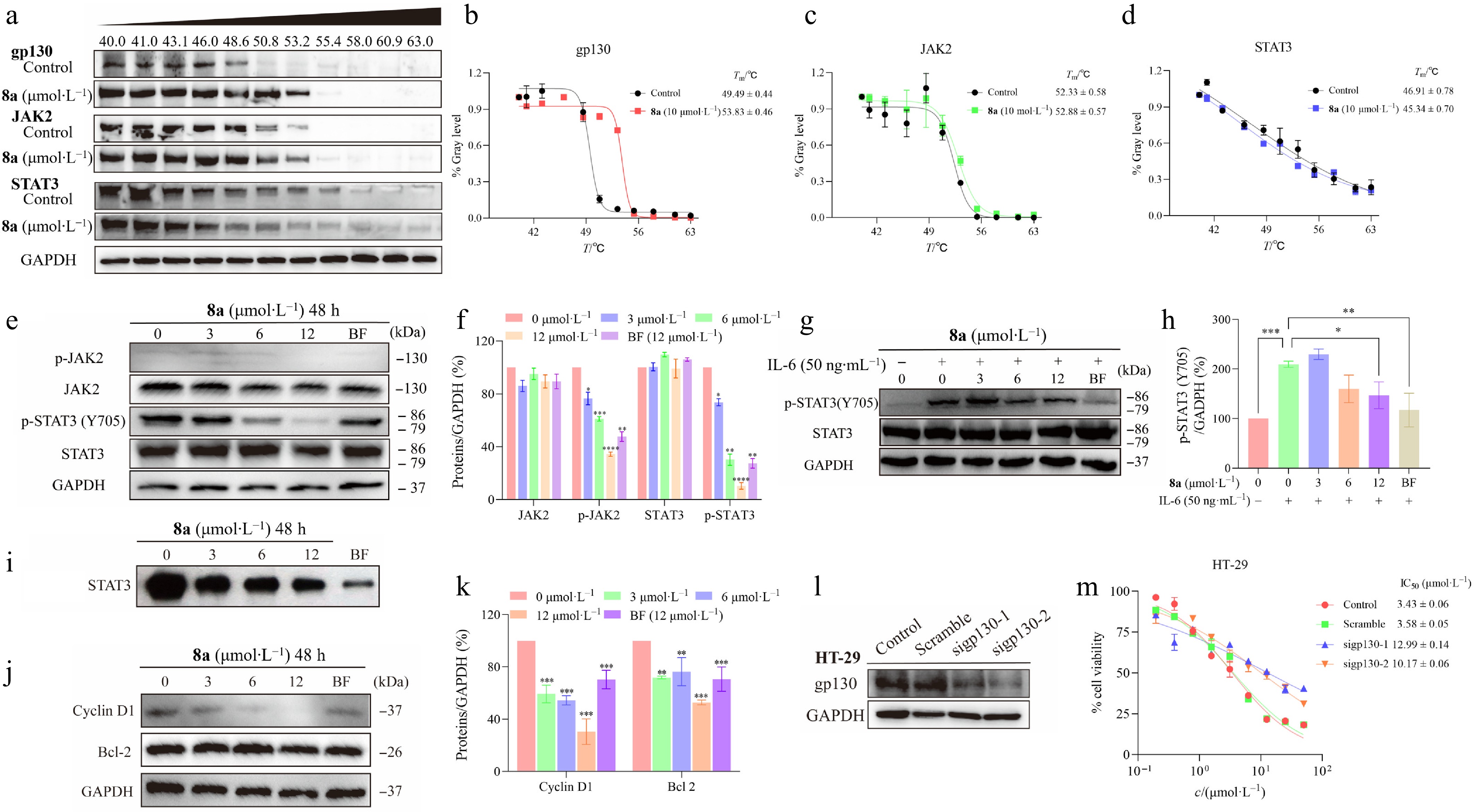
Figure 4.
Compound 8a downregulates the JAK2/STAT3 signaling pathway. (a) CETSA experiments were conducted to assess the impact of compound 8a on the thermal stability of key proteins in the gp130/JAK2/STAT3 signaling pathway, followed by analysis using Western blotting. (b) ImageJ was used to analyze the abundance of gp130 protein in HT-29 cells treated with 8a and heat treatment, and GraphPad was used to fit the Tm curve. The thermal denaturation temperature (Tm) of gp130 increased from 49.49 to 53.83 °C (ΔTm = +4.34 °C). (c) The Tm of JAK2 slightly increased from 52.33 to 52.88 °C (ΔTm = +0.55 °C). (d) The Tm of STAT3 decreased from 46.91 to 45.34 °C (ΔTm = −1.57 °C). HT-29 cells were treated with gradient concentrations of 8a for 48 h. Cell lysates were collected, and the expression levels of relevant proteins were analyzed via Western Blot. GAPDH was used as a control and quantified via ImageJ with a grayscale analysis system. (e), (f) Compound 8a or Bazedoxifene (BF) inhibited the phosphorylation of STAT3 and JAK2 in HT-29 cells. (g), (h) Compound 8a or Bazedoxifene (BF) inhibited the IL-6-induced phosphorylation of STAT3. (i) Compound 8a inhibited STAT3 activation. EMSA analysis of nuclear extracts of equal amounts of total protein containing activated STAT3 from HT-29 cells treated with compound 8a or Bazedoxifene (BF) for 48 h after incubation with an hSIE probe that binds to STAT3. (j), (k) Compound 8a or Bazedoxifene (BF) inhibited the expression of the Bcl-2 and Cyclin D1 downstream genes of STAT3. (l) siRNA was used to knock down gp130 protein in HT-29 cells, and Western blot analysis was performed to assess the expression level of gp130. (m) HT-29 cells with siRNA-mediated knockdown of gp130 were treated with compound 8a to test the change in tumor cell inhibitory activity. Data are presented as the mean ± SEM (n = 3). ** P < 0.01; *** P < 0.001.
We then investigated the effects of 8a with varying concentrations on the JAK2/STAT3 signaling pathway in HT-29 cells. Western blot results indicated that 8a significantly inhibited the phosphorylation of JAK2 and STAT3, but had little effect on their total protein levels (Fig. 4e and f). Notably, 8a has a stronger inhibitory effect on p-STAT3 (Tyr705) at the same concentration (12 μmol·L−1) compared with Bazedoxifene. Studies have shown that IL-6 could induce the phosphorylation of STAT3[30]. We further explored whether 8a could still inhibit STAT3 activation mediated by the gp130 pathway in the presence of free IL-6. As shown in Fig. 4g and h, IL-6 stimulation significantly increased the level of p-STAT3 (Y705), while 8a treatment could effectively reduce its phosphorylation.
Then, we performed an EMSA to assess the effect of 8a on the DNA-binding activity of STAT3. The results demonstrated that 8a inhibited the binding of STAT3 to its target DNA (Fig. 4i). Proteins associated with cell cycle progression, such as Bcl-2 and Cyclin D1, have been demonstrated to be directly associated with STAT3[31,32]. Therefore, we detected their expression. Western blot analysis confirmed that 8a downregulated the protein level of Bcl-2 and Cyclin D1 in a dose-dependent manner (Fig. 4j and k). This functionally verified the inhibition of STAT3 transcriptional activity.
Furthermore, we constructed gp130-knockdown HT-29 cells to demonstrate the dependency of the activity of 8a on the gp130 protein. The results revealed that the inhibitory effect of 8a on the JAK2/STAT3 signaling pathway was significantly attenuated in the HT-29cells with reduced gp130 expression (IC50 increased by 3–4 times).
In summary, this finding confirmed that compound 8a exerts antitumor effects by targeting the gp130 protein, thereby inhibiting the downstream JAK2/STAT3 signaling pathway.
Compound 8a inhibited cell proliferation and induced apoptosis in vitro
-
To further investigate the antiproliferative activity of compound 8a, colony survival tests were performed. HT-29 cells were treated with increasing concentrations of compound 8a (3–12 μmol·L−1) for 48 h and cultured with fresh medium containing 10% fetal bovine serum at 5% CO2 and 37 °C for 2 weeks. After the cells formed colonies, the cells were fixed and stained with crystal violet. The experimental results indicated that compound 8a could inhibit the proliferation of HT-29 cells in a dose-dependent manner (Fig. 5a). Subsequently, the study analyzed HT-29 cells treated with different concentrations of 8a using the Annexin V-FITC/PI double staining method to explore the apoptosis effect of 8a on HT-29 cells. As shown in Fig. 5b and c, 8a dose-dependently induced apoptosis in HT-29 cells. Notably, at the same concentration (12 μmol·L−1), the apoptosis-inducing effect of 8a was superior to that of Bazedoxifene.
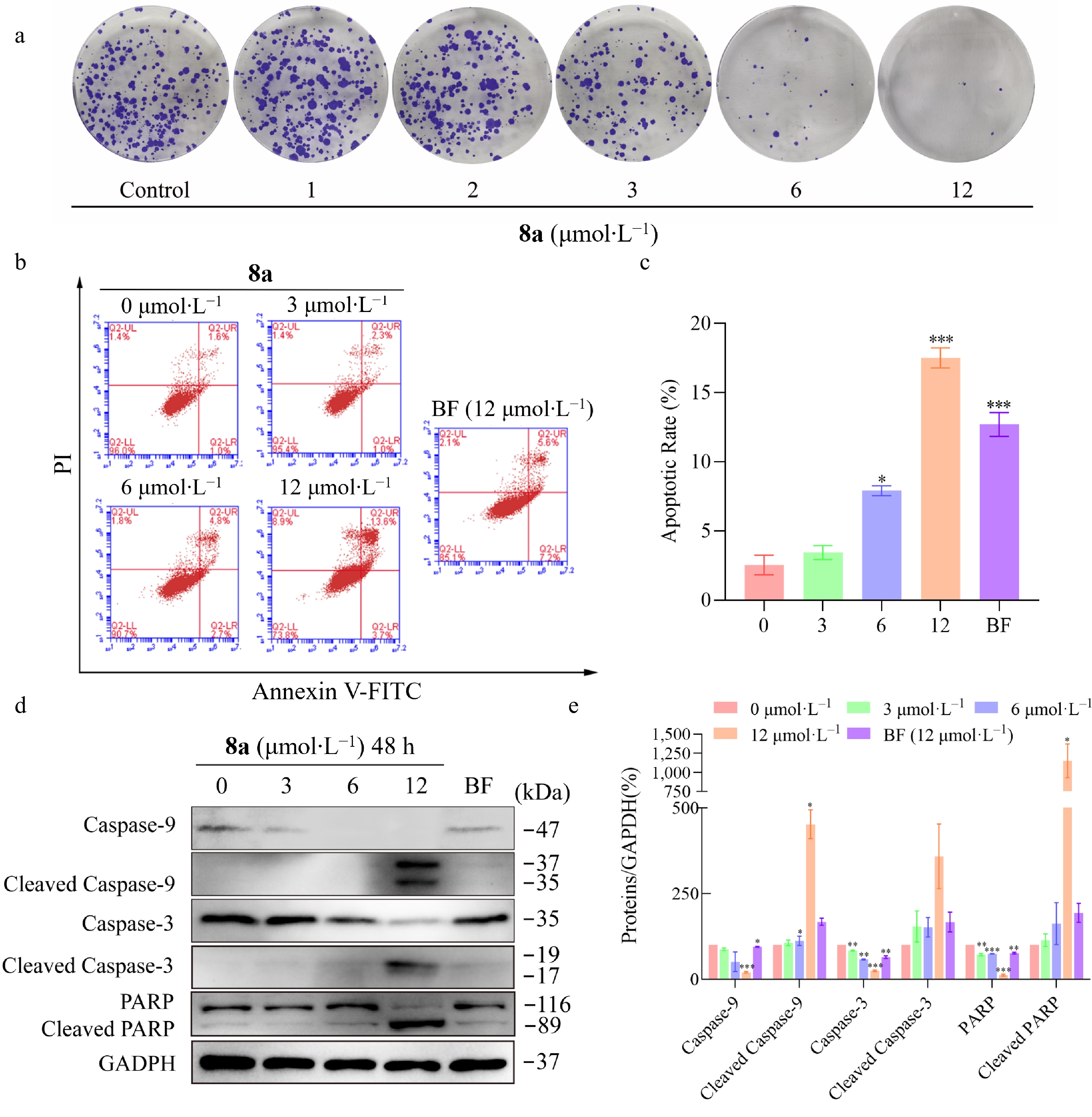
Figure 5.
Compound 8a inhibits HT-29 cell proliferation and induces apoptosis. HT-29 cells were treated with compound 8a or BF at the tested concentrations for 48 h. (a) Compound 8a inhibited the proliferation of HT-29 cells in vitro. (b) Apoptotic effects were detected via Annexin V-FITC/PI staining and flow cytometry. (c) Histograms of the percentage of apoptotic cells (early and late stages of apoptosis). (d) Effects of compound 8a or BF on the protein levels of Caspase-9, Cleaved Caspase-9, Caspase-3, Cleaved Caspase-3, PARP, and Cleaved PARP. The cell lysates were harvested and analyzed via Western blot with the corresponding antibodies. (e) Quantitative analysis of the western blot results via the Image Lab program, with GAPDH serving as the internal control. Data are presented as the mean ± SEM (n = 3). ** P < 0.01 *** P < 0.001.
The Caspase-3, Caspase-9, and PARP are key proteins involved in mitochondria-dependent apoptosis[33]. To investigate the role of this pathway in 8a-induced apoptosis in HT-29 cells, the study examined the expression levels of these proteins treated with 8a by Western blot. The results showed that 8a treatment induced the cleavage of the Caspase-3, Caspase-9, and PARP proteins in HT-29 cells (Fig. 5d and e), suggesting that the apoptosis-inducing effect of 8a mainly depends on the mitochondrial-related caspase signaling pathway.
Compound 8a exhibited a fast clearance rate in vitro
-
The study preliminarily assessed the metabolic stability of the compound in rat liver microsomes. The results indicated that evodiamine exhibited rapid metabolic capability in vitro, with a half-life (t1/2) of 0.99 min and an intrinsic clearance rate of 182.46 μL·min−1·mg−1 (Fig. 6a). In contrast, compound 8a showed significantly improved stability. Its t1/2 was 2.90 min, approximately three times longer than that of evodiamine, while its intrinsic clearance rate decreased to 71.6 μL·min−1·mg−1 (Fig. 6b).
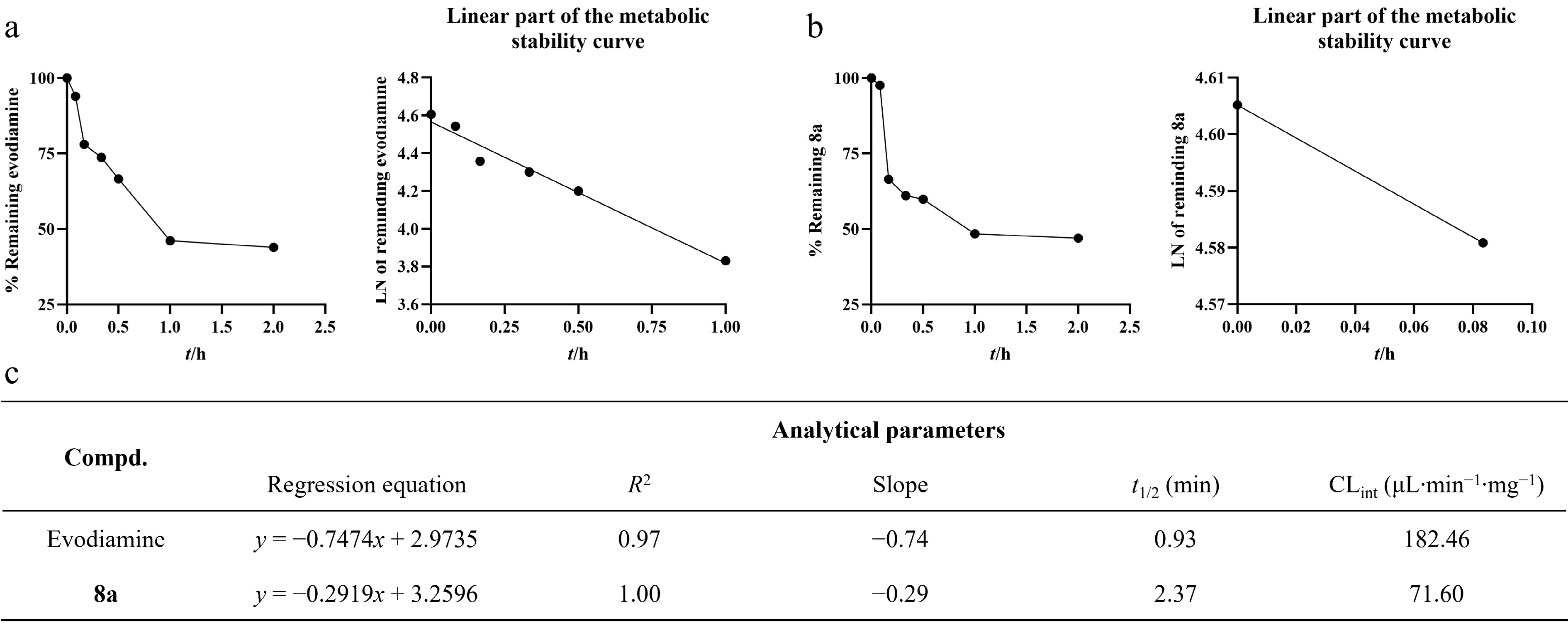
Figure 6.
Metabolic stability of evodiamine and 8a in rat liver microsomes (RLM). (a) The metabolic stability curve of evodiamine in RLM within 2 h and the regression equation of the linear part of the curve. (b) The metabolic stability curve of 8a in RLM within 2 h and the regression equation of the linear part of the curve. (c) Key parameters for the metabolic stability of evodiamine and 8a.
Although liver microsome stability assessment in vitro is a preliminary and important tool for assessing the metabolic characteristics of compounds, its results cannot fully simulate the complex metabolic processes in vivo. In this study, compound 8a demonstrated rapid metabolism, and conducting in-depth pharmacokinetic studies to improve its metabolic stability and exposure is a crucial goal for further development.
Compound 8a inhibited HT-29 xenograft tumor growth in vivo
-
Due to the antitumor activity exhibited by compound 8a in vitro, we further established an animal model of HT-29 xenograft tumor to evaluate its antitumor effect in vivo. HT-29 cells were subcutaneously injected into the right flank of female nude mice to induce solid tumors. Bazedoxifene, a marketed drug regarded as a potential target for gp130, was selected as a positive control. The mice were then randomly divided into three groups and treated with compound 8a (10 and 20 mg·kg−1) or Bazedoxifene (20 mg·kg−1). The results revealed that compound 8a significantly inhibited tumor growth, with a 36.09% reduction in tumor weight in the 10 mg/kg group and a 56.20% reduction in the 20 mg·kg−1 group (Fig. 7a–c). Throughout the experiment, there was no significant decrease in the body weight of the mice (Fig. 7d), indicating that compound 8a had no apparent toxicity.
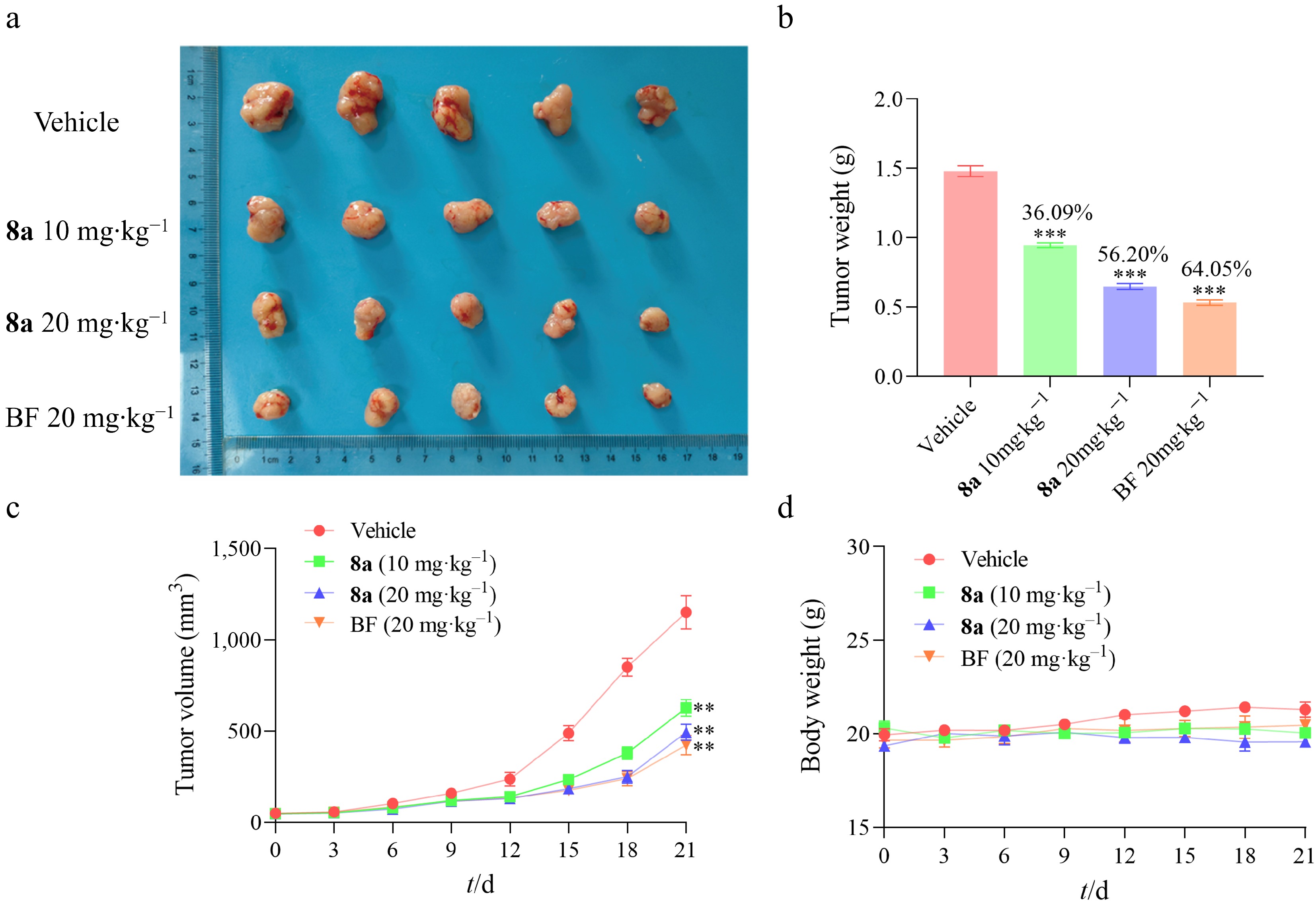
Figure 7.
Compound 8a inhibited the development of HT-29 xenograft tumors in vivo (n = 5). (a) The anatomical diagram of tumors in the experimental group of mice. (b) The comparison of tumor weights in the experimental group of mice was used for the calculation of tumor inhibition rate. (c) Growth curves of mouse tumor volume. (d) Growth curves of mouse body weights. Data are presented as the mean ± SEM (n = 3). ** P < 0.01, *** P < 0.001.
-
The IL-6/IL-6R/gp130 signal is increasingly important as a therapeutic target due to its key role in tumorigenesis. At present, several antibody drugs targeting IL-6 or IL-6R have entered clinical trials. The indications of these drugs mainly focus on inflammatory conditions, and their exploration in antitumor remains relatively limited. However, therapeutic approaches relying on antibody biologics present certain inherent limitations. On one hand, antibody drugs targeting IL-6 may trigger compensatory feedback mechanisms within the signal network due to their potent suppression of IL-6 signaling[34]. On the other hand, while the high specificity of antibody drugs reduces the off-target effects, it also limits the range of signal suppression. For instance, anti-IL-6R antibodies typically only specifically block the signal transduction of IL-6, but are ineffective against the activation pathways of other IL-6 family cytokines that also rely on the gp130 receptor, such as IL-11. In contrast, small molecule inhibitors that directly target the gp130 protein have the potential to block signal transduction depending on this receptor. This may hold a potentially broader and more comprehensive therapeutic strategy.
Artificial intelligence (AI) has become an important technology in modern drug development. To address the challenges brought by the lack of data on gp130 inhibitors, this study attempts to employ a transfer learning strategy to explore the feasibility of discovering compounds with potential inhibitory activity under the conditions of scarce data. Specifically, the study first utilized the STAT3 inhibitor dataset, which has accumulated a large amount of research data, to construct a basic prediction model. Subsequently, the model was fine-tuned using the limited data of gp130 inhibitors. Through activity prediction, ADMET screening, and MCE-18 filtering, we ultimately selected evodiamine for the subsequent experimental verification. Given these results, we conducted structural optimization based on antitumor activity in vitro, and obtained the gp130-targeting compound 8a.
To clarify the mechanism of compound 8a, a series of experimental studies was conducted. The results show that 8a directly binds to the gp130 protein, and the thermal stability of the gp130 protein is significantly improved. It is worth noting that there are no strong direct binding effects between 8a and downstream signals JAK2 and STAT3, demonstrating its selective effect on the gp130 protein. Mechanism studies have shown that the antitumor activity of 8a is mediated by effectively inhibiting the gp130/JAK2/STAT3 signaling axis. Most importantly, the tumor cell inhibitory activity of 8a was significantly reduced in gp130-knockdown HT29 cells. That provides direct evidence for the mechanism of 8a, which depends on the gp130 protein. Furthermore, compound 8a exhibited more stable metabolic characteristics in vitro compared to evodiamine.
Compared to reported gp130 inhibitors, compound 8a has a unique advantage. Its design is based on a rational molecular hybridization strategy that integrates the structural features of evodiamine and Bazedoxifene, resulting in a novel structure. In studies of target affinity, the binding affinity of 8a with gp130 has been extensively validated, providing precise binding data. This addresses the gap in quantitative values for target affinity studies of earlier inhibitors, such as LMT-28, SC144, and GP130-IN-1[11,12,35]. Mechanistically, 8a has been proven to effectively exert its antitumor effects by inhibiting the downstream JAK2/STAT3 signaling pathway mediated by the gp130 protein. The results of functional dependence experiments in cell models indicate that the antitumor effect of 8a is reliant on gp130 expression, compared to the extensive effects of SC144[36,37]. These results collectively support 8a as a gp130-targeted compound with antitumor activity, and provide a solid foundation for future research on gp130 inhibitors.
Although the existing evidence supports the conclusion that 8a targets and inhibits the JAK2/STAT3 pathway through gp130, it is important to recognize the broader biological background of this receptor. As a shared signal transducer for multiple cytokines, including IL-6, LIF, and OSM, the activation of gp130 also recruits SHP2 to initiate the MAPK/ERK pathway, or influences other signaling branches, such as PI3K/Akt and SRC[38−42]. Therefore, targeting and inhibiting the function of gp130 may modulate a broad signaling network beyond the IL-6/JAK2/STAT3 axis. A limitation of this study is that the effect of 8a on signal transduction mediated by other receptor complexes containing gp130 has not yet been verified, such as LIFR-gp130 or OSMER-gp130 complexes. Additionally, the execution of apoptosis may involve a comprehensive signaling network, and the present study cannot completely exclude the contribution of STAT3-independent pathways to 8a-induced apoptosis. Therefore, the study clearly defines compound 8a as an effective gp130 inhibitor that downregulates the JAK2/STAT3 pathway. In future studies, it remains of great significance to verify the inhibitory effect of 8a among cell lines with different gp130 expression levels and clarify its selectivity, potential off-target effects, and in vivo safety.
-
In summary, this study demonstrates that the compound 8a exhibits antitumor activity by targeting the gp130 protein to downregulate the gp130/JAK2/STAT3 signaling pathway. Furthermore, the transfer learning strategy employed in this study has been demonstrated to be applicable for the development of inhibitors targeting gp130, which is characterized by a small amount of data. This provides a practical computational approach for drug development targeting other similar proteins, offering significant implications for broader application and utility.
-
All animal experiments were conducted according to the principles and procedures approved by the Institutional Animal Care and Use Committee (IACUC) of China Pharmaceutical University (License Number: SYXK [SU] 2023–0019, ethics approval number: 2024–05–086).
-
The authors confirm their contributions to the work as follows: study conception and design: Guan J, Xiao L, Liao Y, Richardson DR, Hong J, Yu W; data collection: Guan J, Xiao L, Wang J, Cai G; analysis and interpretation of results: Guan J, Xiao L, Wang J; draft manuscript preparation: Guan J, Xiao L, Wang J, Cai G, Liao Y, Richardson DR. All authors reviewed the results and approved the final version of the manuscript.
-
The data supporting the findings of this study includes a combination of publicly available datasets, literature-curated compound activity data, commercial compound libraries, and model-generated results produced during the current study. The STAT3 inhibitor dataset was obtained from the PubChem Bioassay database (AID 862, https://pubchem.ncbi.nlm.nih.gov/bioassay/862). The candidate compound library was derived from the SelleckChem Natural Products Database (SelleckChem Natural Products Database, www.selleckchem.com/screening/natural-product-library.html). The curated gp130 inhibitor dataset, including molecular structures (SMILES) and binary activity labels, as well as the AI-predicted scores and ranking results for screened compounds, are provided in the Supplementary Materials 1 and 3. All other data generated or analyzed during this study is included in this published article and its supplementary information files.
-
This work was funded by the National Natural Science Foundation of China (Grant No. 82473791) and the Basic Research Program of Jiangsu Province (Grant No. BK20250102). We are also grateful to Yuxuan Hu for his insightful feedback and helpful suggestions for this study.
-
The authors declare that they have no conflicts of interest.
-
accompanies this paper online at: https://doi.org/10.48130/ targetome-0026-0010.
-
#Authors contributed equally: Jie Guan, Lecheng Xiao, Jian Hong
- Supplementary Material 1 SMILES structures and activity labels of 45 literature-reported gp130 small-molecule inhibitors.
- Supplementary Material 2 Figures and tables, NMR spectra, HRMS traces and HPLC traces of synthetic compounds.
- Supplementary Material 3 Model-predicted activity scores and MCE-18 values for 122 ADMET-filtered candidate compounds.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Guan J, Xiao L, Hong J, Wang J, Cai G, et al. 2026. Transfer learning algorithm assisted in the discovery of novel gp130 inhibitors and their application in colorectal cancer treatment. Targetome 2(2): e011 doi: 10.48130/targetome-0026-0010 

Transfer learning algorithm assisted in the discovery of novel gp130 inhibitors and their application in colorectal cancer treatment
- Received: 16 December 2025
- Revised: 05 February 2026
- Accepted: 16 February 2026
- Published online: 20 March 2026
Abstract: The gp130 protein is the common receptor for IL-6 family cytokines, and its aberrant activation significantly promotes tumor progression. However, research on inhibitors targeting gp130 remains limited. Here, we employed an artificial intelligence (AI)-assisted drug design strategy and identified evodiamine as a candidate gp130-targeting scaffold. Based on this, we developed a series of novel indolopyridine-based small molecules. The representative compound 8a effectively downregulates the JAK2/STAT3 signaling pathway by directly targeting the gp130 protein (KD = 2.17 μmol·L−1), disrupting the binding of STAT3 to DNA, and thereby inducing apoptosis. In HT-29 xenograft tumor models, 8a demonstrated 56.20% tumor growth inhibition (20 mg·kg−1). In summary, the compound 8a can be used as a gp130-targeted compound for the treatment of colorectal cancer. Furthermore, the method applied in the research can provide a feasible case for the development of drugs targeting related pathways.
-
Key words:
- Gp130 /
- Bazedoxifene /
- Evodiamine /
- AI-assisted drug design /
- Transfer learning algorithm /
- Colorectal cancer