








-
In recent years, the rapid progress of artificial intelligence (AI) algorithms has been fundamentally reshaping the mode of drug research and development. Traditional experimental methods are often time-intensive and resource-heavy, yet AI-based techniques can efficiently pinpoint potential patterns from vast volumes of chemical and biological datasets, thus greatly boosting the efficiency and success rate of drug development[1]. Within medicinal chemistry research, AI algorithms enable precise prediction of the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of compounds. Such predictive power offers critical insights into lead compound optimization and helps effectively mitigate the risk of failures in the late stages of drug development[2]. For drug target identification, graph neural networks (GNNs) can accurately forecast drug-target interactions (DTIs) through direct learning of molecular topological architectures, outperforming traditional descriptor-driven methods in this aspect[3]. Notably, generative AI models including deep reinforcement learning frameworks have proven capable of designing novel molecular structures with tailored properties, which shifts AI’s role from passive data prediction to active molecular creation[4]. Furthermore, AI excels at processing high-dimensional cell imaging data, mining actionable insights from scientific literature, integrating multi-omics datasets, and even offering novel perspectives for unraveling the complexities of biological systems[5]. Overall, these progressive technological advances have solidified AI's position as a core driver of paradigm transformation in drug research and development, and delivered a powerful impetus for translating basic scientific research into clinical practice.
Natural products, particularly the bioactive ingredients isolated from traditional Chinese medicine (TCM), have long been a vital source for developing innovative drugs, owing to their rich structural diversity and well-characterized biological activities[6]. However, the synergistic mechanisms of action of TCM involve a variety of active components and their corresponding molecular targets, which together form a highly intricate regulatory network. This characteristic makes the traditional single-target research strategy insufficient for the systematic investigation of TCM's pharmacological effects[7]. A core challenge in contemporary TCM research is to systematically decipher the complex interconnections between TCM's chemical components, molecular targets and its overall biological activities, and traditional research methods have an inherent inability to tackle such systemic problems effectively[8]. Notably, breakthroughs in metabolomics, proteomics, and transcriptomics have established a robust database for interpreting the mechanisms of TCM action from a systems biology perspective[9]. At the same time, newly developed experimental technologies such as chemical proteomics and thermal shift analysis are approaching technical maturity, which allows for the direct identification of drug targets throughout the entire proteome[10]. Presently, the combination of these state-of-the-art technologies with AI algorithms has led to the construction of a rigorous methodological system, thereby offering an unprecedented chance to unravel the systemic complexity underlying TCM's pharmacological effects.
This review aims to systematically elaborate on the AI algorithms driving the modern elucidation of TCM from 2012 to 2025, by analyzing the chemicalome, targetome, and bioactivome of TCM systems. The chemicalome is defined as the full set of chemical constituents derived from TCM, including prototype compounds and their metabolites[11]. The targetome encompasses the complete range of biological macromolecules, including primarily receptors, enzymes, and ion channels, with which the TCM chemicalome interacts[12]. The bioactivome denotes the full spectrum of integrated biological activities and phenotypic effects arising from the systemic modulation of the targetome by the TCM chemicalome[13]. Based on the above-defined concepts of the chemicalome, targetome, and bioactivome, this review is structured as follows. First, we demonstrate how AI technology, by leveraging technological progress in mass spectrometry analysis and metabolite identification, enables the comprehensive characterization of the complex chemicalome of TCM. Next, we explore how various algorithms, such as deep learning and GNNs, facilitate the identification of the TCM targetome. In addition, we expound on how AI integrates multi-omics datasets, systematically interprets the bioactivome induced by TCM intervention, and uncovers the multi-pathway synergistic mechanisms of action of TCM. Finally, we explore the potential of AI algorithms in elucidating the holistic mechanisms of TCM, which is achieved by mapping the interactive relationships among the chemicalome, targetome, and bioactivome. This analytical framework highlights that AI acts not only as a tool to improve research efficiency, but also as a foundational method for systematically dissecting TCM systems.
-
Analyzing the in vitro TCM chemicalome acts as a foundational step for identifying its pharmacologically active components[14]. Traditional analytical chemistry, however, encounters considerable challenges in tackling the complex characteristics of herbal medicines. By contrast, AI algorithms possess distinct advantages: they can extract the intrinsic "fragment-structure" correlations from large-scale mass spectrometry datasets, enabling efficient resolution of these challenges[15]. This advancement has spurred technological evolution and transformed research from the traditional, experience-driven manual database querying to proactive prediction and generation.
In terms of applications within this field, AI technology has advanced steadily (Fig. 1, left panel). In the early phases, molecular network algorithms such as Global Natural Products Social Molecular Networking (GNPS) could compute and visualize the similarity of clustered mass spectrum fragments, revealing numerous structural homologies. This addressed the arduous task of manual substance tracing and notably accelerated the discovery of known compound families[16]. Later, integrated computing tools like SIRIUS, designed to identify unknown structures from tandem mass spectrometry, leveraged deep learning to conduct fragmentation tree analysis and structural searches, markedly enhancing the precision of de novo structural analysis for unknown compounds[17].
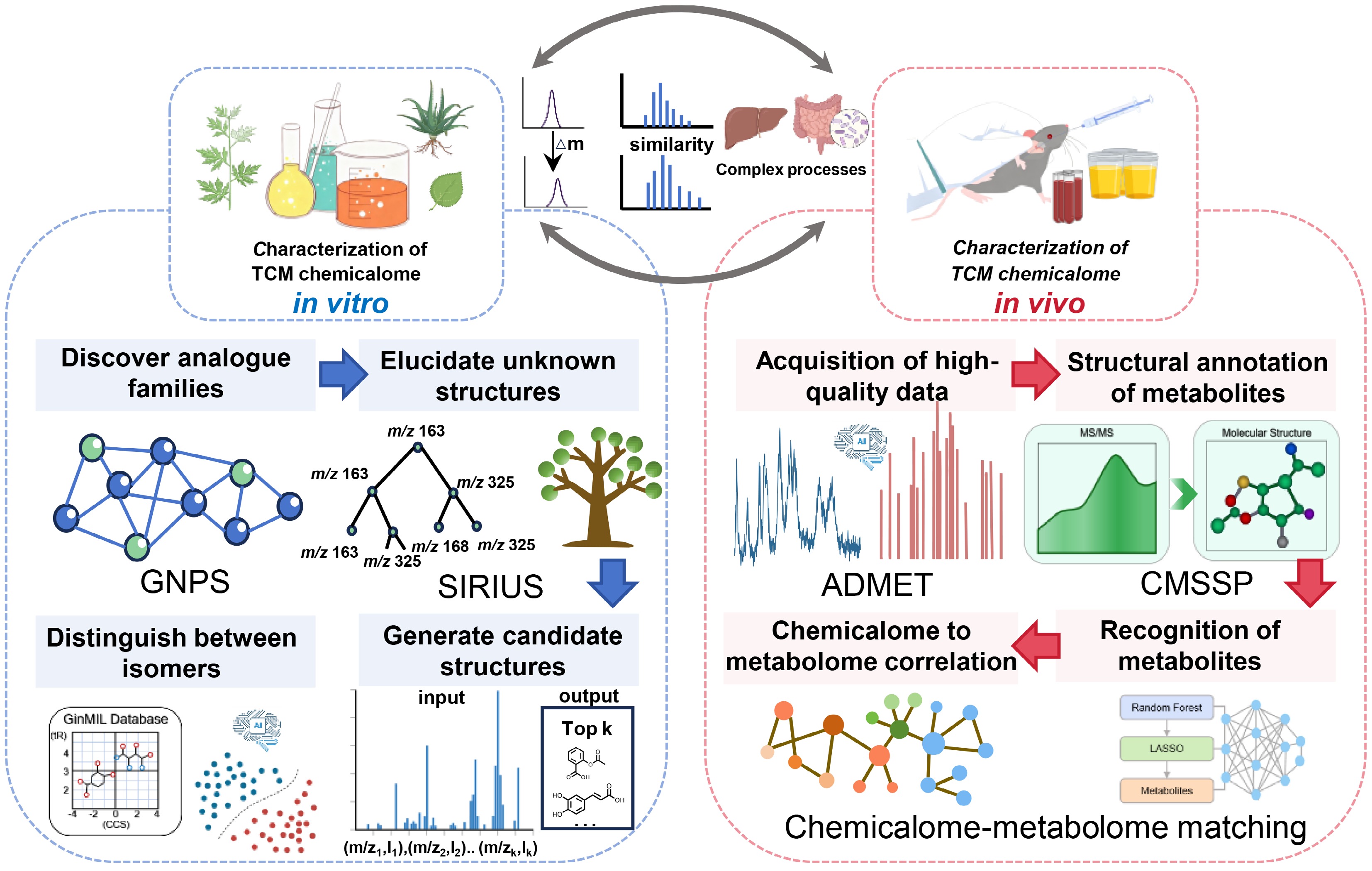
Figure 1.
Schematic diagram for AI-driven revelation of the TCM chemicalome. The diagram delineates comprehensive characterization strategies for the TCM chemicalome, categorized into in vitro (left panel) and in vivo (right panel) approaches. The in vitro workflow integrates GNPS for analog discovery via molecular networking, SIRIUS for structural elucidation using fragmentation tree analysis, and GinMIL for isomer differentiation. The in vivo workflow employs mass spectrometry for high-quality ADMET data acquisition, CMSSP for deep learning-based metabolite annotation, and a chemicalome-metabolome matching approach for the construction of metabolic networks.
Along with the development of neural networks, the reverse design concept of "structure-based mass spectrometry prediction" has been adopted. It has become possible to predict mass spectrometry behavior with high accuracy, which has revolutionized the method of compound verification[18]. Recently, there have been some new breakthroughs at the forefront of technology. For example, tools like mass spectrometry-driven de novo molecular structure generation (MSNovelist) can now deduce possible molecular structures directly from a single MS/MS spectrum. This means that the role of AI has shifted from primarily relying on database comparison to actively generating structures. Importantly, this method no longer depends entirely on physical reference substances[19]. As a result, even in the absence of known compound samples, researchers now have a completely new pathway to identify novel molecules in natural products.
In practical applications, AI has proven capable of addressing complex classification tasks. For instance, researchers integrated deep neural networks (DNNs) with pseudo-targeted metabolomics to differentiate between various types of easily confused ginseng varieties, achieving classification accuracy that outperformed traditional statistical methods by a significant margin. The key strength of DNNs lies in its capacity to automatically discover hidden patterns within large volumes of complex data. Beyond validating the model's high reliability, this study also precisely identified the core chemicalome responsible for the differences among these closely related ginseng types[20]. In another research effort, scientists combined machine learning with ion mobility separation technologies to develop a multi-dimensional information database (GinMIL), which encompasses the full range of ginsenosides. By effectively leveraging retention time and collision cross section data, this technological system has successfully overcome the major challenge faced by traditional mass spectrometry, that is its difficulty in distinguishing ginsenoside isomers[21].
Table 1. Comparative analysis of AI tools for TCM in vitro and in vivo chemicalome characterization.
Tool name Core technologies Key functionalities Advantages Limitations GNPS Molecular network algorithm, MS/MS fragment clustering, unsupervised learning 1. MS/MS fragment similarity visualization;
2. Structural homology mining;
3. Known compound family screening1. Intuitive MS data visualization;
2. Enables rapid natural product tracing;
3. Accelerates discovery of known TCM compound families1. Reliance on existing MS databases;
2. Low structural annotation accuracy for rare TCM components with unique fragments;
3. Clustering affected by low-quality MS dataSIRIUS Deep learning, fragmentation tree analysis, tandem MS database matching, de novo structural analysis 1. Tandem MS-based unknown structure identification;
2. MS fragment decomposition;
3. Structural similarity search1. Enables computational de novo prediction for unknown TCM components;
2. Integrated strategies enable higher annotation accuracy than single-algorithm tools;
3. Supports batch processing of large-scale TCM MS data1. Reduced de novo accuracy for TCM components with multiple chiral centers;
2. Relies on high-quality tandem MS data;
3. Slow analysis for high-diversity TCM samplesMSNovelist MS-driven de novo molecular structure generation, deep neural network, single MS/MS spectrum interpretation 1. Direct molecular structure deduction from single MS/MS spectrum;
2. Reference-free unknown structure generation1. Breaks dependence on physical references;
2. Requires only a single MS/MS spectrum;
3. Improves novel TCM compound discovery efficiency1. Diverse structural candidates require manual verification;
2. Low accuracy for TCM macromolecules;
3. Poor adaptability for rare TCM componentsGinMIL Machine learning, ion mobility separation, multi-dimensional database construction 1. Comprehensive ginsenoside database construction;
2. Ginsenoside isomer differentiation1. Solves the traditional MS bottleneck in TCM isomer distinction (ginsenosides);
2. Multi-dimensional data fusion enables high component identification accuracy;
3. Specialized for saponin-rich TCM targeted analysis1. Ginsenoside-focused;
2. Relies on ion mobility separation equipment;
3. Unable to analyze unknown saponin isomersSwissADME Machine learning, QSAR model, molecular descriptor calculation 1. Rapid ADME indicator assessment for small molecules 1. Fast prediction for high-throughput TCM active component screening;
2. Predicted indicators align with TCM small-molecule in vivo metabolic characteristics;
3. Visualizable results1. Applicable only to small molecules;
2. Low prediction accuracy for rare TCM components with unique structures;
3. Lacks prediction of in vivo metabolic pathways for TCM componentsADMETlab 2.0 Integrated machine learning, multi-task feature learning, large-scale ADMET database 1. Comprehensive prediction of ADMET indicators;
2. Toxicity/metabolic stability prediction;
3. Drug-drug interaction prediction1. Provides extensive ADMET indicators relevant to TCM research;
2. High accuracy;
3. Supports batch/customized prediction;
4. Predicts TCM compound prescription drug-drug interactions1. Basic molecular descriptor knowledge required;
2. TCM metabolite ADMET prediction needs metabolic pathway tools;
3. Lacks specific guidance for lead optimization of TCM componentsCMSSP Contrastive learning model, molecular graph convolution,
MS-structure unified representation space construction1. Metabolite structural annotation;
2. Low-abundance metabolite identification in complex biological matrices;
3. In vivo MS data interpretation1. The MS-structure unified representation space significantly enhances annotation accuracy;
2. Strong anti-interference ability for complex biological matrices;
3. Reduces dependence on in vivo metabolite databases1. Requires large-scale natural product MS-structure paired training data;
2. Slow analysis for TCM in vivo samples with massive metabolites;
3. Sensitive to high-abundance endogenous biological matrix interferencesChemicalome-Metabolome Matching Platform Machine learning, metabolic network construction, chemicalome-metabolome correlation 1. In vivo MS raw data processing;
2. Endogenous background elimination;
3. Parent compound and metabolite distinction;
4. Screening of xenobiotic metabolites1. Integrates in vitro-in vivo matching;
2. Time-series filtering outlines TCM component temporal metabolic characteristics;
3. Constructs complex TCM metabolic networks;
4. Adapts to dynamic biological matrix processing1. High in vitro and in vivo MS data quality requirements;
2. Large-scale metabolic network construction needs high computing resources;
3. Weak automatic identification for rare TCM metabolic pathwaysAI-driven analysis of the in vivo TCM chemicalome
-
Generally, AI has become a core tool for systematically analyzing the complex in vivo chemicalome of TCM (Fig. 1, right panel). Its role has evolved from an auxiliary tool to a core driving force that can identify and discover new structures. It allows for a more thorough understanding of the full range of components actually contained in TCM. In fact, what really matters about TCM is how it affects our bodies after administration. Therefore, many researchers have focused on elucidating the in vivo chemicalome of TCM[22]. The in vivo TCM chemicalome is the collection of parent compounds and their metabolites that exist in the blood and tissues after administration, which constitutes the direct material basis of the pharmacological action. Compared with the in vitro analysis, the in vivo analysis of the TCM chemicalome is extremely difficult because of the low concentration, the unknown metabolic processes, and numerous interferences brought by the complex biological matrix[23].
The application of AI algorithms, online platforms, and related tools has greatly broken through the limitations of traditional analysis[24]. They have thus become the core support for efficient research on the in vivo TCM chemicalome. Moreover, innovations in AI algorithms have significantly improved the accuracy of predicting relevant properties of the in vivo TCM chemicalome. Specifically, among these algorithms, those based on multitask deep featurization use molecular graph convolution technology. They actively learn the core molecular features linked to ADMET properties. This kind of algorithm has raised ADMET prediction accuracy by a breakthrough margin. It has also effectively solved a key shortcoming of traditional methods: they cannot accurately predict novel chemical substances[25]. In addition, researchers have developed a combined classification model for blood-brain barrier permeability with AI technology. Tailored specifically for natural product research, this model has been successfully applied to predict the blood-brain barrier permeability of TCM components and their in vivo metabolites. Comprehensive validation through these experiments confirmed the model's reliability, with its prediction accuracy hitting 81%. Additionally, the model clearly pinpointed the key molecular factors that influence the blood-brain barrier permeability of TCM components[26]. At the same time, a variety of AI-assisted online platforms and tools provide convenient and practical support for analyzing the in vivo TCM chemicalome. SwissADME, in particular, is a free online tool designed for this research field. It can rapidly assess key indicators of small TCM molecules, such as pharmacokinetic properties and drug-likeness[27]. Another notable tool is ADMETlab 2.0, an integrated online prediction platform that can comprehensively and accurately forecast over 70 ADMET-related detection indicators for TCM components and their metabolites[28].
Beyond the ADMET prediction approaches discussed above, combining mass spectrometry with AI algorithms also offers an effective means of analyzing the in vivo TCM chemicalome. Leveraging AI technologies, researchers have developed the Contrastive Mass Spectra-Structure Pretraining (CMSSP) model. This model is capable of building a unified representation space for mass spectra and molecular structures, which significantly boosts the accuracy of metabolite structural annotation. When applied to the analysis of Glycyrrhiza glabra metabolites, the CMSSP model achieved exceptional performance, with its identification accuracy surpassing that of existing methods by a considerable margin. As such, it has become an effective tool for the precise identification of low-abundance metabolites within the in vivo TCM chemicalome[29].
AI can also be integrated with matching techniques for in vitro and in vivo chemicalomes to support the completion of a range of key procedures, such as raw data processing, endogenous background elimination, and distinction between prototype components and their metabolites. Through these procedures, metabolic networks of complex systems can be effectively established. For example, following the administration of Mai-Luo-Ning injection to rats, the metabolites in the animals were successfully linked to their corresponding parent compounds using the chemicalome-metabolome matching approach[11]. For TCM formulas, AI-supported integrated strategies allow for systematic analysis of in vitro and in vivo chemicalomes. These strategies incorporate techniques including mass defect filtering and characteristic product ion filtering, which have been used to successfully identify and match 37 prototypes and 39 metabolites in Huanghou antidiarrhea dropping pills[30], as well as 21 prototypes and 70 metabolites in Chai-Gui decoction[31]. Additionally, these strategies have clarified the tissue distribution patterns of these prototypes and metabolites. Furthermore, AI serves as the foundation for a time-series-dependent global data filtering strategy, which can quickly screen out xenobiotic metabolites in dynamically complex matrices. In a co-culture system of Ginkgo biloba extract and gut microbiota, this strategy identified 107 flavonoid-derived metabolites and clearly outlined their temporal metabolic profiles[32]. Overall, AI algorithms fulfill their roles through diverse approaches: they optimize the analytical workflows of the in vivo TCM chemicalome, integrate multi-dimensional analytical data, and clarify dynamic metabolic characteristics, thereby providing efficient and viable technical support for the systematic analysis of the in vivo TCM chemicalome. The core technologies, key functionalities, advantages, and limitations of the above AI tools are summarized in Table 1.
-
Traditional drug discovery has predominantly followed a single-target paradigm, attributing the therapeutic effect of drugs to their high-affinity binding to a key biological target in order to regulate physiological activities[33]. However, biological systems are essentially constructed by complex networks. Many drugs with clinical value are not effective by acting on a single target, but by simultaneously regulating multiple targets. To systematically describe this multi-target mode of action, the concept of targetome has been introduced[12]. It refers to the entire set of biological targets that a molecule or a complex drug system acts on and regulates in vivo. These targets mainly include enzymes, receptors, ion channels, and proteins involved in key biological processes such as signal transduction, metabolic regulation, transcriptional regulation, and epigenetic modification[34]. The schematic diagram for AI-facilitated discovery of the TCM targetome is shown in Fig. 2.
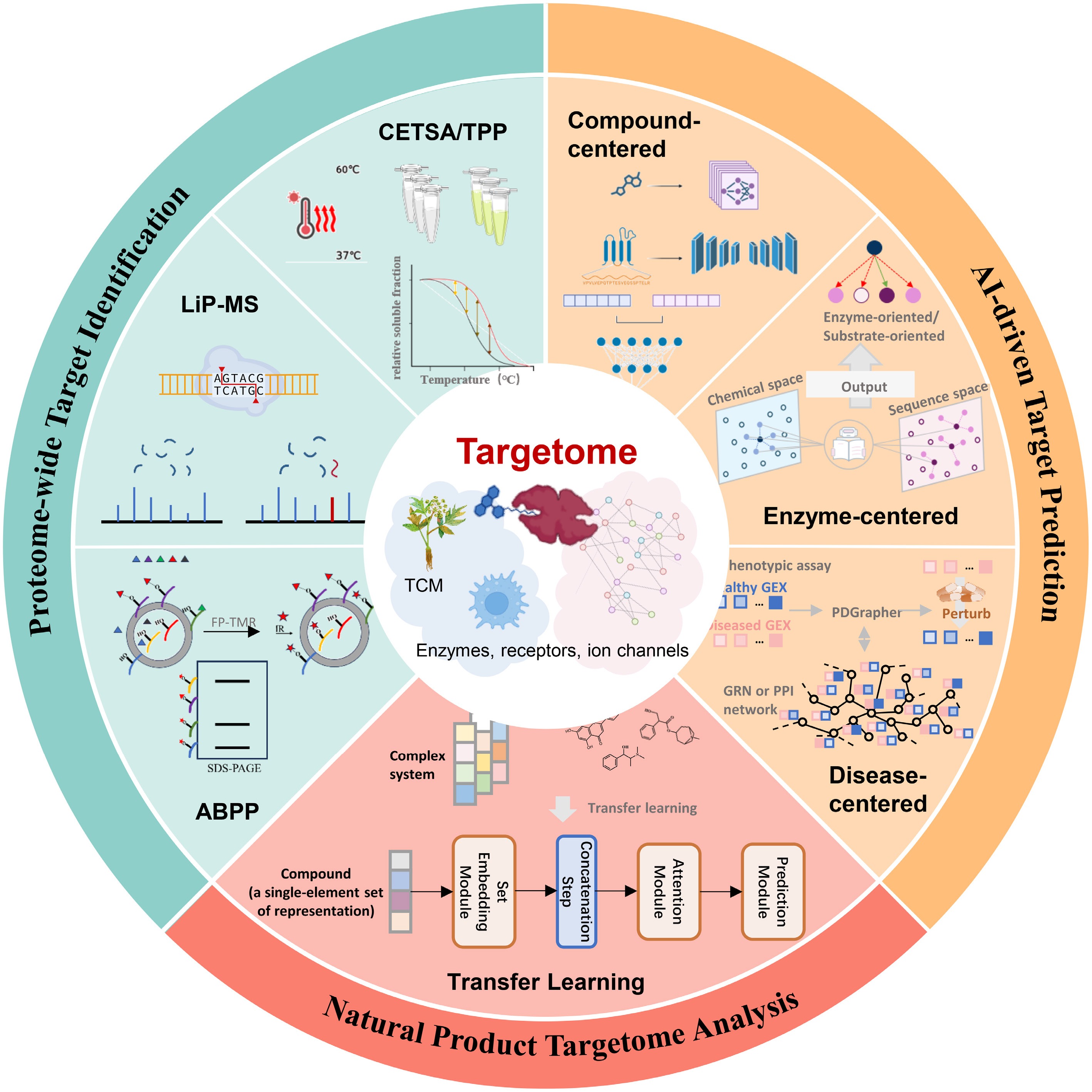
Figure 2.
Schematic diagram for AI-facilitated discovery of the TCM targetome. The diagram integrates three methodological components: AI-driven target prediction by compound-centered, enzyme-centered, and disease-centered methods; proteome-wide target identification by CETSA, TPP, LiP-MS, and ABPP; and natural product targetome analysis by transfer learning models.
AI-facilitated algorithmic strategies for the TCM targetome
-
As a powerful tool for exploring the TCM targetome, AI-enabled algorithms support the systematic analysis of this complex system, which in turn helps clarify the multi-component and multi-target mechanisms of TCM through advancements in compound-target interaction prediction, affinity evaluation, and target deconvolution. GNNs and convolutional neural networks (CNNs) serve as the fundamental basis for these algorithmic strategies: GraphDTA employs molecular graphs to predict compound-target affinity[35], whereas DeepDTA utilizes CNNs to achieve 1D sequence encoding[36]. To further improve the interpretability and generalization ability of these models, attention mechanisms and transformer architectures are integrated: DrugBAN combines bilinear attention with domain adaptation techniques to model local drug-target interactions[37]; AttentionMGT-DTA integrates drug molecular graphs and protein pocket characteristics[38]; TransformerCPI realizes compound-protein interaction prediction using only sequence information for targets without known 3D structures[39]. EviDTI adopts evidential deep learning to conduct uncertainty quantification, thus prioritizing compound-target pairs with high confidence[40].
A series of innovative algorithmic tools further extend the scope of TCM targetome exploration: CATNIP is capable of predicting the compatibility between enzymes and their substrates, which provides insights into the enzyme-mediated mechanisms of TCM[41]; DeepDTAGen adopts a multitask learning approach to simultaneously perform drug-target affinity prediction and target-aware molecule generation[42]; SurfDock applies diffusion models to achieve accurate prediction of protein-ligand complexes[43]. In terms of disease-associated targets, TRESOR incorporates omics data to screen and identify therapeutic targets[44]; PDGrapher is designed to predict combinatorial perturbagens that can reverse disease phenotypes[45]; NetFlow3D elucidates the impacts of mutation-driven target networks[46]. In summary, these AI-enabled algorithms show considerable potential in deciphering the complex targetome of TCM. By enabling precise target identification and detailed interaction profiling, they can provide strong support for interpreting the therapeutic mechanisms of TCM from a target-oriented perspective.
AI-integrated experimental technologies for TCM targetome validation
-
The gap between computational inference and real biological processes still needs to be bridged by AI predictions that rely on high-quality data and experimental verification[47]. Thermal stability detection methods, such as thermal proteome profiling (TPP) and cellular thermal shift assay (CETSA), have become the gold standard for high-throughput target verification. By monitoring ligand-induced changes in protein thermal stability in native cellular environments, these label-free methods can resolve complex proteoform-specific interactions and verify target engagement at the proteome level[48−50]. In order to attain a higher degree of structural resolution, analytical methods based on proteolysis and accessibility do offer complementary research perspectives. Techniques such as limited proteolysis-mass spectrometry (LiP-MS) and LiP-Quant can detect subtle conformational changes induced by ligand binding, as well as changes in the accessibility of residues on the protein surface[51,52]. Additionally, based on activity-based protein profiling (ABPP) and emerging label-free platforms, covalent and high-affinity non-covalent interactions across the entire proteome are mapped via reactive probes or competitive labeling[53,54]. Unlike thermal analysis that focuses on detecting physical changes, these chemical proteomic techniques can directly produce chemical evidence of target occupancy.
The combination of experimental techniques and computational models leads to a "prediction-verification-optimization" closed loop. AI methods can screen and lock high-confidence candidate targets within a huge search space, and experimental methods can effectively eliminate false positives through empirical verification[55]. The high-quality interaction data thus obtained can be used as high-quality ground truth to retrain and optimize AI models, enabling more effective enhancement of model performance[56].
Representative applications of AI-facilitated TCM targetome discovery
-
Natural products have complex structures and the characteristics of multi-target binding, which indeed pose challenges to traditional target identification. In early computational processes, researchers mostly used structural similarity-based inference methods to predict potential targets[57]. In recent years, with the development of AI technology, target prediction has been shifting to a phenotype-driven and task-specific research model. To address the insufficient adaptability of structural descriptors in new scaffold systems, phenotype-driven reverse inference models have begun to mine high-dimensional biological activity data, thereby establishing a correlation between the transcriptome features induced by compounds and protein interaction networks[58].
The integration of deep learning and transfer learning has made it feasible to carry out systematic modeling of multi-component TCM systems, and related frameworks have shown significant value in practical applications. The Set Embedding and Transfer learning model for Complex systems (SETComp), for example, incorporates permutation invariance and treats TCM formulas as unordered molecular sets, rather than as a single entity for isolated processing. This model can efficiently transfer knowledge from single-drug pharmacology to multi-drug combination systems by pre-training on large-scale single-drug transcriptomic data and fine-tuning on natural product datasets. In the rigorous evaluation of complex TCM systems, the model attained a high prediction accuracy, demonstrating AI’s capability to translate the disordered characteristics of TCM systems into molecular hypotheses with practical significance[59]. This strategy marks a transformation in the research paradigm, shifting from predicting the correspondence between isolated compounds and targets to analyzing the overall and systematic regulatory network.
Beyond computational models, key research has illustrated the applicability of chemical proteomic approaches in elucidating the action mechanisms of natural products and facilitating TCM targetome discovery. By combining activity-based probes with quantitative mass spectrometry, researchers have systematically delineated the covalent target profiles of terpenoid compounds (e.g., eriocalyxin B), pinpointed their specific binding interactions in the ubiquitination pathway with high precision, and thus clarified the molecular underpinnings of their anti-proliferative activity[60]. As a valuable complement to covalent capture strategies, emerging label-free analytical platforms now allow for the concurrent characterization of non-covalent binding interactions and target occupancy statuses in living cells. For instance, this analytical method has been successfully applied to identify core RNA metabolism-associated targets of withaferin A in triple-negative breast cancer, which also validates the platform's capacity to resolve high-occupancy binding interactions in complex biological milieus[61].
Furthermore, the integration of fragment-based target research (FBTR) and phenotype-target coupled drug screening (PTDS) methodologies can facilitate a more in-depth understanding of the multi-target binding characteristics of natural products[62,63]. Another representative framework is Herb-CMap, which is specifically tailored for TCM research. It leverages multimodal data to combine gene expression features with protein-protein interaction networks, enabling the identification of active ingredients in complex TCM systems and their corresponding therapeutic targets, thus serving as an effective tool for clarifying the molecular mechanisms underlying TCM systems[64].
Numerous practical studies have validated the value of AI technologies in the targetome discovery of TCM formulas. For the treatment of metabolic-associated fatty liver disease (MAFLD) with Qigui Jiangzhi Formula, AlphaFold3 predicted the structures of key targets AMPK and SIRT1 with high confidence. Combined with molecular docking, these predictions clarified the formula's component binding and its regulatory mechanism through the AMPK/SIRT1-TFEB axis[65]. For XiaoErFuPi granules against functional dyspepsia, an unsupervised machine learning strategy classified its compounds into functional modules, clarifying its multi-target mechanism and identifying potential active components[66]. Additionally, AI-assisted target analysis revealed that Fuzheng Jiedu Decoction inhibits coronaviruses by blocking SARS-CoV-2 Spike protein-ACE2 binding and regulating inflammatory pathways, with caffeic acid and octyl gallate as core active components[67]. Overall, these integrated efforts demonstrate that AI technologies are effective in elucidating the TCM targetome.
-
The exploration of the functional mechanisms of TCM is undergoing a profound transformation driven by AI. By systematically analyzing the causal links between the chemical components of TCM, their corresponding action targets, and the resulting biological activity responses, AI has established a novel methodological system for research into TCM modernization[68]. Within this newly developed system, the bioactivome plays a vital bridging role. It connects the upstream targetome and further converts the interactions between the chemicalome and targetome into dynamic functional changes at both the cellular and molecular levels[69]. When these microscopic alterations are comprehensively integrated, they eventually give rise to the observable pharmacological effects of TCM. As a result, the bioactivome has become the core focus for understanding TCM[70]. AI technologies combine multi-omics data with advanced computational models, aiming to systematically decode this complex network of functional responses. This decoding process enables us to understand the mechanisms underlying the synergistic action of multiple components in TCM[24].
Conceptual analysis of the bioactivome
-
To clarify this concept, we define the bioactivome in this review as the set of biological activities arising from the molecular and cellular events triggered by the action of the TCM chemicalome on the targetome (Fig. 3). These responses are typically multifaceted, combining to form a comprehensive profile of functional activity. This feature can be characterized by multidimensional dynamic indicators: those mainly at the molecular, pathway, and network levels[71]. More specifically, at the molecular level, we can observe quantitative variations in biomolecules, including gene expression levels, proteins and their modification states, as well as metabolite concentrations[72]. At the pathway level, the degree of activation or inhibition of signaling pathways can be assessed through aggregated analyses and special algorithms[73]. At the network level, the topology and dynamic changes of functional interaction networks such as gene coexpression networks and protein-protein interaction networks are described using parameters such as node centrality and module stability[74]. The integration of these multifaceted indicators forms the quantitative basis of the bioactivome. Therefore, a key focus of current research driven by AI is to elucidate the complex connections among various levels within the bioactivome as well as the relationship between the functional level and the final macroscopic therapeutic outcomes[75].
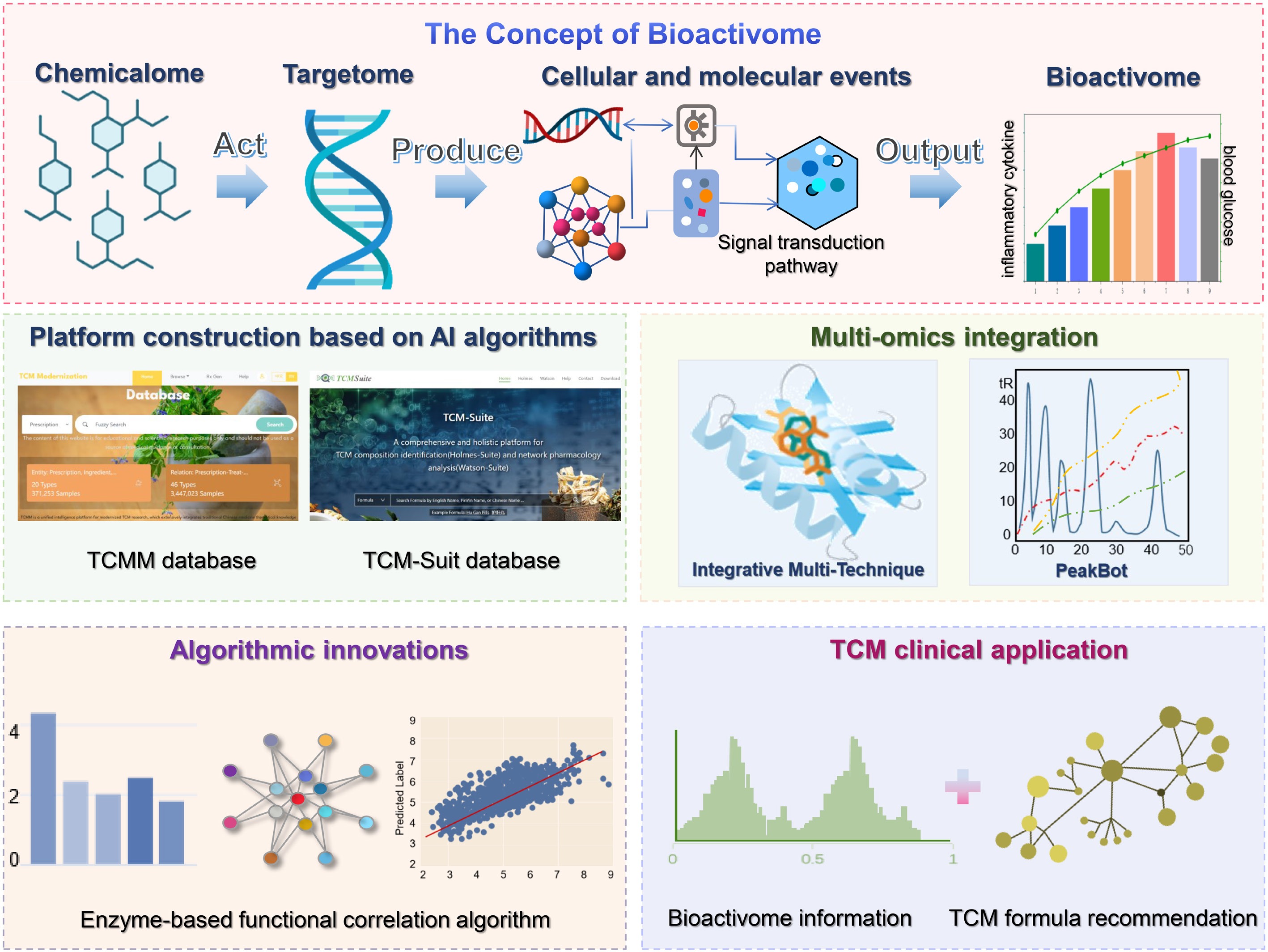
Figure 3.
Schematic diagram for AI-accelerated revelation of the TCM bioactivome. The diagram introduces the concept of the bioactivome, which refers to the set of biological activities derived from the molecular and cellular events induced by the action of the TCM chemicalome on the targetome. Research on the bioactivome mainly focuses on platform construction based on AI algorithms, multi-omics integration, algorithmic innovations, and TCM clinical application.
The targetome research, which is exploring the question of "which biological macromolecules the TCM components will bind to", depicts a static picture of potential interactions. By contrast, the bioactivome research focuses on a distinct question: what cellular and molecular functional changes do these interactions trigger? It uncovers dynamic functional responses within biological systems. For example, a single TCM component of andrographolide may bind to multiple target proteins, in turn inhibiting the activity of key kinases, a molecular-level event that falls within the scope of the bioactivome. This inhibition can then block tumor cell proliferation pathways and activate senescence, which represent pathway- and network-level events of the bioactivome, ultimately resulting in specific pharmacological effects in organisms[76]. In this way, while the targetome defines the potential for TCM components to act, the bioactivome reveals the practical functionality of these actions. As such, it serves as a critical bridge connecting intermolecular interactions to the overall pharmacological phenomena observed in TCM treatments.
AI-driven methodologies for the bioactivome research in TCM
-
Through multi-level and multi-dimensional research methods, the decoding of the TCM bioactivome is being systematically carried out by AI technology[77]. A comprehensive research framework encompassing data infrastructure, algorithm innovation, and clinical application has been constructed. At the data infrastructure level, those knowledge platforms like the TCM modernization (TCMM) and linking traditional Chinese medicine with modern medicine platform (LTM-TCM) have actually laid a solid foundation for the bioactivome research. The TCMM database, which integrates 20 modern Chinese medicine concepts and 46 kinds of biological relationships, contains more than 3.4 million records[78]. Meanwhile, the LTM-TCM platform utilizes biomedical natural language processing technology and carefully organizes the multi-level interactions from symptoms, prescriptions, components to targets, thus also creating a huge network with millions of relations[79]. These platforms effectively connect the TCM theories and the modern medical insights, thus creating an indispensable data foundation for systematically decoding the multi-level functional outputs following TCM intervention.
In the area of algorithms and multi-omics integration, significant progress has been made with the enzyme-based functional correlation (EBFC) algorithm. This algorithm has advanced the traditional statistical correlation analysis to the functional association level. In investigating BX (a drug derived from Psoralea corylifolia L.) for osteoporosis, researchers adopted the approach of function-oriented multi-omics analysis to integrate bone mineral density parameters, metabolic mechanism data and gut microbiome data. This method identified 59 distinct metabolites along with 9 key metabolic pathways, while the EBFC algorithm uncovered how major bacterial species regulate enzymes involved in purine and tryptophan metabolism. Through these findings, researchers have clarified and established a functional link between gut microbiota and host metabolites[80]. Similarly, in research focused on treating chronic kidney disease with the Jian-Pi-Yi-Shen formula, this algorithm has successfully unraveled the metabolite-microbiota-enzyme functional network, enabling TCM to reconstruct the interactions between host metabolism and gut microbiota[81]. It is important to note that the core technical challenge in building a reliable bioactivity graph lies in integrating heterogeneous multi-omics data, which is characterized by high noise levels, multiple origins, and batch effects. Resolving this challenge requires a rigorous methodological framework anchored in robust AI and computational strategies, including standardized data preprocessing, integrated noise-reduction modeling, rigorous validation, and biological interpretation. More importantly, it is essential to ensure that these interpretations are both biologically meaningful and reproducible[82,83].
In TCM clinical practice, AI algorithms have been extensively utilized to overcome the drawbacks of formula application relying on subjective experience, with their primary focus on intelligent prescription recommendation and prediction. FordNet, a system based on deep learning, incorporates bioactivome information for TCM formula recommendation and attains remarkable performance by leveraging large volumes of electronic health records for model training[84]. For personalized prescription recommendation, TCMPR adopts a subnetwork-oriented symptom term mapping approach, which can effectively characterize the features of unrecorded symptoms and thereby improve recommendation accuracy[85]. As for TCMFP, it integrates TCM clinical experience, AI technologies and network science algorithms to screen optimal herbal formulas for specific diseases such as Alzheimer's disease, offering a novel approach to formula optimization[86]. Collectively, these AI-driven methods facilitate the precise treatment of TCM, and accelerate the transformation of TCM research models.
-
While previous discussions have focused on individual research on the chemicalome, targetome, and bioactivome, the next critical step is to explore the cross-links among these three omes. This integration is key to understanding systemic perturbations and demonstrating the efficacy of TCM. These three types of information allow researchers to describe the effects of TCM in organisms in a more systematic manner.
The hierarchical framework of the chemicalome, targetome, and bioactivome
-
The framework that integrates the chemicalome, targetome, and bioactivome can serve as the primary paradigm for characterizing the overall mechanism of TCM. AI stands as a key technology, permeating every stage of this multi-level analytical cascade. The chemicalome constitutes the fundamental material foundation of TCM. Instrumental analytical techniques such as mass spectrometry are combined with AI-powered structural prediction to systematically characterize both the in vitro and in vivo chemicalome of TCM[87,88]. This approach helps tackle the chemical complexity of TCM and identify the biologically relevant substances that engage with living systems. The intermediate layer of these interactions is referred to as the targetome. GNNs and Transformers are AI models that can combine molecular structures with protein sequences to predict high-confidence DTIs[89,90]. These approaches in conjunction with experimental validation methods, such as CETSA and chemical proteomics, are used to construct and validate the polypharmacological networks in which TCM components interact with enzymes, receptors, and signaling proteins[91]. Finally, the bioactivome corresponds to the functional output layer. AI integrates multi-omics data to elucidate the dynamic cellular and molecular responses triggered by targetome engagement. These responses can be changes in biomarkers, regulation of pathways and even creation of synergistic functional modules[92].
Research approaches for chemicalome-targetome-bioactivome interactions of TCM
-
Depending on different research goals, the starting points of AI analysis will vary, and relevant interactions can be roughly categorized into three research directions (Fig. 4). The chemicalome-oriented research starts from the chemical structure of natural products and investigates the effect brought about by the interactions between compounds and biological targets[93]. The targetome-oriented research focuses on targets like receptors and enzymes and seeks to understand how target perturbations spread across molecular networks and translate into changes in system-level functions[94]. AI has made this process much more efficient by providing an interpretable output from target perturbations to system-level output. The bioactivome-oriented research employs a reverse-reasoning strategy. It uses observed system-level phenotypes as clues to retrospectively trace and infer how TCM components collectively shape complex biological responses[95]. In order to achieve the integration of the aforementioned research directions, it is important to use the AI methods. By encoding multi-dimensional features of compounds, targets, and biological activities, researchers can discover new perturbation-response patterns, thereby realizing the integration of the information from the three scales at a computational level[96].
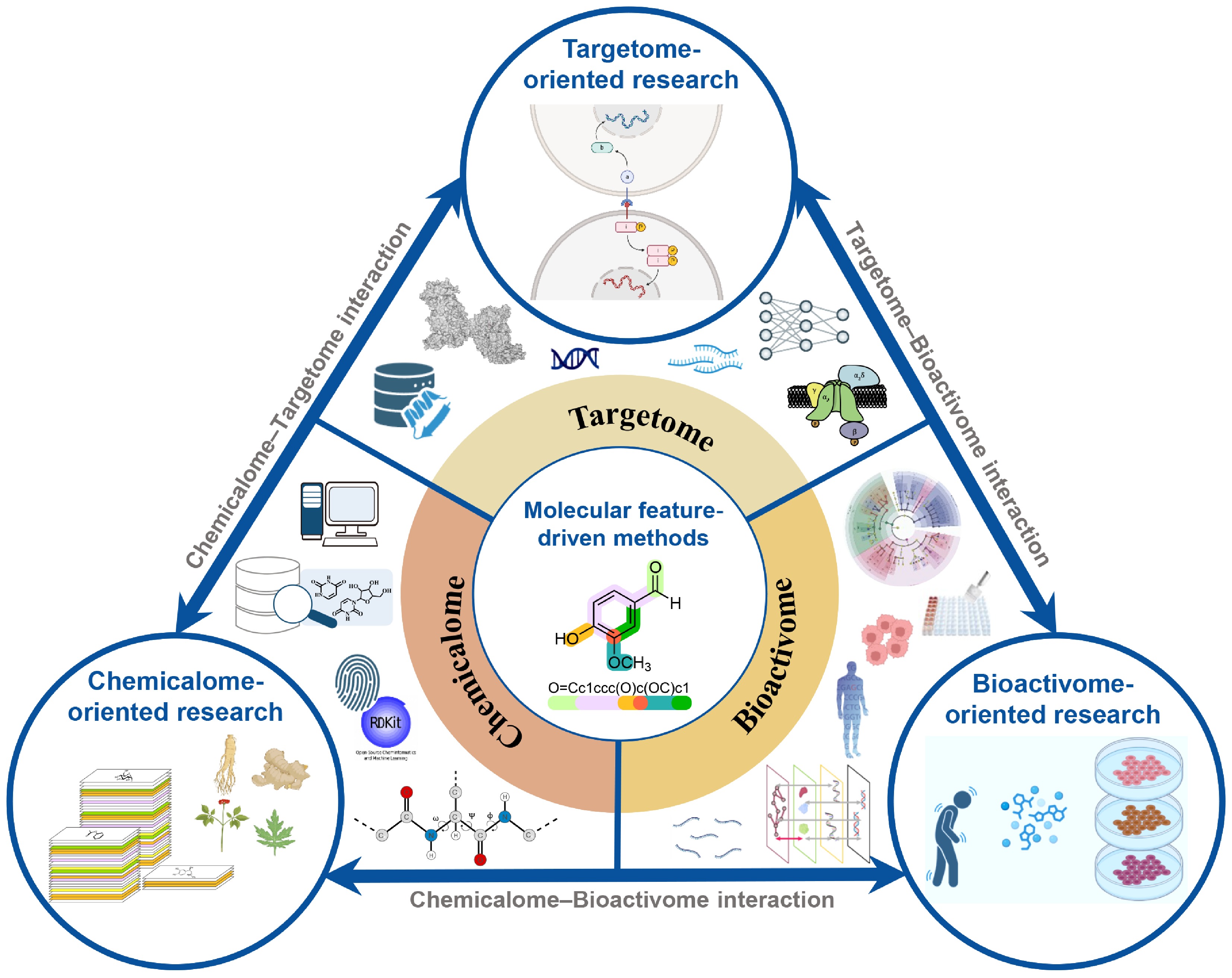
Figure 4.
Schematic diagram for interactions among the chemicalome, targetome, and bioactivome. The framework includes chemicalome-oriented, targetome-oriented, and bioactivome-oriented research to construct their interactions. Molecular feature-driven methods play an important role in this process.
TCM formulas are inherently composed of multiple components, and their therapeutic effects are primarily exerted through mechanisms involving multiple targets and pathways[97]. This inherent complexity makes TCM formulas an ideal system for studying the entire chain of "chemical composition-target action-biological activity", as they perfectly align with our newly proposed concepts of the chemicalome, targetome, and bioactivome. Therefore, via multi-omics, metabolic perturbation mapping, and deep learning, we can better integrate TCM chemicalome, targetome, and bioactivome data into interaction networks. These data-driven approaches will not take the place of traditional pharmacology. Instead, they serve as a valuable complement to it. The two together drive progress in the field by speeding up hypothesis generation, identifying effective component combinations, and enabling the explanation of TCM mechanisms from a network perspective[98,99].
Limitations of the present AI models in integrating the chemicalome, targetome, and bioactivome
-
Although AI demonstrates great capabilities in processing large-scale data, numerous fundamental limitations that need to be addressed still remain when AI is applied to complex biological systems. Additionally, effective methods for dataset partitioning and model performance evaluation are currently lacking. Specifically, traditional model training relies on random splitting approaches, which can lead to structural similarities between molecules in the training and testing sets. The similarities prevent rigorous assessment of the model's capability to handle completely novel chemical structures[100]. Some researchers contend that UMAP (Uniform Manifold Approximation and Projection) clustering, which is based on chemical similarity, better mimics the structural diversity of molecules. However, the dimensionality reduction process inherent to UMAP may lead to the loss of critical structural features. Furthermore, UMAP is particularly effective at preserving local structures rather than global ones, meaning it may fail to accurately reflect overall dataset trends or inter-category relationships in certain scenarios[101,102]. If AI models are unable to achieve effective skeleton hopping, their capacity to explore the unknown chemical space of TCM will be limited.
Additionally, we must confront the long-standing criticisms surrounding AI applications in TCM research: the lack of model interpretability acts as a barrier to elucidating underlying mechanisms. While complex AI models deliver strong predictive capabilities, their decision-making logic remains unclear, as they generate outputs based solely on input data without transparent reasoning. In life sciences research, explaining the rationale behind why therapies work is just as important as predicting their efficacy. We can use AI to pinpoint potential pathways for mechanism validation, but we currently lack logical explanations for how or why these specific pathways are identified. This gap in reasoning creates a disconnect between computational predictions and experimental validation processes, undermining trust in the reliability of AI-driven findings[103].
-
This review clarifies how AI technology is incorporated into TCM research to systematically decode its action pathways within biological organisms. By integrating the chemicalome, targetome, and bioactivome, AI offers significant potential for uncovering the intricate mechanisms that underlie the synergistic effects of TCM, which are characterized by its multi-component, multi-target, and multi-pathway nature. We highlight AI's influence across various domains of TCM research: it transforms the way complex TCM constituents are characterized, supports the prediction of multiple targets, processes large-scale bioactivity data efficiently, and aids in the reverse engineering of TCM mechanisms. For thousands of years, TCM has depended mainly on empirical guidance for medication use, and the complexity of how its components act has long posed a stubborn challenge for modern scientific research. The incorporation of AI technology has further advanced contemporary TCM studies, signifying a major paradigm shift in the field.
Looking ahead, the integration of AI and TCM research points to several critical frontiers for future exploration. Research efforts moving forward should focus on developing explainable and causal AI models, which will allow the field to move beyond mere correlation analysis and uncover authentic therapeutic pathways underlying TCM's therapeutic effects. The next key challenge lies in establishing dynamic and multi-scale models to capture the interactions within and across the three core omics layers. This modeling task encompasses a broad range of biological processes, from the ADME processes of TCM components to the phenotypic changes observed at the whole-organism level. Additionally, generative AI offers tremendous potential for designing new and network-based therapeutic agents that draw on TCM’s accumulated wisdom. Fulfilling this potential depends heavily on two key factors: building high-quality TCM data ecosystems and fostering in-depth interdisciplinary collaboration. Additionally, combining multi-omics profiles with the analysis of TCM's three core domains will pave the way for personalized TCM practice. This advancement will usher in an era of precision herbal medicine, where ancient TCM wisdom is systematically translated into modern therapeutic approaches.
-
Not applicable.
-
The authors confirm contributions to the review as follows: conception and design: Xin G, Song H; draft manuscript preparation: Song H, Liang Z, Zhang X, Yang F; analysis and interpretation: Zheng M, Xin G. All authors reviewed and approved the final version of the manuscript.
-
Data sharing is not applicable to this review as no datasets were generated or analyzed.
-
We gratefully acknowledge financial support from the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0830200 and XDB1260301), the National Natural Science Foundation of China (T2225002 and 82274064), and the Lingang Laboratory (LGL-8888-02).
-
The authors declare that they have no conflict of interest.
-
#Authors contributed equally: Huipeng Song, Zeyuan Liang, Xinru Zhang
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Song H, Liang Z, Zhang X, Yang F, Zheng M, et al. 2026. Artificial intelligence algorithms drive the deciphering of traditional Chinese medicine by analyzing the chemicalome, targetome, and bioactivome. Targetome 2(2): e010 doi: 10.48130/targetome-0026-0002 

Artificial intelligence algorithms drive the deciphering of traditional Chinese medicine by analyzing the chemicalome, targetome, and bioactivome
- Received: 04 December 2025
- Revised: 09 January 2026
- Accepted: 14 January 2026
- Published online: 13 March 2026
Abstract: Artificial intelligence (AI) is reshaping the research paradigm of traditional Chinese medicine (TCM) in a profound way. This review offers a systematic account of how AI algorithms propel the modernization of TCM through the integrated analysis of three core concepts: the chemicalome, referring to the collection of in vitro and in vivo chemical constituents derived from TCM; the targetome, defined as the set of biological macromolecules that engage in interactions with TCM components; and the bioactivome, signifying the range of integrated biological activities and phenotypic outcomes induced by TCM interventions. First, it illustrates how AI enables comprehensive characterization of complex in vitro and in vivo chemicalomes by revolutionizing mass spectrometry analysis and metabolite identification techniques. Next, it examines how AI works in conjunction with experimental technologies to systematically predict the targetome and validate these predictions. Furthermore, the review clarifies how AI deciphers the bioactivome arising from TCM interventions and uncovers mechanisms through the integration of multi-omics datasets. Finally, it explores methodologies for establishing comprehensive interconnections among the chemicalome, targetome, and bioactivome. This analytical framework demonstrates that AI functions not just as a tool to enhance research efficiency, but also as a foundational methodology capable of systematically decoding TCM and linking traditional wisdom with modern science.
-
Key words:
- Traditional Chinese medicine /
- Artificial intelligence /
- Chemicalome /
- Targetome /
- Bioactivome /
- Drug discovery