









-
Hepatic ischemia-reperfusion injury (HIRI) represents a significant pathophysiological process characterized by excessive oxidative stress, metabolic imbalance, and inflammation, which occurs when the liver undergoes a period of inadequate oxygen and nutrient supply, followed by the restoration of blood flow[1]. It is an inevitable complication in various clinical scenarios, such as liver resection or transplantation, which further results in severe outcomes, including systemic inflammatory response syndrome and multiple organ failure during the perioperative period[2]. The pathogenesis of HIRI typically consists of two stages. In the initial ischemic phase, restricted blood flow induces cellular metabolic disturbances, leading to the overproduction of reactive oxygen species (ROS), collapse of mitochondrial membrane potential (MMP), and hepatotoxicity[3]. Reperfusion of blood further amplifies oxidative stress and inflammation by inducing the release of endogenous damage-associated molecular patterns (DAMPs), like high mobility group box 1 (HMGB1), or mitochondrial DNA (mtDNA) and subsequently activating Toll-like receptors (TLRs)[4]. Current clinical strategies to avoid or mitigate HIRI primarily focus on minimizing ischemic time, employing ischemic preconditioning, or timely utilization of pharmacological interventions. However, existing antioxidants along with mitochondrial protectants exhibit limited protective efficacy against HIRI, posing a significant clinical challenge that necessitates urgent attention.
Mitochondria and their associated metabolic processes simultaneously influence complex interactions among oxidative stress, metabolic disorder, and inflammatory responses. We most recently noticed that HIRI triggered the destruction of adenosine triphosphate (ATP) synthesis, explosive ROS accumulation, and mitochondrial dynamics imbalance, leading to an energy crisis in hepatocytes[5]. Previous research emphasized that during the ischemic phase, mitochondrial dysfunction was associated with lipid metabolism disorders, occurring prior to inflammatory events and cell death in HIRI[6]. Interestingly, lipid deposition induced by free fatty acids (FFAs) also stimulated ROS outbreak and oxidative stress in hepatocytes, and exerted a reciprocal effect on mitochondrial function[7]. Furthermore, lipid-ROS accumulation and mitochondrial dysfunction synergetically accelerated the oxidation and release of mitochondrial DNA (mtDNA)[8,9], further establishing a vicious cycle characterized by disorders in lipid metabolism, mitochondrial redox dysfunction, and heightened inflammation[10,11]. Therefore, identifying the key pathways that simultaneously influence mitochondrial lipid metabolism and mtDNA-centered oxidative stress may directly mitigate the deterioration associated with HIRI. Cytosine monophosphate kinase 2 (CMPK2) is predominantly localized in the mitochondria, where it functions as a rate-limiting enzyme responsible for synthesizing deoxynucleoside triphosphates to regulate mtDNA replication[12]. It was reported that the knockdown of CMPK2 could reduce the release of mtDNA into the cytosol[13]. Most recently, CMPK2 was first reported to exhibit significant upregulation in the livers of patients with metabolic liver diseases. Additionally, hepatocyte-specific Cmpk2 deletion substantially suppressed inflammation and subsequent hepatic pyroptosis[14], though its precise function in HIRI development remains undefined.
Numerous natural products, known for broad bioactive properties, high safety profiles, and minimal side effects during long-term use, shows promise in providing new hope for the treatment of HIRI. Preclinical studies have demonstrated that resveratrol, ginsenosides, curcumin, salvianolic acid, and total saponins of Panax notoginseng exhibit antioxidant and anti-inflammatory effects, along with the capability to regulate metabolic disorders in HIRI, whether alone, or in combination with other medications[15,16]. Acteoside (ACT), a phenylpropanoid glycoside widely found in traditional Chinese medicinal herbs such as Cistanches Herba and Rehmanniae Radix has received increased attention due to its antioxidant, anti-inflammatory, and antiapoptotic properties against various liver diseases. Our previous study demonstrated that ACT inhibited the synthesis and transfer of damaged hepatocyte-derived DAMPs to liver sinusoidal endothelial cells (LSECs), thereby reversing the LSEC senescence and improving the liver sinusoidal microenvironment in HIRI[4]. Furthermore, we have recently proved that ACT enhanced the binding affinity of poly (RC) binding protein 2 (PCBP2)-system Xc−, and thus limited the HMGB1-induced hepatocyte ferroptosis, M1 macrophage recruitment, and immune response during HIRI through imposing restrictions on hypoxia-induced factor 1α (HIF-1α) and p300[17]. Although the association between ACT and hepatocyte protective effects has been preliminarily observed, further substantiation is required to elucidate the precise function of CMPK2 in lipid metabolism abnormalities, oxidative stress, and HIRI development, as well as to clarify the specific mode of action of ACT.
By integrally employing an in vivo HIRI mouse model, an in vivo hepatocyte-specific Cmpk2 overexpression model, and an in vitro hypoxia and reoxygenation (HR) model; in conjunction with RNA sequencing (RNA-Seq) of whole liver and single hepatocyte cell types, and a series of molecular biology experiments, we thoroughly investigate how ACT improves redox metabolism and interrupts the CMPK2-centered vicious cycle of lipotoxicity-hyperoxidation-inflammation in HIRI by regulating the TLR9-interferon regulatory factor 1 (IRF1)-CMPK2/dual oxidase 2 (DUOX2)-mtDNA axis. This study not only offers novel insights into the pathological role of CMPK2 in the interaction between mitochondrial lipid metabolism and redox dysregulation during HIRI, but also facilitates the transition of ACT from laboratory settings to clinical practice as a promising new strategy to prevent HIRI.
-
Male C57BL/6J strain mice (8 weeks old, weighing 23 ± 1 g) were obtained from SIBEIFU Biotechnology Co. (Beijing, China) and were housed in sterilized cages under a 12 h light/dark cycle with constant temperature, appropriate humidity, standard diet, and ad libitum access to sterile water. After 1 week of acclimation feeding, mice were equally divided into the following groups (n = 6/group): (1) sham group; (2) HIRI group; (3) HIRI + ACT at low-dose group (25 mg·kg−1); (4) HIRI + ACT at medium-dose group (50 mg·kg−1); (5) HIRI + ACT at high-dose group (100 mg·kg−1); (6) HIRI + N-acetylcysteine (NAC, a positive drug) group (100 mg·kg−1). Different dosages of ACT or NAC were orally administered to relative treatment groups, while phosphate-buffered saline (PBS) was given to the sham and HIRI groups via intragastric administration for 1 week. The dosages and timing of ACT used in the experiment were determined based on prior research findings[17]. After 8 d of administration, isoflurane gas was used to anesthetize all mice. Subsequently, an atraumatic microvascular clip was employed to occlude the hepatic artery and portal vein, achieving 70% hepatic ischemia in the HIRI and different treatment groups. Following a duration of 1 h, the clip was removed to initiate liver reperfusion for a period of 6 h. The same surgical procedure was performed on the sham group without the application of liver vascular clamping. All liver and serum samples were collected following the surgical procedure for further analysis. To investigate the role of CMPK2 in HIRI disease and ACT therapy, mice were equally divided into the following groups (n = 6/group): (1) sham + AAV8-empty vector group; (2) HIRI + AAV8-empty vector group; (3) HIRI + ACT (50 mg·kg−1) + AAV8-empty vector group; (4) HIRI + ACT (50 mg·kg−1) + AAV-CMPK2 group (NM_020557.4). The mice in group four were pre-injected with CMPK2-carrying AAV8 via tail vein, while the mice in other groups were pre-injected with AAV8 empty vector for 2 weeks. Other surgical manipulations and drug administration protocols in this study were the same as those in the first animal batch. The Ethics Committee of the Institutional Animal Care and Use Committee of Beijing University of Chinese Medicine had approved all animal procedures (BUCM-2023112904-4146 and BUCM-2025030306-1093).
Cell culture, treatment, and transfection
-
AML12 cells were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin G/streptomycin under 37 °C and 5% CO2 environment. (1) Cells were pretreated with ACT dissolved in DMSO at serial concentrations of 25, 50, and 100 μmol·L−1 for 8 h and exposed to a hypoxic environment for 16 h and then replaced with an oxygen-enriched environment for 8 h (named as the HR model). After modeling completion, cells were collected for further experiments. (2) Once 70% confluence was reached, cells were transfected with the small interfering RNAs (siRNAs) by an RNA Fit reagent (Cat No. HB-RF-1000, Hanbio, Shanghai, China) for 24 h, incubated with ACT, and then established HR models for another 32 h. The siRNAs against the target gene of Cmpk2 (sequencing 5'-GGCUUCUGAAAUAGCUAAATT-3'), Duox2 (sequencing 5'-GAGGATAAGTCCCGTCTAATGTTTA-3'), and Vamp8 (5'-CAAGACAGAGGACUUGGAATT-3'), were obtained from Hanbio (Shanghai, China). (3) To construct the mtDNA-transfected cell model, the mtDNA from the control, HR, and HR + ACT groups was first extracted and purified by the DNA Blood Mini Kit (Cat No. 51104, QIAGEN, Dusseldorf, Germany). After overnight adherence for 12 h, the culture medium from AML12 cells was replaced with Opti-MEM medium, and then transfected with the extracted mtDNA using the Lipofectamine 3000 reagent (Cat No. L3000001, Thermo, Waltham, USA) following the manufacturer's instructions for 24 h. (4) For the recombinant protein stimulation experiment, after pretreatment with DMSO or ACT for 8 h, AML12 cells were further treated with recombinant CMPK2 protein (100 ng·mL−1, Cat No. CSB-EP688686HU, Wuhan Huamei Biotech, Wuhan, China) for 32 h. At the end of treatments, cells from the above groups were collected for further experiments.
RNA-Seq of mouse liver and hepatocytes, and bioinformatics analysis
-
The RNA in mouse liver of different groups was extracted by Trizol reagent and was quantified by NanoRhatometer@ spectrophotometer (IMPLEN, USA), of which integrity was measured by the Nano Assay (Agilent, Shanghai, China). On the Illumina Novaseq platform, libraries of mouse RNA-Seq were established as previously mentioned[4] after mRNA purification, and cDNA fragment enrichment. For hepatocytes RNA-Seq, total RNA was extracted and quantitated as described above and cDNA was obtained by cDNA reverse transcription kit (Cat NO. R323-01) from Vazyme company (Nanjing, China). The libraries of cell RNA-seq were also established on the Illumina Novaseq platform. Heatmap and Gene Ontology (GO) enrichment analysis was marked by the edge R software. Based on differentially expressed genes (DEGs) and example datasets provided by Gene Set Enrichment Analysis (GSEA) (
www.gsea-msigdb.org/gsea/index.jsp ), the GSEA analysis, circle network, cluster heatmap, volcano plot, 3D area, and polar bar were conducted by Weishengxin (www.bioinformatics.com.cn ). Combining the results of ChEA3 (https://maayanlab.cloud/chea3 ) and the viridis R package, the transcription factors (TF) of genes were predicted. JASPAR (https://jaspar.elixir.no ) was used to predict transcription factor binding sites.Chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR)
-
Collected AML12s were cross-linked with 1% formaldehyde at 37 °C for 10 min and then quenched by glycine solution at room temperature. According to the CHIP assay instructions provided by Beyotime (Cat No. P2078, Beyotime, Shanghai, China), cell lysates were washed three times with ice-cold PBS, lysed in a lysis buffer and sonicated to generate DNA fragments of 200–1,000 bp using a JY92-IIN ultrasonic cell disrupter. Finally, we carried out immunoprecipitation using anti-IRF1 antibody (11335-1-AP, Proteintech). After washing with washing buffer, the chromatin fragments were de-crosslinked and examined by qPCR experiments. The information about used primers was 5'-TCACCCACAGGAACTTCTTG-3' (forward) and 5'-TGCCAGTTTAAGATTCCTAG-3' (reverse) for Cmpk2 promotor 1, 5'-ACGCAGTCAGTGTCAGAGTCTCTC-3' (forward) and 5'-TCTTCCTCCACTGGCTGATACAGG-3' (reverse) for Cmpk2 exon 1, and 5'-GCATAGCACAGCCACCTACGC-3' (forward) and 5'-CACAGTCAGCAGCAGGACCAAG-3' (reverse) for Cmpk2 exon 2.
Drug affinity responsive target stabilization assay (DARTS)
-
AML12 cells were washed with PBS and subsequently lysed using M-PER on ice for 30 min. The cell lysate was then evenly divided into two tubes, which were incubated with either DMSO or ACT (50 μmol·L−1) at 37 °C for 1 h to ensure adequate binding of the target proteins. Finally, different lysate samples were digested with pronase E at ratios of 0, 1 : 10,000, 1 : 3,000, 1 : 1,000, 1 : 300 and 1 : 100, for 30 min. The reactions were terminated by adding loading buffer, preparing the samples for subsequent Western blot analysis. The detailed antibody information was provided in Supplementary Table S1.
Cellular thermal shift assay (CETSA)
-
AML12 cells were incubated with DMSO or 50 μmol·L−1 ACT for 0.5 h and then washed three times with PBS. Following co-incubation, both groups were further divided into eight parts and subjected to sequential heating at increasing temperatures of 37, 40, 45, 50, 55, 60, 65, and 70 °C. After the heating process, three rapid freeze-thaw cycles were applied to lyse cells, followed by Western blot analysis to assess the binding affinity of IRF1 and CMPK2 with ACT.
Surface plasmon resonance (SPR) assay
-
SPR analysis was performed on a Biacore S200 instrument. CMPK2 protein was diluted to 50 μg·mL−1 in sodium acetate buffer and immobilized onto a CM5 sensor chip (Cat No. BR100399, Cytiva, Sweden). The ACT compound was serially diluted and applied in increasing concentrations over the protein-conjugated surface. Finally, interaction kinetics between the drug and protein were analyzed using Biacore X-100 apparatus (Biacore GE, Uppsala, Sweden).
Microscale thermophoresis (MST) assay
-
At the beginning, the target protein CMPK2 was fluorescently labeled using the RED-NHS 2nd generation labeling Kit (Cat No. MO-L011, NanoTemper Technologies, Munich, Germany) and eluted in PBS buffer. Simultaneously, the ACT compound was serially diluted in PBS buffer to generate a gradient concentration. Equal volumes of the protein solution and gradient-diluted ACT were completely mixed, then loaded into premium capillaries for analysis on the MST Monolith NT.115 instrument (NanoTemper Technologies, Munich, Germany) to determine drug-protein binding affinity.
Statistical analysis
-
All experiments were conducted at least three times, and the results were presented as mean ± SD. A one-way ANOVA followed by Tukey's post hoc test was employed for data comparison, and GraphPad Prism 8.1 Software (La Jolla, CA, USA) was used for data analysis. A P-value of less than 0.05 was considered to indicate significant differences. Additional information about the used methods or other details was provided in the Supplementary File 1.
-
We preliminarily indicated that ACT improved HIRI by repairing PCBP2-centered hepatocyte ferroptosis and alleviating LSEC senescence, both of which were inextricably interconnected with the cellular metabolic disorders and redox reactions[4,17]. Here, we conducted animal experiments to further investigate the effects of ACT on mitochondrial lipid metabolism and oxidative death patterns in livers. As shown in Supplementary Fig. S1a, in accordance with the elevated serum transaminase changes, HIRI significantly increased the inflammatory infiltration, sinusoidal congestion, edema, and hepatocyte-centered cell apoptosis, as evidenced by the increased co-staining area of the TUNEL reagent and hepatocyte nuclear factor 4 alpha (HNF-4α) (Fig. 1a and b; Supplementary Fig. S1b–S1d). Notably, ACT dramatically reversed these pathological phenomena associated with ischemic injury and hepatocyte death in livers. Next, we performed RNA-Seq analysis both in livers and in single-cell type hepatocytes isolated from the liver tissue of different groups. This different sequencing data was utilized to complete comparison and correlation analysis, facilitating the screening of the core target cluster. As shown in Fig. 1c, the major changed function annotation in the whole livers of the HIRI groups was involved in ATP synthesis-coupled electron transport, apoptotic process, and inflammatory response, of which the processes were significantly reversed by ACT. Interestingly, we also observed that the similar biological processes of acyl-coenzyme A (Acyl-CoA) metabolic process, DNA metabolic process, lipid metabolic process, and release of cytochrome c from mitochondria and mitochondrial electron transport, were dramatically changed in the HIRI group but not in the ACT-treated groups (Fig. 1d). After comparing and analyzing the data set at the whole-liver and cellular levels, the shared critical targets were the Cmpk2 and myeloid differentiation primary response protein 88 (Myd88) principally (Fig. 1c and d). In addition, we conducted Fig. 1e, and highlighted the crucial processes involving ACT reversing HIRI, which were involved in the endoplasmic reticulum-associated protein degradation pathway, execution phase of apoptosis, fatty acid oxidation, and oxidative phosphorylation. Furthermore, by intersecting the RNA-seq data from the in vivo and in vitro levels, we identified the most essential pathways that primarily included mitochondrion, lipid metabolic processes, and apoptotic process (Fig. 1f). Finally, we performed adipocyte differentiation-related protein (ADRP) staining on liver tissue and hepatocytes, further clarifying that ACT primarily intervened in the lipid metabolism process by rebalancing lipid accumulation and redox events in mitochondria (Supplementary Fig. S2a and S2b).
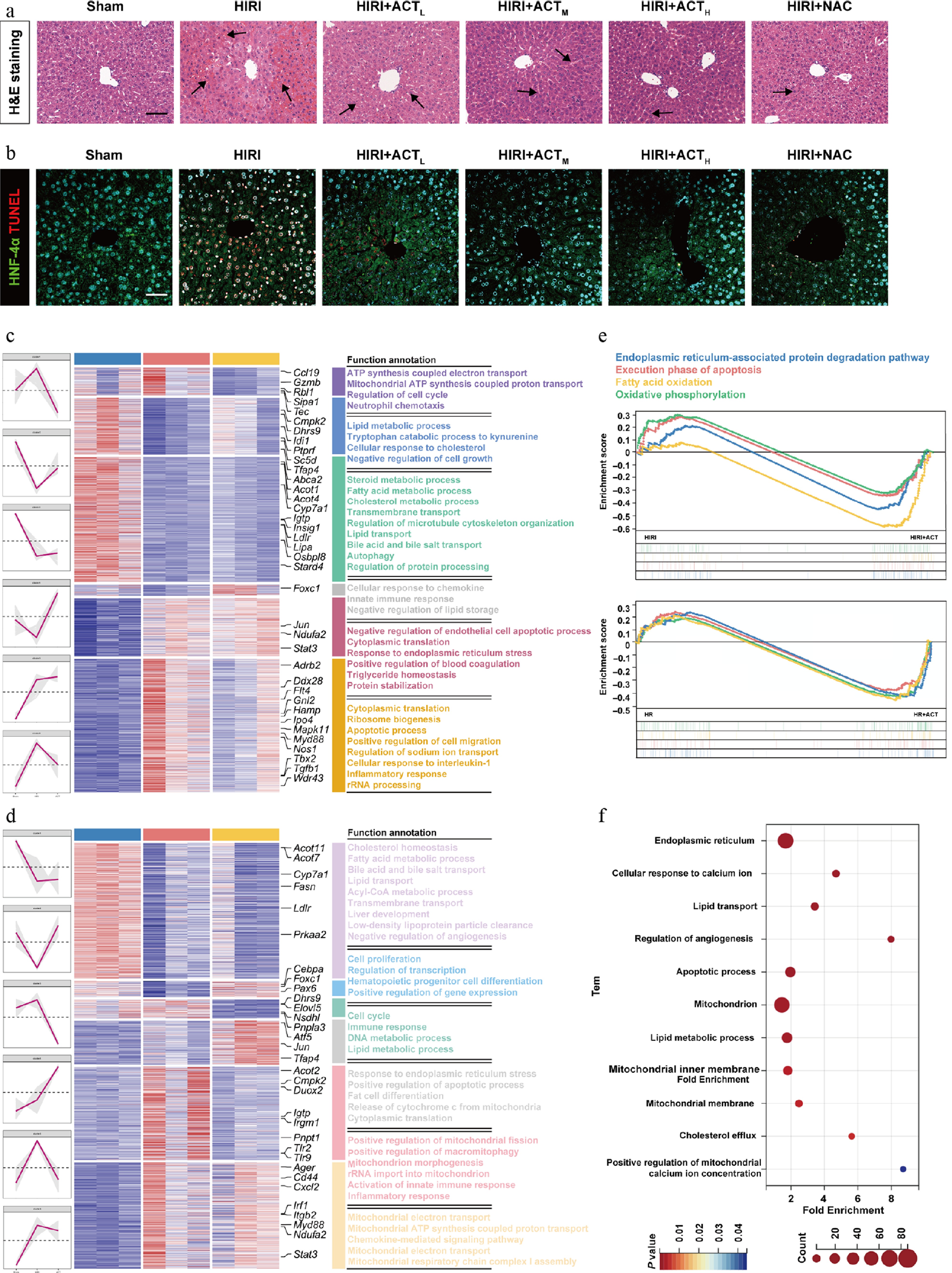
Figure 1.
ACT alleviated liver damage in HIRI mice, and HR-stimulated hepatocytes. (a) Representative images of H&E staining in different groups (arrows indicate regions of liver injury, including inflammatory foci). Scale bar = 100 μm. (b) Representative images of immunohistochemistry staining for HNF-4α and TUNEL in different groups. Scale bar = 100 μm. The heatmap of critical DEGs with red lines indicating different clusters in the (c) whole livers and (d) primary hepatocytes, isolated from different mice groups. (e) GSEA analysis from HIRI and HIRI + ACT groups in mouse livers (upper panel), and primary hepatocytes (below panel). (f) GO enrichment analysis results of the whole liver, and primary hepatocyte DEGs visualized with a bubble diagram.
ACT influences fatty acid synthesis controlled by ACOT2, and reshapes mitochondrial oxidative metabolism
-
Subsequently, a heatmap analysis of RNA-seq data from primary hepatocytes was conducted, focusing on genes involved in lipid synthesis, transport, and metabolism, especially those from the Acyl-CoA thioesterases (ACOTs) family. Hierarchical clustering of DEGs is shown in Fig. 2a, a notable upregulation of Acot2, a target responsible for hydrolyzing Acyl-CoAs to FFAs in hepatocytes but not other ACOTs families, was detected in the HIRI group, instead of the HIRI + ACT group. Consistently, the hepatic ACOT2 expression and its co-distribution with mitochondrial marker outer mitochondrial membrane 20 (TOM20) were all increased in the livers of HIRI mice, but were reversed after ACT treatment (Fig. 2b; Supplementary Fig. S3a). The translational changes of ACOT2 in AML12 cells exhibited the same trend as those in mouse livers (Supplementary Fig. S3b). Due to the intense intervention of ACOT2, the hepatic FFA content was significantly increased following HIRI surgery (Fig. 2c, upper panel), and more importantly, was positively correlated with the changes in Acot2 mRNA expression in mouse livers (Fig. 2d, upper panel). Meanwhile, ACT reversed the accumulation of FFA and broke its positive correlation with Acot2 caused by HIRI surgery. Further analysis revealed a similar trend in both the content of FFA and its correlation with Acot2 mRNA in hepatocytes under HR insult and ACT administration (Fig. 2c and d, lower panels). Furthermore, accumulated FFA usually results in the overproduction of mitochondrial ROS through multiple pathways. As shown in Fig. 2e and f; Supplementary Fig. S3c, ACT mitigated HR-induced ROS accumulation and oxidative stress, as characterized by a reduction in the fluorescent area of DCFH-DA and malondialdehyde (MDA) levels (a typical marker indicating lipid peroxidation), increased glutathione (GSH) and superoxide dismutase (SOD) levels (recognized indicators of antioxidant capacity). Oxidative reactions are crucial for maintaining mitochondrial homeostasis through preventing lipid deposition and facilitating respiration to generate energy. FFA biological processes-related genes involved in the FA synthesis like Acyl-CoA synthetase long-chain family member 1 (Acsl1), FA transport and oxidation-like carnitine palmitoyl transferase 1a (Cpt1a), and Acyl-CoA oxidase 1 (Acox1) are exhibited in Fig. 2g. Notably, changes in genes associated with β-oxidation were particularly significant. Consistently, the downregulated mRNA expression of FA β-oxidation-related genes in mitochondria induced by HIRI or HR were markedly restored by ACT administration (Fig. 2h). Furthermore, the ACT-treated AML12 cells exhibited superior mitochondrial function than the HR group, evidenced by increased levels of ATP, as well as enhanced activities of mitochondrial complex I and IV (Fig. 2i). These results suggest that ACT reduces FFA hyperproduction by decreasing ACOT2 expression, and selectively reactivates ACOX1 to restore oxidative metabolism, thereby reshaping the redox balance in mitochondria.
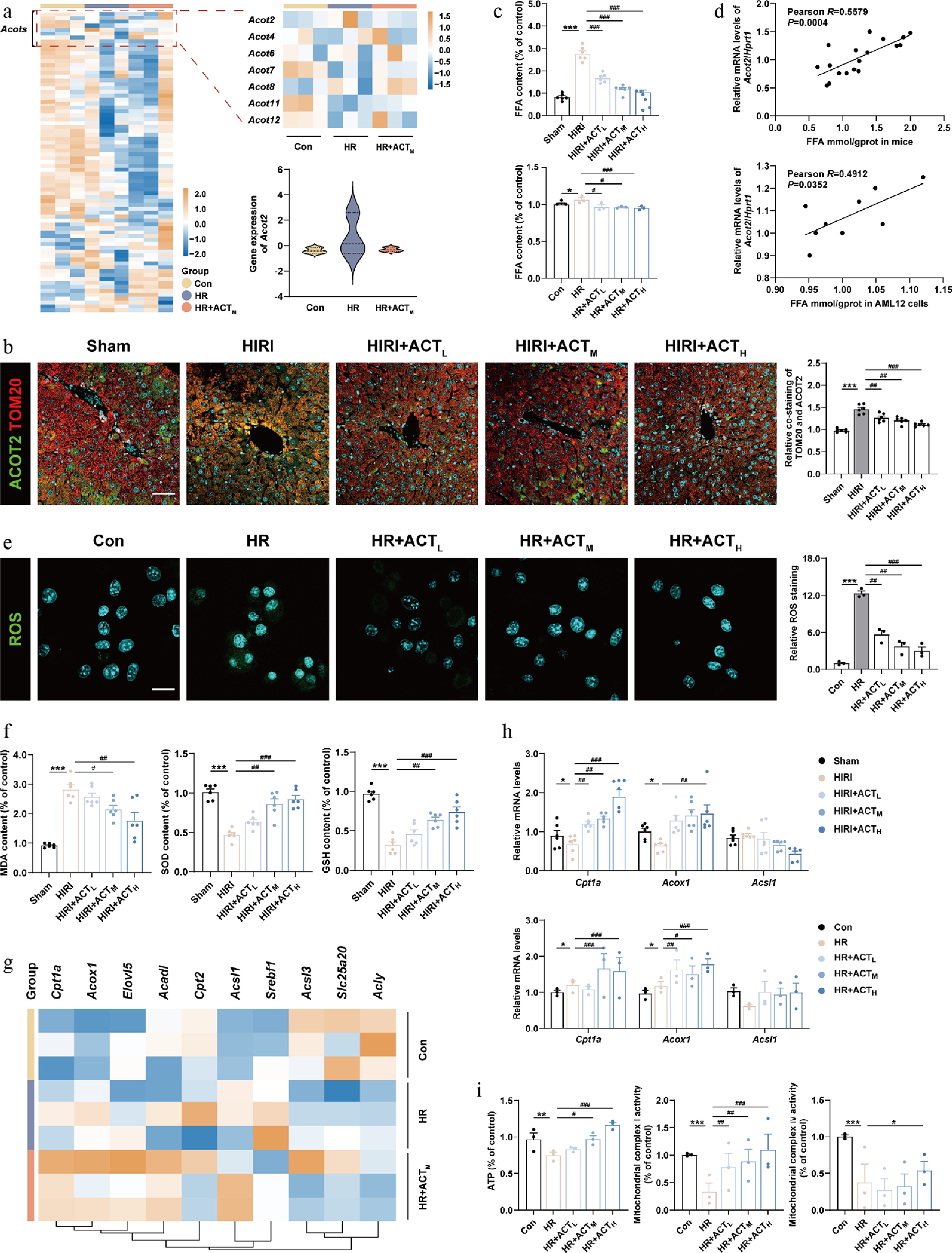
Figure 2.
ACT suppressed ACOT2 expression and subsequently mitochondrial oxidative metabolism disorder. (a) The heatmap of genes involved in mitochondrial metabolism in primary hepatocytes. (b) Representative images of immunofluorescence staining for TOM20 (594 nm), ACOT2 (488 nm), and DAPI (405 nm) in mouse livers. Scale bar = 100 μm. (c) The FFA level and (d) correlation analysis between FFA levels and Acot2 gene expression in mouse livers and AML12 cells. (e) Representative images of immunofluorescence staining for ROS (488 nm) and DAPI (405 nm) in AML12 cells. Scale bar = 20 μm. (f) The level of MDA, SOD, and GSH in mouse livers. (g) The heatmap of genes involved in fatty acid β-oxidation, and Acyl-CoA metabolism in primary hepatocytes. (h) The mRNA levels of Cpt1a, Acox1, and Acsl1 were normalized with Hprt1 in mouse livers and AML12 cells treated with ACT. (i) The level of ATP, and mitochondrial complex Ι and IV activities in AML12 cells. Statistical significance: * P < 0.05, ** P < 0.01, *** P < 0.001, compared with control group; # P < 0.05, ## P < 0.01, ### P < 0.001, compared with the model group (Data are presented as the mean ± SEM, n = 3 for cell experiments, n = 6 for mice experiments).
ACT prevents the production and release of mtDNA in hepatocytes by inhibiting CMPK2
-
Oxidative stress not only triggers mitochondrial dysfunction but also stimulates the continuous release of DAMPs. We conducted a circular network diagram analysis and noticed that the vast majority of DEGs involved in nucleic acid, including Tlr9, Cmpk2, and Irf1, which mainly influenced the recognition, synthesis, and regulation of mtDNA in hepatocytes of the HIRI, and HIRI + ACT groups (Fig. 3a). Next, we focused on investigating the alterations in mtDNA-related oxidative stress and mitochondrial function. A volcano plot of DEGs revealed that the genes involved in the production and response to ROS and mtDNA, including Cmpk2, Tlr2, Tlr9, and Cxcl10, were significantly increased under HR insult but exhibited a decrease following ACT treatment (Fig. 3b). Among these genes which collectively reflected key steps of the mtDNA axis, Cmpk2 showed the most robust and consistent ACT-reversed change. Under high-ROS conditions, mtDNA is particularly susceptible to oxidative damage due to the absence of histone protection and its proximity to the oxidative respiratory chain. This vulnerability leads to an increased production of oxidized mtDNA (ox-mtDNA)[18]. Subsequently, the content of mtDNA and ox-mtDNA were both visualized through co-staining of TOM20 and anti-DNA or 8-OHdG. As shown in Fig. 3c, Supplementary Fig. S4a and S4b, mtDNA and ox-mtDNA content were dramatically reduced by ACT, when compared with HR stimulation. Notably, among various DEGs involved in mtDNA-related biological processes in Fig. 3b, we noticed Cmpk2 was most tightly associated with mtDNA synthesis. Interestingly, the protein levels of CMPK2 in whole hepatic cells and isolated mitochondria were markedly increased following HIRI or HR insult, which were notably diminished by ACT intervention, especially in the mitochondria fractions (Fig. 3d and e). The excessive opening of the mitochondrial permeability transition pore (mPTP), which is mediated by voltage-dependent anion channel 1 (VDAC1), leads to mitochondrial swelling, disruption of cristae, and subsequent rupture of the membrane. Here, the mPTP inhibitor cyclosporine A (CsA), mPTP opening activators including carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP, promoting MMP depolarization), and ATP (stimulating VDAC oligomerization) were employed to check the status of mPTP opening[19]. ACT repressed the VDAC1-mediated mtDNA release and the abnormal opening of the mPTP, as evidenced by downregulated co-localization of anti-DNA and VDAC1, as well as restored Calcein fluorescence intensity (Fig. 3f and g; Supplementary Fig. S4c). Similarly, we found that ACT significantly decreased the content and release of mtDNA into the serum and conditional medium after HIRI or HR insult (Fig. 3; Supplementary Fig. S4d). As depicted in Fig. 3i and Supplementary Fig. S4e, hepatocyte death caused by abnormal MMP was observed in HR-treated but not in ACT-treated hepatocytes, as indicated by the results of JC-1 and TMRE staining.
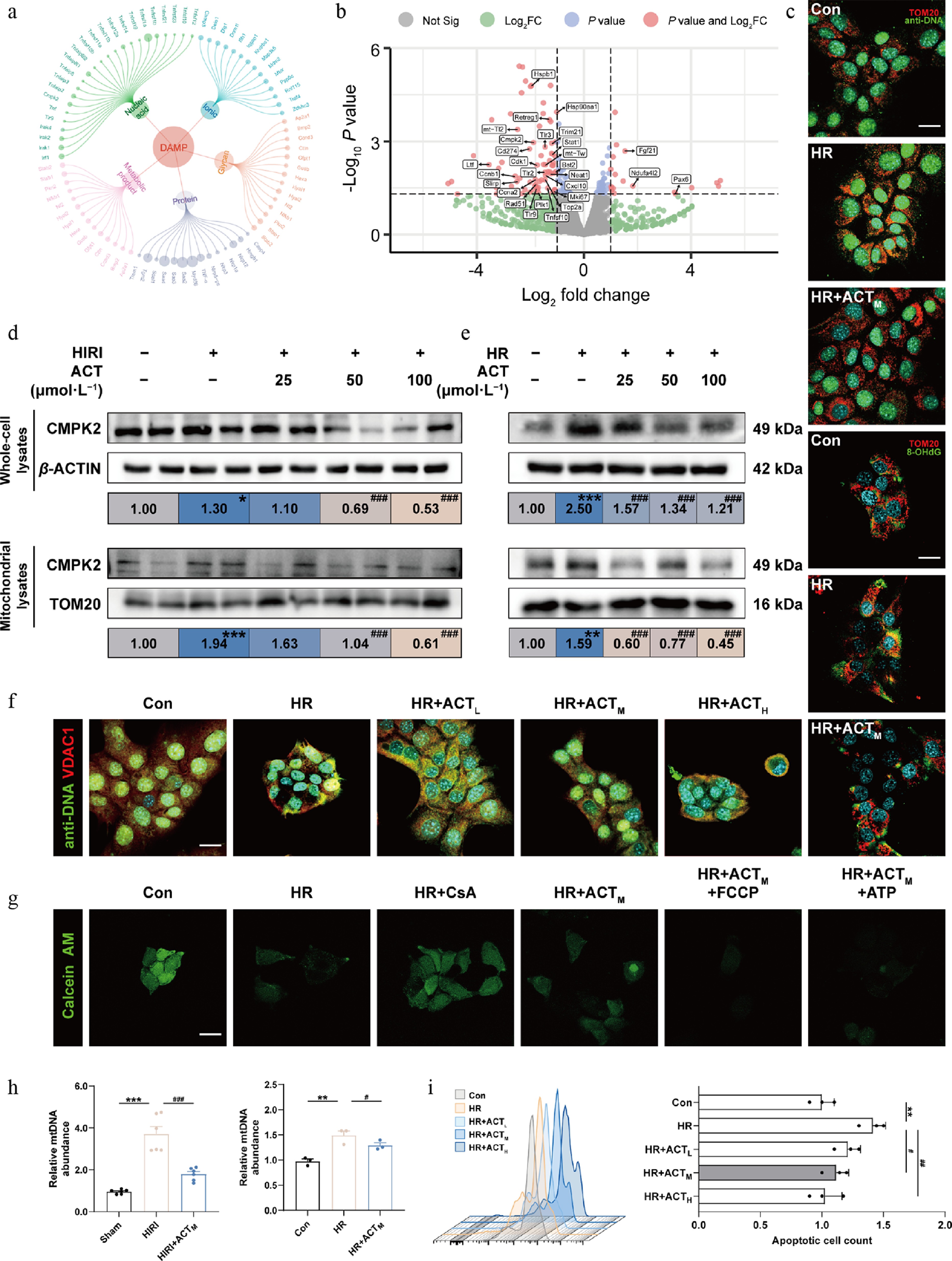
Figure 3.
ACT inhibited the synthesis and release of mtDNA in hypoxic hepatocytes by suppressing CMPK2. (a) Circle network of DAMP molecules. (b) Volcano plots of DEG expression in primary hepatocytes from HIRI, and HIRI + ACT groups. (c) Representative images of immunofluorescence staining for TOM20 (594 nm), anti-DNA (488 nm), 8-OHdG (488 nm), and DAPI (405 nm) in AML12 cells. Scale bar = 20 μm. (d), (e) The protein expression levels of CMPK2 in whole cells and mitochondria of mouse livers and AML12 cells were normalized with β-ACTIN or TOM20. (f) Representative images of immunofluorescence staining for VDAC1 (594 nm), anti-DNA (488 nm), and DAPI (405 nm) in AML12 cells. Scale bar = 20 μm. (g) Representative images of immunofluorescence staining for Calcein AM (488 nm) and in AML12 cells. Scale bar = 20 μm. (h) Left panel, mtDNA level in mouse serum; right panel, mtDNA level in the culture medium of AML12 cells. (i) The flow cytometry results of JC-1 analysis in AML12 cells. Statistical significance: * P < 0.05, ** P < 0.01, *** P < 0.001, compared with control group; # P < 0.05, ## P < 0.01, ### P < 0.001, compared with the model group (Data are presented as the mean ± SEM, n = 3 for cell experiments, n = 6 for mice experiments).
Hypoxia triggers the activation and nuclear translocation of IRF1 through TLR9-MYD88-NF-κB signaling pathway, which is completely blocked by ACT
-
After analyzing the primary response receptor profiling based on hepatocyte-specific RNA-sequencing data, we predicted that several TLR expressions, like Tlr2 were dramatically increased after HR stimulation and decreased after ACT intervention. Although the expression levels of Tlr9 and Tlr7 were not high in the HR group, a significant reduction was observed after ACT administration, particularly with a greater P-value noted for the Tlr9 gene (Fig. 4a). It is noteworthy that TLR9 is predominantly expressed in endosomes and is recognized for its response to mtDNA derived from both intracellular and extracellular sources[20]. While there is currently no literature indicating that TLR2 can directly respond to mtDNA, it may sense other DAMPs triggered by mtDNA[21,22]. Consistent results were observed in both qPCR analysis and multiple immunofluorescence staining, demonstrating the upregulation of TLR2 and TLR9 in both HIRI mouse livers and hepatocytes (marked by hepatocyte markers HNF-4α or CK18) subjected to HR; however, a downregulation was noted following the administration of ACT (Fig. 4b and c; Supplementary Fig. S5a–S5c). As shown in Fig. 4d and Supplementary Fig. S6a, the closure of mPTP caused by CsA markedly decreased the relative fluorescence intensity of TLR2 and TLR9 even under HR conditions; while continuous opening of mPTP induced by FCCP and ATP stimulated the expression of these two receptors, it was no longer reversed by ACT. All TLRs except TLR3 transmit signaling via the MyD88 pathway and activate the nuclear factor-kappa B (NF-κB) transcription factor for exacerbating liver inflammation[23]. The protein levels of TLR9, MyD88, and p-NF-κB p65 were markedly increased in both the HIRI and HR group, which were reduced by ACT treatment (Fig. 4e; Supplementary Fig. S6b). The effect of ACT on reducing TLR2 expression in vitro was not significant. Next, to further define which transcription factors or downstream genes were altered by NF-κB, we utilized the CHEA3 database for prediction and finally noticed IRF1 (Fig. 4f). Recently, NF-κB p65 was reported to bind to the Irf1 promoter region and lead to its transcriptional activation[24], which was further translocated into the nucleus, and functioned as a transcription factor for activating downstream gene networks. As depicted in Fig. 4g and Supplementary Fig. S6c, the fluorescence expression and nuclear localization of IRF1 were significantly downregulated by ACT compared with the HR group. However, this ACT-mediated restrictive translocation effect was completely inhibited under the continuous activation of mPTP (Fig. 4h; Supplementary Fig. S6d).
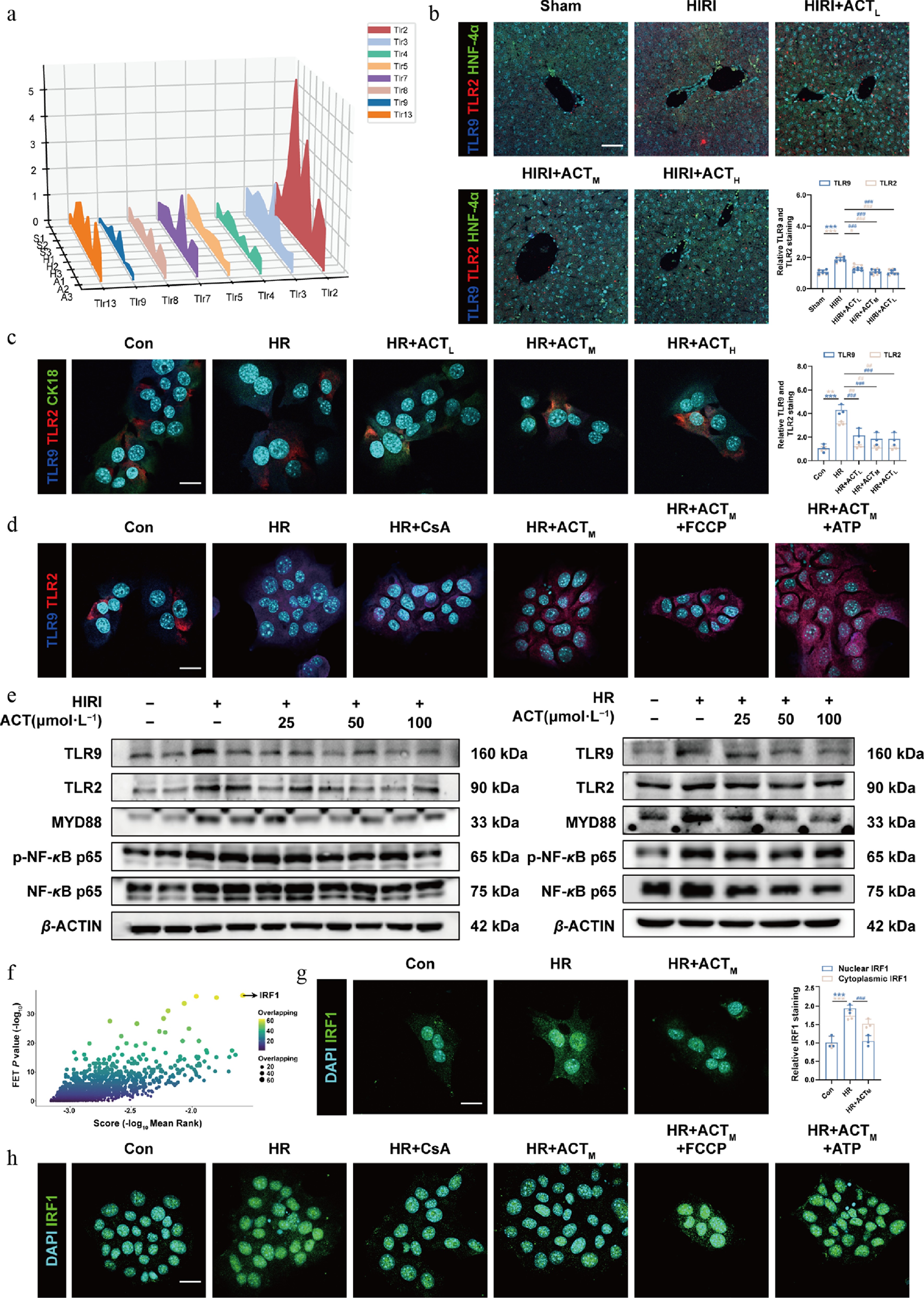
Figure 4.
ACT inhibited the nuclear translocation of IRF1 by regulating the TLR9-MYD88-NF-κB cascade. (a) The three-dimensional graph of gene expression of TLRs in primaryhepatocytes. Representative images of immunofluorescence staining for TLR9 (647 nm), TLR2 (594 nm), HNF-4α (488 nm), CK18 (488 nm), and DAPI (405 nm) in (b) mouse livers, and (c) AML12 cells. Scale bar = 100 and 20 μm, respectively. (d) Representative images of immunofluorescence staining for TLR9 (647 nm), TLR2 (594 nm), and DAPI (405 nm) in AML12 cells. Scale bar = 20 μm. (e) The protein expression of TLR9, TLR2, MYD88, p-NF-κB p65, and NF-κB p65 in the mouse livers and AML12 cells were normalized with β-ACTIN. (f) Screening for transcription factors regulating CMPK2 genes. (g), (h) Representative images of immunofluorescence staining for IRF1 (488 nm), and DAPI (405 nm) in AML12 cells. Scale bar = 20 μm. Statistical significance: *** P < 0.001, compared with control group; # P < 0.05, ## P < 0.01, ### P < 0.001, compared with the model group (Data are presented as the mean ± SEM, n = 3 for cell experiments, n = 6 for mice experiments).
ACT reduces the CMPK2 transcription and subsequent synthesis and release of mtDNA by suppressing IRF1
-
To further demonstrate whether ACT influences the activation of mito-DAMPs-related targets through the inhibition of IRF1 nuclear translocation, we integrated the DEGs from the HIRI + ACT, vs HIRI groups (yellow shaded area) with gene information from the databases of ENCODE (green shaded area), GTRD (blue shaded area) and the CHIP_Atlas (pink shaded area) associated with IRF1 downstream targets (Fig. 5a). We focused on the intersection of 191 genes across four gene sets, which primarily encompassed domains including mtDNA replication, cellular redox homeostasis, and inflammatory reaction. Additionally, we also performed reverse prediction analysis on the key genes among these 191 DEGs, revealing that the major potential transcription factor was IRF1 (Fig. 5b). Combining the above two prediction methods, we selected genes that exhibited significant fold changes for further investigation, including Cmpk2, Hmgb1, and Duox2. The mRNA expression of Cmpk2, Hmgb1, and Duox2 were elevated in the HIRI and HR group, but were reduced by ACT both in mouse livers and hepatocytes (Fig. 5c; Supplementary Fig. S7a). IRF1 can sense a signal from the TLR-MyD88-TRIF pathway, bind to the promoter of CMPK2 and subsequently induce its transcription[25]. Subsequently, to investigate whether ACT influences CMPK2 transcription through IRF1, we utilized the biological prediction website JASPAR to identify the target binding site of IRF1 on CMPK2 (Fig. 5d). Furthermore, our ChIP-qPCR results in AML12 cells confirmed that HR promoted the combination between IRF1 and the promoter or exon 2 regions of Cmpk2 in hepatocytes, which was largely reversed by ACT (Fig. 5e). We then transiently transfected siRNA-targeting Cmpk2, or treated purified CMPK2 protein to validate the indispensable role of CMPK2 in triggering mtDNA leakage and subsequent changes in downstream targets. Notably, the knockdown of Cmpk2 synergistically interacted with ACT, leading to a decrease in mtDNA content (Fig. 5f; Supplementary Fig. S7b), the inhibition of IRF1 nuclear translocation (Fig. 5g; Supplementary Fig. S7c), following transcription of Irf1 and Hmgb1 (Supplementary Fig. S7d). Besides, recombinant CMPK2 proteins efficiently antagonized the therapeutic effect of ACT on mtDNA-TLRs/NF-κB-IRF1 signaling (Fig. 5f and g; Supplementary Fig. S7b and S7c). Furthermore, the SPR assay showed that ACT was able to bind to the IRF1 protein (Fig. 5h) at a specific region: (TRP38 LYS39 HIS40 ALA41 ALA42 PRO74 LYS75 THR76 LYS78 ALA79 ARG82 MET85 ASN86 SER87 LEU88 PRO89 ILE91 GLU92 GLU93 VAL94 LYS95 GLN97 SER98 ARG99 ASN100 LYS101 SER103 SER104 ALA105 VAL106 ARG107) (Fig. 5i, Vina score = −7.4), which might influence where IRF1 binds to the promoter region of CMPK2.
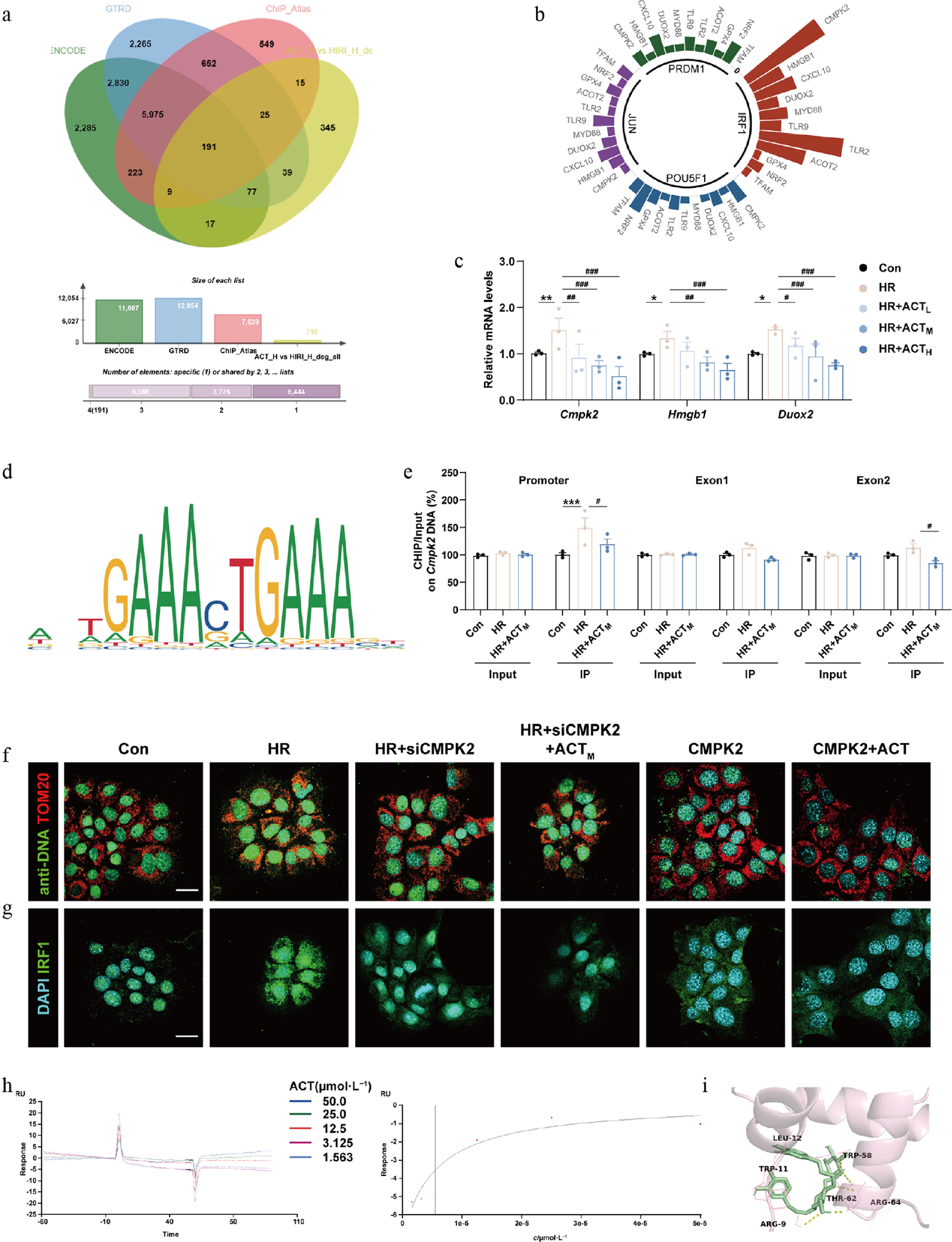
Figure 5.
The inhibition of CMPK2 and ACT administration synergetically reduced the mtDNA synthesis and nuclear translocation of IRF1. (a) The vein diagram of potential IRF1 downstream targets in primary hepatocytes. (b) Prediction analysis of major potential transcription factors. (c) The mRNA levels of Cmpk2, Hmgb1, and Duox2 were normalized with Hprt1 in AML12 cells. (d) The predicted transcription sequence of CMPK2. (e) The IRF1 binding sites on the Cmpk2 promoter and exons were measured by ChIP analysis. Representative images of immunofluorescence staining for (f) TOM20 (594 nm), anti-DNA (488 nm), (g) IRF1 (488 nm), and DAPI (405 nm) in AML12 cells treated with ACT, siCMPK2, or a recombinant CMPK2 protein. Scale bar = 20 μm. (h) The SPR result of ACT binding to IRF1. (i) Molecular docking result of ACT and IRF1. Statistical significance: * P < 0.05, ** P < 0.01, *** P < 0.001, compared with control group; # P < 0.05, ## P < 0.01, ### P < 0.001, compared with the model group (Data are presented as the mean ± SEM, n = 3).
The inhibition of DUOX2 or complete elimination of secreted mtDNA from hypoxic hepatocytes exerts similar anti-HIRI protective effects of ACT
-
Since aberrant production of ROS caused by DUOX2 can exacerbate oxidative stress and hepatocyte damage, we checked and found the increased co-localization of DUOX2 and CK18 in the HR group, which was reversed by ACT (Fig. 6a; Supplementary Fig. S8a). The biological prediction website JASPAR was used to analyze whether DUOX2 was a potential downstream target of IRF1 and predicts the target binding site (Fig. 6b). Additionally, our ChIP-qPCR results in AML12 cells demonstrated that HR facilitated the combination of IRF1 with the promoter or exon 1 regions of Duox2 in hepatocytes, which was significantly reversed by ACT (Fig. 6c). To further prove the involvement of DUOX2 in increased ROS production and subsequent redox damage in AML12 cells, we silenced Duox2 by transfecting siRNA with a specific sequence. The siRNA targeting Duox2 triggered a corresponding decrease in the mRNA levels of Duox2, Cmpk2, Hmgb1, and Cxcl10 (Fig. 6d), and strongly limited ROS generation compared to cells subjected to HR (Supplementary Fig. S8b), which had a synergistic effect with ACT. As expected, after HR insult, the level of ox-mtDNA released into the cytosol (Fig. 6e; Supplementary Fig. S8c) and the nuclear translocation of IRF1 (Fig. 6f; Supplementary Fig. S8d) was increased, but was largely reversed by siDUOX2 with ACT administration. Next, we extracted mtDNA from the culture medium of the different hepatocyte groups, and concentrated purified mtDNA for further cell transfection into another set of hepatocytes. As shown in Fig. 6g and h, the mRNA and protein levels of TLR9 and MYD88, but not TLR2, were upregulated in the mtDNA-HR group compared with the mtDNA-Con group, which were decreased by mtDNA-HR + ACT or totally blocked by DNase I intervention. Furthermore, it is noteworthy that DNase I counteracted the increased transcription and nuclear translocation of IRF1 and subsequent CMPK2 and DUOX2 expression caused by mtDNA-HR transfection (Fig. 6g, i, j; Supplementary Fig. S9a and S9b). These results suggest that the generation and autocrine release of ox-mtDNA may represent the downstream factor of IRF1, and the final oxidative damage driving force, which is also a crucial part of the ACT's protection mechanism against HIRI.
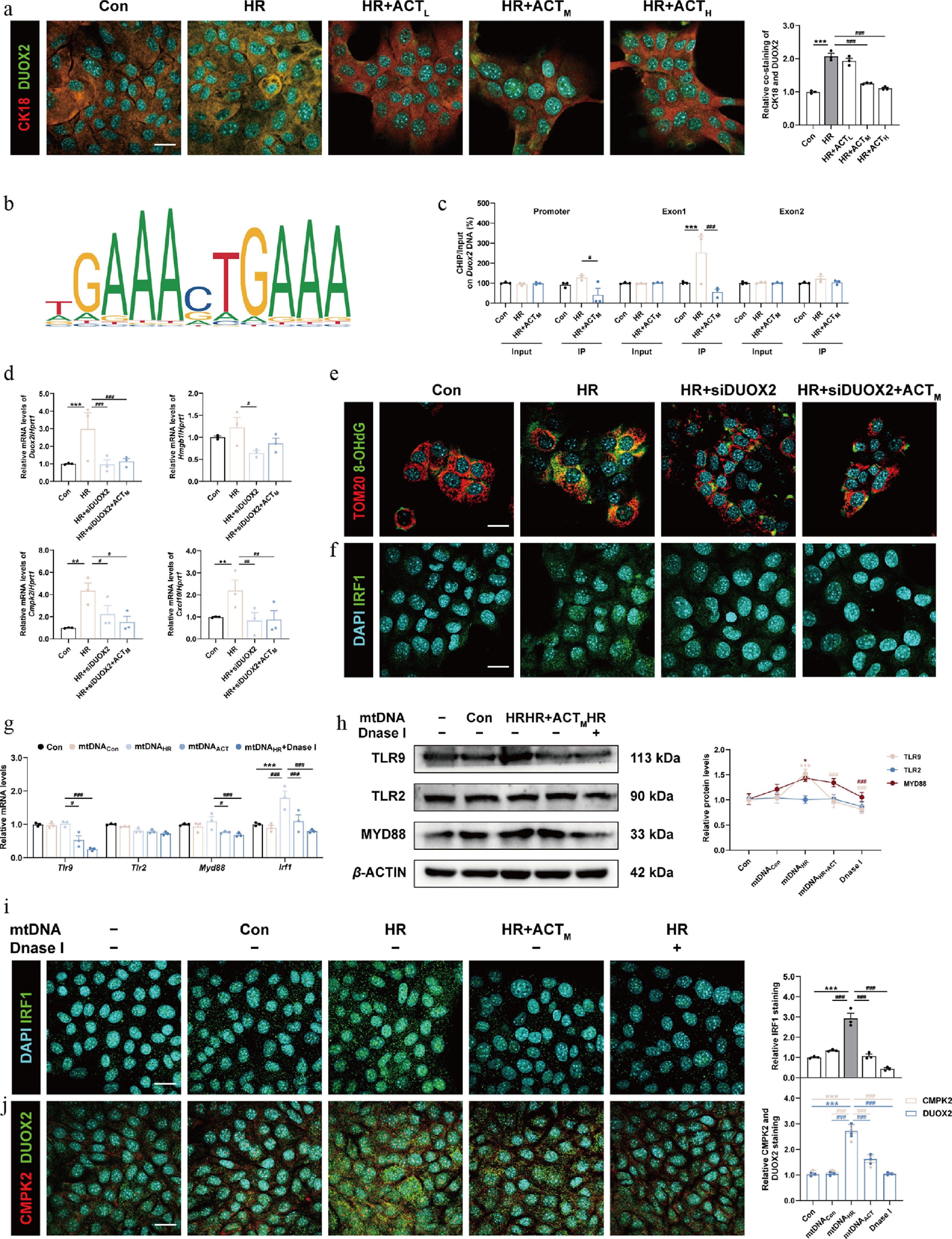
Figure 6.
The inhibition of DUOX2 and ACT administration complementarily reduced the generation and oxidation of mtDNA, which was crucial for the activation of the TLRs-IRF1 pathway. (a) Representative images of immunofluorescence staining for CK18 (594 nm), DUOX2 (488 nm), and DAPI (405 nm) in AML12 cells. Scale bar = 20 μm. (b) The predicted transcription sequence of Duox2. (c) The IRF1 binding sites on the Duox2 promoter and exons were measured by ChIP analysis. (d) The mRNA levels of Duox2, Hmgb1, Cmpk2, and Cxcl10 were normalized with Hprt1 in AML12 cells. Representative images of immunofluorescence staining for (e) TOM20 (594 nm) and 8-OHdG (488 nm), (f) IRF1 (488 nm) and DAPI (405 nm), in AML12 cells treated with siDuox2 and ACT. Scale bar = 20 μm. (g) The mRNA levels of Tlr9, Tlr2, Myd88, and Irf1 were normalized with Hprt1 in AML12 cells transfected with purified mtDNA isolated from different groups. (h) The protein expression of TLR9, TLR2, and MYD88 in AML12 cells were normalized with β-ACTIN. Representative images of immunofluorescence staining for (i) IRF1 (488 nm), (j) CMPK2 (594 nm), DUOX2 (488 nm), and DAPI (405 nm) in AML12 cells. Scale bar = 20 μm. Statistical significance: * P < 0.05, ** P < 0.01, *** P < 0.001, compared with control group; # P < 0.05, ## P < 0.01, ### P < 0.001, compared with the HR or mtDNAHR group (Dataare presented as the mean ± SEM, n = 3).
ACT binds to, and promotes mitophagy-dependent degradation of CMPK2
-
To further investigate the precise mechanism responsible for ACT-mediated degradation of CMPK2, we first applied DARTS and CETSA assays in AML12 cells and found that ACT increased the thermal stability of CMPK2 and inhibited the hydrolysis of CMPK2 caused by pronase, suggesting a potential direct binding between ACT and CMPK2 (Fig. 7a and b; Supplementary Fig. S10a). The interaction analysis from the perspective of molecular docking predicated a strong binding affinity of ACT to the CMPK2 (Supplementary Fig. S10b). To further confirm the interaction between ACT and CMPK2, SPR and MST were employed to determine that CMPK2 was a direct target of ACT (Fig. 7c and d; Supplementary Fig. S10 and S10d). Next, a cycloheximide (CHX) chase experiment showed that the degradation of the CMPK2 protein level was facilitated following ACT treatment (Fig. 7e; Supplementary Fig. S10e). Protein stability is mainly affected by diverse protein degradation manners, including ubiquitin-proteasome or autophagolysosomal degradation pathways. To this end, we treated AML12 cells with MG132 and bafilomycin A1 (BafA1) to inhibit proteasome and lysosome activities, respectively, and found that BafA1, but not MG132, effectively resulted in the accumulation of CMPK2 in the presence of ACT (Supplementary Fig. S11a). Next, Lyso-Tracker (a lysosome-specific fluorescent probe) was used to quantify lysosome biogenesis and CTSB (cathepsin B, a major lysosome hydrolase) was responsible for determining lysosomal function. As shown in Fig. 7f and g, Supplementary Fig. S11b and S11c, the red fluorescence intensity of Lyso-Tracker was increased with ACT treatment in HR condition, which was reversed by BafA1. Meanwhile, ACT promoted an increase in mature CTSB levels in the HR group, but BafA1 blocked the conversion of pro-CTSB to mature CTSB induced by ACT. In addition, we found a higher co-localization of CMPK2 and lysosomal-associated membrane protein 1 (LAMP1) in the HR + ACT group and confirmed that ACT promoted the fusion of CMPK2 to lysosome for degradation, which was reversed by BafA1 (Fig. 7h; Supplementary Fig. S11d).
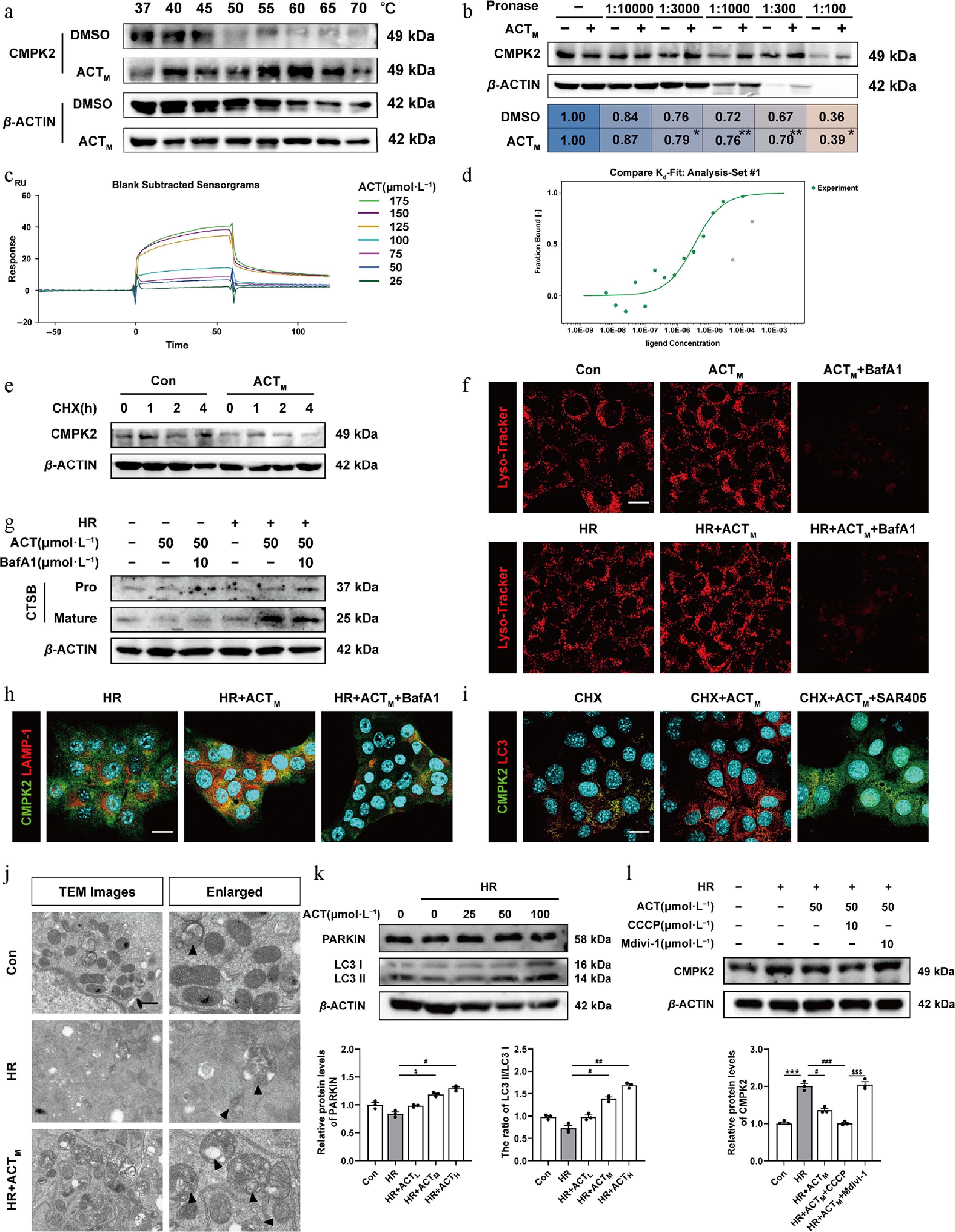
Figure 7.
ACT directly combined to, and induced the degradation of CMPK2 via activating mitophagy. The (a) CETSA, and (b) DARTS assay. The (c) SPR result, and (d) MST analysis of ACT binding to CMPK2. (e) The protein expression of CMPK2 in the CHX chase assay of AML12 cells was normalized with β-ACTIN. (f) Representative images of immunofluorescence staining for Lyso-Tracker (594 nm), and DAPI (405 nm) in AML12 cells. Scale bar = 20 μm. (g) The protein expressions of pro-and mature-CTSB in AML12 cells were normalized with β-ACTIN. (h), (i) Representative images of immunofluorescence staining for CMPK2 (488 nm), LAMP-1 (594 nm), LC3 (594 nm), and DAPI (405 nm) in AML12 cells. Scale bar = 20 μm. (j) Representative TEM images of mitochondria in AML12 cells. Scale bar = 20 μm. The protein expressions of (k) PARKIN, LC3-I, LC3-II, and (l) CMPK2 in AML12 cells were normalized with β-ACTIN. Statistical significance: * P < 0.05, ** P < 0.01, *** P < 0.001, compared with relative control groups; # P < 0.05, ## P < 0.01, ### P < 0.001, compared with the HR group; $^{{\$}{\$}{\$}} $ P < 0.001, compared with the HR + ACTM + Mdivi-1 group (Data are presented as the mean ± SEM, n = 3).
Lysosome-mediated degradation processes mainly include nonselective autophagy and selective autophagy targeting different substrates, P62-dependent ubiquitinated proteins, P62-dependent damaged mitochondria (known as mitophagy), and invading microbes via their interaction with ubiquitination[26]. The fluorescent ratio of LC3/CMPK2 was upregulated following ACT administration but was all inhibited by autophagy inhibitors SAR405 (inhibition of vacuolar protein sorting 34) and 3-MA (inhibition of class III phosphatidylinositol 3-kinases), respectively (Fig. 7i; Supplementary Fig. S11e). The level of CMPK2 was significantly elevated in CHX + ACT + SAR405 or 3-MA groups compared with CHX + ACT group (Supplementary Fig. S11f). Next, we sought to figure out what kind of autophagy-lysosome pathway is responsible for CMPK2 degradation. Transmission electron microscope (TEM) results indicated the formation of autophagosome containing damaged mitochondria in AML12 cells with ACT treatment compared with the HR group (Fig. 7j). Subsequently, the protein levels of PARKIN, LC3, PINK1, and P62 were markedly increased while P62 was decreased after ACT treatment compared with the HR group (Fig. 7k; Supplementary Fig. S11g). Notably, the protein expression changes of mitophagy markers like PARKIN and PINK1 in mouse livers exhibited the same trend as those in AML12 cells (Supplementary Fig. S11h). STX17, SNAP29, and VAMP8 constitute the complex of SNARE proteins that play a key role in the fusion of autophagosomes with lysosomes. Notably, there was a significant change in the level of VAMP8 compared to STX17 and SNAP29 (Supplementary Fig. S12a). Subsequently, we designed three distinct small interfering RNAs (siRNA) for the knockdown of VAMP8, and then screened out the most effective VAMP8 siRNA for a further CMPK2 degradation experiment (Supplementary Fig. S12b). Supplementary Figure S12c illustrated that the knockdown of VAMP8 inhibited the effect of ACT in promoting CMPK2 degradation. Furthermore, we treated AML12 cells with a mitophagy agonist (CCCP) or inhibitor (Mdivi-1) and checked CMPK2 expression changes after ACT treatment under HR insult. The decreasing trend of CMPK2 levels in the HR + ACT group was clearly attenuated by Mdivi-1 administration, but intensified by CCCP (Fig. 7l). Meanwhile, the immunoprecipitation assay confirmed the reduction of CMPK2 binding with both P62 and K63 following ACT administration (Supplementary Fig. S12d). The above results indicate that ACT promoted CMPK2 degradation in hepatocytes by promoting mitophagy, but not influencing the ubiquitin-proteasome degradation pathway.
Hepatocyte-specific CMPK2 overexpression directly counteracts the protective anti-HIRI effects of ACT
-
To further corroborate the role of CMPK2-mediated hepatocyte lipid metabolism injury in HIRI and whether it is unique to ACT therapy, the CMPK2-carrying AAV8 with ZsGreen fluorescein labeling was successfully injected into mice via the tail vein before HIRI surgery and ACT pre-administration (Fig. 8a; Supplementary Fig. S13a). Hepatic function indices (AST and ALT in the serum), along with lipid contents (FFA and total cholesterol [TC] in serum), indicated that liver hypoxic damage and abnormal lipid metabolism were aggravated by CMPK2 overexpression in hepatocytes, which couldn't be alleviated even with the presence of ACT (Fig. 8b and c; Supplementary Fig. S13b). Meanwhile, histological examination and co-staining of TUNEL and HNF-4α in CMPK2-overexpression livers revealed significant signs of extensive inflammatory cell infiltration and hepatocellular necrosis, which was improved with the intervention of ACT to some extent (Fig. 8d and e). Besides, the hepatic level of FFA was downregulated after injection with CMPK2-carrying AAV8 and ACT treatment (Fig. 8f). To further investigate the stimulatory effect of CMPK2 overexpression on hepatic lipid metabolism, the alterations of oxidative stress markers, including ROS, MDA, and GSH, were analyzed in different groups. As shown in Fig. 8g; Supplementary Fig. S13c and S13d, the fluorescence staining of DCFH-DA and the level of MDA were all significantly increased, and the content of GSH was decreased in the HIRI + AAVCMPK2 group and only slightly changed after ACT intervention. Notably, ACT restrained the secretion of mtDNA into the serum compared with the HIRI + AAVCMPK2 group (Fig. 8h). These results suggest that the explosive increase of CMPK in hepatocytes directly stimulates mtDNA production and simultaneously affects ROS accumulation caused by excessive FFA oxidation. Furthermore, we validated the expression changes of several key targets involved in CMPK2-centered signaling pathways among various groups. The changes of CMPK2, TLR9, MYD88, p-NF-κB, and IRF1, but not HMGB1, TLR2, ACOT2, or DUOX2, were all significantly elevated in the HIRI + AAVCMPK2 group, but almost unchanged after ACT pre-administration in mouse livers (Fig. 8i and j; Supplementary Fig. S13e and S13f). Taken together, while ACT can directly bind to, and inhibit the hepatic expression of CMPK2, it is unable to withstand the adverse effects caused by the stimulation of exogenous CMPK2 and the subsequent lipid metabolism disorders under HIRI conditions.
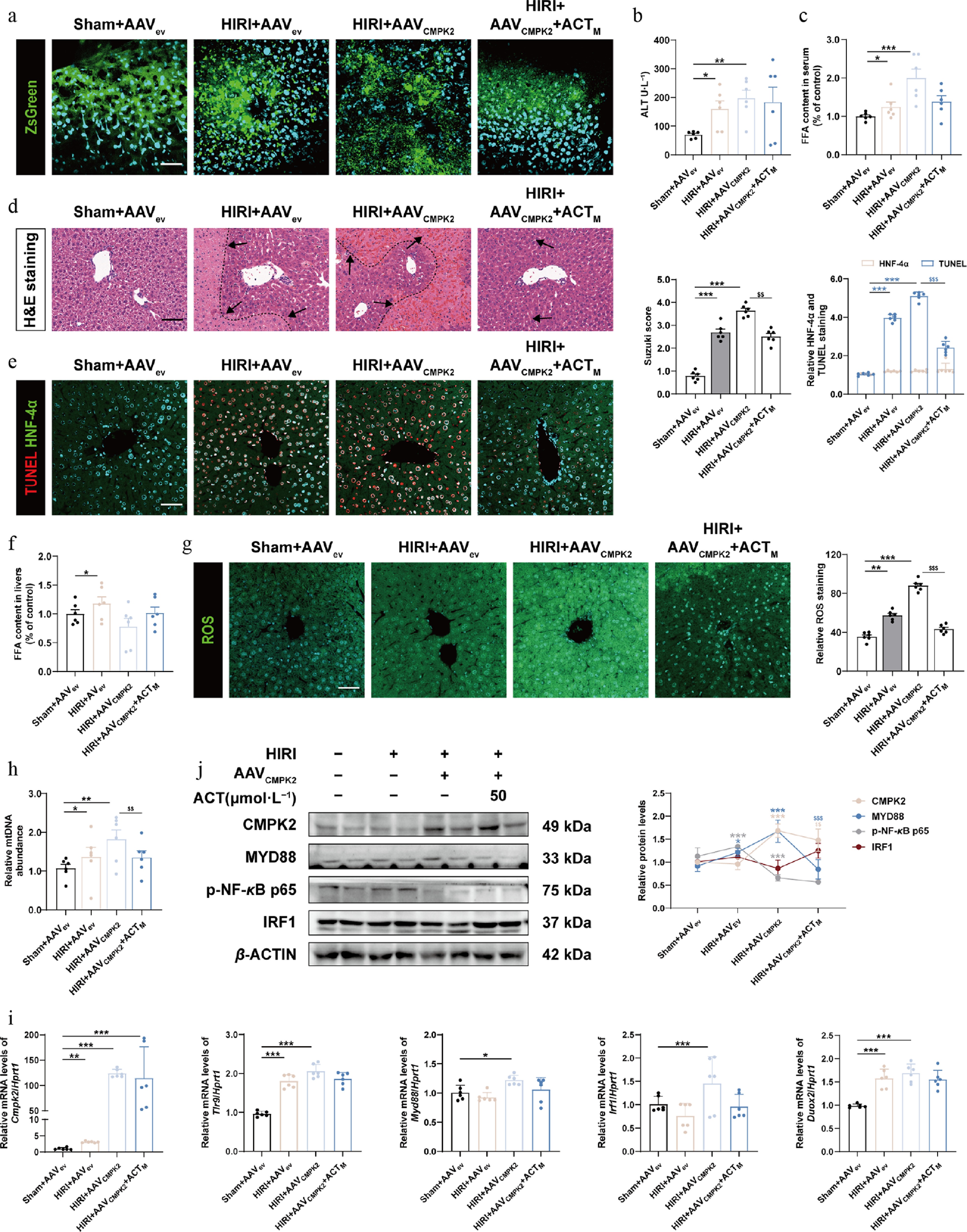
Figure 8.
Hepatocyte specific overexpression of CMPK2 largely blocked the anti-HIRI effects of ACT on mouse livers. (a) Representative images of immunofluorescence staining for ZsGreen (488 nm) and DAPI (405 nm) in mouse livers. Scale bar = 100 μm. The serum level of (b) ALT and (c) FFA in different groups of mice. (d) Representative images of H&E staining were quantified using Image J software. Scale bar = 100 μm. (e) Representative images of immunofluorescence staining for TUNEL (594 nm), HNF-4α (488 nm), and DAPI (405 nm) in mouse livers. Scale bar = 100 μm. (f) The level of FFA in different groups of mouse livers. (g) Representative images of immunofluorescence staining for ROS (488 nm) and DAPI (405 nm) in mouse livers. Scale bar = 100 μm. (h) The serum level of mtDNA in different groups. (i) The mRNA levels of Cmpk2, Tlr9, Myd88, Irf1, and Duox2 were normalized with Hprt1 in mouse livers. (j) The protein expression of CMPK2, MYD88, p-NF-κB, and IRF1 in the mouse livers were normalized with β-ACTIN. Statistical significance: * P < 0.05, ** P < 0.01, *** P < 0.001, compared with Sham + AAVev groups; $^{{\$}{\$}} $ P < 0.01, $^{{\$}{\$}{\$}} $ P < 0.001, compared with the HIRI + AAVCMPK2 group (Data are presented as the mean ± SEM, n = 6).
-
Our previous report indicated that, in the process of alleviating HIRI damage, it is crucial to promptly block redox damage and mitochondrial dysfunction in hepatocytes[17]. Moreover, if the processes of hepatocyte lipotoxicity and death are not immediately halted, they will further mess up metabolic homeostasis in the entire liver and trigger a more severe inflammatory cascade reaction[27,28]. Here, we noted that during HIRI, the abnormal elevation of mitochondrial ACOT2 in hepatocytes promoted the synthesis of FFAs from Acetyl-CoA and subsequently stimulated redox imbalance characterized by an increase in ROS (Figs 1 and 2). Bioinformatics analysis of sequencing data from the whole HIRI liver and hepatocytes indicated a significant upregulation of CMPK2 expression, which was positively associated with mitochondrial dysfunction and disease progression. Importantly, in the ROS-stimulated oxidative environment, CMPK2 further promoted the oxidation and synthesis of a typical DAMP, mtDNA, and stimulated its secretion through the mPTP opening in hepatocytes (Fig. 3). Cell-based autocrine experiments have confirmed that purified mtDNA from oxidized hepatocytes stimulated the transcription of Cmpk2 and Duox2 by activating the TLR9-MYD88-NF-κB-IRF1 signaling pathway in those hepatocytes with low oxidation state (Figs 4 and 5). Mechanistically, ACT combined with IRF1, inhibited its nuclear translocation, and the expression and function of downstream targets, especially for preventing CMPK2-stimulated mtDNA synthesis, while simultaneously reducing ROS level produced by DUOX2, thereby ameliorating the pathological phenomena caused by HIRI or HR (Fig. 6). Additionally, Fig. 7 supported that ACT also promoted the translational degradation of CMPK2 through mitochondrial autophagy rather than through other lysosomal or ubiquitin-mediated degradation. In contrast, hepatocyte-specific Cmpk2 overexpression directly counteracted the protective anti-HIRI effect of ACT in vivo (Fig. 8).
FAs are essential components of cell membranes and serve as important energy substrates that directly influence the homeostasis of mitochondrial function. However, the accumulation of FAs in the liver, particularly within hepatocytes, leads to the overproduction of ROS and various modes of cell death[29]. Here, we found that ACOT2, but not other members of the mitochondrial ACOT family, was abnormally elevated and positively correlated with FFA levels during HIRI, which was significantly reduced by ACT. Interestingly, under hypoxic conditions, the accumulation of FFA mediated by ACOT2 did not enhance fatty acid β-oxidation; instead, it shifted towards exacerbating the production of ROS (Fig. 2). High concentrations of FFA induced mitochondrial uncoupled respiration, leading to proton re-entry without ATP production, which in turn inhibited the respiratory chain[30,31]. Further research suggested that the mechanism might involve FFAs acting as cofactors to enhance the proton permeability of adenine nucleotide translocase 2, thereby accelerating the uncoupling of mitochondrial respiration and the opening of mPTP. This exacerbates the disorder of mitochondrial ROS production, forming a lipotoxicity-hyperoxidation-inflammation vicious cycle[32]. Following ACT administration, the induction of ACOX1 and the downregulation of CMPK2 synergistically restored normal FFA oxidation and entire mitochondrial function. Furthermore, it led to a reduction in FFA production, partially attributed to the decreased levels of ACOT2. However, the damage priority and interaction patterns of CMPK2 and ACOT2 targets under HIRI, as well as potential drug targets, require further investigation.
CMPK2 is necessary for the production of ox-mtDNA fragments within the mitochondria. Once ox-mtDNA was cleaved by FEN1, its fragments escaped into the cytoplasm via mPTP, activating the NLRP3-cGAS-STING-IFN-γ pathway and inducing inflammation[33]. CMPK2 also directly promoted NLRP3 inflammasome activation and macrophage recruitment by ensuing ox-mtDNA-cGAS-STING signaling pathway, establishing it as a critical mediator during the progression of metabolic liver disease[14,34,35]. ACT reduced the CMPK2 transcription caused by HIRI or HR and decreased mtDNA synthesis, while it was simultaneously accompanied by a decrease in co-staining with TOM20 and 8-OHdG probes (Fig. 3). These resulsuggest that ACT inhibits the synthesis, oxidation, and release of mtDNA in hypoxic hepatocytes. Interestingly, ACT did not largely influence the expression of IFN-γ in these conditions, indicating that it may affect other downstream pathways of mtDNA other than cGAS-STING. Owing to its unmethylated CpG motifs, released mtDNA is also reported to activate different molecular signaling papathways, like pattern recognition receptors. Notably, this mtDNA-dependent activation exhibits a certain degree of sensitivity, demonstrating a specific preference for TLR9, which in turn induces a series of inflammatory responses through NF-κB-related pathways[36]. Although both TLR2 and TLR9 were increased under HIRI conditions, ACT significantly reduced the expression of both receptors, with a more pronounced effect observed for TLR9 (Fig. 4e). Additionally, ACT inhibited the TLR9-MYD88-NF-κB pathway, decreased the nuclear translocation of IRF1 and subsequent transcriptional expression of CMPK2 as well as mtDNA synthesis. Notably, we recently discovered that hypoxia enhanced the HMGB1 release, which further increased the nuclear translocation and function of IRF1 through the TLR2 receptor, accompanied by the infiltration of macrophages[4]. This current study further supplied the possibility that HIRI activated IRF1 transcription via different DAMPs, as well as the synergistic antioxidant and anti-inflammatory protective effects of ACT (Figs 3, 4, 6). More importantly, after the administration of ACT, there was a reversal of the excessive accumulation of FFAs, mitochondrial respiration uncoupling, and the opening of mPTP, thereby preventing the efflux-recognition-uptake-activation circuit of mtDNA and forming a protective loop.
Although the basal hepatic expression of CMPK2 is relatively low, we observed a significant increase in its levels within the mitochondria of hepatocytes under hypoxic stimulation, which was dramatically reversed after ACT treatment (Fig. 3d, lower panel). In addition to affecting Cmpk2 transcription, ACT also significantly decreased the CMPK2 protein stability (Fig. 7e) without affecting its mRNA stability (data not shown). Furthermore, the observed differences between the co-treatment of ACT with MG132 or BafA1 indicated that ACT degraded CMPK2 through the autophagy-lysosome pathway and may explain why it promoted the co-localization of CMPK2 and lysosomal marker LAMP1 (Fig. 7h). In addition to influencing other classical autophagy-lysosome degradation pathways, ACT activated the expression of mitophagy-related proteins, particularly PARKIN, PINK1, LC3, and VAMP8. Notably, the accumulation of CMPK2 was markedly observed upon the inhibition of the early stage of autophagy by using SAR405 and 3-MA, the inhibition of mitophagy by using mdivi-1 or silencing VAMP8 in hepatocytes, even in the presence of ACT (Fig. 7l; Supplementary Figs S11f, S12c). These results suggest that ACT specifically promotes the P62-dependent mitophagic degradation of CMPK2, revealing a novel mechanism for the targeted degradation of CMPK. P62 can bridge ubiquitinated substrates, like PARKIN on damaged mitochondria, with autophagosomal membranes, which is dependent on distinct post-translational modifications within the UBA domain[37]. However, we preliminarily found that ACT did not affect the activation of TBK1 and ULK1 (responsible for the phosphorylation of P62 at Ser349 and Ser407 sites), nor TIP60 expression (responsible for the acetylation of P62 at K420 site) (data not shown). After confirming that ACT can bind to CPMK2 via SPR and MST assays, we also predicted potential specific binding site (GLY10, GLY11, PRO12, GLY13, ALA14, GLY15, LYS16, GLY17, THR18, HIS31, SER33, ALA34, and GLY35). Whether ACT assists P62 in executing PARKIN-dependent ubiquitination of the outer mitochondrial membrane and subsequent autophagy by binding to the accumulated CMPK2 in damaged mitochondria, and whether this process is related to specific domains on CMPK2, warrants further investigation.
-
Taken together, our study underscores the significance of CMPK2-driven abnormal redox metabolism in the pathogenesis of HIRI and highlights its potential as a novel therapeutic target for combinatorial intervention with natural bioactive compounds like ACT.
-
All experiments involving animals were conducted according to the ethical policies and procedures approved by the Institutional Animal Care and Use Committee of the Beijing University of Traditional Chinese Medicine (Approved project: BUCM-2023112904-4146 and BUCM-2025030306-1093).
-
The authors confirm contributions to the work as follows: analysis and interpretation of results, draft manuscript preparation: Luo R, Yang Y; formal analysis: Luo R; methodology: Luo R, Zhang Y, Wang L, Dou Y; visualization: Che X, Kang W; validation: Chen R; investigation: Liu R; study conception and design, writing, review & editing: Li X. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
-
This work was supported by grants from the Scientific Research Innovation Capability Support Project for Young Faculty (ZYGXQNJSKYCXNLZCXM-H4); National Natural Science Foundation of China (Grant No. 82274186 to Xiaojiaoyang Li); National High-Level Talents Special Support Program to Xiaojiaoyang Li.
-
The authors declare that there are no conflicts of interest.
-
accompanies this paper online at: https://doi.org/10.48130/targetome-0026-0011.
-
#Authors contributed equally: Ranyi Luo, Yun Yang
- Supplementary Table S1 The information of used antibodies.
- Supplementary File 1 Supplementary methods to this study.
- Supplementary Fig. S1 The injured hepatic function caused by HIRI process was alleviated by ACT.
- Supplementary Fig. S2 ACT improved lipid accumulation in HIRI livers.
- Supplementary Fig. S3 ACT mitigated mitochondrial damage and oxidative stress under hypoxia condition.
- Supplementary Fig. S4 ACT inhibited the synthesis and release of mtDNA.
- Supplementary Fig. S5 ACT downregulated the expression of TLR9 and TLR2.
- Supplementary Fig. S6 ACT repressed the nuclear translocation of IRF1.
- Supplementary Fig. S7 Inhibition of CMPK2 and ACT treatment coefficiently reduced the nuclear translocation of IRF1 and its downstream targets.
- Supplementary Fig. S8 The inhibition of DUOX2 combined with ACT administration decreased the generation, oxidation and downstream pathways of mtDNA.
- Supplementary Fig. S9 Dnase I and ACT treatment counteracted the activation of IRF1 triggered by mtDNA transfection.
- Supplementary Fig. S10 ACT promoted CMPK2 degradation in hepatocytes.
- Supplementary Fig. S11 ACT promoted CMPK2 degradation by activating mitophagy.
- Supplementary Fig. S12 ACT facilitated the fusion of autophagosome lysosome via inducing VAMP8.
- Supplementary Fig. S13 Hepatocyte-specific overexpression of CMPK2 significantly blocked the hepatoprotective effects of ACT against HIRI.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Luo R, Yang Y, Che X, Chen R, Zhang Y, et al. 2026. Acteoside mitigates hepatic ischemia-reperfusion injury by targeting CMPK2-intervened redox metabolism. Targetome 2(2): e013 doi: 10.48130/targetome-0026-0011 

Acteoside mitigates hepatic ischemia-reperfusion injury by targeting CMPK2-intervened redox metabolism
- Received: 06 January 2026
- Revised: 03 February 2026
- Accepted: 23 February 2026
- Published online: 31 March 2026
Abstract: Hepatic ischemia-reperfusion injury (HIRI) is an inevitable complication after liver surgery and liver transplantation. Cytosine monophosphate kinase 2 (CMPK2) plays an essential role in controlling mtDNA synthesis and redox metabolism, yet its contribution to HIRI and potential therapies remain undefined. Through integrative sequencing techniques and various molecular biology experiments, we demonstrated that in the initiation stage of HIRI, excessive Acly-CoA promoted the Acyl-CoA thioesterase 2-dependent synthesis and accumulation of free fatty acids in the mitochondria, thus facilitating ROS production in hepatocytes. In response to oxidative stress, CMPK2 stimulated the synthesis and oxidation of mtDNA, which was further released from opening mPTP, and activated the TLR9-MYD88-NF-κB-IRF1 pathway in an autocrine manner. We then demonstrated that acteoside (ACT) significantly protected CMPK2-mediated redox metabolism and following HIRI both in vivo, and in vitro. Mechanistically, ACT inhibited IRF1 nuclear translocation to impose the transcription of both Cmpk2 and Duox2, preventing ROS production and mtDNA leakage. Furthermore, ACT binds to, and promotes the mitophagy-dependent degradation of CMPK2. Notably, specific overexpression of CMPK2 in hepatocytes interposed the therapeutic benefits of ACT. Collectively, our findings establish CMPK2 as a key driver of redox dysregulation in HIRI, and underscore ACT as a multitarget therapeutic agent for clinical translation.
-
Key words:
- Hepatic ischemia-reperfusion injury /
- CMPK2 /
- Acteoside /
- mtDNA /
- IRF1