








-
Natural products (NPs), derived from diverse biological sources including plants, microorganisms, marine organisms, fungi, and animals have long been regarded as an irreplaceable treasure trove for drug discovery and development[1]. For centuries, they have served as the foundation of traditional medicine systems worldwide, and their significance in modern pharmacology remains unparalleled, statistics from the World Health Organization (WHO) and the US Food and Drug Administration (FDA) indicate that approximately 60% of clinically approved anticancer drugs and 70% of anti-infective agents are either NPs, their derivatives, or synthetic analogs inspired by natural scaffolds[2−4]. The two most important characteristics distinguishing the NP from other synthetic molecules are broad structural diversity and bio-compatibility[5,6].
NPs are structurally intricate and chemically diverse, including polyketides, terpenoids, anthraquinones, alkaloids, and polyphenols. These molecules have distinctive functional groups, for example, the peroxide bridge of artemisinin, the anthraquinone core of emodin, and the polyphenolic scaffold of resveratrol. This molecular precision enables effective interaction with biological targets from proteins and nucleic acids to lipid membranes. NPs also contain stereogenic centers shaped by evolutionary pressures. This evolutionary refinement underlies their continued importance as privileged scaffolds in drug discovery[1,7].
Although NPs have enormous therapeutic potential, their clinical translation is somewhat blocked by the challenge of target identification[8]. Without defined targets, optimizing pharmacokinetics, minimizing off-target effects, mapping structure-activity relationships (SAR), or predicting drug interactions becomes largely speculative[8,9]. This difficulty stems from four inherent properties of NPs:
Structural complexity and instability. NPs are structurally complex and contain multiple reactive groups, including polyphenols, peptides, and polyhydroxylated alkaloids. Some NPs show poor solubility or become unstable under physiological conditions. These properties make probe design and binding experiments difficult[5,7].
Low abundance bioavailability. NPs often act at nanomolar to micromolar levels in vivo. Their low concentrations reduce the sensitivity of conventional methods[7].
Polypharmacology. Unlike synthetic drugs designed for single targets, NPs usually modulate several proteins or signaling pathways simultaneously. These overlapping effects make it hard to delineate the contribution of individual targets.
Indirect mechanisms. Some NPs do not bind proteins directly, but by modulating gene expression, metabolic flux, or cellular microenvironments. This obscures the connection between molecular mechanism and phenotypic effect[8,9].
In recent years, the progress in chemical biology, omics-technologies, computational biology, and structural biology has provided powerful tools to address this challenge. The following sections systematically review the current strategies for identifying targets of NPs and highlight their principles, applications, advantages, and limitations.
-
Protein libraries including cell lysates, purified proteomes, and recombinant protein collections have emerged as foundational tools for unbiased NP target discovery, addressing a major challenge in drug development: figuring out which proteins in the cell interact with natural bioactive compounds. Protein library-based methods enable large-scale, unbiased screening of NP-protein binding events, leveraging intrinsic changes in protein physicochemical properties (e.g., stability, protease sensitivity, oxidation resistance, solubility) induced by ligand binding, eliminating the need for cumbersome chemical modification of NPs or preselection of candidate targets.
Thermal shift assay (TSA-CETSA-TPP)
-
Proteins tend to unfold, denature, and precipitate when temperature increases. Ligand binding can perturb the energy state of the target protein and consequently alter its thermal stability. Such effects result from shifts in Gibbs free energy associated with enthalpic and entropic contributions. Within the free energy landscape model, unfolded conformation resides at the highest energy state, while the native structure is at the minimum of the energy funnel[10]. Ligand-bound proteins are less likely to denature or aggregate at the same temperature than unbound proteins[11]. This stabilization is due to the lower energy state of ligand-protein complexes, which require a greater energy barrier for unfolding[12]. Protein stability is commonly quantified by the melting temperature (Tm), defined as the temperature at which 50% of the protein population transitions from its folded state to an unfolded state[13,14].
These principles underpin the thermal shift assay (TSA), also called differential scanning fluorimetry (DSF)[14−16]. In a typical DSF experiment, purified proteins are incubated with environmentally sensitive fluorescent dyes in multi-well plates[17]. The protein begins to unfold as the temperature increases, and the previously buried hydrophobic regions become exposed. These regions interact with the dye and then generate a sharp fluorescence signal. Changes in fluorescence are used to determine the melting temperature (Tm) and to assess ligand binding[18,19]. This assay is rapid, sensitive, and well suited for screening protein-ligand interactions. Notably, it only works for purified proteins[20].
Molina and colleagues adapted this approach for intact cells and established the cellular thermal shift assay (CETSA) in 2013[11]. CETSA is based on the principle that the ligand binding changes the thermodynamic stability of proteins within their native environment, so it works with living cells, cell lysates, and tissue samples. After compound treatment, protein samples are heated to different temperatures. Ligand-bound proteins remain stable, while unbound ones are denatured and degraded. The soluble protein fraction is then analyzed via western blot or mass spectrometry to confirm the target engagement (Fig. 1a). CETSA requires relatively high compound concentrations and offers limited throughput, making it more suitable for target validation than for de novo target discovery[20].
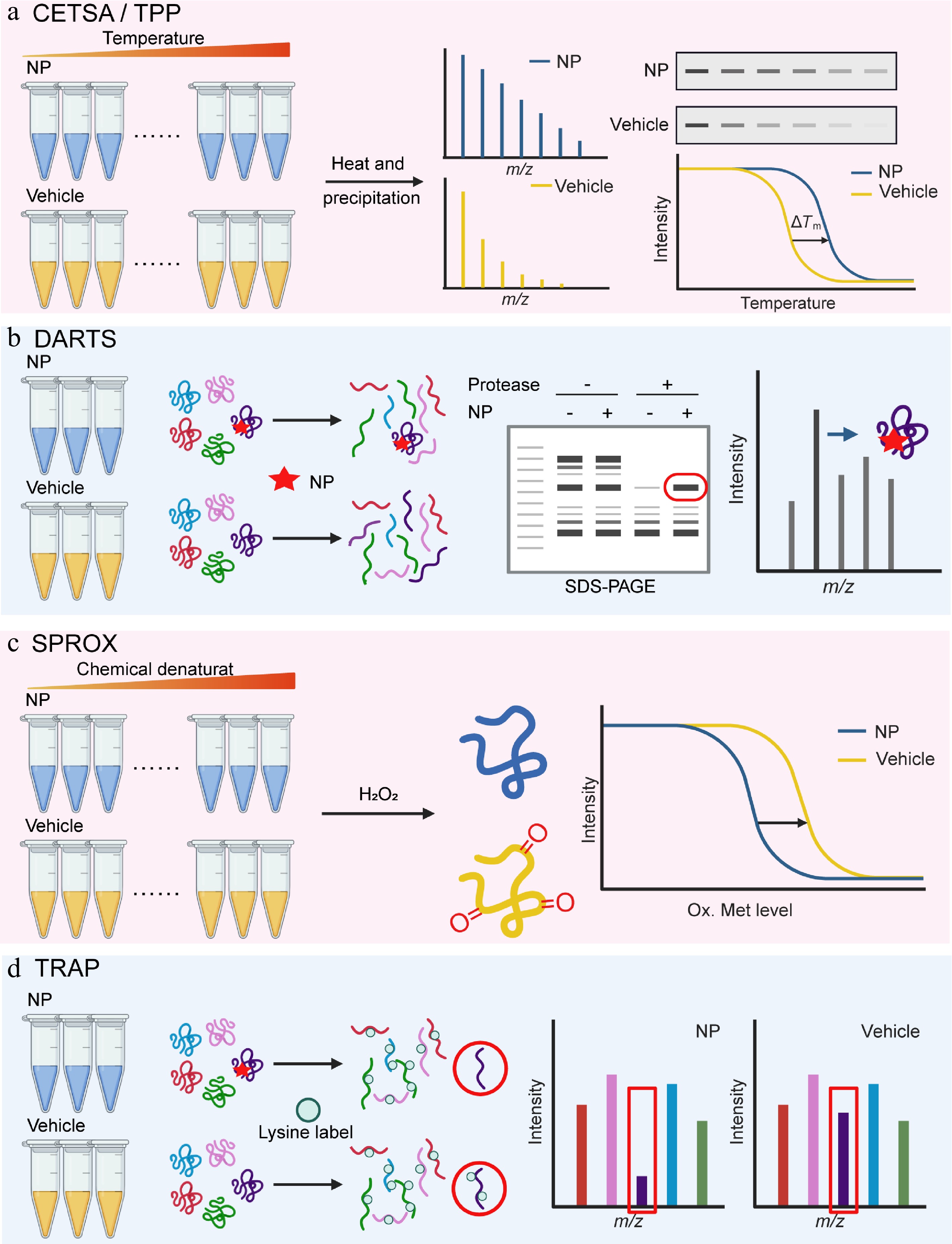
Figure 1.
Natural product target identification based on protein libraries. (a) Thermal shift assay (TSA-CETSA-TPP), (b) drug affinity responsive target stability (DARTS), (c) stability of proteins from rates of oxidation (SPROX), and (d) target-responsive accessibility profiling (TRAP).
To address the low sensitivity and insufficient throughput of CETSA, and expand its application in target/off-target identification and biomarker discovery, Savitski et al. developed thermal proteome profiling (TPP) technology based on CETSA in 2014[21]. TPP is often considered synonymous with MS-CETSA (mass spectrometry-CETSA). MS-CETSA is an advanced variant of CETSA that replaces western blot with mass spectrometry[12,22,23]. It can simultaneously detect thermal stability changes in thousands of proteins and enables high-throughput quantitative assessment of drug binding affinity and mechanisms of action. Concretely, NPs and solvent controls are incubated with living cells or cell lysates, respectively, then heated at different temperatures. Untreated soluble protein samples are subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) detection and analysis. After data normalization, melting curves are plotted to identify proteins with altered stability (Fig. 1a). Franken et al. found that TPP can unbiasedly analyze direct or indirect binding between NPs and target proteins within the 37–67 °C range[24]. Direct binding is detectable by incubating drugs with cell extracts, while downstream targets can be identified by comparing TPP results from intact cells and cell extracts.
Savitski and his team initially applied TPP for the target validation of bacterial NPs, specifically identifying over 50 target proteins of Stellatin[21]. Several recent studies have successfully applied TPP to elucidate the molecular mechanisms of bioactive natural compounds. For instance, the phenanthroindolizidine alkaloid PF403 was found to exert anti-glioma activity through direct binding to nicotinamide phosphoribosyl transferase (NAMPT), a key enzyme in NAD+ biosynthesis. PF403 inhibits NAMPT enzymatic activity, leading to NAD+ depletion in glioma cells[25]. In another study, the flavonoid neoeoricitrin was shown to promote osteogenic differentiation of human dental pulp stem cells and enhance bone regeneration in vivo. TPP revealed BECLIN1, an essential regulator of autophagy, as the direct binding partner of neoeoricitrin[26]. Gambogic acid was characterized as a selective inducer of pyroptosis in prostate cancer cells. TPP identified canopy FGF signaling regulator 3 (CNPY3) as the direct target of gambogic acid. Gambogic acid promotes the recruitment of the delactylase sirtuin 1 (SIRT1) to CNPY3, resulting in the removal of lysine lactylation of CNPY3 and subsequent lysosomal destabilization that triggers pyroptotic cell death[27].
TPP offers advantages such as good stability, a high number of identified proteins, and being a broad-spectrum protein identification technology that does not require antibody-based incubation. However, it has drawbacks, including long experimental time, high cost, and limited detection of membrane proteins.
Drug affinity responsive target stability (DARTS)
-
DARTS is a label-free, small-molecule target discovery technology based on chemical proteomics, first proposed and investigated by Lomenick et al.[28]. The research team hypothesized that the binding of drugs to target proteins stabilizes the protein structure, endowing it with resistance to protease hydrolysis. To verify this conjecture, they selected the FKBP12 protein, known to bind the inhibitors rapamycin and FK506, as the research object, hydrolyzed it with subtilisin, and observed the hydrolysis resistance of FKBP12 upon binding to these ligands via SDS-PAGE[28]. This study confirmed that proteins become more stable after drug binding and established the systematic target identification technology, DARTS.
In DARTS experiments, protease-mediated hydrolysis outcomes need to be detected by specific methods, such as SDS-PAGE, gel staining techniques (e.g., Coomassie brilliant blue staining), as well as two-dimensional electrophoresis (2D-PAGE) and gel-based or non-gel mass spectrometry (LC-MS/MS). By comparing the drug group with the control group, obvious differences are identified to screen candidate targets for subsequent validation (Fig. 1b). DARTS has been widely used due to its simple operation and short time consumption.
DARTS has been widely used to identify targets of NPs. Matrine, a natural alkaloid isolated from Sophora flavescens, was found to promote axonal regeneration and functional recovery in a mouse model of a spinal cord injury. DARTS, coupled with mass spectrometry (DARTS-MS), identified heat shock protein 90 (HSP90) as a direct binding target of matrine[29]. Similarly, the limonoid walrobsin A from Walsura robusta exhibited strong anti-inflammatory and antioxidant effects in septic acute kidney injury. DARTS-MS identified G protein-coupled receptor 75 (GPR75) as the direct target of WA in renal macrophages[30]. Moreover, the flavonoid acacetin from Acacia farnesiana was found to target LAMTOR1 (late endosomal/lysosomal adaptor, MAPK and MTOR activator 1) to activate autophagy and alleviate metabolic dysfunction-associated fatty liver disease (MAFLD)[31]. The same strategy also identified targets for other NPs, including tanshinone I[32], astragaloside II[33], and the marine alkaloid chagosensine C[34].
DARTS offers several advantages. It does not require chemical modification of NPs. This feature allows direct identification of binding proteins for both purified compounds and complex extracts. DARTS-MS strategies can also detect weak interactions because the workflow does not include washing[35,36]. However, it has inherent limitations. Low-abundance proteins may be missed because they are difficult to observe in SDS-PAGE and LC-MS analyses. Since complex protein mixtures are applied, nonspecific bindings may be captured, and further biological validation is required. Moreover, experimental variables influence the outcome. The choice of protease and the type of cell lysate can affect target recognition and reduce accuracy.
Stability of proteins from rates of oxidation (SPROX)
-
To investigate the local conformational dynamics and the stability of native protein structures, Ghaemmaghami and colleagues developed a technique that monitors amide groups by hydrogen/deuterium (H/D) exchange in unpurified proteins. The rate of amide H/D exchange reflects the thermodynamic stability of proteins and protein-ligand complexes[37]. This concept was later adapted to establish the SPROX technique[38].
SPROX is a stability-based approach for detecting ligand-target interactions. Different from DARTS, which monitors proteolytic changes, SPROX measures the oxidation of methionine residues. The method is based on the principle that ligand binding stabilizes the target protein and reduces its susceptibility to oxidation[39]. A typical SPROX workflow includes the following key steps: protein samples are first divided into two groups, a ligand-treated group and a solvent control. A gradient of chemical denaturants, such as guanidine hydrochloride, is added to both groups to induce controlled unfolding and the mixtures are incubated to reach equilibrium; an oxidant such as hydrogen peroxide (H2O2) is then introduced to oxidize exposed methionine residues for a defined time. After the oxidation reaction is quenched, the proteins are digested into peptides and analyzed for oxidation of methionine-containing peptides by LC-MS/MS (Fig. 1c).
By comparing the oxidation extent of methionine residues between the ligand and control groups, SPROX analyzes the thermodynamic properties of protein folding or unfolding reactions. It can detect and analyze multiple target proteins interacting with drugs and quantify the binding affinity. In specific experiments, a key consideration factor is the concentration of the denaturing agent and the oxidation conditions. SPROX relies on monitoring the variation of the methionine oxidation rate with the concentration of the modifier. Therefore, it is crucial to accurately prepare concentration gradients of chemical modifiers, such as guanidine hydrochloride. Similarly, the concentration or reaction time of hydrogen H2O2 can alter the degree of methionine oxidation, leading to greater differences between samples. In addition, methionine residues are susceptible to unintended oxidation during the sample preparation, which can introduce artifacts. To alleviate this situation, it is recommended to use isotope-labeled H2O2. By using H218O2 to control the oxidation step, researchers can distinguish between the oxidation required for specific experiments, and the background that may occur during the treatment process[40].
SPROX has proven effective for target identification of NPs. Dearmond et al. used the method with resveratrol and identified six new targets beyond the known cytoplasmic dehydrogenase, including elongation factor 3A (EF-3A), ribosomal proteins S9-A and L8-A, and the nascent polypeptide-associated complex subunit alpha (NACα), and NADPH-dependent aldo-keto reductase and 2-dehydropantoate 2-reductase[41]. The natural anticancer compound manassantin A was analyzed by iTRAQ-SPROX, which identified 28 proteins with altered thermodynamic stability after treatment, providing insights into its mechanism of action[42]. SPROX, combined with isobaric mass tagging was applied to quantify the binding affinity of the natural antibiotic geldanamycin to endogenous HSP90 directly in MCF-7 cell lysate without protein purification[43].
Despite its promising application potential, SPROX has several limitations. SPROX analysis applies only to peptides that contain methionine residues. Some methionines show similar oxidation behavior with different treatments, which reduces confidence in identifying ligand-protein interactions. This technique does not provide precise information about ligand-binding sites[42,44]. Moreover, the requirement for chemical denaturation and oxidizing conditions prevents its application in living cells.
Solvent-induced protein precipitation (SIP)
-
Organic solvents are known to denature and precipitate proteins, and ligand-bound proteins often show greater resistance to organic solvents. Based on this principle, SIP was developed to identify targets of small-molecule ligands by distinguishing ligand-protein complexes from unbound proteins based on their different solubilities in organic solvents[45].
Zhang et al. introduced SIP as an approach to identify drug targets and off-target proteins[46]. Different from heat-induced protein precipitation, SIP uses a mixed organic solvent (50% acetone: 50% ethanol: 0.1% acetic acid, A.E.A) to reduce the dielectric constant and disrupt protein hydration. The workflow of SIP includes centrifugation to collect supernatants, preparation of bottom-up proteomic samples, and stable isotope dimethyl labeling of light and heavy peptides. The proteome coverage obtained with SIP is comparable to that of conventional bottom-up proteomics and exceeds that achieved by SPROX.
SIP has been applied in both target validation and off-target analysis for NPs. Kurarinone, a prenylated flavonoid derived from Sophora flavescens showed neuroprotective effects in a mouse model of Parkinson's disease (PD). Combined SIP with TPP analysis identified soluble epoxide hydrolase (sEH) as a principal target of kurarinone[47]. Studies using TPP and SIP also identified the target protein of shikonin, which acts as a potential inhibitor of the IKKβ/NEMO complex[48].
Notably, SIP can also evaluate drug affinity and, as mentioned, enables target discovery without chemical modification of drugs. However, SIP requires the addition of organic solvents, which prevent its application in living cells and restricts it to in vitro studies using cell lysates[46].
Limited proteolysis-mass spectrometry (LiP-MS)
-
Limited proteolysis is a classical biotechnology method, and both the DARTS method and pulse proteolysis methods essentially rely on this technology. In 2004, Fontana et al. proposed a 'native structure-unfolded structure' equilibrium theory to explain the process of limited proteolysis[49]. According to this theory, some unfolded regions of proteins (such as flexible loops) have higher structural variability, thereby providing greater accessibility for the binding of protease active sites.
Limited proteolysis has long been used to study protein structure. However, due to the lack of effective separation and detection methods, this approach was limited to the study of purified single proteins, especially focusing on the intact remaining proteins after proteolysis rather than the proteolytic products. The aforementioned DARTS and pulse proteolysis methods analyze the remaining proteins in samples after short-term digestion, which only provide protein-level information while ignoring domain-related information about cleavage sites.
Building on previous studies, the Picotti team creatively proposed a two-step digestion-based limited proteolysis method in 2014, combining it with mass spectrometry-based quantitative proteomics to study protein structure. This technique was named LiP[50]. The LiP workflow includes two key digestion steps for native proteins extracted under non-denaturing conditions. Limited proteolysis is first performed with a non-specific protease such asproteinase K or thermolysin at low concentration for a short time. This treatment produces large protein fragments, which are further transferred to denaturing conditions to quench the initial digestion. Trypsin is used to complete digestion with peptides of a suitable length for bottom-up proteomic analysis. The classical quantitative proteomic analysis is carried out on treatments and controls derived from the same source. The highly sensitive selected reaction monitoring (SRM) strategy is commonly used to measure differences in peptide abundance and to reveal structural changes in proteins. Differences in peptide abundance between treated and control samples indicate altered cleavage sites and reveal potential binding regions.
Several technical considerations are essential for reliable LiP-MS analysis[51]. Reproducibility depends on strict regulation of digestion time, temperature, and protease-to-protein ratio. Variability in these parameters can alter peptide patterns and structural differences. In addition, over-digestion destroys structural information by cleaving stable domains. The goal is 'limited' protein hydrolysis, which means only cutting flexible and exposed areas[51]. The cutting specificity, optimal pH value, temperature, and buffer conditions of each protease are different, so the choice of protease strongly affects the final result[52,53]. Trypsin is considered the 'gold standard' protease in bottom-up proteomics due to its high specificity, primarily cleaving the C-terminus of lysine (K), and arginine (R) residues. Many traditional LiP MS workflows rely on trypsin; however, high specificity may also lead to the loss of some structural information. Protease K exhibits broader cleavage activity and effectively digests flexible or unfolded regions, making it sensitive to subtle conformational changes. Widespread specificity can also generate complex peptide mixtures that require careful data analysis[51,54].
Piazza and colleagues developed LiP-small molecule mapping, LiP-SMap, to screen for small-molecule targets in complex proteomic extracts[55]. Chen et al. used this method for the anti-obesity hyperforin and confirmed dihydrolipoamide S-acetyltransferase (DLAT) as the direct molecular target[56]. Piazza's group later developed LiP-Quant, which combines dose titration with machine learning to identify targets, map binding sites, and quantify affinities. Using this approach, they identified the targets for several natural fungicides, including fostriecin, staurosporine, and FK506[57].
LiP-MS achieves a resolution of approximately 12 amino acids for identifying small-molecule binding sites, supplementing lacunae left by CETSA/TPP and DARTS regarding protein structure and site information in technologies[56,58,59]. This strategy detects structural changes in a near-physiological environment, and can simultaneously measure the structural fingerprints of all MS-detectable proteins in samples at the proteome-wide scale. Yet, LiP-MS has certain limitations. For example, to identify binding sites, peptides at the binding interface must be detected by MS, requiring high sequence coverage of the target protein. Additionally, if small molecules induce structural changes outside the binding region, the interaction can be detected, but the binding site cannot be accurately localized[59].
Target-responsive accessibility profiling (TRAP)
-
In addition to inducing stability changes, ligands can alter the accessibility of protein residues in their target proteins to covalent labeling reagents after binding. This is due to steric hindrance from direct engagement or binding-induced allostery. In 2023, Tian et al. developed the TRAP method[60]. When a ligand binds to its target protein, the reactive lysine residues in the binding region become inaccessible to covalent labeling due to steric hindrance or allosteric effects. TRAP uses isotope-labeled formaldehyde (CD2O) and borane-pyridine complex (BPC) to label protein lysines via demethylation. This process creates a mass shift for target protein detection, so that the researchers can screen candidate target proteins by comparing changes in proteome labelability between ligand-treated samples and untreated samples (Fig. 1d).
The team successfully identified Acyl-CoA synthetase long-chain family member 4 (ACSL4), an enzyme that is necessary for ferroptosis, as the target of the NP silibinin by using TRAP in living cells[61]. SIL inhibits ACSL4 enzyme activity, thereby alleviating ACSL4-mediated ferroptosis. Another example is celastrol, an NP that improves metabolic syndromes by blocking resistin-induced inflammation. Zhu et al. used TRAP to identify cyclase-associated protein 1 (CAP1) as the target of celastrol[62]. Their binding of celastrol to CAP1 inhibits the cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA)-nuclear factor kappa-B (NF-κB) signaling pathway, and thereby ameliorates the high-fat diet-induced metabolic syndrome in mice fed a high-fat diet. In addition, Researchers also found that cycloastragenol was identified as a direct target of cathepsin B, thereby reducing MHC-I degradation on tumor cells, and enhancing CD8 T cell-mediated antitumor immunity[63].
However, TRAP depends on labeling reactive lysine residues in proteins[60]. It can detect targets only when metabolite binding changes the accessibility of lysine residues. If the binding site of a protein has no reactive lysines, or if metabolite binding does not change lysine accessibility, TRAP fails to detect the target. This problem occurs in some membrane proteins and small peptides without lysine residues, so the detectable target types are limited. Cell lysis can also lower the true positive rate of ligand-target interactions.
Peptide-centric local stability assay (PELSA)
-
The PELSA is a limited proteolysis method similar to DARTS and LiP-MS. Its core principle is that ligand binding can alter the thermodynamic stability of proteins, thereby affecting their sensitivity to proteolysis.
Traditional limited proteolysis methods use small amounts of non-specific proteases to generate large protein fragments[49,50]. PELSA uses a high concentrations of trypsin to produce small peptides directly from native proteins. These peptides form under native conditions, so their abundance reflects the local stability of the protein[64]. When a ligand binds to its target protein, the binding region becomes more stable and possesses lower energy that fails to access to the active pocket of trypsin, whereas the non-binding regions stay flexible and are rapidly digested into small peptides. Through shotgun proteomics analysis, the target protein and the ligand-binding site are characterized by an obvious reduced abundance, and the position of the corresponding peptide. PELSA has successfully identified the target proteins for lapatinib and rapamycin, and determined the binding sites and affinities with their targets[64]. Recently, PELSA recognized ubiquitin-conjugating enzyme E2 C (UBE2C) as the functional target of natural terpenoid Halorotetin B, and revealed Halorotetin B inhibits UBE2C to induce M-phase arrest and tumor cell senescence[65]. A key advantage of PELSA is that it requires no chemical modification of the ligand so that its native structure and binding activity can be kept. PELSA can also directly provide binding-site information with high sensitivity when compared with TPP and LiP-MS.
Protein microarrays
-
Most drug targets are proteins, so protein and peptide microarrays are important for drug discovery. Protein microarray technology allows fast identification of protein-ligand interactions. It uses photolithography or inkjet technology to place human recombinant proteins on a solid surface to form a protein microarray chip. Interactions between human proteins and their ligands can be characterized by specific detections such as fluorescence scanning. Protein microarrays allow thousands of protein biochemical reactions to occur in parallel, and are quantitatively characterized at the same time. In a typical experiment, the test drug molecule is incubated with the protein microarray to bind to specific targets, labeled with fluorophores, and then scanned for fluorescence signals. The signal changes show which proteins interact with the test molecule. Label-free methods are gaining attention for detecting protein-ligand interactions. For example, imaging ellipsometry directly measures changes in the molecular layer on a surface after binding and characterizes the binding kinetics without labeling[66]. However, label-free methods usually need higher concentrations for detectable signals than labeled methods. Mass spectrometry-based methods like desorption electrospray ionization mass spectrometry (DESI-MS), combined with protein microarrays, provide a simple and fast way to screen small ligands that bind to proteins[67]. Mass spectrometric imaging of bead arrays (MALDI-MSI) can also screen drugs across the proteome and detect both on-target and off-target binding[68].
Notably, this technology has been successfully applied to identify targets of NPs and bioactive compounds. Grincamycins are a class of angucycline glycosides isolated from the Streptomyces strain and show strong antitumor activity. Wang et al. used a protein microarray to identify isocitrate dehydrogenase 1 (IDH1) as the target of grincamycins B[69]. Chen et al. screened 17,950 proteins for binding to doxorubicin using protein microarrays, and confirmed the interaction between DOX and UMP/CMP kinase 1 (CMPK1)[70]. Researchers also utilized this strategy to identify ubiquitin-specific protease 7 (USP7) as the direct target for eupalinolide B to exert anti-neuroinflammatory effects[71]. Reticulon-4 (RTN4/Nogo) is an important regulatory factor of the endoplasmic reticulum (ER) membrane and has been identified as a target of α-mangostin. Its function is to trigger pyroptosis by facilitating the removal of the ER membrane[72].
The main advantage of protein microarray technology is high-throughput screening. This method helps researchers identify potential targets of NP more quickly and provides useful information in the early stages of target identification. However, it also has limitations. For example, immobilized proteins must remain stable and fold correctly, and experimental results need to be reproducible across different batches of samples[73,74]. Some proteins contain only partial domains, which may result in missed or underestimated ligand-protein interactions. Furthermore, ligand-protein binding observation does not always reflect behavior in living systems, so subsequent validation is necessary. Efforts to address these issues include optimization of surface chemistries and development of more reliable immobilization strategies. The ProteomeTools project aims to generate a comprehensive synthetic human proteome to standardize protein array content and improve reproducibility[75]. High-resolution micro-confocal Raman spectrometry combined with photo-affinity microarrays is also being explored to examine how bioactive NPs recognize their targets in greater detail[76].
-
Traditional target identification approaches commonly use cell lysates or purified protein systems to reduce the complexity of intact cells. However, the simplified system cannot fully recapitulate features of the living environment, such as dynamic subcellular localization, native protein conformations, and transient protein-protein interactions (PPIs). By contrast, live-cell models maintain cellular integrity and physiological homeostasis, making it possible to observe real-time interactions between NPs and their targets.
There are many difficulties in identifying targets in living cells. Interactions between NPs and their targets are often non-covalent and transient, which makes them difficult to capture. Additionally, introducing other functional groups during probe construction may alter the activity or binding properties of NPs. To overcome these difficulties, researchers have developed many innovative strategies that minimize interference while increasing sensitivity, enabling the identification of NP targets in living cells.
Click chemistry strategy
Copper-catalyzed click chemistry (CUAAC)
-
With the emergence of the bioorthogonality concept and the expansion of reaction types, bioorthogonal reactions have been incorporated into the design of probe molecules, simple bioorthogonal groups are introduced into drugs or NP molecules to prepare molecular probes[77]. Among the various bioorthogonal reactions developed, the copper-catalyzed azide-alkyne cycloaddition (CuAAC) stands out as the most powerful. In 2002, the research teams of Sharpless and Meldal found that the cycloaddition between azides and alkynes, when catalyzed by copper ions, exhibits an extremely rapid rate, over 1 × 106 times faster than without metal catalysis[78]. The CuAAC reaction not only stands as a pivotal example of bioorthogonal chemistry, but has also inspired in-situ target identification research due to its applicability in living systems: the active probes target and label the proteome or capture target proteins, followed by Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC), catalyzing the covalent crosslinking between their terminal azide/alkynyl groups and reporter groups (e.g., biotin bearing complementary azide/alkynyl groups) to form triazoles. Finally, avidin-modified solid-phase microspheres are used to enrich the target proteins, which are then identified by mass spectrometry (Fig. 2a). This CuAAC-based strategy is efficient, sensitive, and specific, holding significant application value in drug target discovery.
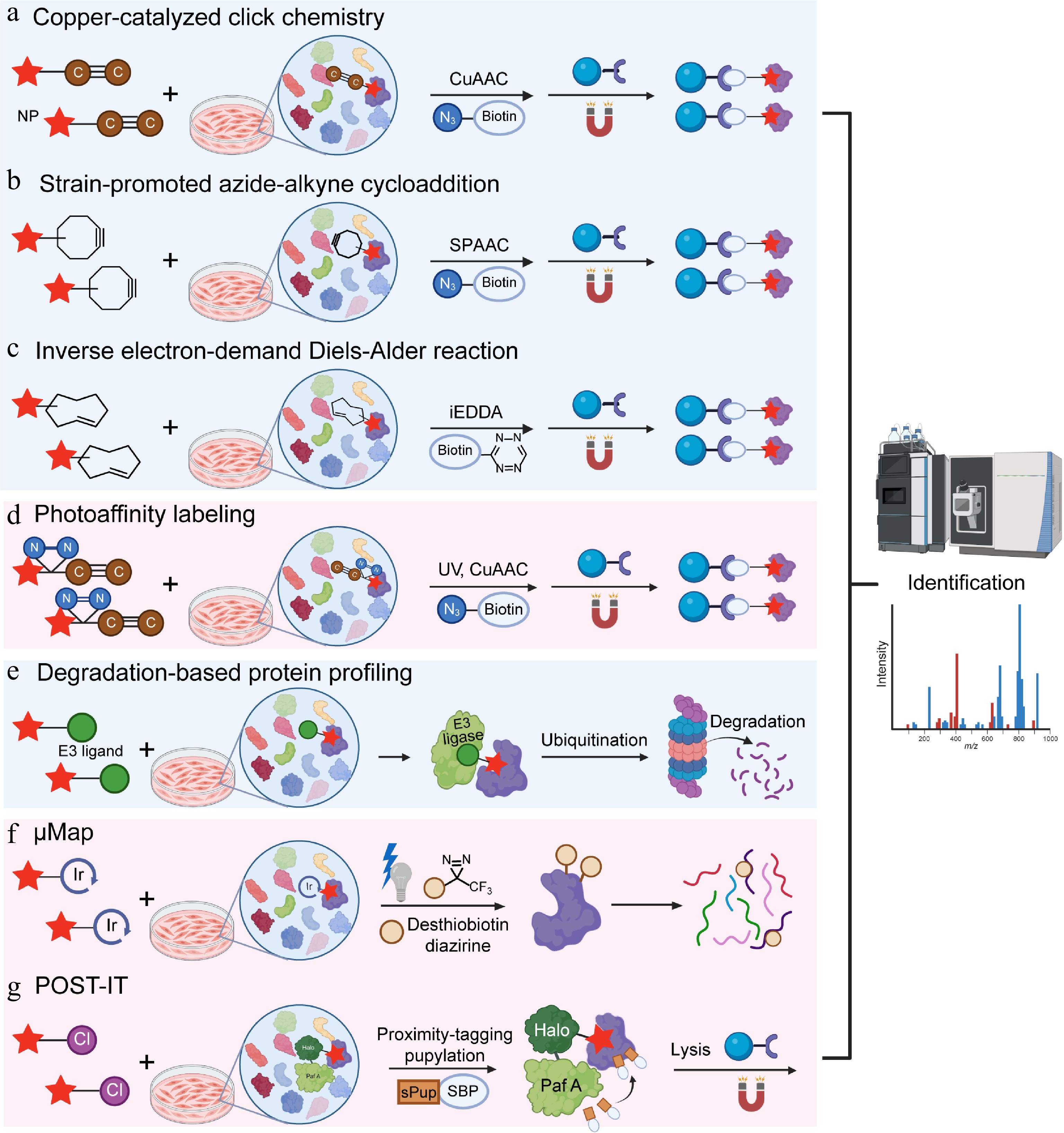
Figure 2.
Natural product target identification based on in-situ models. (a) Copper-catalyzed click chemistry (CUAAC), (b) strain-promoted azide-alkyne cycloaddition (SPAAC), (c) inverse electron-demand Diels-Alder reaction (iEDDA), (d) photoaffinity labeling (PAL), (e) degradation-based protein profiling (DBPP), (f) μMap, and (g) pup-on-target for small molecule target identification technology (POST-IT).
Using this approach, a series of chemical probes derived from bioactive compounds of NPs have been synthesized and employed for proteome-wide target identification via click chemistry. For example, Naamidine J, an ascidian-derived alkaloid, exerts potent anti-inflammatory activity in macrophages. Using a Naamidine J-based photoaffinity probe to capture its direct protein targets in live macrophages, the authors identified the chromosome segregation 1-like protein (CSE1L) as its primary functional target[79]. In another example, through the synthesis of a forskolin-based probe, researchers established that transglutaminase 2 (TMG2) serves as the direct molecular target of forskolin. Forskolin engages a previously undisclosed allosteric site, triggering a large-scale structural rearrangement that shifts TMG2 from its autoinhibited 'closed' conformation into an enzymatically active 'open' conformation[80].
However, the introduction of copper catalysts into biological systems and organisms may cause toxicity issues. Biological molecules also contain numerous copper chelation sites, which may reduce the activity of the catalyst[81].
Strain-promoted azide-alkyne cycloaddition (SPAAC)
-
Although CuAAC is valued for its fast and clean reactivity, the cytotoxicity caused by copper catalysts limits its application in living systems. At the molecular level, copper(I) ions participate in Fenton-like redox cycling and generate hydroxyl radicals via reaction with endogenous hydrogen peroxide[82,83]. These highly reactive oxygen species (ROS) trigger a cascade of cellular damage, including oxidative DNA damage, mitochondrial dysfunction, and ultimate apoptosis or necrosis. To mitigate these limitations, alternative bioorthogonal reactions have been introduced, among which SPAAC is a widely adopted example.
SPAAC, also known as copper-free click chemistry, was reported by Agard and colleagues as a copper-free version of cycloaddition[84]. Unlike CuAAC, SPAAC is a copper-free cycloaddition driven solely by ring strain, dibenzocyclooctyne (DBCO) is the most widely used strained cyclic alkyne for SPAAC, with a second-order rate constant of around 0.1 M−1·s−1[85]. The experimental principle is to integrate strained alkyne into NPs to synthesize probes while retaining the binding ability of NPs to target proteins. After introducing live cells, the probe can bind to the target protein under natural conditions. After achieving equilibrium through sufficient incubation, cells were lysed under mild conditions to preserve the protein-probe complex. An azide biotin reagent is added in the next stage, and the strain-induced ring strain in the cyclooctyne drives a rapid cycloaddition with the azide, forming a stable triazole linkage without requiring metal catalysts or harsh conditions. This enables the target to be covalently linked to a high-affinity tag, facilitating further enrichment and identification.
SPAAC has been applied to the target identification of NPs, particularly for artemisinin. Using this method, researchers identified hundreds of artemisinin-alkylated proteins in Plasmodium falciparum and revealed that the artemisinin broadly disrupts essential parasite pathways[86]. Notably, the Ismail research group designed and compared two click chemistry methods, CuAAC and SPAAC. Experimental results demonstrated that SPAAC was more sensitive, highlighting that SPAAC can be a better choice than CuAAC when researchers need to avoid copper-related cytotoxicity and improve detection sensitivity[86].
Inverse electron-demand Diels-Alder reaction
-
To successfully perform the ligation step and minimize impacts on the stability and homeostasis of cells or organisms, bioorthogonal reactions must meet strict requirements[87]: (i) highly selective reactions between two components without interfering with abundant biomolecules and metabolites in the surrounding environment; (ii) efficient and rapid reactions in aqueous environments under physiological conditions (pH 6–8, temperature 37 °C, and exposure to O2); (iii) biocompatibility of all reaction components with no toxicity to cells or organisms; (iv) small functional groups to avoid potential disturbances to labeled biomolecules. Tetrazine (Tz) and trans-cyclooctene (TCO) meet the requirements of click chemistry. As a third-generation click reaction, it offers a much faster second-order rate constant (> 103 M−1·s−1) than both CuAAC and SPAAC[88], further expanding the scope of click chemistry applications[89]. The Tz-TCO reaction, often termed an inverse electron-demand Diels-Alder (iEDDA) reaction, involves the highly reactive Tz reacting with the strained trans-cyclooctene to form a dihydropyridazine intermediate, which then undergoes retro-Diels-Alder to yield a stable bicyclic product and nitrogen gas[90]. The strategy has two key steps: chemical labeling of a molecule of interest with a bioorthogonal handle, and subsequent specific detection or enrichment using the Tz-TCO click reaction (Fig. 2c). The Weissleder group modified small-molecule with TCO and combined this step with iEDDA chemistry. Using this approach, they validated the interaction of taxol (paclitaxel) and its target, tubulin, in living cells, and demonstrated the feasibility of in situ imaging of intracellular protein targets via bioorthogonal turn-on probes[91].
Tz-TCO click chemistry also has a drawback. Tz and TCO are slightly larger than azides or simple alkynes. This larger size may limit their use, as modifying NPs with such bulky reactive groups could alter the original properties of the parent molecules.
Photoaffinity labeling (PAL)
-
Most bioactive NPs bind to target proteins through non-covalent interactions[28]. These interactions are often weak and unstable, so they are hard to capture. It is not appropriate to use simple probes in this situation. PAL can solve this problem by forming a covalent link between the NP and the target protein[92].
This covalent binding is achieved by adding a photoreactive group to the probe. A classic photoaffinity probe contains three parts: the NP (ligand), a photoreactive group, and a reporter group. NPs are responsible for recognition and specific binding to targets. Ultraviolet (UV) light then activates the photoreactive group into a highly reactive intermediate, such as a carbene or nitrene species. Highly reactive intermediates form covalent bonds with nearby amino acids in the binding pocket of the target protein, effectively capturing transient binding and low-affinity target proteins. The reporter group assists in the enrichment and detection of target proteins. An alkyne-modified probe can react with a biotin moiety through copper-catalyzed chemistry. This enables selective enrichment via streptavidin pull-down assays and subsequent identification by mass spectrometry-based proteomics (Fig. 2d). The most common photoreactive groups are aromatic azides, diazirines, and benzophenones[93]. Upon specific wavelengths of light, they generate reactive intermediates (nitrenes, carbenes, and biradicals, respectively) to covalently bind to nearby proteins.
The key advantage of PAL is its ability to form covalent bonds through photoactivation, thereby fixing weak or temporary interactions between NPs and targets. This feature is especially valuable for detecting membrane proteins, low-abundance signaling molecules, and short-lived protein complexes[94]. The challenge of PAL is that probe synthesis may alter the activity of NPs. Adding photoreactive groups and reporter tags such as biotin changes the molecular weight, lipophilicity, and three-dimensional structure of the NP[95]. These changes may disrupt key pharmacophore features and reduce binding affinity.
PAL has identified targets for many NPs. For example, using a photoaffinity probe in living cells, researchers found novel fructose-1,6-bisphosphate (FBP)-binding proteins, including mitochondrial enzyme aldehyde dehydrogenase 2 (ALDH2). They revealed a new glucose-sensing pathway via the FBP-ALDH2-ROS axis[96]. Another study identified the natural diterpenoid 12-deoxyphorbol 13-palmitate as a potent anti-fibrotic agent. This compound was confirmed to directly target apolipoprotein L2 (APOL2)[97]. Tetrandrine directly targets the lysosomal integral membrane protein-2 (LIMP-2) and inhibits its role in cholesterol and sphingosine transport[98]. Using a custom-designed photoaffinity probe to mimic artemisinin’s structure and function, researchers systematically mapped both covalent and noncovalent interactions between artemisinin and parasite proteins during different developmental stages[99].
Degradation-based protein profiling (DBPP)
-
DBPP is a novel strategy combining proteolysis-targeting chimera (PROTAC) technology, quantitative proteomics, and mass spectrometry. It is primarily designed for the systematic identification of multiple targets of NPs[100]. Traditional methods depend on direct binding between proteins and small molecules, whereas DBPP focuses on chemically induced PPIs. Theoretically, this design allows degradation of proteins that bind weakly or moderately. A ternary complex including the target protein, PROTAC molecule, and E3 ligase is formed and can then be pulled down to identify targets via immunoprecipitation-mass spectrometry (IP-MS) experiments (Fig. 2e).
Numerous studies have verified the feasibility of this strategy. Celastrol was used as a probe and identified multiple novel targets such as checkpoint kinase 1 (CHK1), O-GlcNAcase (OGA), DNA excision repair protein ERCC-6-like (ERCC6L), and known targets, including inhibitor of nuclear factor κB kinase subunit β (IKKβ), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PI3Kα), and cellular inhibitor of PP2A (CIP2A)[100]. Chen et al. clarified RNA exonuclease 4 (REXO4) as the direct target by designing an evodiamine-based PROTAC[101]. Researchers synthesized an artemisinin-based PROTAC named AD4, combined it with TMT quantitative proteomics and identified the PCNA clamp-associated factor (PCLAF) as a potential target of artemisinin in acute lymphoblastic leukemia[102].
DBPP overcomes the traditional requirement for strong protein small molecule interactions. It fundamentally works by forming ternary complexes that stabilize fleeting protein-probe interactions. By amplifying functional affinity and residence time, it can capture protein targets missed by classical methods, thereby expanding the landscape of druggable targets.
DBPP has certain limitations in design and skill requirements. PROTAC molecules need to match both the E3 ligase ligand and NP ligand. As a result, researchers must perform extensive synthesis and repeated screening to find effective degraders. This process increases costs and time. The method also depends heavily on high-resolution mass spectrometry platforms, which require advanced platforms and skilled operations.
Target identification based on proximity labeling technology
μMap
-
UV-based PAL strategies have several problems, including protein degradation, low cell permeability, and background interference. Diazine probes are inefficient because more than 99% of the generated carbenes react with water rather than the target protein. The MacMillan group developed a proximity labeling technique, μMap, to solve these problems. Through light-driven Dexter energy transfer and drug conjugation, μMap improves the enrichment of target proteins, and can label membrane proteins with fewer exposed amino acid residues. Active drug molecules are conjugated with a photocatalytic iridium metal complex to target the proteins of interest. Upon blue light at around 450 nm, the iridium metal complex activates nearby diazirine probes through Dexter energy transfer and enables efficient enrichment of target proteins[103] (Fig. 2f). Researchers applied this photocatalytic small-molecule target ID platform to analyze targets of several drugs, including taxol, JQ1, dasatinib, MK-8666, and SCH58261[103].
However, light below 500 nm penetrates tissue poorly because of scattering and absorption by endogenous chromophores, which limit its application in vivo. MacMillan and colleagues developed μMap-RED, a red-light-activated labeling approach that uses a Sn(IV) chlorin e6 photocatalyst to activate phenylazide-biotin probes. Through μMap-RED, they analyzed red blood cell surface proteins directly in whole mouse blood, representing a major advancement in light-based proximity labeling methodologies for complex tissue environments[104]. Although this method has not yet achieved in situ target identification in living animals, it provides new insights for other researchers pursuing this goal.
Pup-on-target for small molecule target identification technology (POST-IT)
-
POST-IT is a proximity labeling system designed to identify small-molecule protein targets in live cells, and in vivo[105]. The method uses a modified HaloTag fused to an engineered PafA ligase via an optimized linker. This design allows covalent tagging of nearby proteins when a HaloTag ligand (HTL) linked to the small molecule binds to its targets. Through systematic mutagenesis, key limitations of native PafA, such as self-pupylation, poly-pupylation, and depupylase activity were overcome, and its specificity and reliability were increased correspondingly. The HTL-small molecule derivative recruits the POST-IT fusion protein to its target. PafA then attaches a mutated pup tag (sPupK61R) onto lysine residues of nearby proteins. This step labels true interacting proteins in their native environment (Fig. 2g). Critically, this method works without cell lysis, so it preserves transient or context-dependent interactions that may be lost in traditional pull-down assays. Researchers identified known targets and discovered SEPHS2 as a novel binder of dasatinib and VPS37C as a target of hydroxychloroquine. Additionally, the POST-IT has been demonstrated in live zebrafish embryos[105].
-
HiMAP-seq is a very high-throughput method for gene expression profiling. Unlike conventional approaches that analyze samples individually, HiMAP-seq uses a pooled-sample strategy. This strategy allows researchers to measure thousands of genes across thousands of samples in one experiment. This method does not require mRNA purification or reverse transcription, so it reduces bias and saves time and cost. HiMAP-seq uses unique molecular identifiers (UMIs) to measure gene expression accurately and shows remarkable performance with high mapping rates (over 91% for both Illumina read 1 and read 2), minimal index misassignment, and strong reproducibility. Moreover, gene expression results from HiMAP-seq are highly consistent with RNA-seq and qPCR results, with correlation values (R values) from 0.85 to 0.95.
Researchers leveraged HiMAP-seq to build the chemical-induced gene signatures (CIGS) dataset that contains 319 million gene expression events from 13,221 compounds (including 1,865 traditional Chinese medicine-derived NPs) tested in two human cell lines (MDA-MB-231 and HEK293T), across 93,644 perturbations. This dataset has been successfully applied to decipher the mechanisms of unannotated NPs. For instance, ligustroflavone was identified as a novel BRD4 inhibitor through signature similarity analysis and experimental validation, such as molecular docking, surface plasmon resonance (SPR), and CETSA. Another compound, 2,4-dihydroxybenzaldehyde, was characterized as a ferroptosis inhibitor targeting KEAP1 to protect against cisplatin-induced acute kidney injury in vivo. The CIGS dataset links the disease signatures with NPs screening and target identification, serving as a valuable resource[106]. Overall, HiMAP-seq offers a powerful, cost-effective tool for in situ target identification of NPs by capturing functional gene expressions in human cells, and advances the discovery of novel bioactive NPs and their molecular targets.
NPs target identification based on bioinformatics and AI
-
Bioinformatics integrates disciplines such as computer science, statistics, mathematics, and biology, and plays an important role in modern drug research. Analyzing biological data can help researchers quickly screen drug targets, understand molecular mechanisms, and promote new drug development.
Artificial intelligence (AI) has become a transformative tool for helping NP target recognition by omics data mining, NP structure analysis, target analysis, and biological activity prediction. Specifically, AI can identify potential targets by analyzing complex biological data and associate them with disease-related molecular pathways, supplementing targets that traditional methods may have missed[107].
AI-driven target identification applies multi-omics data analysis, including genomics, epigenetics, transcriptomics, proteomics, and metabolomics, to reveal complex biological interactions and patterns that are difficult to detect with traditional analysis[108,109]. The principle is that artificial intelligence uses omics data to construct a network and identify coexpression modules between genes, proteins, and metabolites for biological analysis[110]. Network based biological analysis generally involves a three-stage workflow: start by mapping out molecular expression and correlation, then run gene set enrichment to identify functionally relevant modules, and finally zoom in on key genes that could serve as therapeutic targets or biomarkers. Researchers studied the Bufonis venenum-derived compound bufalin, which exhibits anti-cancer activity but an unclear molecular mechanism. The integrated AI-based prediction with molecular docking and molecular dynamics simulations found several possible targets, including CYP17A1, ESR1, mTOR, AR, and PRKCD. Experimental validation via SPR, biotin pulldown, and TSA confirmed that bufalin directly binds to the estrogen receptor alpha (ERα), encoded by ESR1. Molecular docking showed a selective interaction at Arg394 of Erα, and molecular dynamics simulations further revealed that bufalin acts like a 'molecular glue' that strengthens the interaction between ERα and the E3 ubiquitin ligase STUB1 to promote proteasomal degradation of ERα[111].
Target-based screening of NPs represents an importantly complementary strategy for investigating the targets of NPs and NPs-derived drug discovery, particularly in cases where the molecular mechanisms are well defined. Common biochemical and biophysical techniques used to characterize interactions between NPs and their molecular targets include SPR, TSA, enzyme activity measurements, X-ray crystallography, and cryo-electron microscopy. In recent years, computational approaches such as molecular docking and machine learning have further enhanced the identification of potential lead compounds, improving screening efficiency and facilitating early insights into binding modes. Yu et al. used target-based screening on NPs from Artemisia annua, and identified three potential GPX4 activators, including artemisinin, patuletin, and kaempferol. Molecular dynamics simulations showed that these compounds form stable complexes with GPX4[112]. Another study developed a semi-automated microplate method to quickly build and screen a library of 2,720 celastrol derivatives for inhibition of peroxiredoxin 1 (PRDX1). This process identified new celastrol analogues with stronger potency and better selectivity for PRDX1 than other peroxiredoxin isoforms[113].
Machine learning-based biological network analysis is useful for handling large and complex datasets. It helps researchers find reliable targets for more effective treatments. Some common approaches used to identify new anti-cancer targets include classification, clustering, and neural networks[107]. Recently, scientists have used this method to more accurately segment tumors and identify biomarkers for specific tumor types by combining information on gene expression, protein expression, and gene mutations across the entire genome.
The AI-driven target discovery process combines multi-omics data analysis, machine learning, and target selection criteria. First, different types of omics data are collected and brought together. This gives a broader picture of how biological systems work and what goes wrong in diseases. Then, AI and machine learning tools are used to study the data. These tools help scientists better understand complex biological interactions and find hidden patterns, which improves their ability to make predictions[114].
Beyond target discovery, AI is also used to predict the potential mechanisms of bioactive compounds. For example, Reker et al. developed a cascaded self-organizing map algorithm to predict targets by comparing their physicochemical and pharmacodynamic characteristics with those of known drugs[115].
Conclusions and future perspectives
-
Over the past decade, the field of NP target identification has advanced considerably. Chemoproteomic platforms, including CETSA, DARTS, and protein microarrays, now routinely map NPs-protein interactions. More recently, some cutting-edge technologies such as in-situ click chemistry, and AI have shown great potential for identifying the targets of NPs in more complex systems. This review outlines the major strategies currently used to identify the molecular targets of NPs. Applications of these strategies to representative natural compounds are summarized in Table 1, and their defining features, strengths, and limitations are compared in Table 2.
Table 1. Representative example of natural product target Identification.
Natural product Activity Target Method used Mechanisms of action Ref. Acacetin Lipid metabolism regulation LAMTOR1 DARTS Modulates autophagy via the LAMTOR1-MTORC1-AMPK signaling pathway. Alleviates metabolic dysfunction-associated fatty liver disease [31] Artemisinin Against malaria Multiple parasite proteins SPAAC Targets across glycolysis, hemoglobin degradation, antioxidant defense, and protein synthesis pathways [86] Artemisinin Against malaria Targets across different stages of the parasite PAL Interferes with the protein synthesis, glycolysis, and oxidative homeostasis pathways of parasites [99] Artemisinin derivatives Antitumor PCLAF DBPP Degrades PCLAF and activates the p21/Rb axis [102] α-Mangostin Antitumor RTN4/Nogo Protein microarray Induces proteasome degradation of RTN4 through recruiting E3 ligase UBR5 and enhances the pyroptosis phenotype [72] Bufalin Antitumor ERα (ESR1) AI + Docking + MD simulation Enhance the interaction between ERα and the E3 ligase STUB1, thereby promoting proteasomal degradation of ERα [111] Celastrol Anti-inflammation CAP1 TRAP Ameliorates metabolic syndrome via cAMP-PKA-NF-κB pathway [62] Celastrol Antitumor, anti- inflammation, alleviating of metabolic disorders Multi-targets including CHK1, OGA, ERCC6L, IKKβ, PI3Kα, and CIP2A DBPP Inhibits PI3K/AKT pathway, NF-κB pathway, and induces G2/M phase arrest [100] Cycloastragenol Antitumor Cathepsin B TRAP Enhances CD8 T cell-mediated antitumor immunity [63] 12-deoxyphorbol 13-palmitate Anti-liver fibrosis APOL2 PAL Disrupts APOL2-SERCA2-PERK-HES1 signaling and alleviates fibrosis [97] 2,4-dihydroxybenzaldehyde Alleviation of acute
kidney injuryKEAP1 HiMAP-seq Inhibits KEAP1 and downstream and NRF2-GPX4 signaling axis [106] Eupalinolide B Anti-neuroinflammation USP7 Protein microarray Inhibits USP7 to cause a ubiquitination-dependent degradation of KEAP1 and further induces an NRF2-dependent transcription activation of anti-neuroinflammation genes [71] Evodiamine Antitumor REXO4 DBPP Degrades REXO4 to induce cell death through ROS [101] Forskolin Osteoporosis TGM2 CuAAC Allostericlly activates TGM2 to improve mitochondrial dynamics and ATP production for osteoblast differentiation [80] Fructose-1,6-bisphosphate (FBP) Signaling molecule ALDH2 PAL Inhibits ALDH2 activity and results in cellular ROS upregulation accompanied by mitochondrial fragmentation [96] Gambogic acid Antitumor CNPY3 TPP Facilitates the recruitment of SIRT1 to remove lysine lactylation of CNPY3 and disrupts lysosomal proteins [27] Geldanamycin Antitumor HSP90 SPROX Binds to HSP90's N-terminal ATP binding domain and its ATPase activity [43] Grincamycin B Antitumor IDH1 Protein microarray Inhibits IDH1 to disrupt the cellular redox balance and 2-oxoglutarate homeostasis, thereby triggers ROS accumulation and eventually causes cell death [69] Halorotetin B Antitumor UBE2C PELSA Induces M phase cell cycle arrest [65] Hyperforin Anti-obesity DLAT LiP-SMap Promotes thermogenesis by stimulating AMPK and PGC-1α via a Ucp1-dependent pathway [56] Kurarinone Alleviation of Parkinson's disease sEH SIP + TPP Suppresses sEH to stabilize the level of epoxyeicosatrienoic acids and inhibits GSK3β [47] Ligustroflavone Antitumor BRD4 HiMAP-seq Inhibits BRD4 and down-regulates MYC, NFKB1 and TP53 [106] Manassantin A Antitumor 28 targets (e.g., filamin A and elongation factor 1α) SPROX Induces conformational change in filamin A to interrupt filamin A-HIF1α interaction, thereby inhibits HIF1α [42] Matrine Alleviation of spinal cord injury HSP90 DARTS Enhances the chaperon activity of HSP90, leading to axonal growth [29] Naamidine J Alleviation of acute lung injury CSE1L CuAAC Inhibits SP1 nuclear translocation and suppresses macrophage inflammation [79] Neoeriocitrin Osteogenesis and bone regeneration BECLIN1 TPP Stabilizes BECLIN1 by inhibiting ubiquitination-mediated degradation, thereby increasing autophagy [26] PF403 Antitumor NAMPT TPP Inhibits the catalytic activity of NAMPT, leading to a decrease in the NAD+ concentration [25] Shikonin Antitumor IKKβ/NEMO complex TPP + SIP Destabilizes the IKKβ/NEMO complex to suppresses NF-κB signaling and impairs cell proliferation [48] Silibinin Hepatoprotection ACSL4 TRAP Inhibits ACSL4 enzymatic activity, thereby mitigating the ACSL4-mediated ferroptosis [61] Tetrandrine Antivirus and antitumor LIMP-2 PAL Binds LIMP-2's ectodomain to inhibit lysosomal cholesterol and sphingosine transport [98] Walrobsin A Alleviation of acute kidney injury GPR75 DARTS Inhibits GPR75 to alleviate macrophage oxidative stress and the inflammatory microenvironment [30] Table 2. Comparative summary of natural product target identification methods.
Method Principle Key features Advantages Limitations TSA/CETSA/TPP Ligand binding alters protein thermal stability
Tm shift detected via fluorescence (TSA), immunoblot (CETSA), or MS (TPP)– Label-free
– TSA: purified proteins
– CETSA: cells/lysates
– TPP: proteome-wide MS– No modification
– CETSA: live-cell compatible
– TPP: high-throughput, unbiased– TSA: pure protein only
– CETSA: low throughput
– TPP: costly, long runtime, membrane protein under-representationDARTS Drug binding confers resistance to proteolytic digestion – Label-free
– SDS-PAGE/MS readout– No probe synthesis
– Works with crude extracts
– Detects low-affinity interactions– False positives
– Low-abundance proteins missed
– Protease choice criticalSPROX Ligand stabilizes protein, reducing methionine oxidation rates; quantitated by MS – Chemical denaturation gradient
– MS detection of oxidized/non-oxidized Met peptides– Quantitative affinity data – Requires Met-containing peptides
– No binding site informationSIP Organic solvents precipitate free proteins; ligand-bound proteins remain soluble. – Mixed organic solvent
– MS readout– Good proteome coverage
– No probe modification– In vitro only
– Organic solvent may disrupt some interactionsLiP-MS Ligand-induced conformational changes alter protease accessibility; MS quantifies differential peptides. – Two-step proteolysis
– Peptide-level resolution– Binding site mapping (~12 aa)
– Proteome-wide
– Near-physiological conditions– Requires high sequence coverage
– Cannot localize distal allosteric sitesTRAP Ligand binding changes lysine accessibility to reductive dimethylation – Isotope-coded formaldehyde labeling
– MS readoutNo protein modification – Requires reactive Lys near binding site PELSA Local stabilization upon ligand binding hinders tryptic cleavage; reduced peptide abundance pinpoints binding region – High-concentration trypsin digestion – No modification
Direct binding site information
– High sensitivity– May need optimization for each protein
– Sequence coverage dependentProtein microarrays Immobilized recombinant proteins probed with labeled or unlabeled NP – High-density chip
– Fluorescence/MSI/DESI-MS readout– Ultra-high throughput
– Parallel screening of thousands of proteins– Improper protein folding and truncated fragments
– Non-physiological in vitro environment
– High experimental costCuAAC Azide/alkyne-modified NP; Cu(I) catalyzes cycloaddition with biotin tag for pulldown – Bioorthogonal probe
– Affinity enrichment– Fast, specific, sensitive – Copper cytotoxicity
– Chelation inhibits catalysisSPAAC Strain-promoted cycloaddition; no catalyst – Copper-free
– Ring-strain driven– No metal toxicity
– Live-cell compatible– Slower kinetics (~0.1 M−1·s−1)
– Larger functional groupsiEDDA Tetrazine (Tz) + trans-cyclooctene (TCO) inverse electron-demand Diels-Alder – Extremely fast – Ultrafast kinetics
Bioorthogonal– Bulky Tz/TCO may alter NP properties PAL UV irradiation activates photoreactive group; covalent crosslinking to target – Probe: NP + photoreporter + tag
– UV-induced carbene/nitrene– Captures transient/weak interactions
– Membrane and low-abundance proteins– Probe synthesis may alter activity
– UV damageDBPP NP-PROTAC conjugate recruits E3 ligase; target degradation – Ternary complex formation
– Quantitative proteomics– Detects moderate/weak binders
– Direct pulldown evidence– Complex design
– Costly, platform-dependentμMap Visible-light photocatalyst (Ir) conjugated to NP; Dexter energy transfer activates diazirine probe nearby – Photocatalytic proximity labeling
– 450 nm blue light– No UV damage
- High membrane protein enrichment– Poor tissue penetration (blue light) POST-IT HaloTag-PafA fusion recruited by HTL-NP; pupylation of proximal lysines in live cells/animals – Prokaryotic pupylation system
– Non-diffusive labeling– Live-cell and in vivo
– Preserves transient interactions– Requires genetic fusion
– Engineering of PafA ligaseHiMAP-seq Multiplexed RNA-seq with UMIs; generates Chemical-Induced Gene Signatures (CIGS) for mechanism inference – Pooled sample barcoding
– No mRNA purification– Ultra-high throughput
– Cost-effective, reproducible– Indirect target inference
– Requires validationAI/
BioinformaticsIntegrates multi-omics, network models, machine learning; predicts targets from chemical structure and expression data – Computational pipelines
– Deep learning, network pharmacology– Accelerates hypothesis generation
– Reveals polypharmacology– Prediction accuracy depends on training data
– Requires experimental confirmationDue to the complex chemical structure of NPs, their biological functions vary with cell permeability, subcellular localization, and target binding kinetics. This leads to a decrease in the reproducibility of NP target identification and makes standardization difficult across laboratories. Researchers need to move away from relying on a single affinity pull-down method toward an integrated validation framework that combines biophysical, biochemical, and cellular techniques to confirm real target engagement and minimize false positives. Affinity-based pull-down, though widely used, suffers from artifacts introduced by probe immobilization or tag conjugation, which can alter the structure and binding behavior of NPs[116,117]. Label-free approaches such as the CETSA provide additional validation by measuring ligand-induced thermal stabilization of target proteins in intact cells or lysates, thereby confirming target binding under physiological conditions without chemical modification of the NPs[20]. To ensure reproducibility, each validation step must incorporate strict controls. Researchers should use inactive analogs to exclude nonspecific binding and perform competition experiments with known ligands to confirm binding-site specificity. Genetic perturbations such as CRISPR knockout should be used to prove functional relevance[118]. Statistical thresholds such as fold-change greater than 2 and P-value less than 0.05 should be applied consistently in proteomic analysis.
Future target discovery methods for NPs will focus on multidisciplinary integration and technological innovation for higher resolution, higher throughput, and more systematic approaches that better reflect real physiological conditions. Chemical proteomics will continue to evolve with upgraded mass spectrometry technologies that offer enhanced resolution and sensitivity, and it will enable the detection of low-abundance targets, weak interactions, and even transient binding events[119,120]. Novel 'smarter' chemical probes will become common, such as bifunctional probes combining PAL and click chemistry to improve labeling efficiency and specificity, cleavable probes that release native NP-target protein complexes via light, enzymes, or chemical reagents for precise binding site analysis, and bioorthogonal traceless labeling strategies that minimize interference with NP activity[121,122].
High-throughput genomics screening is poised to become more widespread. Genome-wide CRISPR screening, in particular, bypasses the need for chemical modification and offers a fast track to identifying the targets of NPs. The field will shift from a 'single target' to a systematic 'multi-target network' view. Integrated multi-omics data will be used to construct 'compound-target-pathway-phenotype' interaction networks through bioinformatics to explain the holistic regulatory effects of traditional Chinese medicine formulas and NPs[123,124]. Parallel target fishing technologies will enable the capture of entire protein complexes instead of single proteins to explain mechanisms at the level of functional macromolecular machines.
Moreover, AI and big data will support precise prediction and data mining. Deep learning models that integrate NPs' chemical structures, known target databases, omics screening results, and information will predict potential targets with high-accuracy and help narrow the scope of experimental validation. AI-optimized virtual screening and molecular docking will enable large-scale prediction of binding possibilities and modes between NPs (and their analogs) and the entire proteome, while standardized large databases and inferential knowledge graphs integrating NPs, targets, diseases, and side effects will provide high-quality data for AI models[125]. Importantly, future target identification will prioritize in situ, dynamic, and real-environment analysis: in situ chemical proteomics in living cells or animal tissues will preserve native intracellular environments (e.g., protein conformation, subcellular localization, interaction partners) to discover more physiologically relevant targets, and technologies for monitoring binding kinetics (association/dissociation rates) will clarify dynamic changes under physiological and pathological conditions, critical for understanding drug efficacy-timing relationships[122].
For clinical translation, target identification is essential for advancing NPs-derived agents into therapeutic development. For example, β-tubulin was established as the target of paclitaxel. Subsequent structural characterization of the paclitaxel-binding site on β-tubulin inspired the semisynthetic modification of the paclitaxel scaffold, and led to second-generation analogues such as docetaxel with enhanced target interaction and improved clinical performance. More broadly, target identification acts as a critical link between discovery and clinical application of NPs by elucidating molecular mechanisms, enabling structural optimization, and guiding clinical application strategies. Future translational efforts are expected to integrate target-based approaches and multi-omics with clinical samples to uncover biomarkers, predict treatment responses, and ultimately guide precision medicine. These technologies will also be extended to more complex biological settings, including studies of NPs-microbiota interactions and investigations into how NPs traverse the blood-brain barrier in neurological disorders.
Ultimately, target identification of NPs will bring together chemical proteomics, functional genomics, network pharmacology, and AI to uncover the mechanisms in real and dynamic cellular environments. These advances will accelerate NPs-based drug development, deepen understanding of complex biology, and provide new strategies for severe diseases such as cancer, infections, and neurological diseases.
-
Not applicable.
-
The authors confirm their contributions to the review as follows: conceptualization: Wang L, Zeng K, Tu P; writing of this manuscript: Zhu Y, Wang L, Zhang J. All authors reviewed the manuscript and approved for publication.
-
Data sharing is not applicable to this article as no datasets were generated or analyzed.
-
This work was supported by the National Natural Sciences Foundation of China (No. 82505116).
-
The authors declare that they have no conflict of interest.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhu Y, Zhang J, Tu P, Zeng K, Wang L. 2026. Recent advances in the target identification strategies of natural products: an overview from 2014 to 2025. Targetome 2(2): e015 doi: 10.48130/targetome-0026-0015 

Recent advances in the target identification strategies of natural products: an overview from 2014 to 2025
- Received: 27 January 2026
- Revised: 04 March 2026
- Accepted: 17 March 2026
- Published online: 15 April 2026
Abstract: Natural products (NPs) are indispensable in drug discovery. Their complex structure, diverse functions, and natural biological compatibility enable potent interactions with cellular macromolecules. Unlike synthetic drugs specifically designed to target therapeutic molecules with well-defined mechanisms of action, the discovery of NPs usually involves exploring natural sources, isolating active compounds, and validating their efficacy. As a result, the precise molecular targets and pharmacological mechanisms of NPs often remain unclear. Additionally, the intrinsic properties of NPs, such as structural lability, low bioavailability, and activity at sub-micromolar concentrations, limit the effectiveness of conventional target identification approaches. Over the past decade (2014–2025), pharmacological research on NPs has shifted toward identifying their direct targets, with significant progress made in chemicobiological, biophysical, and computational techniques. In this review, recent advances in target identification of NPs are summarized, with attention to methodologies that leverage protein libraries, in-situ models, and emerging technologies like bioinformatics. Future directions are also proposed to accelerate NP-based drug development through more efficient, targeted identification.
-
Key words:
- Natural products /
- Target identification /
- Proteomics /
- Click chemistry /
- Proximity labeling /
- Bioinformatics