









-
The common drug development paradigm consists of phenotype-based drug discovery (PDD) and target-based drug discovery (TDD). In the long history of human drug development, most drugs have been discovered through PDD, especially before 1980[1,2]. The common paradigm of PDD is evaluating whether compounds exhibit satisfactory efficacy in specific pathological models. However, with advances in molecular biology and the success of targeted therapies, TDD has become a dominant paradigm in modern drug development[3,4]. Selecting molecular targets based on disease understanding is a critical step in TDD. The drug development pipeline typically spans 12 to 15 years, with less than 10% of therapeutic drugs entering Phase I trials failing to reach approval[5,6]. A major contributing factor is the insufficient identification and validation of effective therapeutic targets[7,8]. Therefore, improving strategies for target discovery is essential for both understanding disease biology and enhancing drug development success rates. There is an ancient Chinese saying, 'A journey of a thousand miles begins with a single step'. For target-based drug development, the discovery and confirmation of new targets is the first, most crucial step in this long journey.
In the contemporary drug discovery process, large-scale omics data from clinical samples have become a major source for identifying potential therapeutic targets. High-throughput technologies, particularly transcriptomics and proteomics, can generate extensive lists of differentially expressed genes or proteins associated with disease states. However, translating these large candidate lists into a small number of truly promising targets remains a significant challenge[9]. Typically, omics data contain hundreds to thousands of differentially expressed genes or proteins. Without effective prioritization strategies, it is difficult to make the optimal choice of potential new drug targets.
Computational tools based on sophisticated algorithms can integrate rich biological information, greatly enhancing the process of target and drug discovery[10]. As an ancient Chinese saying goes, 'A workman must first sharpen his tools if he is to do his work well'. For target discovery based on omics data, efficient and reliable computational methods are like such sharp tools. However, existing computational approaches for target prioritization often rely on statistical associations, functional enrichment, or network-based proximity to known disease-related genes. While these methods are useful, they tend to prioritize well-studied proteins with existing literature evidence, potentially overlooking novel targets that lack prior disease annotations. This limitation is particularly important in the context of first-in-class drug discovery, where originality and unexplored targets are essential.
Previously, we have developed an automated classification tool named OTTM (Omics and Text-driven Translational Medicine), mainly used for drug repositioning. OTTM can efficiently screen candidate proteins from omics data and prioritize those that have not been reported to be associated with the disease of interest, but have corresponding approved, or clinical drugs[11]. Testing about 20–30 drugs, which are selected by OTTM from omics data, can efficiently discover effective approved or clinical drugs that have never been reported to be effective for the disease of interest. This strategy enables rapid experimental validation through compound testing. However, by design, OTTM focuses on targets with known drugs, and does not address the large proportion of candidate proteins that lack any active compounds.
To address this limitation, it is necessary to develop a complementary computational strategy that focuses on identifying original therapeutic targets without known drugs. Thus, here we report the development of a new computational tool named OPTAR (Omics and Pocket Analysis-based Target Assessment and Ranking), which is specifically designed for discovering novel target proteins derived from omics data. Compared with OTTM, OPTAR specifically focuses on proteins that lack approved or clinical drugs. In addition, both OTTM and OPTAR exclude candidate proteins that have already been reported to be associated with the disease of interest through systematic literature mining. Importantly, OPTAR further introduces a disease correlation inference strategy based on protein-protein interaction (PPI) networks, in which candidate proteins are ranked according to the extent to which their interacting partners are associated with the disease. Moreover, OPTAR incorporates a structure-based binding pocket assessment module using AlphaFold-predicted protein structures to evaluate the potential druggability of candidate targets.
Taken together, OTTM and OPTAR form a complementary framework for target discovery from omics data, addressing both drug repurposing and de novo target identification. In this study, starting from differentially expressed genes derived from hepatocellular carcinoma (HCC) omics data, we applied OPTAR using the keywords 'hepatocellular carcinoma' and 'cell cycle', where the latter was selected due to its central role in cancer progression. Candidate genes were prioritized based on their intersection and subsequent network-based evaluation. From the top-ranking candidates, we identified several proteins for experimental validation, demonstrating the feasibility of OPTAR as a computational framework for discovering novel and potentially druggable therapeutic targets.
-
For literature assessment in OPTAR, all PubMed abstract information was extracted from 1,219 XML files (from pubmed24n0001 to pubmed24n1551). For drug availability assessment in OPTAR, the mapping table of proteins and corresponding drugs was extracted from the downloaded data of the TTD database[12]. OPTAR inputs a list of differentially expressed genes or proteins uploaded by users, as well as a keyword used as the disease type of interest. For interacting protein (IP) assessment in OPTAR, human PPI data were extracted from the STRING database. On this basis, OPTAR uses the following formula to calculate the disease correlation score of each candidate protein:
$ \text{Score}\mathit{_{{IP}}}=\dfrac{\mathrm{Hit}\_\mathit{{{Num}}}}{\mathrm{PubMed}\_\mathit{{{Num}}}} $ $ \text{Total Score}=\sum\limits_1^n\text{Score}\mathit{_{IP}} $ For each IP, the score is defined as: the number of abstracts containing disease-related keywords divided by the total number of PubMed abstracts associated with the IP. Subsequently, the total score of the candidate protein was calculated by summing the scores of all its IPs.
On common personal computers, it takes about 1 h for a single CPU core to run OPTAR, depending on the frequency of keywords in the PubMed database, and the performance of the CPU. The first version of OPTAR, with source code written in C++, provides users with executable programs. Subsequent versions will be written in Python with open-source codes. In addition to the literature mining and IP evaluation modules, OPTAR also has an independent drug binding pocket assessment module that uses the AlphaFold structures of human proteins.
Web server of OPTAR
-
The executable programs of OPTAR are available from its web server (
http://otter-simm.com/optar.html ). In the input list of OPTAR, each line of symbols represents a candidate protein or gene. Examples of input protein lists are provided in the downloaded compressed files and on the Help page of the OPTAR web server.Cell culture
-
Human HCC cell lines HepG2, and Huh7 were purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd. Cells were authenticated by STR profiling. Cells were maintained at 37 °C in a humidified incubator with 95% air, and 5% CO2. HepG2 cells were cultured in 87% MEM (with NEAA) (ZQ-300) supplemented with 10% fetal bovine serum (ZQ500-S), 1% L-alanyl-L-glutamine (CSP004), 1% sodium pyruvate (CSP003), and 1% penicillin–streptomycin (CSP006). Huh7 cells were cultured in DMEM high glucose (ZQ-100) supplemented with 10% fetal bovine serum (ZQ500-A), 1% penicillin-streptomycin (CSP006), 1% L-alanyl-L-glutamine (CSP004), and 1% sodium pyruvate (CSP003).
siRNA design and transfection
-
siRNAs targeting UBE2J1, KDELR3, and VTI1A were custom designed and synthesized by MCE (Supplementary Table S1). Transfection was performed using CALNP™ RNAi in vitro (D-Nano Therapeutic). Cells were seeded simultaneously into 12-well plates (for knockdown validation by RT-qPCR) and 96-well plates (for CCK-8 assay). After cell attachment, transfection complexes were prepared according to the CALNP™ protocol to achieve a final siRNA concentration of 300 nmol·L−1, using the following mixture per transfection unit: 20 μmol·L−1 siRNA (3 μL), Reagent A (21 μL), Reagent B (6 μL), combined with 170 μL serum-free medium. For 96-well plates, 10 μL transfection complex was added per well together with 90 μL serum-free medium. For 12-well plates, 100 μL transfection complex was added per well, together with 900 μL serum-free medium. A non-targeting siRNA (si-NC) served as the negative control.
RNA extraction and RT-qPCR
-
At 24 h post-transfection, total RNA was extracted using FastPure®Cell/Tissue Total RNA Isolation Kit V2 (Vazyme, RC112). cDNA was synthesized using HiScript IV All-in-One Ultra RT SuperMix for qPCR (Vazyme, R433). qPCR was performed using ChamQ Universal SYBR qPCR Master Mix (Vazyme, Q711). GAPDH was used as an internal reference gene, and relative mRNA levels were calculated using the 2−ΔΔCᴛ method, consistent with the reference workflow. Primer sequences are provided in Supplementary Table S2 (Primers for RT-qPCR).
Western blotting
-
Cells were harvested for SDS-PAGE and Western blot analysis on NC membranes. After blocking with 5% nonfat milk in TBST for 1 h, membranes were incubated with the primary antibodies overnight at 4 °C, followed by secondary antibodies for 1 h at room temperature. ECL detection systems were applied to develop signals. The following antibodies were used: Caspase 3/P17/P19 polyclonal antibody (Proteintech, 19677-1-AP), Cleaved PARP (Asp214) antibody (Cell Signaling Technology, 9541), and α-tubulin polyclonal antibody (Proteintech, 11224-1-AP).
Cell viability assay (CCK-8)
-
HepG2 cells were seeded into 96-well plates at a density of 5 × 103 cells per well. After 24 h of culture, the cells were treated with compounds at various concentrations (25 and 50 μmol·L−1). After 48 h post-transfection or compound treatment, cell viability was assessed using Cell Counting Kit-8 (Meilunbio, MA0218). We added 100 μL of serum-free medium containing 10% CCK-8 reagent to each well, and the plates were incubated for an additional hour. For each well, 10 μL CCK-8 reagent was added together with 90 μL serum-free medium, followed by incubation for 1 h. Absorbance was measured at 450 nm using a microplate reader, following the typical CCK-8 readout approach.
Wound-healing assay
-
HepG2 and Huh7 cells were seeded in six-well plates and transfected with the indicated siRNAs. After the cells reached near-confluence, a linear wound was generated using a sterile pipette tip. Detached cells were removed by washing with PBS, and a serum-free medium was added. Images were captured at 0 and 48 h under a microscope. The cell migration ability is quantified and statistically analyzed using the cell migration rate. The cell migration rate = (initial wound width − wound width after 48 h)/initial wound width × 100%.
Transwell invasion assay
-
The Transwell invasion assay was conducted using a coated culture chamber (Falcon, 353097) coated with 1 : 8 diluted Matrigel (Corning), which was equipped with a membrane with an 8 μm pore size. After 48 h of siRNA transfection, 5,000 cells were added to the upper chamber of each cell culture chamber, while the lower chamber was filled with a medium containing 10% FBS as a chemotactic agent. After 24 h, the non-migrating cells on the upper surface were removed, and the migrating cells on the lower surface were fixed. After staining with crystal violet, the cells were photographed under a microscope and counted.
Cell cycle analysis by flow cytometry
-
After transfection with si-NC or si-UBE2J1, HepG2 and Huh7 cells were harvested, fixed in 70% ethanol, and stained using a DNA content staining solution according to the manufacturer's instructions (Vazyme, AC101). Cell-cycle distribution was analyzed by flow cytometry, and the percentages of cells in G0/G1, S, and G2/M phases were quantified.
Public database analysis of UBE2J1 expression in HCC
-
UBE2J1 expression in HCC and normal liver tissues was analyzed using GEPIA2 (
https://gepia2.cancer-pku.cn ), an online platform based on transcriptomic data from The Cancer Genome Atlas (TCGA), and the Genotype-Tissue Expression (GTEx) project. The 'LIHC' dataset was used to compare UBE2J1 expression between tumor and normal liver tissues. Expression values were presented as log2 (TPM + 1).Virtual screening
-
The Natural Product Library for HTS purchased from TargetMol (Cat. No. L6000) was used for virtual screening. Proper three-dimensional conformations were generated for 4,654 natural compounds from this library using the LigPrep module of the Schrödinger software package. The AlphaFold structure of UBE2J1 was downloaded from UniProt, and prepared using the Protein Preparation Wizard module of Schrödinger. After all missing hydrogen atoms were added, binding sites were detected using the SiteMap module of Schrödinger. Using the binding site consistent with the position identified by OPTAR, grids for subsequent molecular docking were generated using the Receptor Grid Generation module of Schrödinger. Then molecular docking-based virtual screening was carried out using the Glide module of Schrödinger. Finally, 30 compounds with the higher docking scores, and the binding modes matching the pocket were selected for subsequent SPR-based binding detection.
Surface plasmon resonance (SPR) assays
-
SPR binding assays were performed on a Biacore T200 instrument (GE Healthcare) at 25 °C. The recombinant Human UBE2J1 was purchased from MedChemExpress (HY-P703625). The protein was coupled with the CM5 chip at 100 μg·mL−1 in 10 mmol·L−1 sodium acetate (pH 4.0). After immobilization, the system was equilibrated for 1 h. Each compound was serially diluted in PBS buffer with 0.05% Tween-20 and injected over the chip at a flow rate of 30 μL·min−1. Each injection was associated with the sensor chip for 120 s and dissociated for 180 s. All data were processed using the Biacore T200 evaluation software (v1.0).
Statistical analysis
-
Data are presented as mean ± SD from three biological replicates per group. Statistical analysis and plotting were performed using GraphPad Prism 10.1.2; the data were analyzed by one-way ANOVA and an unpaired t-test. P < 0.05 was considered significant.
-
The development of OPTAR is aimed at complementing our previously developed computational tool OTTM, for the discovery of potential therapeutic targets from omics data of clinical samples and other sources. Both OTTM and OPTAR are based on a shared conceptual framework that classifies candidate genes or proteins derived from omics data into four categories based on two binary criteria: drug availability and prior disease association. Category 1 includes FDA-approved or clinically tested drugs whose target proteins have been previously reported to be associated with the disease of interest. Category 2 comprises FDA-approved or clinically tested drugs whose target proteins have not been previously linked to the disease of interest. Category 3 consists of proteins that lack FDA-approved or clinically tested drugs, but have been reported to be associated with the disease of interest. Category 4 encompasses proteins that lack FDA-approved or clinically tested drugs, and have no prior reported association with the disease of interest (Fig. 1a).
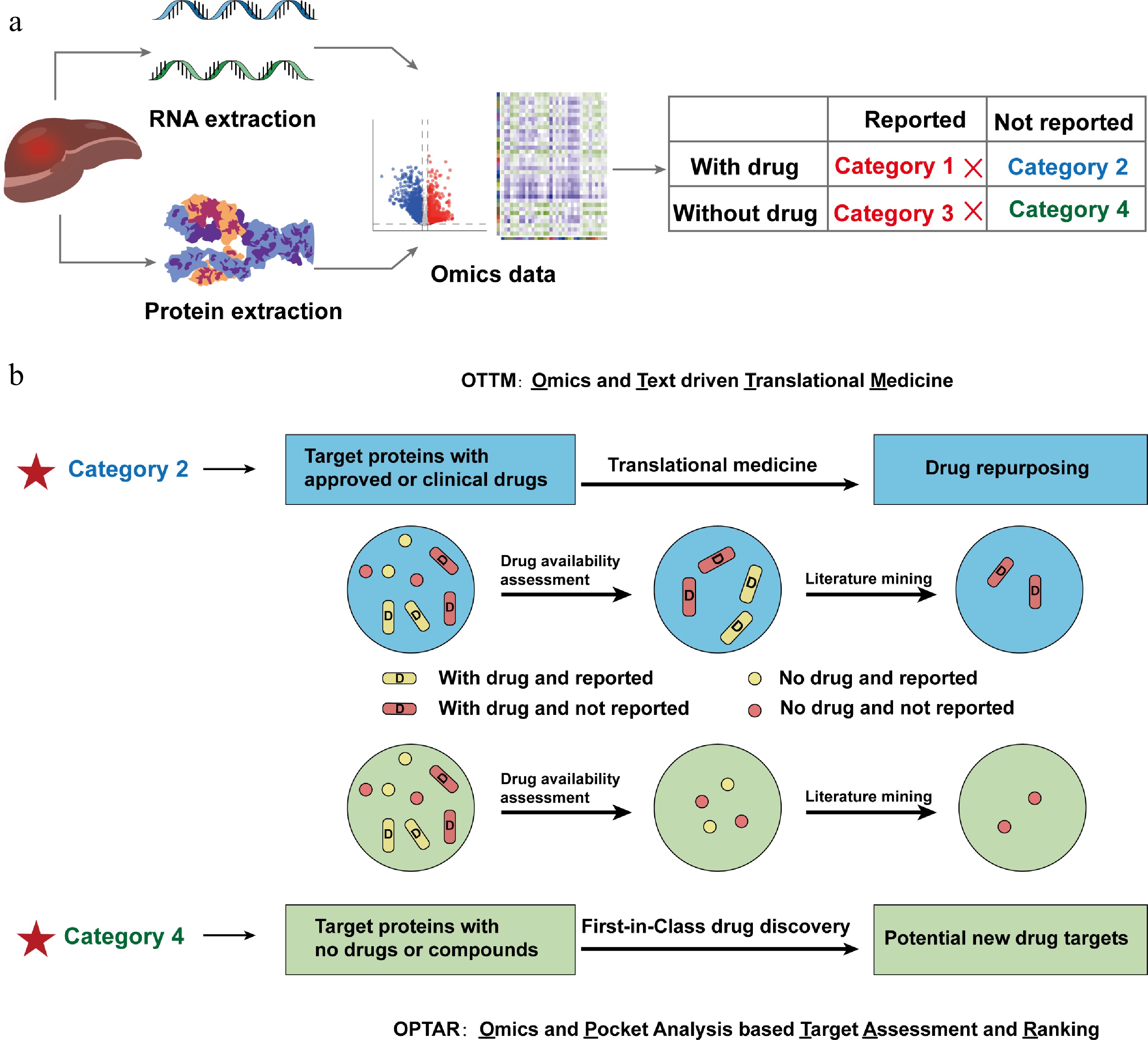
Figure 1.
Schematic diagram illustrating the development principle of a new computational tool OPTAR. (a) Proteins or genes from omics data are classified into four categories according to whether their target proteins have FDA-approved or clinically tested drugs, and whether they have been previously reported associated with the disease: Category 1, with approved or clinical drugs and have been previously reported associated with the disease; Category 2, with approved or clinical drugs and have not been reported associated with the disease; Category 3 lack approved or clinical drugs and the proteins have been previously reported associated with the disease; Category 4 lack approved or clinical drugs and have not been reported associated with the disease. (b) Comparison of the screening workflows of previously reported OTTM and OPTAR. Both tools are based on the classification system described above: OTTM first performs drug availability assessment to retain Categories 1 and 2, then employs literature mining to exclude Category 1, ultimately identifying Category 2 targets (existing drugs with potential new indications). OPTAR first performs drug availability assessment to retain Categories 3 and 4, then uses the same literature mining approach to exclude Category 3, ultimately identifying Category 4 targets (novel targets without existing drugs and no reported disease association).
OTTM aims to identify all candidate proteins or genes belonging to Category 2, whereas OPTAR aims to identify all candidate proteins or genes belonging to Category 4. In the workflow of using OTTM, drug availability assessment is first performed to identify FDA-approved or clinically tested drugs, retaining only Categories 1 and 2. Subsequently, the literature mining step is then performed by checking all PubMed abstracts with the specified disease type to filter for target proteins that have not been previously reported to be correlated to the disease of interest, retaining only Category 2. Similarly, OPTAR begins with a drug availability assessment to identify proteins lacking FDA-approved or clinically tested drugs, retaining only Categories 3 and 4. Then, using the same literature mining approach, OPTAR excludes the candidate proteins or genes belonging to Category 3. Ultimately, OPTAR identifies target proteins that have no known drugs, and have not been previously reported correlated to the disease of interest, retaining only Category 4 (Fig. 1b). This classification framework provides the conceptual basis for distinguishing between targets suitable for drug repurposing and those representing unexplored therapeutic opportunities.
Connections and differences between OPTAR and OTTM
-
OTTM and OPTAR are complementary computational tools designed to leverage omics data for different objectives in drug discovery. OTTM primarily focuses on identifying candidate proteins with existing drugs, facilitating drug repositioning and enabling rapid experimental validation through compound testing. In contrast, OPTAR is specifically designed to identify novel therapeutic targets that lack approved or clinical drugs, thereby supporting the discovery of first-in-class targets. Although these two tools have different purposes, they share common technical modules, including drug availability assessment and literature mining. Both OTTM and OPTAR systematically excludes candidate proteins that have already been reported to be associated with the disease of interest through comprehensive PubMed abstract mining, ensuring a level of novelty in the identified targets.
Despite these shared components, the two tools differ in two key aspects. First, in terms of drug availability, OTTM retains candidate proteins with corresponding drugs, whereas OPTAR focuses exclusively on proteins without any approved or clinical drugs or known active compounds. In this situation, once any active compound is discovered from large-scale virtual or experimental screening, it will be the first inhibitor or agonist for the validated new target protein selected by OPTAR. Therefore, the recommended usage of OPTAR is in combination with gene knockout validation and large-scale compound screening with protein expression.
Second, in terms of disease relevance, OTTM does not explicitly infer disease-target relationships computationally, but relies on downstream experimental validation. However, OPTAR must infer the correlation between proteins and diseases, because typically, when faced with the same differential expression list, OPTAR needs to evaluate approximately nine times more proteins or genes than OTTM. Thus, OPTAR introduces a disease correlation inference strategy based on PPI networks, allowing prioritization of candidate proteins whose interacting partners are enriched for disease associations. Taken together, OTTM and OPTAR form a complementary framework that enables both drug repurposing and de novo target discovery from omics data.
Workflow for using OPTAR
-
The OPTAR framework consists of three main modules: (1) literature mining, (2) disease correlation inference, and (3) binding pocket assessment. First, candidate genes or proteins derived from omics data are filtered to exclude those with prior reported associations to the disease of interest through systematic PubMed mining. Second, disease correlation is inferred using PPI networks, where candidate proteins are prioritized based on the extent to which their interacting partners are associated with the disease. Third, the structural druggability of candidate proteins is evaluated through binding pocket assessment using AlphaFold-predicted structures. For experimental validation, high-ranking candidate targets identified by OPTAR can be subjected to gene perturbation assays, such as knockdown experiments, to assess their functional roles.
Once the correlation between proteins and diseases is experimentally confirmed, high-throughput screening of compounds can be conducted via expression and purification of target proteins. If the target protein has a functional regulatory pocket for small-molecular compound binding, and a high enough number of compounds are screened, compounds with good activity can likely be discovered. Finally, in vivo efficacy evaluation is conducted using animal models to evaluate the therapeutic efficacy and safety of active compounds of target proteins discovered by OPTAR. This workflow can not only prove the therapeutic potential of the target protein discovered by OPTAR, but also provides candidate compounds for drug development. If the properties of the candidate compounds are satisfactory in all aspects, the candidate compound is promising to enter the stages of clinical trials (Fig. 2a).
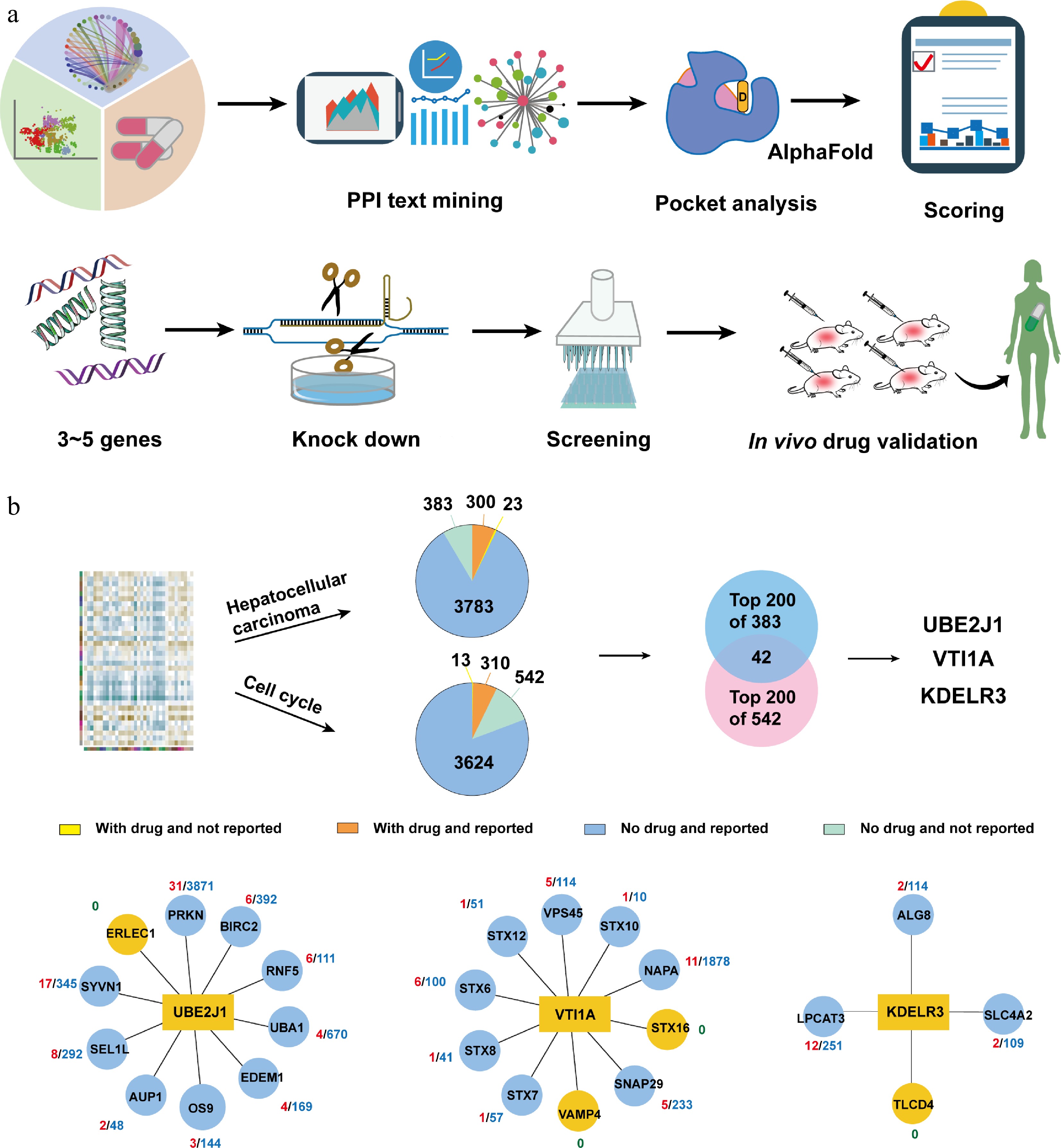
Figure 2.
Using OPTAR for potential target discovery of hepatocellular carcinoma. (a) Typical workflow of using OPTAR combined with experimental validation. Category 4 candidate targets undergo literature mining in interacting proteins to infer disease correlation. High-scoring targets are subjected to binding pocket assessment with AlphaFold-predicted structures using the pocket analysis module of OPTAR, followed by knockdown validation. After protein expression and high-throughput screening, active compounds were subjected to in vivo efficacy and safety evaluation. (b) Target discovery against hepatocellular carcinoma using OPTAR. Starting from the omics data previously used by OTTM, OPTAR generated two protein lists using 'hepatocellular carcinoma' and 'cell cycle' as keywords. Then 383 and 542 proteins without drugs or reported relevance to the disease were identified, respectively. The intersection of the top-ranking 200 proteins from each list contains 42 proteins. Finally, three candidate genes UBE2J1, VTI1A, and KDELR3 were selected for subsequent knockdown experiments, considering the results of pocket assessment and whether recombinant proteins are commercially available. Protein-protein interaction networks were plotted with reference to the STRING database.
This integrated workflow enables systematic progression from omics-derived candidates to experimentally validated and potentially druggable targets. OPTAR is free for all academic users, and the online server provides download links for the local version of programs and the user manual, with the address at
http://otter-simm.com/optar.html .Development of the literature mining module of OPTAR
-
The goal of OPTAR is to discover new targets that may be effective for a certain disease but lack drugs or active compounds from omics data. It achieves this goal through three steps, including literature mining, IP analysis, and drug binding pocket analysis. For the first step of literature mining, OPTAR performs exhaustive text mining in all available PubMed abstracts to determine whether a protein or gene has been previously reported to be correlated to a certain disease. OPTAR scans all PubMed abstracts for the name of disease type, such as HCC, with the names of each protein or gene from the input list. Once the name of a protein or gene appears in the same PubMed abstract as the disease type of interest, OPTAR excludes the protein or gene for further evaluation.
Development of disease correlation inference module of OPTAR
-
For the second step of the IP assessment, OPTAR evaluates the 'disease correlation' via text mining in all PubMed abstracts for all physical IPs of each candidate protein or gene. Using the PPI data from the STRING database, OPTAR determines the IPs for each candidate protein, and scans the keywords of disease types in all PubMed abstracts for these IPs. For all the candidate proteins subjected to this evaluation, the more IPs reported to be associated with the disease of interest, the higher score OPTAR gives for this candidate protein. For example, both protein A and protein B have 10 physical IPs. If eight out of 10 IPs for protein A are reported to be correlated to a designated disease, while two out of 10 IPs for protein B are reported to be correlated, protein A will have much higher score given by OPTAR than protein B.
Development of binding pocket assessment module of OPTAR
-
For the third step of the binding pocket assessment, OPTAR evaluates small-molecular compound binding pockets for each candidate protein via three phases, using their AlphaFold-predicted three-dimensional structures. First, the AlphaFold structure of each protein is placed in a three-dimensional matrix filled with equidistant lattice points. Secondly, the lattice points exposed outside the protein are removed. Third, discontinuous lattice areas are removed, and only the contiguous lattice areas of appropriate size are retained. Inspired by the statistical properties analysis of successful drug targets, the ideal binding pocket for drug-like small-molecular compounds should neither be too large, nor too small, and the number of hydrogen bond donors and hydrogen bond acceptors is appropriate.
Target discovery using OPTAR for HCC
-
To assess the reliability of OPTAR, here we used the same input list as in the previously published OTTM; namely, omics data from clinical samples of HCC. By using the keywords 'hepatocellular carcinoma' and 'cell cycle' for queries, two pie charts were generated, containing 383 and 542 proteins, respectively, that lack FDA-approved or clinical testing drugs, and have not been previously reported to be associated with the disease. Taking the intersection of the top-ranking 200 proteins from the two lists yielded 42 proteins. Subsequently, from these 42 proteins, the STRING database was used to explore protein interaction networks, followed by scoring and ranking. Three candidate proteins, UBE2J1, VTI1A, and KDELR3 were chosen for further validation. Subsequently, gene or protein interaction networks were explored using the STRING database, followed by scoring and ranking to select 3–5 core node genes. Then, three candidate genes UBE2J1, VTI1A, and KDELR3 came to our attention due to their high scores. The reason why OPTAR gives these three genes higher scores is that most of their IPs have been reported associated with liver cancer. Specifically, nine out of 10 IPs of UBE2J1 have been previously reported relevant, eight out of 10 IPs of VTI1A have been previously reported relevant, and three out of four IPs of KDELR3 have been previously reported relevant (Fig. 2b and Supplementary Table S3).
In addition to the disease correlation inference based on PPI literature mining, we also fully considered the pocket assessment by OPTAR for their protein structures. OPTAR utilizes AlphaFold-predicted protein structures of candidate targets to identify suitable small-molecular compounds binding pockets via three stages: placing protein structures in a grid matrix, removing grid points outside the protein, and removing discontinuous grid points (Fig. 3a). Finally, only continuous grid regions of expected size for ideal drug binding were retained (Fig. 3b). Finally, these three candidate genes, UBE2J1, VTI1A, and KDELR3 were selected for subsequent experiments, considering the results of pocket assessment and whether recombinant proteins are commercially available.
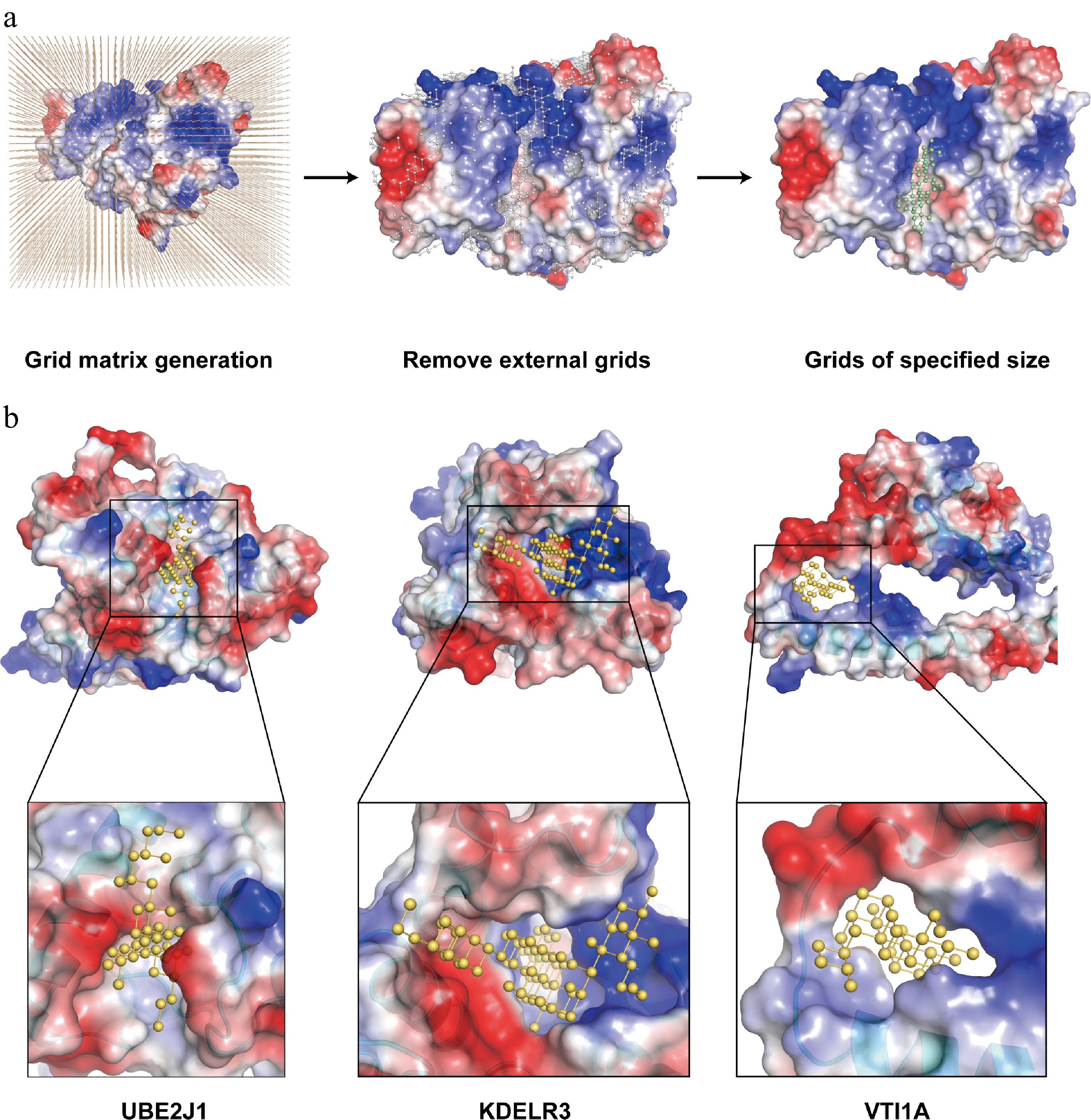
Figure 3.
Development of the binding pocket assessment module of OPTAR. (a) OPTAR utilizes AlphaFold-predicted three-dimensional structures of candidate proteins to identify suitable binding pockets for small-molecular compounds through three stages: placing protein structures in a three-dimensional grid matrix, removing grid points outside the protein, and removing discontinuous grid areas. Finally, only continuous grid regions of expected size for ideal drug binding were retained. Representation of compound-binding pockets identified by OPTAR for (b) UBE2J1, (c) KDELR3, and (d) VTI1A. Protein surface is colored according to electrostatic potential distribution (red indicates negative potential, blue indicates positive potential, and white indicates neutral). Predicted small-molecule binding sites are shown as yellow ball-and-stick models. Zoomed views illustrate the expected size and shape of potential active compounds.
Validation of knockdown efficiency of UBE2J1, KDELR3, and VTI1A, and their effects on phenotypes of HCC cells
-
To investigate the functional roles of UBE2J1, KDELR3, and VTI1A in HCC, HepG2 and Huh7 cells were transiently transfected with specific siRNAs targeting each gene. RT-qPCR analysis at 24 h post-transfection showed that the mRNA expression levels of all three target genes were significantly reduced in the knockdown groups compared with the negative control (si-NC) group, confirming efficient gene silencing (Fig. 4a). Consistently, Western blot analysis performed at 48 h post-transfection further verified the reduction of KDELR3, UBE2J1, and VTI1A protein levels in both cell lines (Fig. 4b).
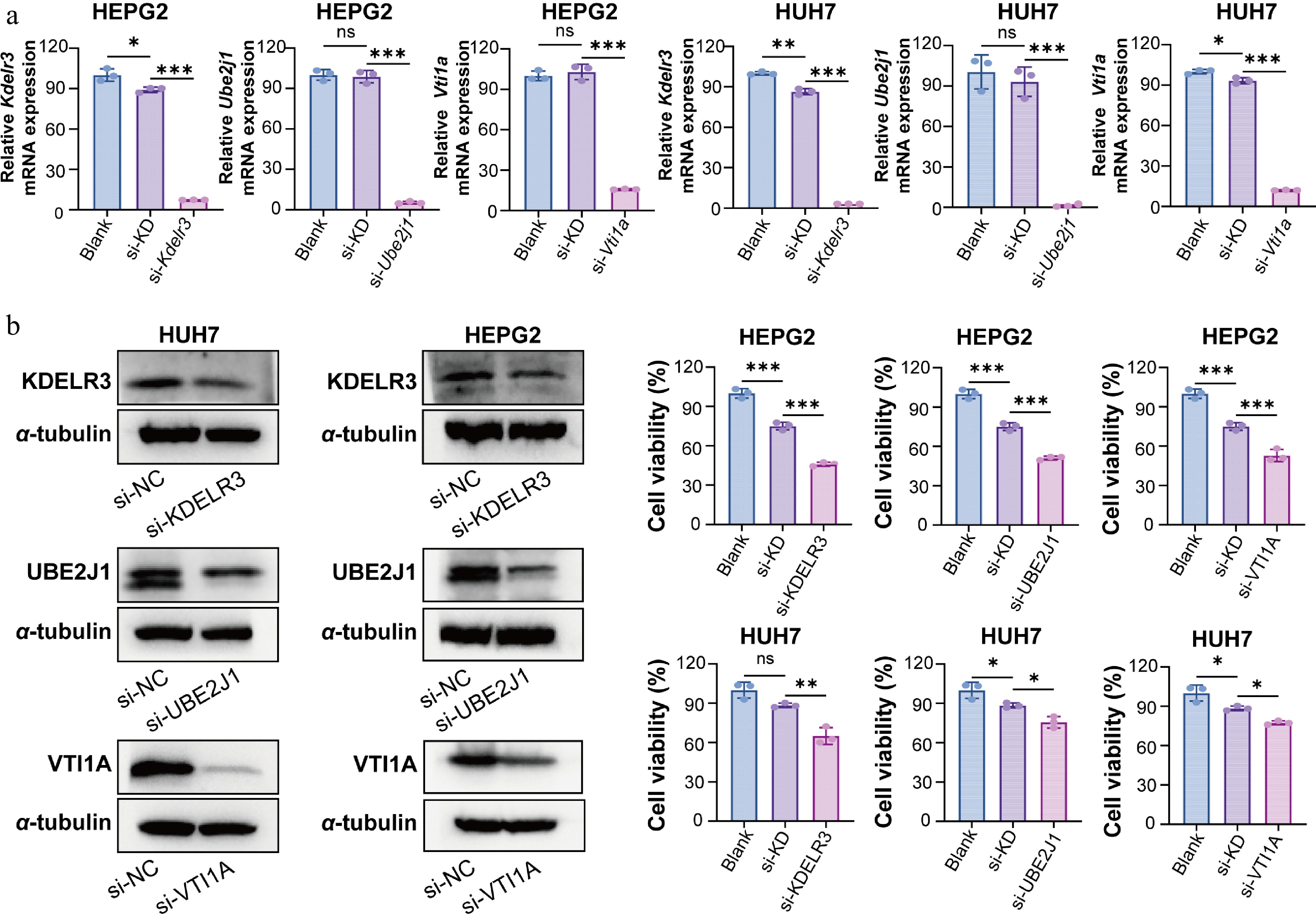
Figure 4.
Validation of knockdown efficiency of UBE2J1, KDELR3, and VTI1A and their effects on HCC cell viability. (a) RT-qPCR analysis was performed at 24 h post-transfection to evaluate knockdown efficiency at the mRNA level. Using GAPDH as an internal control, normalization was performed using 2−ΔΔCᴛ. (b) Western blot analysis was performed at 48 h post-transfection to verify the protein-level silencing of KDELR3, UBE2J1, and VTI1A in HepG2 and Huh7 cells. α-tubulin was used as the loading control. (c) Cell viability was assessed by CCK-8 assay at 48 h post-transfection. Knockdown of KDELR3, UBE2J1, and VTI1A significantly reduced the viability of HCC cells compared with the control group. Data are presented as mean ± SD (n = 3). Statistical significance is indicated as: ns, not significant; * P < 0.05; ** P < 0.01; *** P < 0.001.
To determine the effects of gene silencing on HCC cell function, a series of phenotypic assays was subsequently performed. CCK-8 assays at 48 h post-transfection showed that knockdown of KDELR3, UBE2J1, or VTI1A, significantly decreased the viability of both HepG2 and Huh7 cells compared with the si-NC group (Fig. 4c). In addition, wound-healing assays revealed that silencing of these three genes markedly suppressed the migratory ability of both cell lines (Fig. 5a). Furthermore, transwell invasion assays demonstrated that knockdown of KDELR3, UBE2J1, or VTI1A significantly inhibited the invasive capacity of HepG2 and Huh7 cells (Fig. 5b). Together, these results indicate that UBE2J1, KDELR3, and VTI1A are functionally involved in maintaining the proliferation, migration, and invasion of HCC cells.
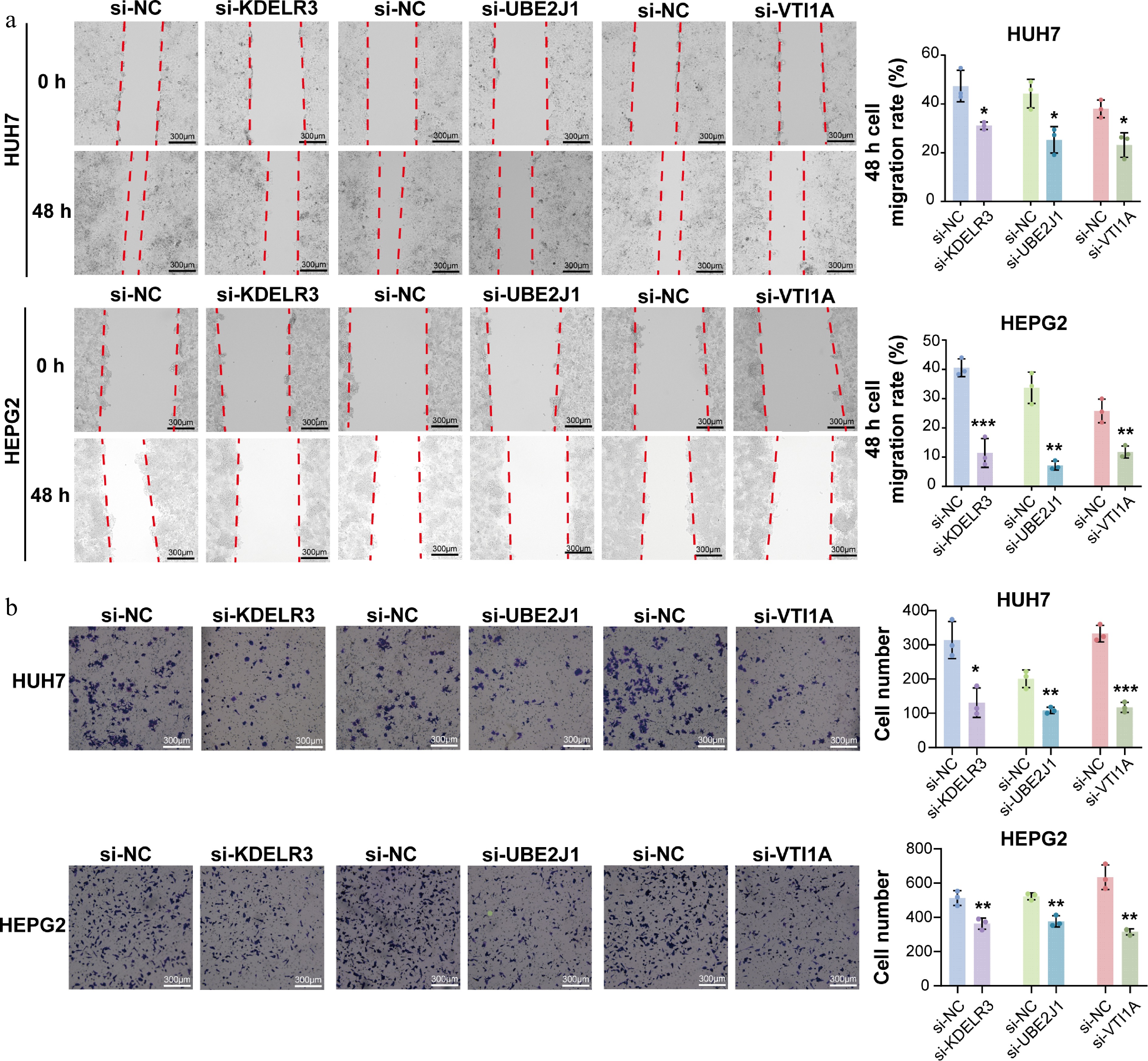
Figure 5.
Knockdown of UBE2J1, KDELR3, and VTI1A suppresses migration and invasion of HCC cells. (a) Wound-healing assays were performed in HepG2 and Huh7 cells transfected with siRNAs targeting KDELR3, UBE2J1, or VTI1A. Knockdown of each target gene markedly inhibited the migratory ability of both cell lines compared with the corresponding si-NC group. (b) Transwell invasion assays were performed to evaluate the invasive capacity of HepG2 and Huh7 cells after gene silencing. Knockdown of KDELR3, UBE2J1, or VTI1A significantly reduced the invasive ability of both cell lines. Data are presented as mean ± SD (n = 3). Statistical significance is indicated as: ns, not significant; * P < 0.05; ** P < 0.01; *** P < 0.001.
Further characterization and compound virtual screening of UBE2J1
-
Due to difficulties of expression and commercial unavailability for purchase, KDELR3 and VTI1A were unable to enter the phase of active compounds discovery. Therefore, we selected UBE2J1 for further characterization and subsequent compound screening. Public database analysis showed that UBE2J1 expression was significantly elevated in LIHC tumor tissues compared with normal liver tissues (Fig. 6a). In addition, Western blot analysis after UBE2J1 knockdown showed decreased Cyclin D1 expression and increased levels of Cleaved caspase-3 and Cleaved PARP in both HepG2 and Huh7 cells (Fig. 6b), suggesting that UBE2J1 may be involved in regulating cell-cycle progression and apoptosis in HCC cells. Flow cytometric analysis further demonstrated that silencing UBE2J1 reduced the proportion of cells in the G0/G1 phase, increased the proportion of cells in the S phase, and caused no significant change in the G2/M population in both cell lines (Fig. 6c and d). These findings provided additional evidence supporting the functional relevance of UBE2J1 in HCC, and further justified its selection for subsequent virtual screening and active compound discovery.
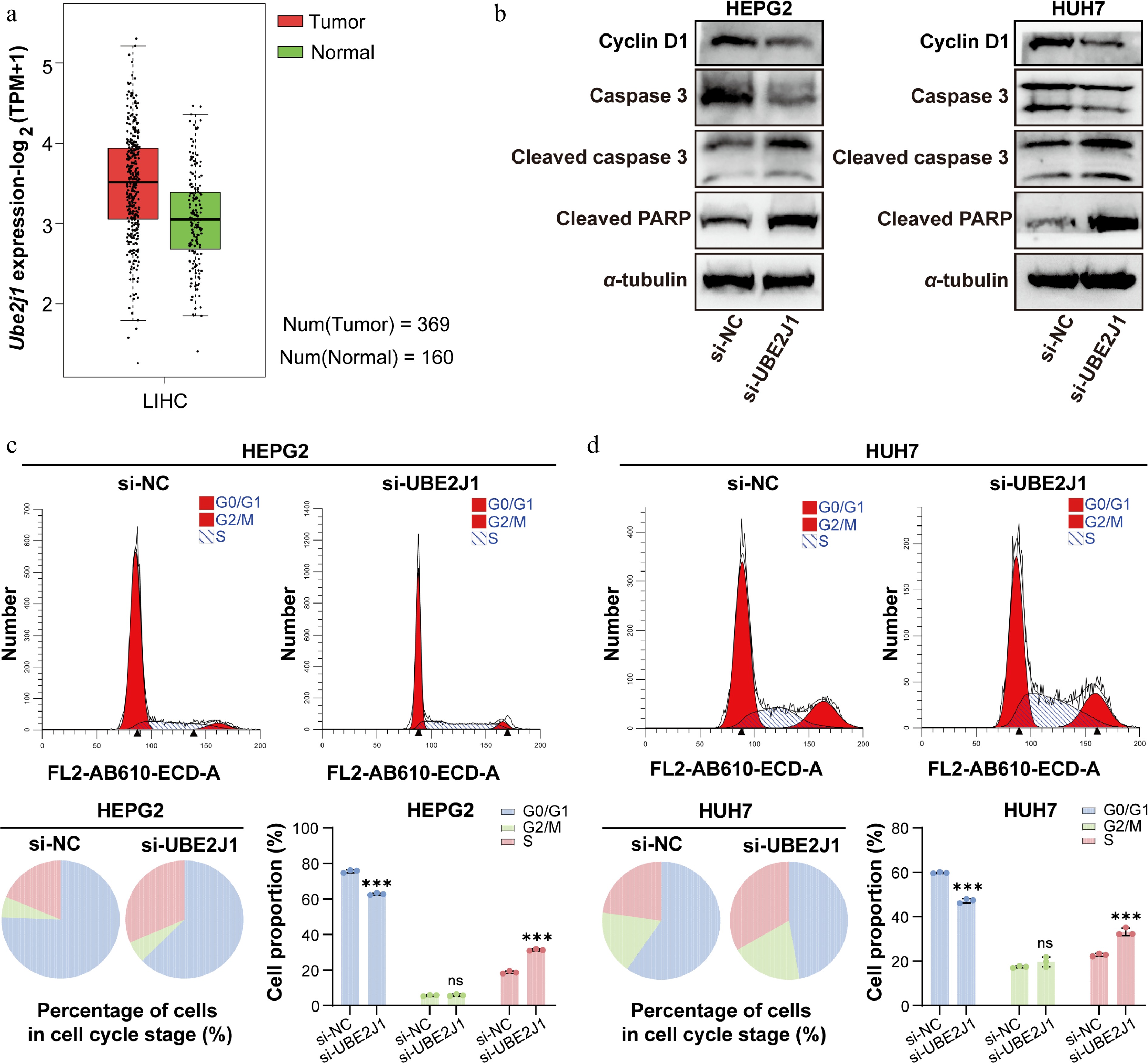
Figure 6.
Clinical relevance analysis of UBE2J1 expression, and the impact of UBE2J1 knockdown on HCC cells as assessed by Western blot and flow cytometry. (a) UBE2J1 expression in LIHC tumor and normal tissues based on public database analysis. (b) Western blot analysis of Cyclin D1, Caspase 3, Cleaved caspase 3, and Cleaved PARP in HepG2 and Huh7 cells transfected with si-NC or si-UBE2J1. α-tubulin was used as a loading control. Flow cytometric analysis of cell-cycle distribution in (c) HepG2, and (d) Huh7 cells after UBE2J1 silencing. Representative histograms, pie charts, and quantification of cells in G0/G1, S, and G2/M phases are shown. Data are presented as mean ± SD (n = 3). Statistical significance is indicated as: ns, not significant; *** P < 0.001.
Then we chose UBE2J1 for virtual screening, using 4,654 traditional Chinese medicine (TCM) compounds from a library. The top-ranking 10% candidates (about 450 compounds) were selected according to docking scores. Among them, 30 candidate compounds, most of which are active TCM ingredients, were selected according to chemical structure diversity for SPR binding detection. After binding detection using SPR with recombinant UBE2J1 protein, 22 compounds were found to possess direct binding with successfully fitted binding curves. Among them, Echinacoside and Butein showed the most potent binding affinity with UBE2J1 protein, with their KD values 1.49 ± 0.16 and 1.62 ± 0.18 μmol·L−1, respectively (Fig. 7a–c). Several other TCM active compounds also showed noticeable binding affinities less than 5 μmol·L−1, such as Forsythoside E, Ginsenoside F3, Icaritin, Orientin, and Piceatannol (Supplementary Fig. S1 and Supplementary Table S4). TCM active compounds from different sources and various skeletons have strong binding affinity with UBE2J1 proteins, suggesting that it is a potential new target with ideal binding pockets which can be effectively regulated by exogenous small-molecular compounds.
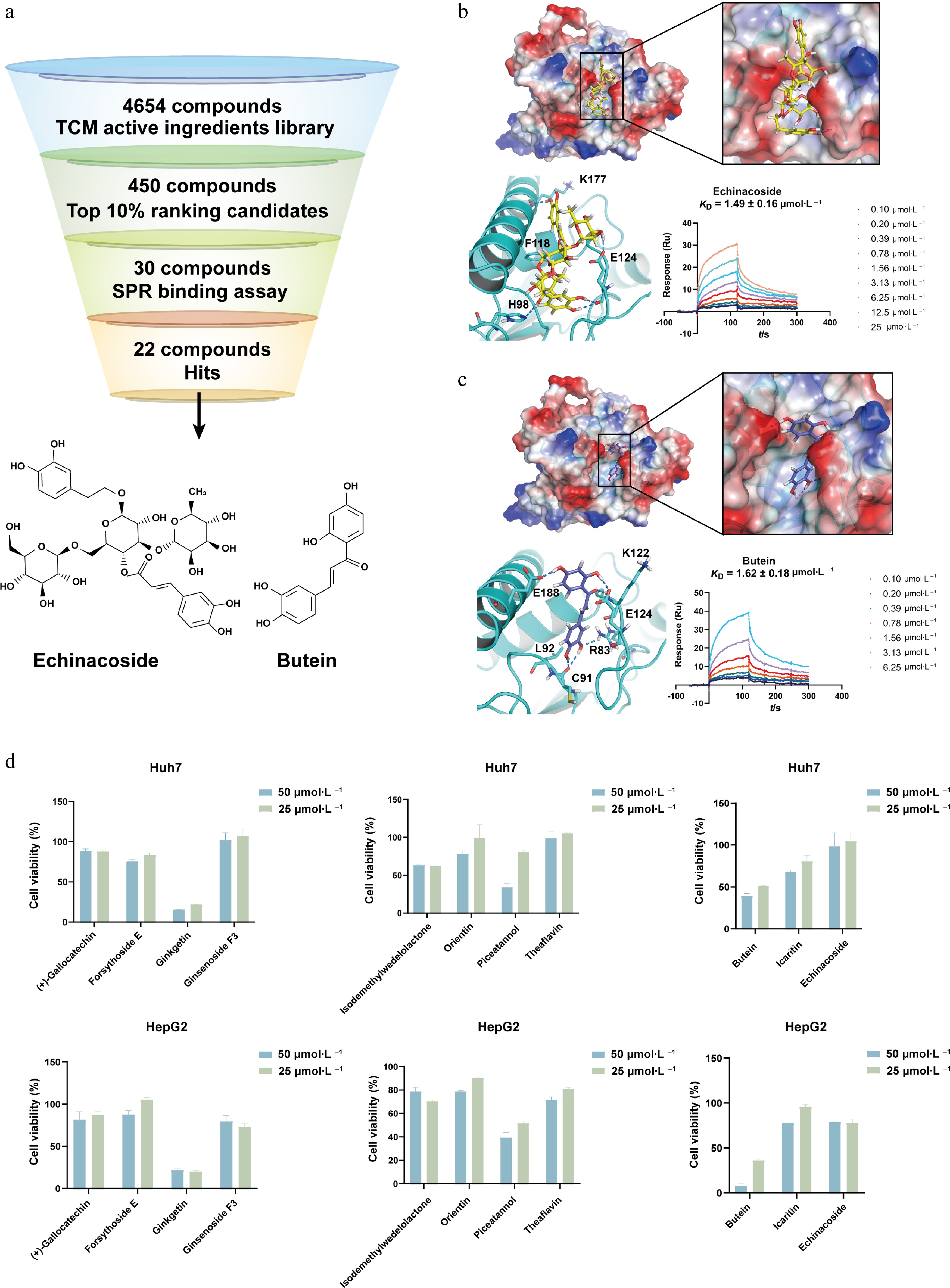
Figure 7.
Virtual screening and cellular activity assessment of UBE2J1-binding compounds. (a) Workflow for compounds virtual screening targeting UBE2J1. A total of 4,654 TCM compounds from a library were used for virtual screening. Top-ranking 10% candidates (about 450 compounds) were selected, and further screening by chemical structure diversity yielded 30 candidates for SPR binding detection. Finally, 22 compounds were identified as hits, exhibiting detectable direct binding to the UBE2J1 protein. The chemical structures of the compounds with the most potent binding affinities, Echinacoside and Butein, are shown below the workflow diagram. Molecular docking and SPR binding detection of Echinacoside and Butein. Electrostatic surface representations show the predicted binding pockets of UBE2J1 with (b) Echinacoside, and (c) Butein, respectively. Zoomed views illustrate predicted binding modes of Echinacoside and Butein, respectively. The measured KD values by SPR are 1.49 ± 0.16 μmol·L−1 for Echinacoside, and 1.62 ± 0.18 μmol·L−1 for Butein, respectively. (d) Measurement of the cell inhibition effect of UBE2J1 binding compound on the proliferation of Huh7 and HepG2 cells. The data are expressed as mean ± standard deviation (n = 3).
Compounds targeting UBE2J1 suppress cell proliferation of HCC
-
The compounds with a binding affinity less than 5 μmol·L−1 to UBE2J1 were subjected to an inhibition assessment against the proliferation of HCC Huh7 and HepG2 cells using the CCK-8 assay. The results showed that most compounds exhibited inhibitions against the proliferation of HepG2 cells, with Butein demonstrating the most potent activity (Fig. 7d). Compared with other compounds, treatment with 50 μmol·L−1 Butein for 48 h markedly suppressed HepG2 cell viability to below 10%. For Huh7 cells, Butein treatment achieved an inhibition rate of approximately 40%. The cellular inhibition of Echinacoside is less than expected, probably because it has too many phenolic hydroxyl groups and too large a molecular weight which makes it difficult to cross the cell membrane.
-
Despite major advances in high-throughput multi-omics technologies, which provide a comprehensive view of intrinsic molecular landscapes underlying disease states, translating these complex datasets into a small number of biologically meaningful and therapeutically actionable targets remains a significant challenge. Our previously reported tool, OTTM, focuses on differentially expressed proteins with approved or clinical drugs, enabling efficient drug repositioning. However, a substantial proportion of differentially expressed genes or proteins identified from omics data lack known active compounds, limiting the applicability of such approaches in discovering truly novel targets[13−16].
In this context, OPTAR was developed to prioritize potentially druggable, yet previously unexplored target proteins from omics data. Compared with conventional target prioritization approaches, which often rely on direct statistical associations or known disease annotations, OPTAR adopts a complementary strategy. Specifically, it excludes proteins with prior reported associations to the disease of interest, and instead infers disease relevance based on the literature evidence of their interacting partners. This design aims to balance the requirement for novelty with the need for biological plausibility. In addition, OPTAR incorporates a binding pocket assessment module based on AlphaFold-predicted protein structures, providing an additional layer of evaluation for the potential druggability of candidate targets.
Applying OPTAR to the omics data from clinical samples of liver cancer, three proteins UBE2J1, KDELR3, and VTI1A were identified and subsequently validated using siRNA knockdown cellular assays, supporting their potential functional relevance in this disease context. Finally, we have chosen UBE21J for virtual screening and identified potent candidate inhibitors, suggesting that UBE2J1 protein may possess suitable binding pockets for small-molecule compounds. Although the other two proteins, KDELR3 and VTI1A, did not enter the stage of compound screening, it can be seen that the proteins selected by OPTAR have a high success rate in the cellular experiments verifying their phenotypes. Once the expression and purification difficulties of these two proteins are resolved, it is highly likely to discover their first-in-class active compounds via large-scale screening. While these findings support the feasibility of the OPTAR framework, they should be interpreted with appropriate caution, as further validation, particularly in vivo studies, will be required to establish therapeutic relevance.
Existing target prediction methods are largely data-driven and tend to prioritize genes based on their direct association with disease-related datasets. While effective, such approaches may favor well-characterized targets and potentially overlook less-studied proteins[17,18]. By contrast, OPTAR is designed to explore candidate targets lacking direct literature evidence but supported indirectly through interaction networks. This strategy may provide an alternative perspective for identifying novel targets, although its performance depends on the completeness and accuracy of current PPI data and literature resources[19−21]. Assuming that proteins exert their effects through synergy rather than acting alone, OPTAR deliberately starts from candidate targets lacking literature-supported associations, it then infers disease correlation via literature mining on IPs. Thus, OPTAR can balance the originality expectation of target discovery and the success rate of subsequent experimental verification.
The binding pocket assessment module in OPTAR introduces a structure-based consideration into the target prioritization process. Proteins predicted to contain suitable binding pockets may be more amenable to subsequent compound discovery efforts, whether high-throughput virtual screening or high-throughput experimental screening is carried out. Because the goal of OPTAR is not only to discover new proteins that are truly related to disease progression, but also to discover corresponding active compounds that are expected to be developed into approved drugs in the future. The introduction of this new algorithm in OPTAR is expected to eventually increase the success rate of discovering active inhibitors or agonists in subsequent compound screening. However, the accuracy of such predictions relies on the quality of structural models and the assumptions underlying pocket identification algorithms. Therefore, experimental validation remains essential to confirm compound binding and functional effects.
Despite the promising results, several limitations should be noted. OPTAR relies on literature mining of IPs; its performance is inevitably limited by the quality and integrity of current PPI knowledge. As the PubMed baseline dataset is updated annually, OPTAR is also updated annually. Thus, the candidate proteins recommended by OPTAR for subsequent experimental validation should be manually checked, confirming the lack of reported evidence between these proteins and the disease of interest. Moreover, since our experimental validation currently lacks assessment of compound efficacy in animal models, in vivo studies will be required to further confirm the therapeutic efficacy of targeting UBE2J1 for the treatment of HCC.
In summary, OPTAR, together with OTTM, provides a complementary framework for target discovery from omics data, addressing both drug repurposing and de novo target identification. The successful identification of UBE2J1 as a potential target protein against the proliferation of HCC demonstrates the reliability of OPTAR as an efficient computational tool for the transformation from omics data into promising druggable targets. Future studies integrating additional data sources and experimental validation are important to further evaluate and refine this framework. Then OPTAR is expected to be widely used in the discovery of first-in-class therapeutic target proteins from omics data.
We gratefully acknowledge the financial supports from the National Key R&D Program of China (NO. 2022YFC3400500), the Strategic Priority Research Program of the Chinese Academy of Sciences (NO. XDB0830301), the National Natural Science Foundation of China (NO. 81903538), the Shanghai Municipal Health Commission Medical New Technology Project (NO. 2025ZZ2060), Shanghai Municipal Education Commission AI for Science Project (NO. 301-0406), Shanghai Oriental Talent Plan Youth Project (NO. QNJY2025170), and Science and Technology Department of Guizhou Province (grant number [2024]015).
-
Not applicable. The present study included only in vitro cell experiments and did not involve human subjects, clinical samples, or animal experiments.
-
The authors confirm their contributions to the work as follows: cell-based experiments and manuscript writing: Yuan X; figure preparation: Zhou S; compound screening: Yu J; reference collection and organization: Wang M; manuscript checking and revision: Luo C; development of the computational tool and the writing of the corresponding methodological and related content: Zhang H. All authors reviewed the results and approved the final version of the manuscript.
-
The data generated or analyzed during this study are included in this published article and its supplementary information files. Additional data related to this study are available from the corresponding author upon reasonable request.
-
The authors declare that they have no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/targetome-0026-0017.
-
#Authors contributed equally: Xiao Yuan, Siyu Zhou, Jiayi Yu
- Supplementary Table S1 List of siRNA used for transfection.
- Supplementary Table S2 Primers for RT-qPCR.
- Supplementary Table S3 OPTAR output protein list.
- Supplementary Table S4 List of 30 compounds for UBE2J1 selected for binding detection via virtual screening.
- Supplementary Fig. S1 SPR binding curves and fitted KD values for 20 UBE2J1-binding compounds identified by virtual screening.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yuan X, Zhou S, Yu J, Wang M, Luo C, et al. 2026. OPTAR: a computational tool for target discovery based on disease correlation inference from literature of interacting proteins. Targetome 2(2): e018 doi: 10.48130/targetome-0026-0017 

OPTAR: a computational tool for target discovery based on disease correlation inference from literature of interacting proteins
- Received: 06 March 2026
- Revised: 17 April 2026
- Accepted: 20 April 2026
- Published online: 30 April 2026
Abstract: The identification of novel therapeutic targets remains a major bottleneck in target-based drug discovery, particularly when mining large-scale omics data. Although transcriptomic and proteomic profiling generate extensive lists of disease-associated candidates, prioritizing truly novel and druggable targets, especially those lacking active compounds, requires the assistance of advanced computational strategies. Here, we report the development of a new computational tool named OPTAR (Omics and Pocket Analysis-based Target Assessment and Ranking) for identifying promising new target proteins from omics data. These new target proteins are expected to have no active compounds, and have not previously been reported to be correlated to the disease of interest. OPTAR applies a multi-layer filtering and ranking workflow, including automated literature-based exclusion of known disease-associated proteins, drug availability screening, and algorithm-driven disease correlation inference, enabling systematic de novo target discovery. From the hepatocellular carcinoma (HCC) omics data used by the previously reported tool OTTM (Omics and Text-driven Translational Medicine), OPTAR identified high-ranking candidate proteins at the intersection of 'hepatocellular carcinoma' and 'cell cycle' lists. Functional verification indicated that silencing of UBE2J1, KDELR3, and VTI1A in HCC cells inhibited cell viability and reduced the migration and invasion abilities of HCC cells. Furthermore, it was found that UBE2J1 was upregulated in HCC tissues, and its knockdown induced apoptosis-related changes, and cell cycle disorder. Together, these findings establish OPTAR as a reliable and efficient computational tool for promising therapeutic target discovery with high originality from omics data.