









-
Metabolites play crucial roles in various biological processes in physiology or pathology. As the main natural small-molecule compounds in prokaryotes and eukaryotes, metabolites have a high degree of structural and functional diversity[1,2]. With the development of metabolomics, a large number of novel metabolites have been discovered, and their biological functions are constantly being re-annotated and expanded[1,3]. However, compared with the mature studies of protein-protein and protein-DNA interactions, the systematic identification of metabolite-protein interactions is still relatively insufficient. This gap greatly limits our understanding of metabolite functions.
In recent years, chemical proteomics has become a key technology to systematically and accurately reveal metabolite-protein interactions. Thanks to continuous breakthroughs in mass spectrometry technology, new chemo-proteomic methods have been developed for identifying these interactions[4]. These methods use labeled or unlabeled metabolites to capture their binding proteins, which greatly facilitates the discovery of new functions of classical metabolites. For example, the application of classical neurotransmitters outside the central nervous system has been gradually confirmed[5−8]. Chemical proteomics methods have constructed the metabolite-protein interactome, showing that metabolites can target multiple proteins and produce global effects, or only act on a few proteins and produce restricted effects[9,10]. Although most chemical proteomics technologies belong to biophysical methods, the means to directly correlate metabolite-protein interactions with specific cellular phenotypes are still limited[11]. In addition, the concentration, localization, and activity of metabolites may change significantly under different nutritional states and disease conditions[12]. Therefore, it is urgent to develop new strategies to integrate multi-source biomedical information, such as cellular localization, concentration, functional pathways, and phenotypes, so as to promote the accurate identification of functional targets for endogenous metabolites.
Previously, we developed a computational tool called OTTER for identifying natural product targets with known pharmacological activities[13]. However, endogenous metabolites face more challenges in target identification: the binding affinity between endogenous metabolites and proteins varies greatly, and there are more biological factors to be taken into account, such as disease relevance and subcellular localization. To address these challenges, we propose a hypothesis: if an endogenous metabolite is significantly associated with or consistent with a protein at the level of multidimensional biological characteristics, the protein is likely to be a potential target of the metabolite. In other words, endogenous metabolites should have a consistent 'fingerprint' with their protein targets. Based on this assumption, we report the development of a new method based on bioinformatics and deep learning called DeepETD (Deep learning-based Endogenous metabolites Target Discovery). This method aims to improve the identification ability of functional targets of endogenous metabolites through advanced data integration and deep learning technology.
Compared with traditional compound-protein interaction prediction methods[14,15], DeepETD fully utilizes comprehensive biomedical data. The model integrates multi-source biomedical information and represents the biological characteristics of metabolites and proteins (including subcellular localization, cellular phenotype, and disease association information[16,17]) as unified feature fingerprints. Subsequently, the model uses a deep learning algorithm with an attention mechanism to process high-dimensional and noisy data[18]. The attention mechanism assigns weights to the inputs from different data sources, which significantly improves the prediction performance of metabolite-protein interactions. All prediction results are stored in the web server named EMTDD (Endogenous Metabolites Target Discovery Database). Users can easily query the potential target information of endogenous metabolites predicted by DeepETD.
-
A dataset containing positive and negative samples of endogenous metabolite-protein interactions was constructed using a binding affinity cutoff strategy. Endogenous metabolite protein interaction pairs with an IC50 value of ≤ 100 nmol·L−1 were designated as positive samples, whereas those with an IC50 value of > 100 nmol·L−1 were considered as negative samples. The multidimensional biological information used to build the dataset was collected from multiple authoritative databases and literature resources, including the Human Metabolome Database (HMDB)[19], the IUPHAR/BPS Guide to PHARMACOLOGY, and the BindingDB database[20]. Disease association information and cellular phenotype data were integrated from the Disease Ontology (DO) database and the Human Phenotype Ontology (HPO) database[21,22]. To enrich the dataset, this study systematically searched all PubMed abstracts to extract the cellular localization, associated diseases, and cellular phenotype information of each metabolite and protein.
The keywords of cellular localization are selected from the commonly used subcellular localization terms in the literature. For each entity, the top five cellular localization terms, the top ten associated diseases, and the top five cellular phenotypes were screened according to the occurrence frequency, and the dataset was constructed based on these characteristics. Each sample contains characteristic information of metabolites and proteins, specifically covering their cellular localization, associated diseases, and cellular phenotypes. After the positive and negative samples are combined, they are divided into a training set and a validation set according to the proportions of 80% and 20%. All features were converted into numerical feature vectors using either one-hot encoding or embedding layers.
Construction of biological feature fingerprints
-
For each metabolite and protein, three core biological characteristics are defined: associated disease types, cellular phenotypes, and subcellular localization. Feature extraction is based on large-scale text mining technology by calculating the co-occurrence frequency of specific biological terms (diseases, phenotypes, etc.) and target objects (metabolites or proteins) in PubMed abstracts. If the two frequently co-exist in the same abstract, they are considered to be highly relevant. Subsequently, these co-occurrence frequencies are quantified and encoded as high-dimensional feature fingerprints, which are used as model inputs to characterize the potential association between metabolites and proteins in a multidimensional biological context.
Model architecture
-
An attention-based neural network model was developed to predict the interaction between endogenous metabolites and proteins. The model comprises the following layers. Embedding layer: The discrete input features (diseases, phenotypes, and subcellular localizations) are mapped into continuous vector representations through the embedding layer. The embedding dimensions of disease, phenotype, and subcellular localization were set to 32, 16, and 16. The model sets two embedding layers for the input features of metabolites and proteins, respectively. These embedding layers map the original features to the low-dimensional space, reducing the dimensions to 64, 32, and 16 in turn. Attention mechanism: To optimize the feature weights, an independent attention layer is introduced after the embedding layer. This layer contains two fully connected layers and a Tanh activation function for outputting attention weights. By using these attention weights to weigh and sum the embedding vectors, the global representation of protein features is obtained. Feature concatenation layer: The feature representations of metabolites and proteins are concatenated to form a comprehensive feature vector.
Deep neural network: The combination vector is processed through two fully connected layers with hidden layer sizes of 256 and 128, respectively. To prevent overfitting, the LeakyReLU activation function and a dropout layer with a rate of 0.3 were added between the fully connected layers to ensure the generalization performance of the model when dealing with unseen data. Finally, the features are mapped to a scalar through an additional fully connected layer, and the sigmoid activation function is applied to output the predicted probability of the interaction.
Model training and optimization
-
The model was trained using the binary cross-entropy loss function. The Adam optimizer was employed to update the parameters. The learning rate was set to 0.001. Training was conducted over 20 epochs. Each epoch includes forward propagation to calculate the loss and backpropagation to update the parameters. To prevent overfitting and improve the generalization ability of the model, the early stopping strategy was adopted. If the area under the receiver operating characteristic curve (AUC-ROC) on the validation set did not improve for 10 consecutive epochs, the training was stopped, and the best-performing model was retained. The AUC-ROC value and accuracy were used as evaluation metrics in both the training and validation stages. When the validation set reached the optimal AUC-ROC value, the model parameters were saved for subsequent testing. Ten-fold cross-validation was used to evaluate the generalization ability and robustness of the model on different data sets. To ensure the reproducibility of the results, the random seed was set to 42 at the beginning of training, and the randomness control was maintained during data loading, model initialization, and training. The experiment was conducted in a computing environment equipped with NVIDIA GPUs and implemented based on the PyTorch deep learning framework.
Disease-context-aware target prediction
-
For each metabolite, disease-associated contexts were identified according to the co-occurrence frequency between metabolites and disease terms extracted from PubMed abstracts. The top 10 most frequently associated diseases were selected as the disease contexts for prediction. Under each disease context, candidate proteins were filtered to retain only proteins associated with the corresponding disease. Metabolite-protein pairs were then input into DeepETD for interaction prediction, and the predicted interaction probabilities were used as the final interaction scores under the corresponding disease context.
Web-server of the endogenous metabolites target discovery database (EMTDD)
-
Using the optimized DeepETD model, potential targets for 3,382 endogenous metabolites recorded in the Human Metabolome Database (HMDB) were predicted across 10 highly relevant diseases. Relevant data were used to build a publicly accessible web server called EMTDD. The server is available at
http://otter-simm.com/EM/EMTDD.html .Microscale Thermophoresis (MST)
-
MST experiments were performed using a Monolith NT.115 (Blue/Red) instrument (NanoTemper Technologies GmbH, Germany). The target protein with an EGFP fluorescent label was overexpressed in HEK293T cells, and cell lysates were used for the experiment. HEK293T cells were transfected with plasmids using EZ Trans Lipo (Life-iLab) and lysed 48 h after transfection. A 5 μL cell lysate was mixed with 5 μL of endogenous metabolites (Testosterone, Nature-Standard, ST05380100; Leukotriene B4, MedChemExpress, HY-107608) at different concentrations. Then the samples were incubated for 10 min at room temperature. Using nanoblue excitation, the MST power was set to medium. The fitting was performed using the Kd model method incorporated by the MO Affinity Analysis v2.3 software (NanoTemper Technologies).
Molecular docking
-
Molecular docking was performed using Schrödinger and displayed by PyMOL. The reported ACAT1 crystal structure (PDB ID: 2IBY) and the AlphaFold-predicted structures of PRMT2 and NR2F6 were used as the initial conformation, and the ProteinPreparation module was used to optimize the protein. After all missing hydrogen atoms were added, binding sites were detected using the SiteMap module. Testosterone and Leukotriene B4 were prepared using the LigPrep module, and the Standard Precision docking mode was selected. The docking result with the highest score was chosen to generate a visual 3D structure diagram using Pymol software.
-
Target identification of endogenous metabolites is crucial for understanding their roles in physiological and pathological processes, but due to the complexity of bioinformatics data, this field still faces many challenges. To address this, we propose a core hypothesis: an endogenous metabolite has consistent multidimensional biological characteristics with its protein target. Therefore, proteins that are highly similar to a given metabolite in these dimensions are likely to be a functional target (Fig. 1a). In other words, endogenous metabolites and their corresponding protein targets have similar 'fingerprints'. When considering describing a metabolite or a target protein biologically, three questions were commonly asked: (1) where is it located; (2) which cellular process, mechanism, or phenotype is regulated; and (3) what kind of disease is correlated. The three questions correspond to three levels of biological features from subcellular levels to tissue levels, which have biological connections. Therefore, we basically incorporated three main features for each sample, which are subcellular locations, cellular phenotypes, and correlated disease types. The process is known as fingerprint matching (Fig. 1b).
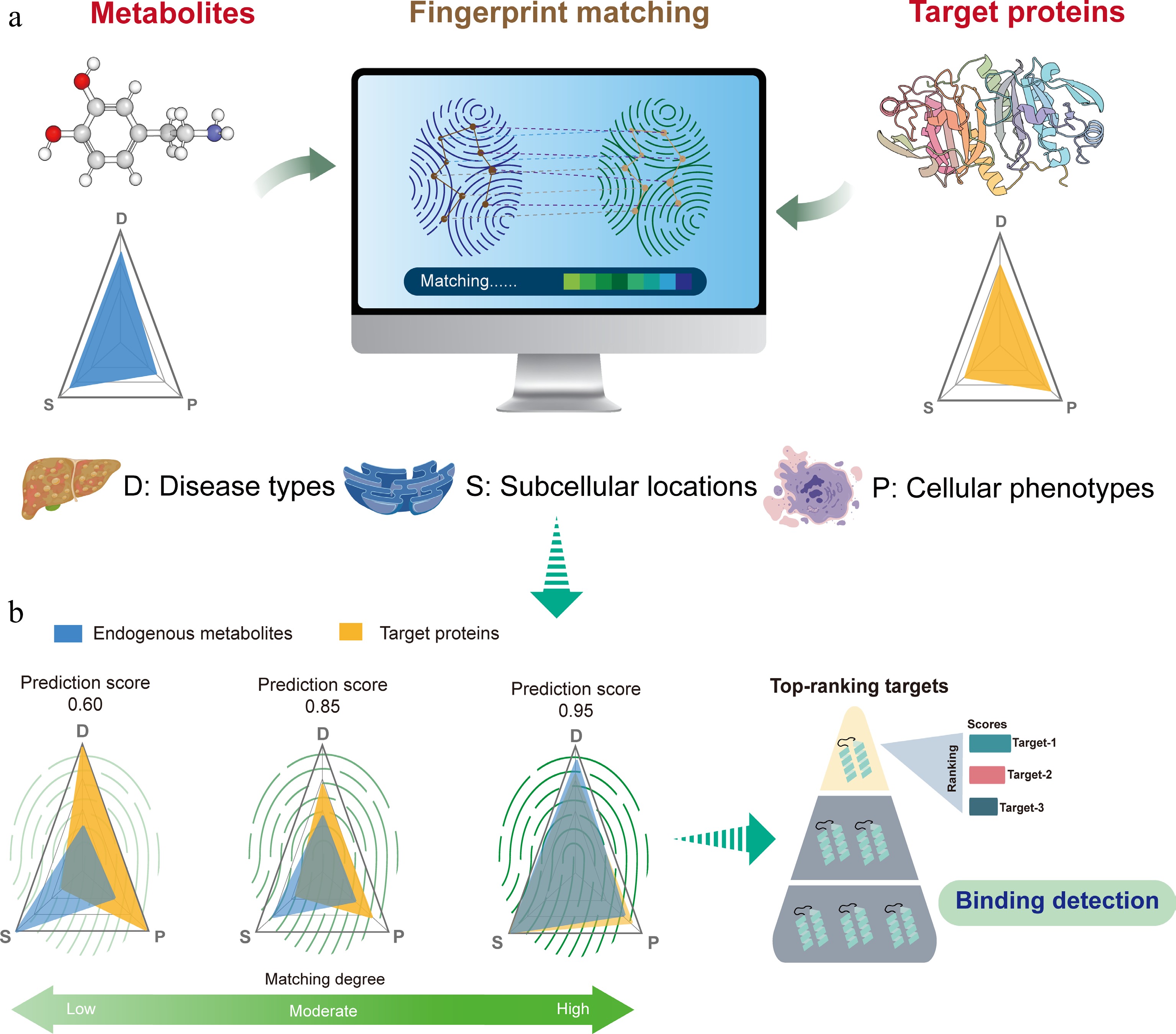
Figure 1.
Hypothesis of biological similarities of metabolites and its potential targets guides the development of DeepETD. (a) Biologically similar features were demonstrated: if an endogenous metabolite exhibits significant correlation or consistency with a certain protein at the multidimensional biological feature level (associated diseases, cellular phenotypes, and subcellular localization), then this protein may serve as a potential target for the metabolite. (b) The match of biologically similar features guides the development of DeepETD: the model represents the biological characteristics of metabolites and proteins as unified characteristic fingerprints; the potential association/binding probability (fingerprint matching) between metabolites and proteins was quantified through deep learning.
PubMed-based feature fingerprint construction and dataset generation
-
A binding affinity cutoff strategy was applied to construct the dataset. To generate the positive and negative samples for our model, protein-metabolite pairs with an IC50 value equal to or less than 100 nmol·L−1 were defined as positive samples, whereas those with an IC50 value greater than 100 nmol·L−1 were considered as negative samples (Fig. 2a). By focusing on high-affinity interactions, this strategy improves the reliability and biological significance of the positive samples used for model construction. Interaction data were sourced from the Human Metabolome Database (HMDB)[19], the IUPHAR/BPS Guide to PHARMACOLOGY (GtoPdb), the ChEMBL[23], and the BindingDB[20]. Overall, the dataset contains a total of 15,872 samples, including 2,565 positive samples and 13,307 negative samples. The final dataset was randomly divided into a training set and a validation set in a ratio of 4:1 to ensure the reliability of model evaluation.
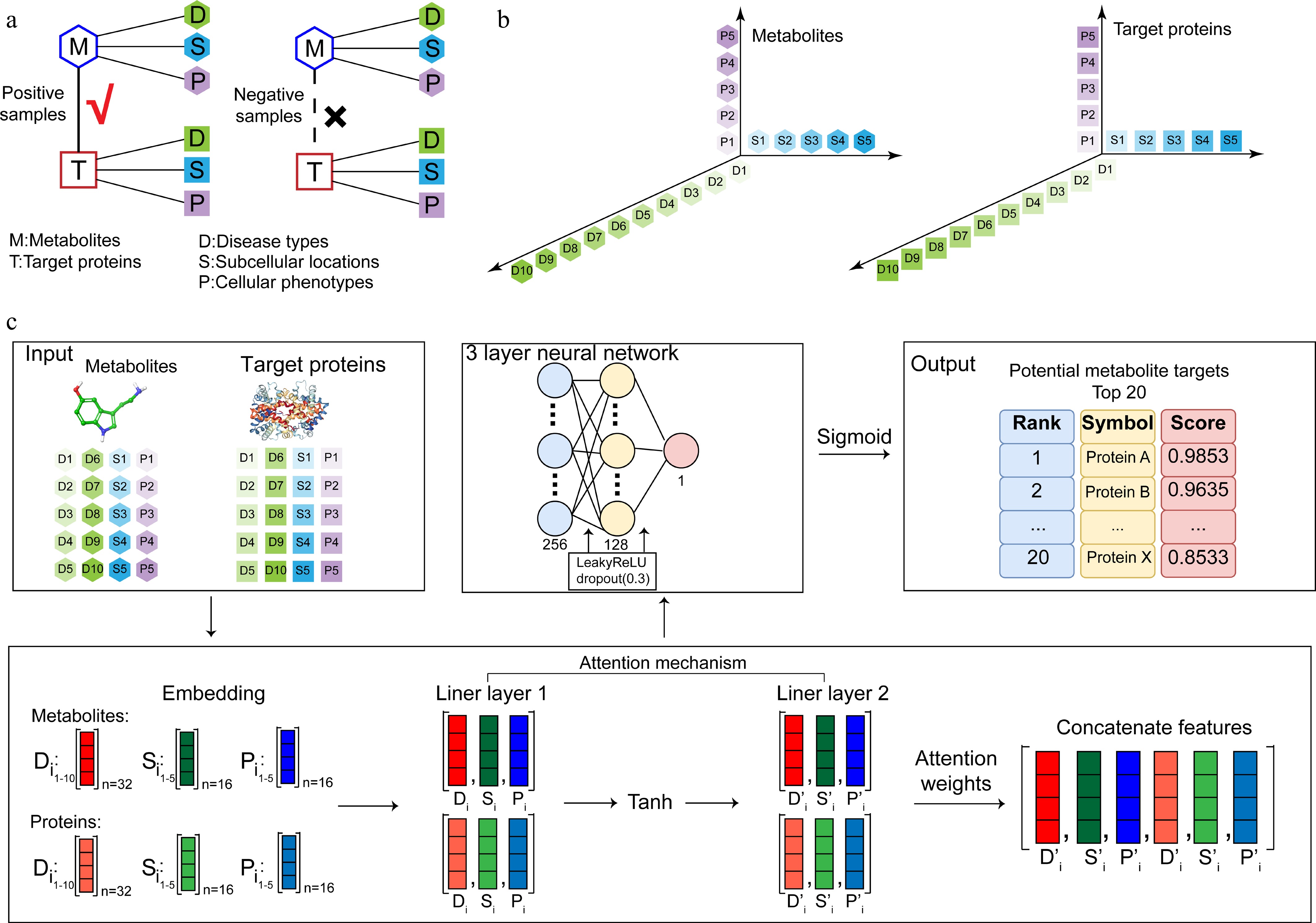
Figure 2.
Dataset construction and model workflow of DeepETD. (a) Training dataset construction: a dataset containing positive and negative samples of interactions between endogenous metabolites and target proteins. (b) Multidimensional feature matrix representation of metabolites and target proteins. (c) Model design: the input features (associated diseases, cellular phenotypes, and subcellular localization) are encoded through the embedding layer. The encoded features are processed by the attention mechanism, followed by the feature concatenation layer. The model employs a fully connected layer to output the potential binding probability scores between endogenous metabolites and target proteins, and generates a list of the top 20 potential target proteins for subsequent experimental verification.
These computational inputs are derived from the transformation of biomedical texts. We defined three core biological characteristics of metabolites and proteins: disease types, cellular phenotypes, and subcellular localization. Feature extraction was mainly based on the co-occurrence analysis of PubMed abstracts: if a biological term (such as a specific disease or phenotype) and a metabolite or protein name frequently appear in the same abstract, the feature is judged to be highly related to the object. By constructing multidimensional fingerprints, this approach provides rich biological information for DeepETD, enabling it to accurately predict potential target proteins on a whole-proteome scale (Fig. 2b).
Framework of the DeepETD model
-
The model architecture of DeepETD is constructed to efficiently process high-dimensional and noisy data of metabolites and their potential targets to output highly accurate target prediction results. Its core components include an embedding layer, an attention mechanism layer, a feature concatenation layer, and a deep neural network (Fig. 2c). Specifically, the model first encodes the input disease, phenotype, and subcellular localization features through the embedding layer. We set the embedding dimension of disease features to 32 and the embedding dimension of phenotypic and subcellular localization features to 16.
To enhance the ability of the model to capture key biological information, we introduced an independent attention mechanism after the embedding layer. The attention layer calculates the attention weights through a fully connected layer and a Tanh activation function, and uses the Softmax function to normalize. This process gives different weights to features of different dimensions. This mechanism enables the model to automatically focus on the features that are most relevant to the interaction prediction, thus effectively filtering the noise.
After attention weighting, the metabolite features and protein features are concatenated into a comprehensive feature vector. This vector is then input into a deep neural network consisting of three fully connected layers. The leaky ReLU activation function is used for nonlinear transformation between network layers, and Dropout regularization is introduced to prevent overfitting. The Dropout rate is set to 0.3 to improve the generalization ability of the model. Finally, the model outputs a probability score between 0 and 1 through the sigmoid function, realizing the quantitative prediction of the likelihood of metabolite-protein interaction.
DeepETD is effective for target prediction of metabolites
-
To evaluate the prediction performance of DeepETD, we tested the model using the constructed training dataset. The prediction accuracy of the model was 0.9274 on the training set and 0.8249 on the validation set. These results show that the proportion of true positives and true negatives is high, and there is strong overall accuracy. The area under the receiver operating characteristic curve (AUC-ROC) values for the training set and the validation set reached 0.9771 and 0.8470, respectively, demonstrating the robust performance of the model. Meanwhile, we analyzed the recall performance of the model to reflect the model's ability to correctly identify positive samples under imbalanced data conditions. The model achieved a recall value of 0.6570 on the validation set. These results indicate that the model retained the ability to identify positive samples and did not exhibit severe bias toward predicting negative samples (Fig. 3a and b; Supplementary Fig. S1a). In addition, we performed 10-fold cross-validation on the endogenous metabolite protein interaction dataset. The results consistently showed high prediction accuracy, further supporting the robustness of the model (Fig. 3c).
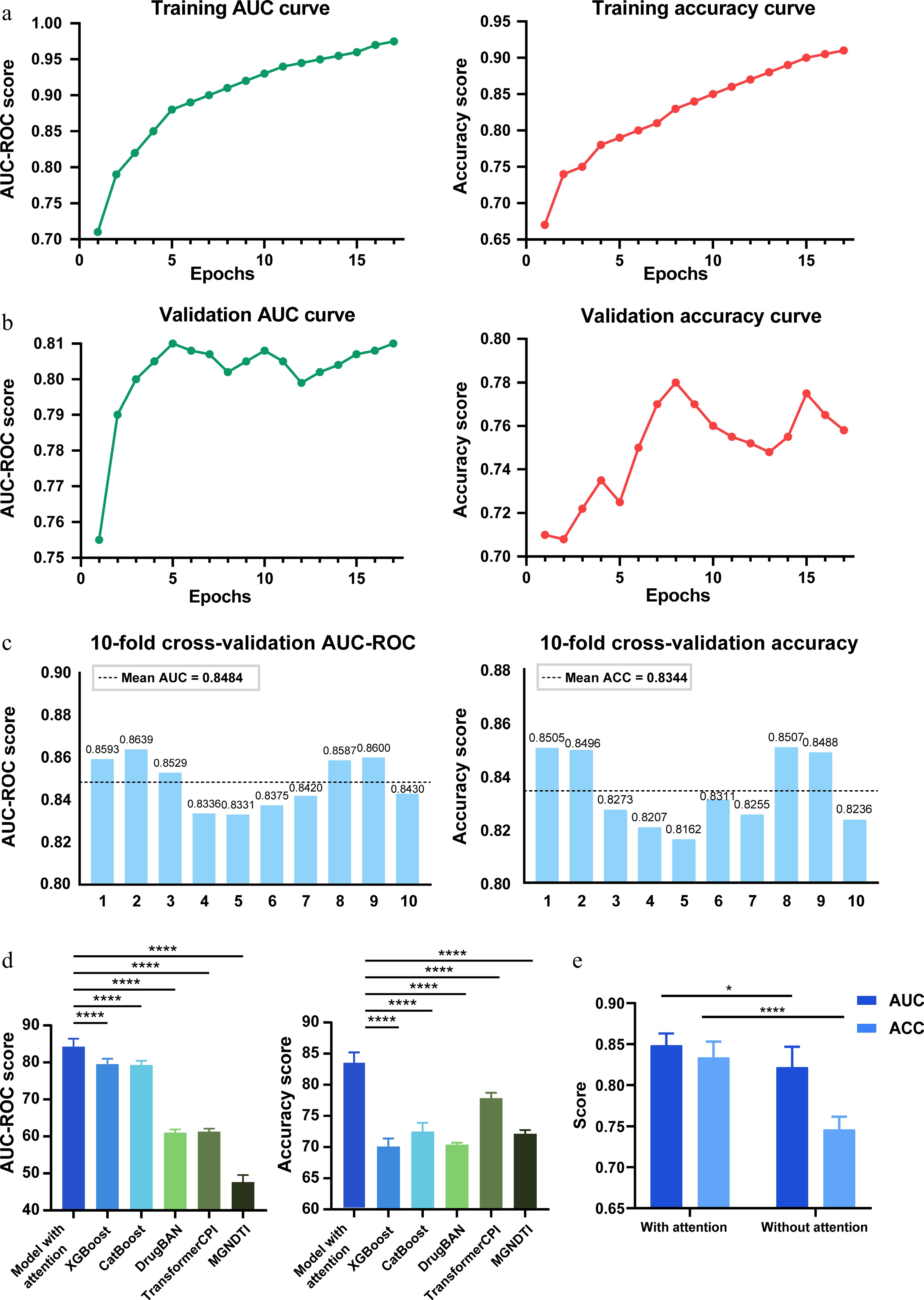
Figure 3.
Model training and performance evaluation. (a) Performance of the DeepETD model on the training set. The AUC-ROC curve and accuracy results demonstrate the predictive capability of DeepETD. (b) Performance of the DeepETD model on the validation set. (c) Evaluation of model stability using 10-fold cross-validation. Most AUC values fall within the range of 0.80 to 0.88, with an average of 0.8484 and a standard deviation of 0.0137 Similarly, most accuracy values lie between 0.80 and 0.86, with a mean of 0.8344 and an approximate standard deviation of 0.0145. (d) Performance comparison of DeepETD with baseline models (XGBoost/CatBoost) and deep learning models (DrugBAN/TransformerCPI/MGNDTI). DeepETD showed better predictive performance than the other models. **** P < 0.0001; one-way ANOVA. (e) An ablation study was conducted to evaluate the contribution of the attention mechanism to model performance. Results from models with and without the attention layer demonstrate that the attention mechanism significantly improves prediction accuracy. **** P < 0.0001, * P < 0.05; one-way ANOVA. Data are presented as the mean ± SEM, n = 5.
To verify the necessity of the attention mechanism, we conducted ablation experiments, and the comparison results showed that, compared with the model without the attention mechanism, the full version of the DeepETD model significantly improved the AUC-ROC and accuracy. This finding shows that when dealing with multidimensional biomedical data, assigning weights to different data dimensions through the attention mechanism is crucial for predicting the results (Fig. 3e). To further explore which biological features were mainly utilized by the attention mechanism, we conducted additional ablation analyses by individually removing disease, phenotype, and subcellular localization features from the full model. The result showed that removing disease-related features caused the largest decrease in model performance. Phenotype or subcellular localization features led to a relatively smaller impact when removed (Supplementary Fig. S1b). These findings suggest that disease-associated biological features contribute most to metabolite-protein interaction prediction.
We further compared the DeepETD model with traditional machine learning methods, including XGBoost and CatBoost, as well as several recent deep learning-based DTI/CPI prediction models, including DrugBAN, TransformerCPI, and MGNDT. The comparison results showed that DeepETD using the attention mechanism achieved higher AUC and accuracy. In particular, DeepETD exhibited stronger capability in distinguishing positive from negative metabolite-target interactions, suggesting improved generalization performance for endogenous metabolite-target prediction tasks. This result verifies that the DeepETD framework is more effective and applicable in predicting metabolite targets compared with both conventional machine learning approaches and representative deep learning frameworks (Fig. 3e).
Overall, the DeepETD model shows strong accuracy and reliability in metabolite target prediction. By introducing the attention mechanism, the model can automatically focus on key predictive features, thus improving the prediction performance of the model and the interpretability of the results.
The enrichment of disease-related targets of well-known metabolites by DeepETD
-
We performed a Top-K enrichment analysis to evaluate the global target prioritization capability of DeepETD. All metabolite-protein candidate pairs in the validation set were ranked according to their prediction scores, and the overall positive interaction rate in the validation set was 16.16% (513/3,175). The top-10 predictions achieved a Precision@10 of 0.92 ± 0.10 with an enrichment rate of 5.73 ± 0.77×, while the top-20 predictions achieved a Precision@20 of 0.88 ± 0.07 with an enrichment rate of 5.47 ± 0.56× (Supplementary Fig. S1c). These findings indicate that DeepETD can effectively prioritize disease-associated metabolite targets at a global level.
To further verify the accuracy of DeepETD in actual prediction scenarios, we selected two representative endogenous metabolites with clear known targets, dopamine[24,25] and estradiol[26−29], as verification cases. The results showed that, within the list of top 20 potential targets for dopamine in the context of Parkinson's disease, the model accurately identified multiple known receptors. Similarly, the validated targets of estradiol in the context of breast cancer were also precisely ranked at the top of the candidate list (Fig. 4a−f).
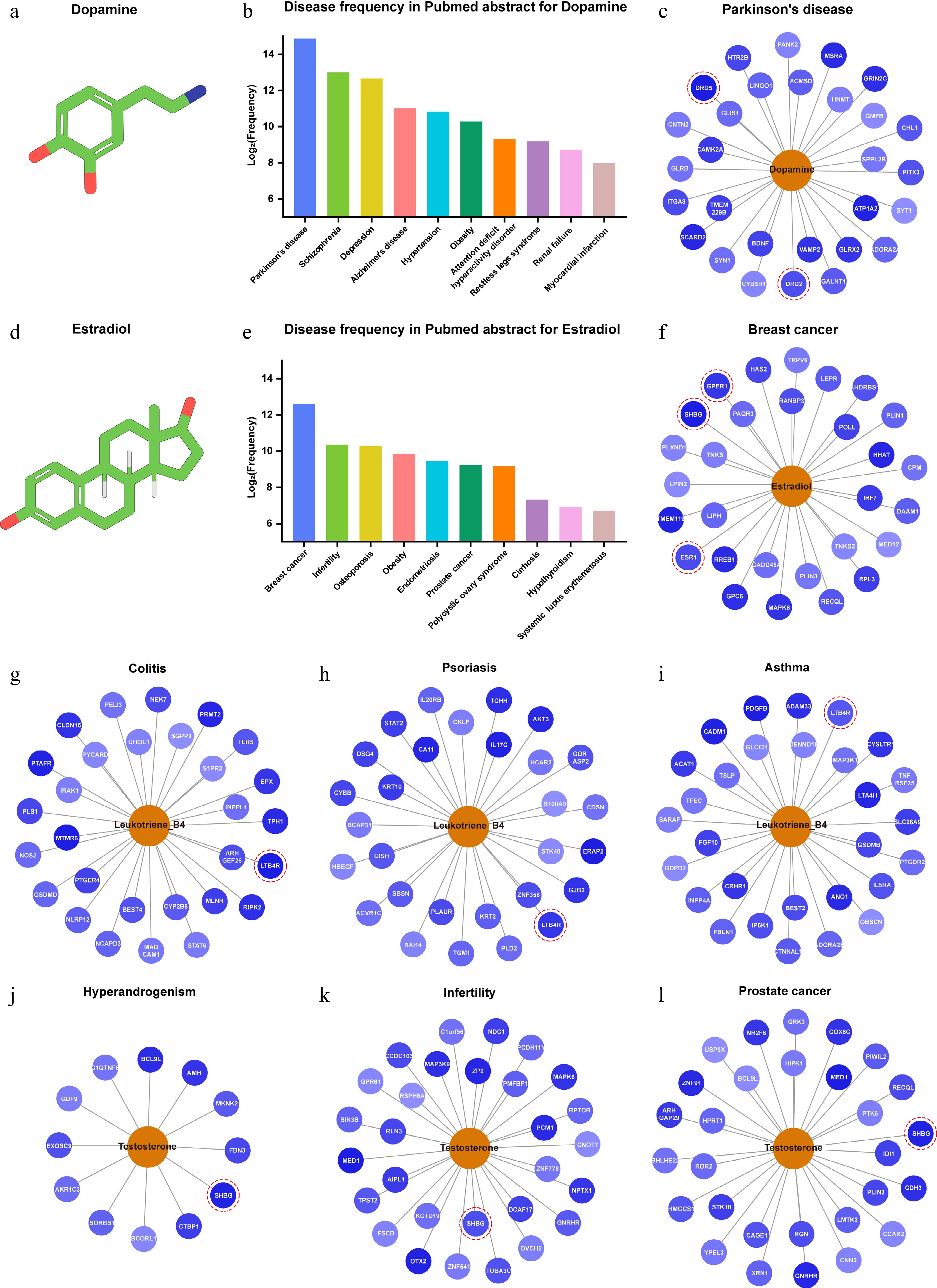
Figure 4.
DeepETD successfully enriched disease-related targets of common metabolites. The standard output format of DeepETD via divergence plot and bar graph (taking dopamine and estradiol as examples). Bar chart: displaying the top 10 associated diseases with the highest frequency of appearance in endogenous metabolite literature. Divergence plot: showing the fingerprint matching strength between endogenous metabolites and their top 20 predicted target proteins. (a) Chemical structure of dopamine. (b) Co-occurrence frequency of dopamine-related diseases in PubMed abstracts (top 10). (c) Successful prediction of multiple known targets of dopamine in the context of Parkinson's disease: DRD2 and DRD5. (d) Chemical structure of estradiol. (e) Co-occurrence frequency of estradiol-related diseases in PubMed abstracts (top 10). (f) Successful prediction of multiple known targets of estradiol in the context of breast cancer: GPER1, SHBG, and ESR1. (g)–(l) Successful identification of verified targets for a variety of metabolites in multiple disease contexts: LTB4R and SHBG.
In addition, we conducted the target prediction of multiple metabolites in different disease contexts. DeepETD has successfully predicted multiple known and experimentally verified targets that have key regulatory roles in the disease process (Fig. 4g−l)[30−36]. This result proves that the model can not only process basic biochemical data but also accurately capture metabolite-protein associations in a complex pathological context. The successful validation on disease-related metabolites proves that DeepETD has high model sensitivity and accuracy. These results support the wider application of DeepETD in discovering new functional targets.
Construction of the endogenous metabolites target discovery database (EMTDD)
-
To better promote the target discovery of endogenous metabolites, we applied DeepETD to a large-scale target prediction task and built a comprehensive endogenous metabolite target discovery database (EMTDD).
We collected 3,382 endogenous metabolites that have been qualitatively and quantitatively characterized from the Human Metabolome Database (HMDB). Using the trained DeepETD model, we scanned and scored these metabolites on a proteome-widescale. For each metabolite, the candidate proteins are ranked based on the fingerprint matching score (interaction probability), and the top 20 potential targets are reported. At present, the database covers the potential target prediction results of 3,382 endogenous metabolites in 10 different disease contexts, with a total number of 33,820 entries (Fig. 5a). All prediction results are stored on the server and are accessible at
http://otter-simm.com/EM/EMTDD.html (Fig. 5b). Users can easily query the potential functional targets of specific metabolites through the database, and view the details of diseases, phenotypes and literature co-occurrence frequency (Fig. 5c). In the standardized output of this database, the top 10 high-frequency diseases related to metabolites are displayed in abar chart. The top 20 target proteins with the highest prediction scores and their matching intensities are visually displayed through the divergence plot (Fig. 4b, c and e, f).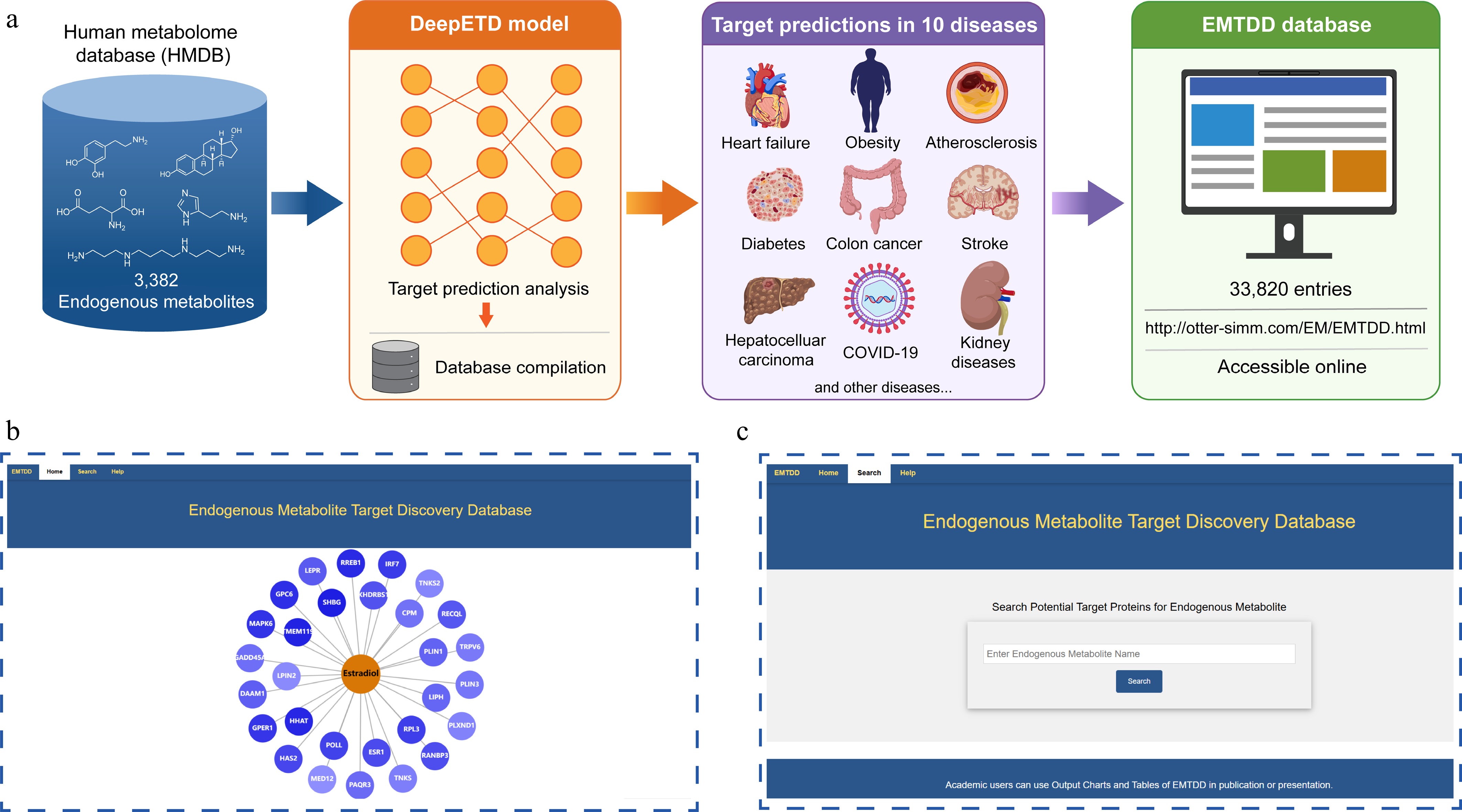
Figure 5.
Construction of the Endogenous Metabolites Target Discovery Database (EMTDD). (a) Data source: collection of 3,382 endogenous metabolites from the Human Metabolome Database (HMDB). Database component: the database encompasses 10 associated diseases with high-frequency co-occurrence of each metabolite in PubMed, along with corresponding potential target prediction results totaling 33,820 entries. (b), (c) EMTDD web interface display: the Homepage and Search function.
Compared with conventional metabolite-related databases, EMTDD performs disease-context-aware target prediction, enabling the identification of metabolite-protein interactions under the most relevant pathological conditions for each metabolite. Moreover, with the support of the DeepETD framework, EMTDD integrates disease associations, phenotypic features, and subcellular localization information to improve the biological relevance of predicted targets. For the result presentation, EMTDD provides intuitive visualization and standardized output formats. Users can conveniently interpret disease relevance and target matching intensity. All statistical graphs and corresponding datasets are freely available for download. The establishment of EMTDD provides potential data support and a computational tool for subsequent target discovery and mechanism exploration of endogenous metabolites.
Experimental validation of endogenous metabolite-target binding predicted by DeepETD
-
To evaluate the reliability of DeepETD, we selected two representative endogenous metabolites for experimental verification: testosterone and leukotriene B4 (LTB4). According to the prediction results, four candidate protein targets were selected for each metabolite. EGFP-tagged plasmids were transiently transfected into HEK293T cells, and then cell lysates were used for MST to determine whether endogenous metabolites were bound to target proteins. The positive interaction results are shown in Fig. 6, while the experimental results of candidate targets that failed to show concentration-dependent binding responses are summarized in the Supplementary Fig. S2.
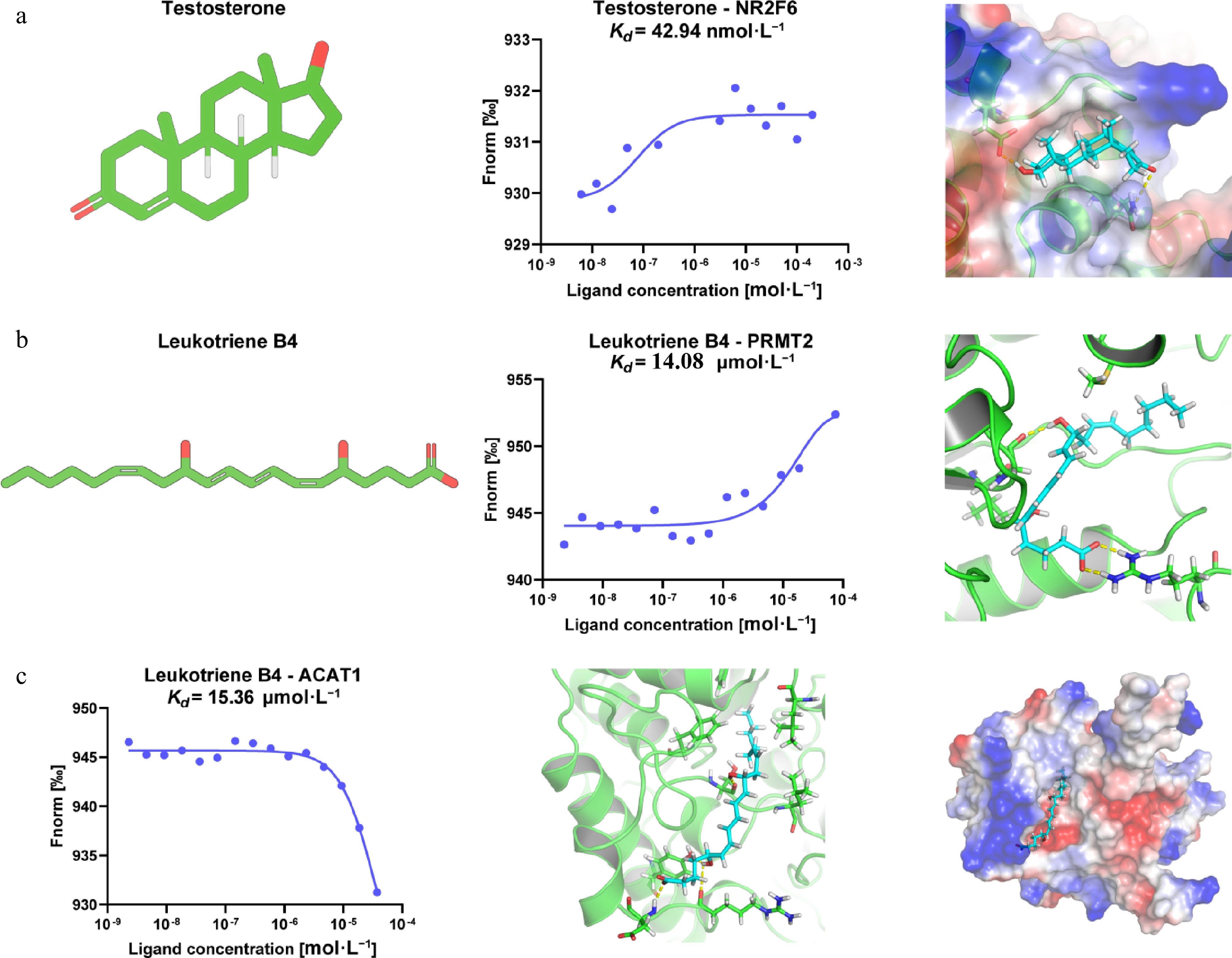
Figure 6.
Experimental validation of endogenous metabolite-target binding predicted by DeepETD. (a) Validation of the interaction between testosterone and NR2F6. Left: chemical structure of testosterone. Middle: MST binding assay demonstrating direct binding of testosterone to NR2F6. Right: molecular docking analysis shows that testosterone may occupy the predicted ligand-binding pocket of NR2F6 and form favorable interactions with surrounding amino acid residues. (b) Validation of the interaction between leukotriene B4 (LTB4) and PRMT2. Left: chemical structure of LTB4. Middle: MST analysis confirms the direct binding of LTB4 to PRMT2, with the dissociation constant Kd of 14.08 μmol·L−1. Right: the molecular docking analysis elucidates the predicted binding mode of LTB4 in the binding pocket of PRMT2 and its interactions with surrounding residues. (c) Validation of the interaction between LTB4 and ACAT1. Left: MST binding curve shows direct binding of LTB4 to ACAT1, with the dissociation constant Kd of 15.36 μmol·L−1. Middle and right: the molecular docking analysis shows the binding conformation of LTB4 within the ACAT1 pocket and the electrostatic potential of the protein-ligand complex.
Testosterone is a major androgen steroid hormone, which is synthesized from cholesterol through a series of enzymatic reactions. It mainly comes from Leydig cells of the testis, and a small amount is produced by the adrenal gland and ovary. Recent studies have found that testosterone can interact with a variety of protein targets and participate in inflammatory response and energy metabolism. Therefore, identifying the potential target proteins of testosterone is of great significance for further understanding its physiological and pathological functions[37]. Among the four tested candidate targets for testosterone, NR2F6 showed a concentration-dependent binding curve with a dissociation constant Kd of 42.94 nmol·L−1 (Fig. 6a), indicating the presence of high-affinity interactions. On the contrary, other candidate targets did not show binding signals capable of fitting under experimental conditions (Supplementary Fig. S2a−c). Molecular docking analysis further showed that testosterone could be accommodated in the predicted ligand-binding pocket of NR2F6, where it interacted with surrounding residues, supporting the direct interaction determined by MST.
LTB4 is a bioactive lipid metabolite produced by arachidonic acid through the 5-lipoxygenase (5-LOX) pathway. LTB4 is one of the most potent proinflammatory lipid factors in vivo and can mediate neutrophil chemotaxis, activation, and inflammatory cell recruitment through receptors BLT1 and BLT2. Due to its central role in inflammatory responses, exploring new targets of LTB4 can help discover its unexplored mechanism of action[38]. For LTB4, we also selected four predicted targets for verification. An MST experiment confirmed that LTB4 directly binds PRMT2 and ACAT1. The dissociation constant Kd for the interaction between LTB4 and PRMT2 is 14.08 μmol·L−1 (Fig. 6b), while the dissociation constant Kd for the interaction with ACAT1 is 15.36 μmol·L−1 (Fig. 6c). The remaining predicted targets did not show detected binding with LTB4 in the MST analysis (Supplementary Fig. S2d, e). Molecular docking analysis showed that LTB4 could be stably accommodated in the binding pocket of PRMT2 and ACAT1, establishing hydrophobic interactions and potential hydrogen-bonding interactions with residues from the target proteins. Notably, although the concentration-dependent MST response of the LTB4-ACAT1 interaction was clearly observed, the highest ligand concentration achievable was limited by the stock concentration and solubility of LTB4. Consequently, the binding curve did not fully reach the saturation plateau, and the fitted Kd value should be regarded as an approximate estimate rather than a determined dissociation constant.
-
Endogenous metabolites are not only basic metabolic intermediates in organisms, but also play a leading role as key signaling regulatory molecules. In recent years, there has been growing academic interest in their novel biological phenotypes beyond the traditional metabolic functions. In addition, artificial intelligence has been deeply integrated with traditional computer-aided drug discovery (CADD) technology to improve the efficiency and hit rate of small molecule target prediction through machine learning and deep learning algorithms[39]. Inspired by our previous research on target prediction and bioinformatics-based analysis[13,40], we developed DeepETD, a metabolite target prediction model that integrates multiple biological datasets. The model is based on bioinformatics and deep learning, and the attention mechanism is introduced to significantly improve the prediction accuracy. Compared with traditional machine learning methods, our attention-based model shows superior performance. It can not only accurately predict known interactions, but also effectively identify potential novel targets. Using DeepETD, we established a database called EMTDD, which contains target predictions of 3,382 human-annotated endogenous metabolites. This open-source database can be used as a valuable resource for further functional research.
The existing computational target prediction methods largely rely on omics data and chemical structures[14,15,41]. However, for endogenous metabolites, large amounts of biomedical data, including disease association, cellular phenotypes, and subcellular localization, contain valuable biological information[19]. Relying only on structural information can overlook the key biomedical information, thus limiting the accuracy and reliability of predictions. In addition, due to the diverse functional phenotypes of metabolites, screening suitable and clean cell models to obtain high-quality omics data for gene-specific target prediction often faces many technical challenges. In contrast, DeepETD effectively integrates multidimensional biomedical knowledge and accurately captures the common features between metabolites and proteins, thus enhancing the identification ability of potential targets of endogenous metabolites.
Despite the encouraging results, this study has certain limitations that are worthy of further discussion in future work. First, the excellent performance of DeepETD largely depends on the availability of the biological information related to metabolites and proteins. However, for many newly discovered or unannotated metabolites, such as secondary metabolites of microbes, the ability of the model to predict their targets may be limited due to the lack of sufficient biological data. In addition, several limitations exist in the sample construction. Although the binding affinity cutoff strategy improved the reliability of positive and negative sample labeling, the use of a fixed cutoff may introduce label ambiguity. The interactions with similar binding affinity can be assigned to different categories. Furthermore, protein-metabolite pairs with IC50 values greater than 100 nmol·L−1 may still have weak biological interactions, which could introduce false-negative samples in the dataset. Future studies may further improve dataset quality by incorporating continuous affinity values or experimentally validated non-interacting pairs. Another limitation is the imbalance between the positive and negative samples, which may still affect model robustness and generalization ability. Future studies will further explore more effective strategies for handling imbalanced datasets, such as weighted loss functions or data augmentation approaches.
Experimental validation is a key step to evaluate the accuracy of target discovery tools. In this study, we selected two endogenous metabolites, testosterone and leukotriene B4, for validation. The successful validation of these chemically and functionally distinct metabolites suggests that DeepETD may have broad applicability for endogenous metabolite target discovery. MST experiments using HEK293T cell lysates overexpressing candidate targets confirmed the direct binding of testosterone to NR2F6 and LTB4 to PRMT2/ACAT1, further indicating the reliability of DeepETD. Another observation is that not all computationally predicted targets have been experimentally validated. This difference is expected because protein abundance, folding status, post-translational modifications, cofactor requirements, and limitations inherent to lysate-based binding assays all affect the detectability of interactions. Future studies will expand the scope of experimental validation and adopt complementary validation methods such as surface plasmon resonance (SPR), thermal proteomic analysis (TPP), and in vivo models to more accurately explore the interactions between metabolites and targets and further evaluate the reliability of DeepETD prediction.
In conclusion, the DeepETD model based on bioinformatics and attention mechanisms not only expands our understanding of the biological functions of metabolites but also creates new opportunities for biomedical research and clinical application. In addition, DeepETD can be integrated into the phenotype-based drug discovery workflows, expanding the potential space for therapeutic target identification[42,43]. By identifying endogenous metabolite-target interactions associated with specific diseases or patient groups, this method can provide information for the design of targeted diagnosis and therapeutic strategies and ultimately promote precision medicine.
-
Not applicable. The present study included only in vitro experiments and did not involve human subjects, clinical samples, or animal experiments.
-
The authors confirm their contributions to the work as follows: development of the computational tool and the writing of the corresponding methodological and related content: Yang X, Xu Z, Min X; manuscript writing: Xu Z, Wang X; experimental validation and figure preparation: Wang X, Yuan X; reference collection and organization: Xu Z, Zhu K, Xiao W; manuscript checking and revision: Zhang H, Xu H, Luo C. All authors reviewed the results and approved the final version of the manuscript.
-
The code of DeepETD is available at https://github.com/AIDDHao/DeepETD. The data generated or analyzed during this study are included in this published article and its supplementary information files. Additional data related to this study are available from the corresponding author upon reasonable request.
-
We thank the staff members of the Large-scale Protein Preparation System at the National Facility for Protein Science in Shanghai, for providing technical support and assistance in data collection and analysis. We gratefully acknowledge the financial supports from the National Key R&D Program of China (2022YFC3400500 to Cheng Luo), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0830301 to Cheng Luo), the National Natural Science Foundation of China (81903538 to Hao Zhang), the Science and Technology Department of Guizhou Province (CXPTXM[2025]021 and KXJZ[2025]014 to Cheng Luo), the Applied Basic Research Foundation of Yunnan Province (202501BC070005 to Cheng Luo), the Shanghai Municipal Health Commission Medical New Technology Project (2025ZZ2060 to Hao Zhang), the Shanghai Oriental Talent Plan Youth Project (QNJY2025170 to Hao Zhang), the Shanghai Science and Technology Committee Computational Biology Special Project (YDZX20233100004032 to Cheng Luo), and (24JS2830200 to Heng Xu), the Lingang Laboratory (LG-QS-202204-07 to Hao Zhang), and the Shanghai Municipal Education Commission AI for Science Project (301-0406 to Hao Zhang).
-
The authors declare that they have no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/targetome-0026-0024.
-
#Authors contributed equally: Zhixuan Xu, Xiaomin Wang, Xiaobo Yang
- Supplementary Fig. S1 Performance evaluation of DeepETD.
- Supplementary Fig. S2 Negative MST validation results for predicted targets.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Xu Z, Wang X, Yang X, Yuan X, Zhu K, et al. 2026. DeepETD: a novel deep-learning based model for endogenous metabolite target discovery. Targetome 2(3): e024 doi: 10.48130/targetome-0026-0024 

DeepETD: a novel deep-learning based model for endogenous metabolite target discovery
- Received: 07 April 2026
- Revised: 31 May 2026
- Accepted: 03 June 2026
- Published online: 17 June 2026
Abstract: Metabolites are involved in almost all fundamental biological processes. The identification of their protein targets is crucial for elucidating non-canonical signaling roles and evaluating their therapeutic potential. In recent years, due to the continuous development of chemical proteomics, atypical phenotypic functions of classic metabolites have been continuously discovered. However, the discovery of endogenous metabolites for their disease-related functions is still progressing slowly. To accelerate the identification of disease-related targets for endogenous metabolites, we propose a new hypothesis: endogenous metabolites and their molecular targets are expected to have similar disease associations. Following this hypothesis, here we report the development of a novel deep-learning model called DeepETD, which integrates bioinformatics data and introduces an attention mechanism to predict functional targets of specific metabolite phenotypes. Using this model, we constructed a publicly accessible database named EMTDD containing potential targets for 3,382 common human endogenous metabolites. Overall, this study presents a new computational method and resource for endogenous metabolite target discovery as an important supplement to experimental methods such as chemical proteomics.
-
Key words:
- DeepETD /
- Metabolite target discovery /
- EMTDD /
- Testosterone /
- Leukotriene B4