









-
Drug discovery remains slow, costly, and failure-prone, with many programs failing because early target hypotheses are not supported by sufficient genetic, mechanistic, structural, safety, or clinical evidence before entering expensive experimental and clinical stages[1]. Modern drug discovery increasingly depends on genomics, transcriptomics, proteomics, metabolomics, structural biology, phenotypic screening, literature, electronic health records, and clinical trial data[2−6]. However, these resources differ in identifiers, evidence standards, assay conditions, temporal coverage, and curation bias. As a result, target-disease associations, druggability, safety, and translational evidence are often difficult to connect in a coherent and auditable manner (Fig. 1a). Conventional machine-learning models can rank candidate targets or compounds, but their predictions often lack explicit evidence paths for mechanism tracing, toxicity interpretation, causal assessment, and experimental follow-up.
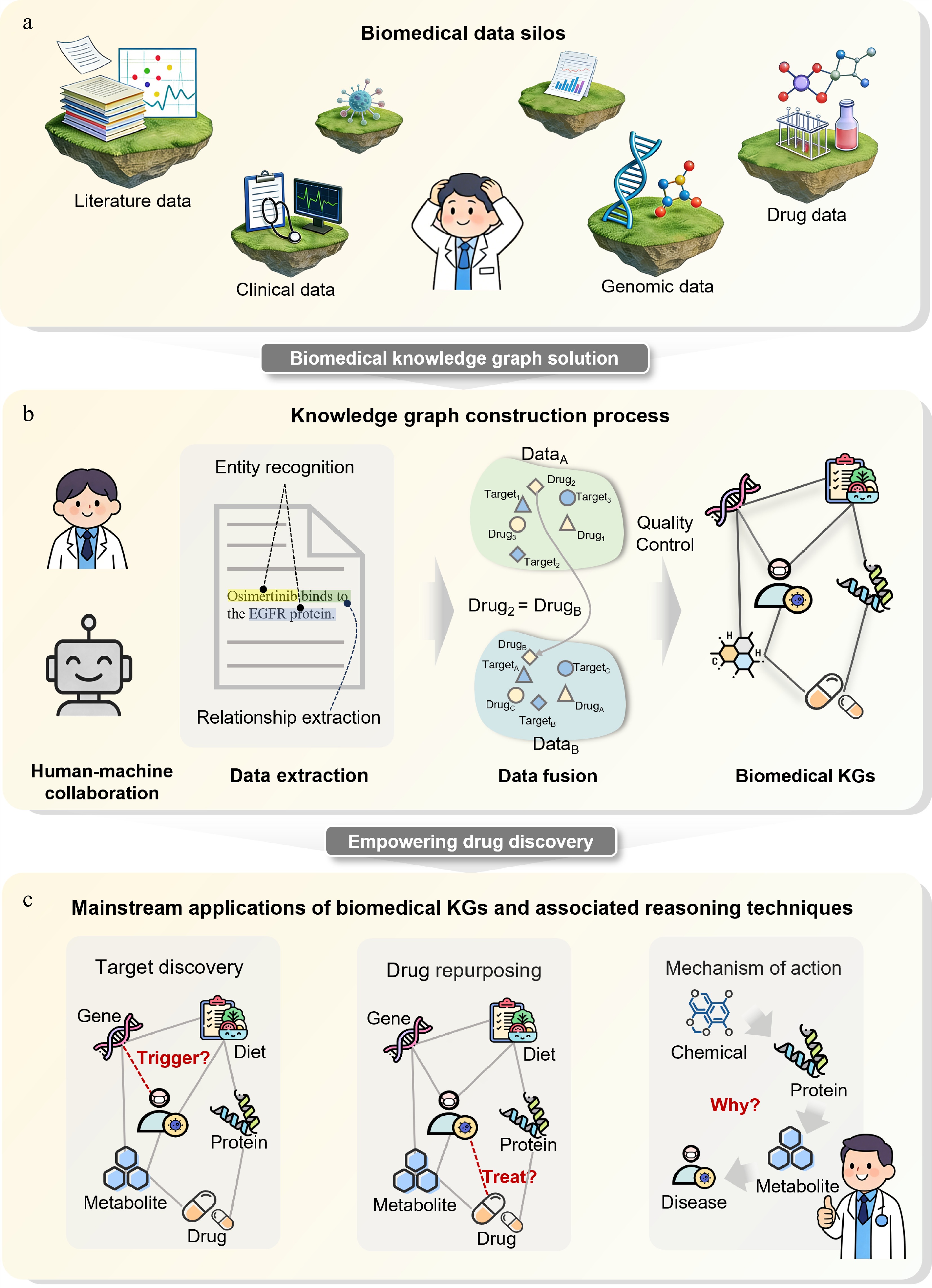
Figure 1.
Biomedical knowledge graphs break down data silos and promote drug discovery. (a) Biomedical data silos. Different data sources are isolated; real-world data, including literature, clinical records, genomic data, and drug-related information are heterogeneous and fragmented, hindering cross-semantic and cross-hierarchical integration. (b) Knowledge graph construction pipeline. Human-machine collaboration is used to extract entities and relationships from multi-source data, followed by integration and quality control to form a biomedical knowledge graph. (c) Major applications of biomedical knowledge graphs. Knowledge graph-based reasoning enables drug target discovery, drug repositioning, and mechanistic interpretation.
Biomedical knowledge graphs (KGs) offer a structured way to partially address this evidence-fragmentation problem by representing biomedical entities and their typed relationships as a computable semantic network (Fig. 1b)[7]. By linking drugs, genes, proteins, diseases, phenotypes, pathways, structures, assays, adverse events, and clinical evidence, KGs provide explicit context for target prioritization, druggability assessment, mechanism interpretation, drug-target interaction prediction, repurposing, and safety evaluation[8]. Knowledge graph reasoning (KGR) further supports rule-based, embedding-based, path-based, graph-neural, transformer-based, and LLM-assisted inference over these relationships[9]. Nevertheless, KG-derived predictions should be interpreted as evidence-supported hypotheses that require benchmark-aware evaluation and experimental or clinical validation, rather than as direct proof of therapeutic efficacy (Fig. 1c).
This review focuses on how biomedical KGs support target identification and translational decision-making in drug discovery. This review first introduces KG concepts, data resources, construction pipelines, quality control, and LLM-assisted extraction. The analysis then compares major KGR paradigms, quantitative benchmarks, leakage risks, validation strategies, and structure-augmented multimodal reasoning. Subsequent sections discuss applications in target discovery, druggability assessment, mechanism interpretation, drug-target interaction prediction, molecular design, multi-parameter lead optimization, ADMET and safety assessment, drug repurposing, combination therapy, biomarkers, precision medicine, and dynamic KGs for resistance analysis. Finally, current limitations and future directions are summarized.
-
The term 'knowledge graph' was formally proposed by Google in 2012[10]. Mathematically, a knowledge graph is a labeled directed graph structure used to model knowledge and relationships between entities[11].
Biomedical knowledge graphs take entities such as genes, proteins, compounds, diseases, phenotypes, and pathways as nodes, and semantic relations, including targeting, treatment, pathogenesis, and participation, as edges. They integrate molecular-level information (genes, proteins), mesoscale processes (cellular functions, tissue physiology), and macroscale phenomena (disease phenotypes, clinical manifestations), enabling cross-scale integration and providing a computational basis for understanding complex biological mechanisms and cross-scale reasoning.
Data resources
-
High-quality biomedical knowledge graphs rely on extensive and reliable multi-source data resources, which focus on different biological scales. This paper provides a practical classification based on the core features and typical applications of each resource, rather than a strict semantic division (Supplementary Table S1).
The molecular layer is the basis of the knowledge framework and provides a large number of core entities. In the case of ChEMBL[12], such databases contain information on small-molecule chemical structures and target binding activity; UniProt[13] and Ensembl[14] are important sources of knowledge about gene function and protein function. The interaction layer mainly describes the interaction relationship at a higher level. As for STRING[15], it provides the interaction network between proteins. The pathway layer records the signal transmission process, and the representative resources are KEGG[16] and Reactome[17]. The phenotypic layer links the molecular changes with the phenotypic characteristics and biological outcomes of the disease, and DisGeNET[18] integrates the disease-related genes and variation information. The clinical layer involves medical terminology, diagnosis and treatment intervention, and drug use data. DrugBank[19] collects the core clinical information of drugs and records the molecular mechanisms and clinical decisions.
Based on these data, researchers have constructed many biomedical knowledge graphs, and each has its own emphasis on the breadth and depth of data integration. As exemplified by Pharos[20], it concentrates on druggable genes. There are also some special KGs for specific scenarios such as drug target interaction[21], COVID-19[22], and precision medicine[23]. To compare the construction objectives, data coverage, and typical application scenarios of these KGs, representative resources are summarized in Supplementary Table S2.
KG construction pipeline
-
The knowledge graph follows the bottom-up construction process and is divided into three stages: knowledge extraction, knowledge fusion, and quality evaluation (Fig. 1b).
Knowledge extraction
-
Knowledge extraction mainly includes named entity recognition (NER) and relation extraction (RE). The task of NER is to recognize biomedical entities from text. Early methods highly relied on experts to formulate rules and dictionaries[24], and their generalization ability was limited. Subsequently, traditional machine learning methods such as conditional random fields (CRFs)[25] and support vector machines (SVMs)[26] improved the flexibility and generalization ability of the model[27]. Deep learning further reduces the burden of artificial feature design. Pre-trained language models such as BERT[28] and BioBERT[29] significantly improve the accuracy and generalization ability of NER by better capturing context semantics[30].
RE is to further extract the semantic relationship between entities after identifying entities, but it is often limited by the lack of annotation data. Therefore, such tasks usually adopt a weak supervision method, use unmarked data or an external knowledge base, and reduce the labeling cost through distant supervision[31].
The traditional pipeline mode is prone to cascade errors. In recent years, researchers have begun to use the end-to-end deep learning method to jointly extract entities and relationships[32], so as to better model the complex interaction between the two.
Knowledge fusion
-
Knowledge fusion mainly solves the differences and conflicts in multi-source data integration, and its core tasks include ontology matching and entity alignment. Different biomedical databases are usually built independently by different institutions, so there are often semantic differences and conceptual gaps. Ontology matching[33] aims to correspond to the conceptual systems in different data sources. Taking DrugBank's drug level and ChEMBL's compound level as examples, the difficulty lies in the inconsistency between classification criteria and conceptual granularity. Entity alignment[34] identifies and merges nodes that actually point to the same object for different names or identifiers of the same biological entity in different databases to achieve entity normalization and ambiguity resolution. Early methods mainly relied on string matching and standard thesaurus mapping, and now the alignment method based on embedding[35,36] has gradually become the mainstream.
Quality assessment and optimization
-
In order to make the knowledge graph reliably serve the downstream drug discovery, it is necessary to carry out systematic quality assessment and optimization[37]. The evaluation mainly focuses on accuracy and consistency. The former determines whether the triplet conforms to the facts, and the latter checks whether there is a logical conflict. At present, there are many automatic verification methods, but automatic denoising can only deal with common errors. Due to the high accuracy requirements of biomedical data, expert manual review is still irreplaceable.
The knowledge graph also has the problem of incompleteness, but the completion of the knowledge graph in drug R&D and application is usually the final stage.
LLM-driven construction
-
The large language model has strong natural language understanding, generation, and context learning capabilities, so it can be used for the automatic construction of a biomedical knowledge graph[38] and has obvious advantages over traditional machine learning and early pre-training language model methods[39−41]. In the process of knowledge extraction, the outstanding advantage of LLM lies in its learning ability of zero-shot and few-shot learning ability. Through the design of prompts or instructions, the model can also complete named entity recognition and relation extraction in the absence of annotation data[42,43], and can directly convert complex biomedical literature into structured triples, potentially reducing intermediate pipeline errors, but only when the extracted triples are subsequently verified[44]. In knowledge fusion, the fine-tuned LLM is easier to identify the same standard concept behind different expressions, showing great potential in zero-shot scenarios and long-tail concept alignment tasks[45,46]. In addition, a large amount of prior biomedical knowledge implied in the model parameters[47] can also provide assistance in KG refinement, quality control, and link prediction[38].
However, LLM-based KG construction still faces a measurable hallucination risk. Because LLMs do not naturally obey graph-structural, ontological, or evidence constraints, hallucinated entities or relations may be converted into false biomedical triples and then propagated to downstream drug-target, disease-gene, or drug repurposing models. In a systematic evaluation of 12 BioNLP benchmarks, LLaMA 2 13B under zero-shot prompting generated 506 hallucinated responses on ChemProt, approximately 3% of 16,943 instances, and 102 hallucinated responses on HoC (32%); GPT-4 also remained below PubMedBERT on BC5CDR-chemical NER and below BioBERT on ChemProt and DDI2013 RE[48].
Two practical safeguards are therefore required. First, pure zero-shot extraction should be avoided when possible because one task-specific example reduced HoC hallucinations from 102 to 0 and inconsistent responses from 69 to 23[48]. Second, LLM-generated triples should be treated as candidate evidence rather than curated facts, and should be inserted into biomedical KGs only after schema checking, ontology/entity grounding, evidence tracing to sentences or PMIDs, and expert or automated validation.
-
This review classifies KGR methods into five core paradigms according to evidence representation and inference modes: symbolic reasoning, representation learning, path- and graph-structure-aware reasoning, Transformer-based graph reasoning, and KG-LLM integrated reasoning. These paradigms reflect a gradual shift from explicit rule-based inference to latent representation learning, multi-hop structural reasoning, global graph context modeling, and language-guided hybrid reasoning. Structure-augmented multimodal KG reasoning is discussed as a cross-cutting evidence layer instead of a standalone paradigm, linking graph topology to protein structures, binding pockets, variants, conformational states, and ligand compatibility. Typical quantitative benchmarks and evaluation criteria are summarized, along with prevalent evaluation flaws and translational validation challenges that affect the application of benchmark results to real-world drug discovery.
Symbolic reasoning
-
Symbolic reasoning derives new knowledge using explicit first-order logic rules (Fig. 2a). For drug repositioning, a rule can be formulated: if drug A inhibits target T and target T drives disease D progression, drug A may treat D. Such rules yield clear causal chains, valuable in interpretable drug R&D.
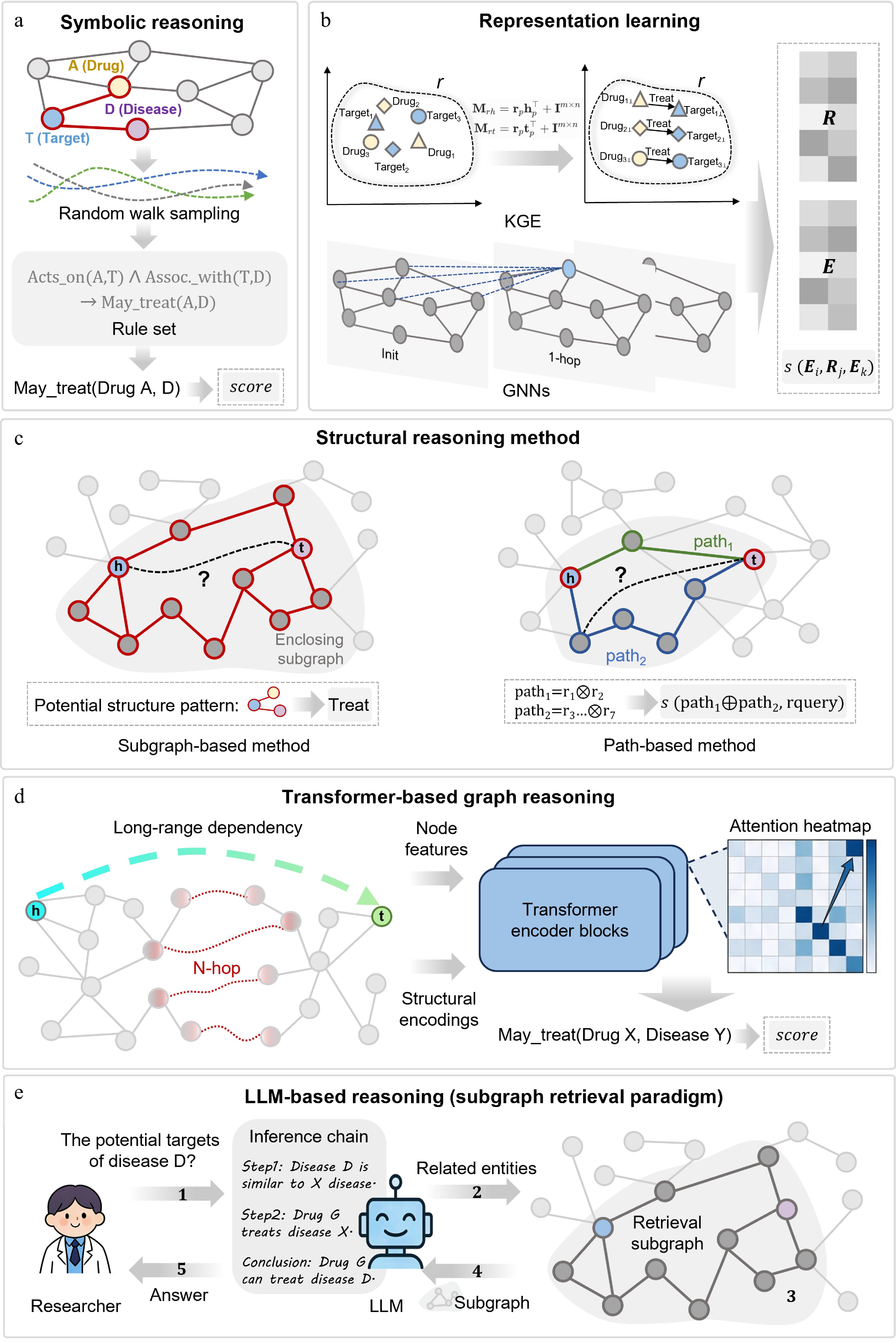
Figure 2.
Mainstream knowledge graph reasoning paradigms for target and drug discovery. (a) Symbolic reasoning. Performs inference via random walk sampling and first-order logic rules with high interpretability. (b) Representation learning. Maps entities and relations to low-dimensional vector spaces via KGE or GNNs and scores triples. (c) Path and structural reasoning. Infers links by mining enclosing subgraphs or multi-hop path patterns. (d) Transformer-based graph reasoning. Integrates node features and structural encoding into self-attention to capture long-range dependencies. (e) LLM-KG integrated reasoning. Retrieves structured subgraphs as context to guide LLMs for traceable logical inference. Structure-derived features such as pockets, residues, conformational states, variants, and ligand poses can serve as multimodal evidence layers across these paradigms.
Representative algorithms include AMIE+[49] and AnyBURL[50]. AMIE+ uses top-down search under the partial completeness assumption, while AnyBURL relies on bottom-up path sampling. Though logically rigorous, both suffer from a rule-space explosion in large graphs.
Differentiable rule learning methods combine symbolic structures with parametric learning. NeuralLP[51] models rule application as differentiable matrix multiplication with attention; DRUM[52] uses bidirectional RNNs to share parameters and avoid overconfidence in erroneous rules. These methods improve scalability and robustness to noise.
However, strict rule matching limits generalization. Even with soft matching, performance heavily depends on graph completeness, severely restricting utility in sparse biomedical graphs.
Representation learning
-
Knowledge graph embedding (KGE) models represent triple semantics via vector algebra or geometry, divided into translational, semantic matching, and geometric transformation methods (Fig. 2b).
Translational models, represented by TransE[35], model relations as vector translations but struggle with complex drug-target associations; later models introduce relation-specific projections to address this. Semantic matching models use bilinear transformation or tensor decomposition: DistMult[53] performs well on symmetric relations but lacks directionality, while ComplEx[54] uses complex embeddings for asymmetric relations such as gene regulation. Geometric models, including RotatE[55], model relations as rotations in complex space, supporting diverse relation patterns.
In biomedicine, entity functions depend on local network topology. Traditional KGEs underutilize high-order structure, leading to the adoption of graph neural networks (GNNs) such as R-GCN[56], which propagates relation-aware information.
Representation learning performs well in practice, but traditional KGEs fail to generalize to unseen nodes, and GNNs suffer from over-smoothing that erases fine-grained biological differences.
Path and structural reasoning
-
Path and structure reasoning is centered on the multi-hop connectivity pattern between entities and mainly includes explicit path features, reinforcement learning path search, subgraph inductive reasoning, and neural path reasoning. The core criterion distinguishing this class of methods from representation learning methods is whether they actively leverage graph structural information (Fig. 2c). Early methods directly enumerate multi-hop paths or local subgraphs and convert them into discrete features for link prediction[57], which is intuitive and interpretable, but is prone to a combination explosion in large-scale sparse graphs. Reinforcement learning path reasoning models link prediction as a sequential decision problem[58]. However, due to the graph sparsity and long-tail features, it often faces the problems of sparse rewards and a too-large search space.
Subgraph reasoning extracts the enclosing subgraph around the target entity pair and completes the prediction with the help of relationship-aware GNN[59,60], which can be generalized to unseen entities, but the computational cost is high. Neural path reasoning implicitly models multi-hop dependency through differentiable message passing[61,62], taking into account structural expression, representation learning, and strong interpretability, which is more suitable for biological hypothesis generation. Overall, this direction is moving from explicit enumeration to neuro-implicit modeling.
Transformer-based graph reasoning
-
Many biological processes have long-distance and cross-level connections, which are not easy to capture in terms of remote gene regulation, protein allosteric effects, and cross-pathway coupling. When the layers of the graph neural network are deepened, the problems of over-smoothing and over-compression often occur, so the modeling ability of remote biological association is limited. In contrast, transformers can directly establish the connection between any node by relying on the self-attention mechanism, so it has a better chance of breaking through the restriction of local message delivery (Fig. 2d).
However, the standard transformer is not naturally suitable for graph data. Therefore, researchers have proposed a variety of improved methods with structural bias. As exemplified by Graphormer, this model explicitly encodes node centrality and shortest path distances and incorporates structural information into the attention mechanism[63]. GPS and other frameworks combine local graph convolution and global transformer to achieve a balance between local structure modeling and long-range dependency capture[64]. Recent studies have begun to explore the state space model (SSM), represented by Graph-Mamba, which reduces the computational overhead with linear complexity and retains strong long-range dependency modeling capability[65].
LLM-KG integrated reasoning
-
The core goal of the combination of LLM and KG is to connect the ability of structural constraints with the ability of semantic understanding[38]. The earliest research mainly transformed structured knowledge into text[66], but the expression ability of complex multi-hop structures is limited. Subsequent studies mapped graph topology to semantic space through structure coding[67], but the intermediate reasoning process was still not transparent enough.
A graph retrieval enhancement model is an important development in this direction. It relies on a knowledge graph to build a structured context and uses factual evidence to constrain language model generation[68] (Fig. 2e). In addition, the multi-agent collaboration framework can also alleviate the instability of single-model reasoning. It reduces false associations and semantic drift through interactive debate, adaptive graph search, self-reflection, and reverse reasoning[69]. Overall, such LLM + KG frameworks have gone beyond the task of traditional knowledge graph completion and are more suitable for complex inductive reasoning in drug discovery and precision medicine. These five paradigms mainly model graph topology, semantic relations, and evidence paths. For drug discovery, however, target relevance is not enough: tractability also depends on three-dimensional structure, pocket accessibility, conformational dynamics, variant effects, and ligand compatibility. Structure-augmented KG reasoning should therefore be viewed as a cross-cutting multimodal evidence layer rather than a sixth independent paradigm.
Multimodal structure-augmented KG reasoning
-
Structure-augmented reasoning is not a separate paradigm, but a multimodal evidence layer shared by the paradigms above: rules, embeddings, subgraph models, graph transformers, and KG-LLM systems. Disease-gene-drug paths suggest relevance; structure indicates whether the target has an accessible, ligandable, variant-sensitive, and stable pocket.
Sources include AlphaFold2[70], RoseTTAFold[71], ESMFold[72], and AlphaFold3[73] for protein or complex models, plus Fpocket[74], SiteMap[75], and MDpocket[76] for pocket geometry, druggability, and dynamics. Because cryptic allosteric pockets may be transient[77], annotations should keep source, conformational state, and confidence.
A structure-aware KG should add structural nodes (structures, residues, variants, pockets, conformational states, ligands, conformers), edges (protein-has-structure, structure-has-pocket, variant-perturbs-pocket, pocket-binds-ligand, ligand-compatible-with-pocket), and attributes (pLDDT/PAE, pocket descriptors, MD occupancy, docking pose, assay/literature evidence). This enables disease-gene-protein-pocket-ligand paths.
Existing work shows feasibility: MKGE adds SMILES text and molecular graphs to KG embeddings[78], and BioBLP combines protein sequences, drug SMILES, disease text, and missing attributes[79]. Extending these designs to pockets, residues, and conformational states would connect KG reasoning with structure-based drug discovery.
Without structure, predictions may be biologically plausible but chemically weak. Topology-only models can over-rank targets with buried or ligand-incompatible pockets and miss targets enabled by cryptic pockets[80]. Graph-only links remain hypotheses; links also supported by pocket geometry, dynamics, ligand compatibility, and experiments have stronger mechanistic credibility.
Benchmarking and comparative interpretation of KGR paradigms
-
After defining the major paradigms and the need for structure-aware evidence, subsequent comparisons focus on the evaluation of KGR methods. Table 1 provides a comprehensive overview of three link prediction benchmarks: FB15k-237, Hetionet, and ogbl-ppa, as they offer the most widely reported metrics across various paradigms (more benchmarks can be obtained from Open Graph Benchmark[81]). MRR measures how highly the correct entity or relation is ranked, whereas Hits@K measures whether the correct answer appears among the top K candidates. Filtered ranking is commonly used in KGC to avoid penalizing other known true triples.
Table 1. Representative quantitative benchmarks for knowledge graph reasoning paradigms.
Dataset/benchmark Model Paradigm MRR Hits@1 Hits@10 Hits@100 FB15k-237[82]: general
multi-relational KGC benchmarkNeural-LP[51] Symbolic reasoning 0.237 0.173 0.361 NR TransE[35] Representation learning 0.312 0.212 0.51 NR RotatE[55] 0.338 0.241 0.533 NR TuckER[83] 0.358 0.266 0.544 NR CompGCN[84] Path and structure-aware reasoning 0.355 0.264 0.535 NR NBFNet[61] 0.415 0.321 0.599 NR HittER[85] Transformer-based graph reasoning 0.373 0.279 0.558 NR KnowFormer[86] 0.43 0.343 0.608 NR SimKGC[87] Hybrid KG-LLM reasoning 0.338 0.252 0.511 NR SAT[88] 0.354 0.319 NR NR SRP-KGC[89] 0.431 0.353 0.585 NR Hetionet[90]: biomedical
KG filtered tail predictionAnyBURL[50] Symbolic reasoning 0.322 0.229 0.553 NR Noisy-or rule aggregation[91] 0.472 0.377 0.642 NR TransE[35] Representation learning 0.205 0.099 0.444 NR ComplEx[54] 0.25 0.152 0.47 NR RotatE[55] 0.257 0.185 0.403 NR CompGCN[84] Path and structure-aware reasoning 0.292 0.172 0.543 NR MINERVA[92] 0.37 0.264 0.593 NR PoLo[93] 0.43 0.337 0.641 NR HittERnc[85] Transformer-based graph reasoning 0.453 0.316 0.74 NR ogbl-ppa[81]: large-scale biological protein
association link
predictionCommon neighbor[94] Symbolic reasoning NR NR NR 0.2765 Resource allocation[95] NR NR NR 0.4933 Matrix factorization[81] Representation learning NR NR NR 0.3229 SEAL[96] Path and structure-aware reasoning NR NR NR 0.488 NGNN + SEAL[97] NR NR NR 0.5971 Neural common neighbor[98] NR NR NR 0.6119 GraphGPT[67] Transformer-based graph reasoning NR NR NR 0.7655 Downstream biomedical tasks often require task-specific metrics. Binary classification tasks such as drug-target interaction, disease-target association, drug repurposing, adverse-event prediction, and combination-synergy classification commonly use AUROC, AUPRC, F1-score, precision, recall, specificity, balanced accuracy, and Matthews correlation coefficient (MCC). Ranking-oriented screening tasks often report recall@K, precision@K, enrichment factor, BEDROC, MRR, or Hits@K. Binding-affinity and dose-response prediction tasks may use RMSE, MAE, Pearson or Spearman correlation, and concordance index. Therefore, Table 1 should be interpreted as a representative KGC/link-prediction comparison, not as a complete evaluation framework for all downstream drug-discovery tasks.
The metrics support several paradigm-level observations. Symbolic reasoning remains competitive when rule-like graph regularities are informative: Resource Allocation reaches a Hits@100 of 0.4933 on ogbl-ppa, and Noisy-or rule aggregation achieves an MRR of 0.472 and Hits@10 of 0.642 on Hetionet. Representation learning provides scalable baselines, but richer structural models are often stronger; for example, TuckER reaches an MRR of 0.358 on FB15k-237, whereas NBFNet improves this to 0.415. Path and structure-aware methods are particularly useful when multi-hop topology is informative, as shown by PoLo (pruned) on Hetionet (MRR = 0.430, Hits@10 = 0.641). Transformer-based graph reasoning further improves long-range or relation-aware modeling, with KnowFormer reaching an MRR of 0.430 on FB15k-237 and HittERnc reaching an MRR of 0.453 and Hits@10 of 0.740 on Hetionet. Hybrid KG-LLM models show promising general KGC results, represented by SRP-KGC on FB15k-237 (MRR = 0.431, Hits@10 = 0.585), but their biomedical utility still requires leakage control, task-specific validation and prospective evidence.
Quantitative analysis also has certain limitations, as high MRR, Hits@K, AUROC, or F1 may still reflect dataset-specific shortcuts rather than prospective biological utility. The five paradigms are therefore further compared from a practical drug-discovery perspective, focusing on use cases, strengths, biomedical limitations, and validation strategies (Table 2).
Table 2. Practical comparison of KGR paradigms for target and drug discovery.
Reasoning paradigm Core methodology Key applications in drug discovery Critical comparison in biomedical scenarios Symbolic reasoning Logical rule learning, differentiable rule reasoning, random walk path sampling Target identification, mechanism of action
analysis, high-confidence association predictionThese methods are most useful when transparent evidence chains and expert-auditable hypotheses are required. They can be competitive on biomedical link-prediction benchmarks, but their coverage may be limited in sparse or noisy KGs, and rule search can become expensive in large heterogeneous graphs. Representation learning Knowledge graph embedding (KGE), relation-aware message passing, attention-based feature aggregation Drug-target interaction prediction, disease-target association identification, molecular property
predictionThese models scale well to large biomedical KGs and provide strong graph-fitting baselines for candidate ranking. Their main limitations are limited mechanistic interpretability, sensitivity to negative sampling and random splits, possible hub-node or literature-density bias, and over-smoothing in deep GNNs. Path and structure-aware reasoning Explicit path enumeration, reinforcement learning for path search, subgraph inductive reasoning, neural implicit path modeling Multi-hop mechanistic inference, cold-start target/molecule prediction, drug repurposing path
miningThese models are attractive when the task requires interpretable multi-hop evidence or generalization to sparsely annotated diseases and drugs. Their reliability depends strongly on KG completeness, path quality, disease-split design and candidate-universe definition. Transformer-based graph reasoning Structural encoding + self-attention, spectral feature embedding, local convolution + global transformer, linear-complexity state space models Long-range target regulation prediction, cross-pathway association inference,
large-scale KG reasoningThese models are conceptually suitable when distant graph context, cross-pathway information or multi-source molecular features are expected to influence prediction. However, biomedical benchmark evidence is often task-specific rather than evaluated under a shared KG benchmark protocol, and performance may be inflated when similar drugs, homologous proteins or related diseases appear across train and test splits. Hybrid KG-LLM reasoning Retrieval-augmented generation, subgraph prompting, multi-agent collaborative reasoning, chain-of-thought graph reasoning Complex mechanism analysis, low-resource drug discovery, interpretable clinical translational reasoning These methods are promising for natural-language evidence synthesis and explanation-rich biomedical reasoning. However, their evaluation should include evidence of faithfulness, path validity, calibration, prompt robustness, and hallucination control rather than predictive accuracy alone. Benchmarking and validation of biomedical knowledge graph reasoning models
-
The evaluation of biomedical KGs is affected by negative-sample design, data splitting, and benchmark bias. Uniform random negative sampling often creates biologically implausible or trivially false entity pairs, which lowers task difficulty and may inflate performance. More realistic strategies, including type-constrained, relation-aware, degree-aware, and hard-negative sampling, better reflect real discovery settings but also make prediction more challenging. Because unobserved biomedical relations are usually unlabeled rather than truly negative, negative sets may contain undiscovered true associations. Evaluation protocols should therefore respect entity and relation ontologies, avoid impossible combinations, and report the positive-to-negative ratio, resampling strategy, and whether different models use the same negative set.
Data splitting can also cause leakage. Random train-test splits may share inverse or duplicated edges, ontology parent-child relations, similar drugs or proteins, overlapping disease hierarchies, literature co-occurrence, or similar entity neighborhoods across splits. These shortcuts allow models to infer test links without learning generalizable biological mechanisms. Temporal splits, target- or disease-disjoint splits, scaffold-based drug splits, relation holdout, and cold-start entity splits are more suitable for testing real-world generalization. For target identification and drug discovery, time-cutoff-based prospective evaluation is especially important because it asks whether models trained on historical knowledge can predict later evidence.
Benchmark bias is another major concern. Public databases are enriched for well-studied targets, common diseases, approved drugs, and high-degree entities. Models may therefore reproduce historical research attention rather than discover new mechanisms, and may exploit shortcuts from ontology structure, gene-family similarity, chemical analogs, literature frequency, or curation patterns. This partly explains why many biomedical KG models perform well on internal benchmarks but fail in prospective validation.
Accordingly, metric and model selection should match dataset scale, relation heterogeneity, entity-type constraints, graph topology, and the biomedical task. Resources such as Open Graph Benchmark, OpenBioLink, KG-Hub, and PyKEEN support reproducible comparison. However, MRR and Hits@K only measure ranking quality, not translational value. Prospective validation should train models on data available before a defined time point and test predictions against later literature, experiments, or clinical progress. Low-throughput assays, perturbation screens, and expert review remain necessary to confirm biological relevance, feasibility, and safety.
-
In the early stage of drug discovery, researchers need to screen candidate targets from a large number of genes, proteins, pathways, and disease-related molecules. Traditional target discovery mostly relies on single experimental results or manually collated disease-gene relationships, with limited sources of evidence. Knowledge graphs can integrate literature, omics, genetics, protein–protein interactions, pathways, phenotypes, drugs, and clinical annotations. PrimeKG, DisGeNET, and TarKG have demonstrated this multi-level integration.
Early network methods mainly relied on network proximity and disease modules. Candidate targets are usually evaluated according to their distance from known disease-associated genes or proteins[99,100] (Fig. 3a). This method can find candidate targets in disease-related network regions, but can only provide evidence of association. With the development of knowledge graph representation learning and graph neural networks, disease-target discovery has gradually turned to multi-relational reasoning and graph completion. KDGene[101], MiGCN[102], KG2ML[103], Progeni[104], TarIKGC[105], and other methods can capture the complex relationships among diseases, genes, proteins, drugs, pathways, phenotypes, and literature evidence.
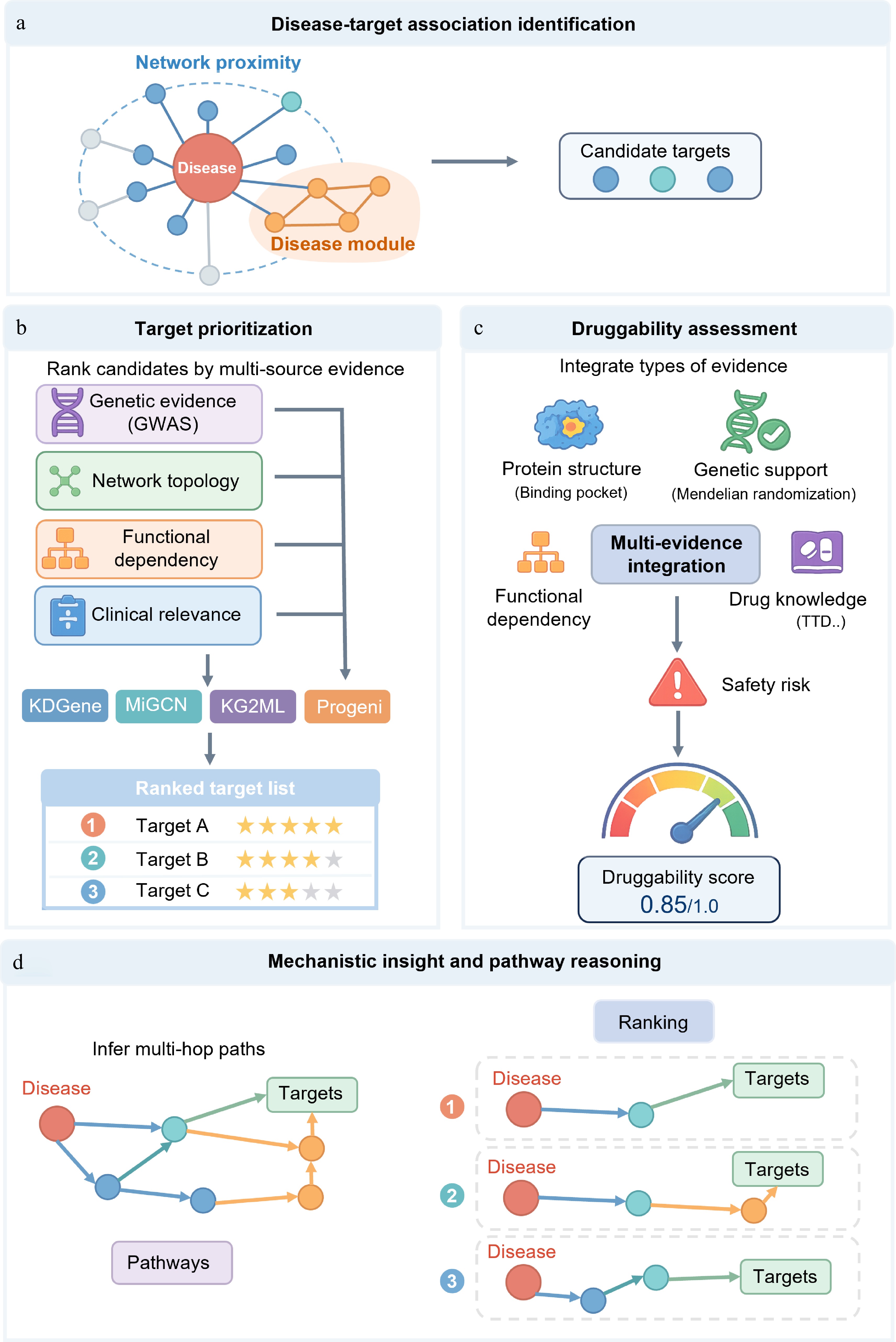
Figure 3.
A knowledge graph-based framework for disease-related target discovery, prioritization, druggability assessment, and mechanism analysis. (a) Identification of disease-related targets. Candidate targets associated with disease-related molecules are identified using network proximity, disease module expansion, and multi-relational knowledge graph representation learning. (b) Target prioritization. Candidate targets are ranked by integrating genetic evidence, network topology, functional dependencies, and clinical relevance. (c) Druggability assessment. The development potential of targets is evaluated by combining structural suitability, genetic support, functional networks, existing drug knowledge, and safety risks. (d) Mechanism elucidation and pathway inference. Infer and rank multi-hop pathways connecting diseases to candidate targets within the context of disease-related networks and pathways to reveal potential molecular mechanisms.
Target ranking needs to pay attention to the level of evidence. Literature co-occurrence, network proximity, and knowledge graph link prediction can propose candidate targets, but they are vulnerable to publication bias, hub node effect, and incomplete database. GWAS signals, colocalization analysis, Mendelian randomization, and Open Targets Genetics[106] can provide stronger genetic support. CRISPR screening, RNAi perturbation, Perturb-seq, loss-of-function experiments, and dependency mapping analysis[107] can further determine whether the perturbed target changes the disease-related phenotype.
At present, target ranking increasingly focuses on multidimensional evidence, including disease relevance, genetic support, functional dependence, tractability, safety, tissue specificity, and clinical relevance (Fig. 3b). Open Targets Genetics[106], resources and models such as dependency map analysis[107], GraphEvo[108], KGDRP[109], and PubMed Knowledge Graph 2.0[110] illustrate that joint evaluation of multiple evidences can improve the reference value of target ranking. However, diseases or proteins with more research may have higher scores due to more literature, more annotations, and more connections; less studied targets may also be underestimated due to insufficient evidence. Therefore, target ranking also needs to combine structural tractability, ligandability, tissue distribution, safety, and experimental validation results.
Target druggability assessment
-
After the candidate targets are sorted, the druggability and tractability need to be further evaluated (Fig. 3c). Disease relevance alone is not sufficient to support target development. A therapeutic target should not only participate in the disease process but also be effectively regulated by small molecules, antibodies, peptides, RNA drugs, or targeted protein degradation technology. Target tractability emphasizes safe and effective regulation, while druggability pays more attention to the development of drug-like small molecules or biological agents[111,112].
Structural biology is the core of drug evaluation. In the context of small molecule discovery, major considerations include binding pockets, molecular volume, cavity depth, hydrophobicity, electrostatic complementarity, conformational flexibility, and allosteric sites[113]. For antibody or protein drugs, greater emphasis is placed on extracellular accessibility, membrane topology, epitope exposure, and tissue-specific expression. Structural resources such as PDB[114], AlphaFold2/3, ESMFold, and RoseTTAFold can provide important information, but the predicted structures need to be interpreted carefully when the confidence is low, the key conformation is missing, or there is a ligand-induced conformational change.
Knowledge graphs can transform structural information and tractability into nodes and edges. A protein can be linked to a protein structure, binding pocket, ligand-binding events, mutation annotation, known inhibitors, tissue expression, essentiality score, and safety risks. Pocket descriptors such as fpocket score, SiteMap volume, DoGSiteScorer score[115], hydrophobicity, flexibility, and allosteric potential can also be added to the knowledge graph. This structure-aware representation helps to distinguish 'disease-related targets' from 'therapeutically tractable targets'. DrugnomeAI[116] and DrugTar[117] also show that gene, network, protein language model, and Gene Ontology information can jointly improve the druggability prediction performance.
Safety is also part of the tractability assessment. If the target is important for normal tissue function or widely expressed in key organs, regulating it may bring target-related toxicity. Therefore, tissue expression, human genetic constraints, essential gene status, pathway redundancy, and disease background specificity need to be considered when sorting targets.
Mechanism interpretation and pathway reasoning
-
After the candidate targets are sorted, it is also necessary to explain how they participate in the disease mechanism (Fig. 3d). Knowledge graphs can put candidate targets into disease-related biological processes, signaling pathways, cellular phenotypes, and molecular cascades, and form traceable relationship paths and subgraphs[118]. With the increase of multi-omics data, mechanism analysis needs cross-level integration. Similarity network fusion has been used for disease typing and identification of key regulatory factors[119]. Breast cancer research also shows that integrating molecular, pathway, and clinical information is helpful in discovering regulatory factors and disease-driving pathways[120]. GNNRAI further combines multi-omics data with biomedical knowledge to improve the interpretability of disease prediction[121].
Path-based knowledge graph reasoning can connect candidate targets with disease biology. Metapath, random walk, shortest path, subgraph interpretation, and neural path reasoning can identify the relationship chain among diseases, targets, pathways, phenotypes, drugs, and molecular functions. BioPathNet improves link prediction ability through learning path representation and shows key reasoning paths[122].
The text-enhanced knowledge graph model can also connect the graph relationships with semantic evidence of the literature, thereby improving the interpretability[123]. However, a disease-target pathway can only explain that the mechanism may be reasonable and cannot directly prove causality. Incomplete databases, uneven literature coverage, duplicate pathway annotations, and hub node interference may affect pathway interpretation.
Perturbation evidence can test mechanistic hypotheses. CRISPR screening, RNAi experiments, single-cell perturbation, drug perturbation, transcriptomics, and pathway activity analysis can determine whether downstream pathways or disease-related phenotypes change after regulating candidate targets. For COVID-19 research, virus-host interaction networks are adopted to reconstruct infection-related molecular networks and identify potential intervention targets[124]. MMiKG and MicrobiomeKG also support mechanism-related pathway discovery by integrating microbial, host, and phenotypic information[125,126].
-
Early DTI predictions mainly relied on chemical similarity, molecular fingerprints, target annotation, and handcrafted descriptors. When the target has a known ligand, this kind of method has a certain effect; however, in data-sparse or cold-start scenarios, the effect is easily limited. With the development of deep learning, DTI prediction has gradually turned to representation learning. DeepDTA[127] and DeepAffinity[128] use protein sequences and SMILES sequences to predict binding affinity. GraphDTA represents compounds as molecular graphs, thus capturing richer chemical topology information[129] (Fig. 4a).
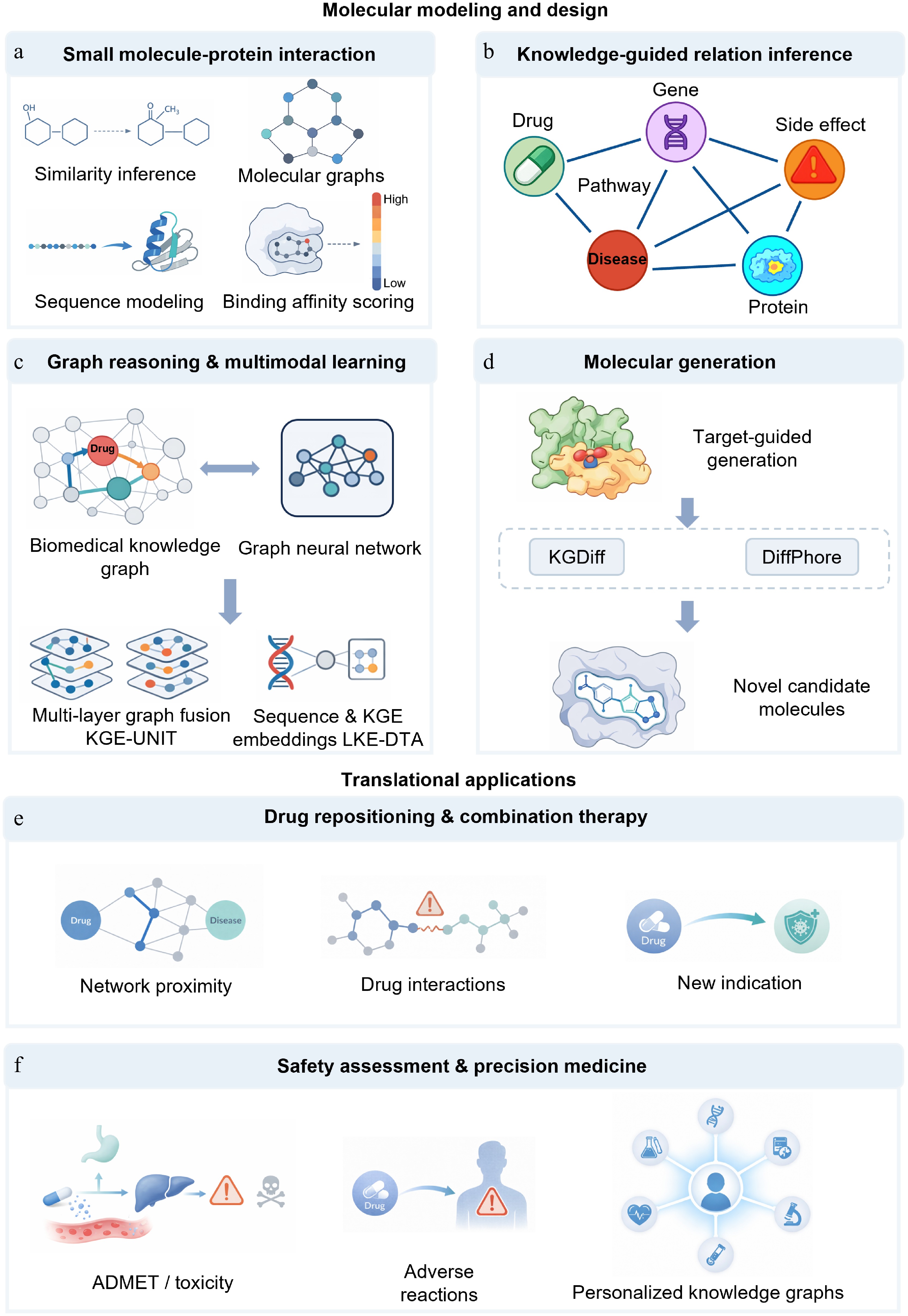
Figure 4.
Knowledge graph-based prediction of small-molecule-target interactions, molecular design, and translational applications. (a) Prediction of small-molecule–protein interactions. This has evolved from predictions based on chemical similarity and ligand-protein pairs to deep learning-based affinity prediction using molecular graph and protein sequence features. (b) Knowledge-guided relationship inference. Integrating heterogeneous entities such as drugs, proteins, diseases, pathways, genes, and side effects into a knowledge graph enables models to shift from local pairwise judgments to relationship inference within a biomedical context. (c) Graph reasoning and multimodal learning. With the introduction of graph reasoning, multi-task knowledge sharing, and multimodal representation, drug-target identification has evolved from simple relationship completion to joint modeling within complex biological contexts. (d) Molecular generation. Knowledge graphs are not only used for candidate screening but also provide conditional guidance for molecular generation and lead optimization, shifting design from rule-driven to information-driven approaches. (e) Drug repurposing and combination therapy. Knowledge graphs support drug reuse, synergistic effect prediction, and drug interaction modeling, extending candidate discovery to system-level inference based on multi-source evidence and contextual constraints. (f) Safety assessment and precision medicine. Knowledge graphs can integrate ADMET, toxicity, adverse reactions, and patient multimodal data for safety risk assessment and precision medicine analysis.
It is still not enough to rely only on chemical structures and protein sequences. The prediction results may also be affected by target family, pathway background, disease relevance, drug category, off-target spectrum, and experimental assay evidence. A knowledge graph can put drugs, proteins, diseases, pathways, phenotypes, and experimental data into the same relationship space (Fig. 4b). DTINet[130] and TriModel[131] illustrate that heterogeneous network integration helps to improve DTI prediction. It is recommended that the systems-level model and the multi-relational graph completion method can also improve the prediction effect in the weak supervision and cold-start scenarios through the information propagation between related entities.
In recent years, research has put more emphasis on the joint modeling of chemistry, protein, semantics, and graph context. DTKGIN improves DTI recognition ability in sparse settings[132]. KGE-UNIT shows that cross-task knowledge sharing can improve the effect of relationship prediction[133]. CBKG-DTI extracts complex network features through hierarchical embedding and knowledge distillation[134]. LKE-DTA further integrates protein sequences and biomedical knowledge by combining the sequence semantic representation of large language models with the embedding of knowledge graphs[135] (Fig. 4c). It should be noted that the predicted drug-target edge may only reflect the similarity, common annotation, literature co-occurrence or database curation rules, which still need to be verified by molecular docking, molecular dynamics, biophysical experiments, biochemical experiments, and cellular target engagement assays.
Structure-aware knowledge graph for small molecule discovery
-
Three-dimensional structure is a key factor in the discovery of small molecules. The physical binding depends on the geometric complementarity between the compound and the binding site as well as the physicochemical complementarity. It is difficult to judge whether the ligand can enter the pocket, form effective interactions, avoid steric clashes, or stabilize the functional conformation based on sequence similarity or graph proximity alone.
The structure-aware knowledge graph can connect chemical nodes, target nodes, protein structure nodes, binding pocket nodes, experimental nodes, mutation nodes, and disease nodes. As exemplified by target proteins, they can be linked to PDB experimental structures, predicted structures generated by AlphaFold2/3, ESMFold or RoseTTAFold, binding pockets, known ligands, drug-resistance mutations, allosteric sites, and bioactivity assays. Pocket volume, depth, hydrophobicity, electrostatic properties, flexibility, ligandability, and allosteric potential can also be added to the graph as features. Tools such as fpocket, SiteMap, and DoGSiteScorer[115] can provide relevant descriptors. This structural representation can distinguish 'biologically relevant targets' from 'chemically tractable targets'. Because the predicted structure is affected by flexible regions, alternative conformations, ligand-induced fit, membrane environment, and disordered regions, the structure-aware model should also incorporate structural confidence and combine docking, molecular dynamics, or experimental validation.
KG-guided molecular generation
-
After the ranking of candidate targets and potential interactions, drug discovery enters the phase of molecular generation and lead optimization. Knowledge graphs can connect target biology, pathway safety, off-target risk, structure-activity relationship, ADMET properties, toxicity signals, and disease background, provide constraints for molecular design, and make molecular generation pay more attention to therapeutic relevance than chemical novelty.
Previous studies have used external knowledge to guide molecular generation and reinforcement learning to focus the model on target-specific chemical space[136]. KGDiff introduces protein-ligand binding information into the diffusion denoising process. DiffPhore guides 3D conformation generation through pharmacophore matching and supports virtual screening and lead discovery[137] (Fig. 4d). KGG added chemical knowledge at the orbital level to graph self-supervised learning, showing potential in low-data scenarios[138]. However, the generated molecules may be chemically unreasonable, difficult to synthesize, unstable, difficult to purify, or approximate known compounds. High predicted binding affinity also does not necessarily translate into selectivity, cellular activity, permeability, metabolic stability, or safety.
Lead compound optimization and multi-parameter optimization
-
Lead compound optimization is not to improve the target binding ability alone, but to find a balance between activity, selectivity, solubility, permeability, metabolic stability, hERG risk, CYP-mediated metabolism or CYP inhibition, toxicity, and synthetic accessibility. Multi-parameter optimization (MPO) has become an important strategy to improve the quality of candidate compounds and reduce the risk of late failure[139].
Knowledge graphs can connect compounds, targets, off-target proteins, ADMET endpoints, metabolic enzymes, transporters, toxic phenotypes, and disease pathways, thus providing constraints for multi-parameter optimization. In the case of transporter protein knowledge graphs, the integration of AutoInt_KG and MolGPT_KG enables drug-transporter relationship prediction and transporter information-guided molecular generation[140]. Graph-based molecular Pareto optimization uses graph representation and the non-dominated sorting genetic algorithm for Pareto frontier optimization[141]. ChemMORT uses deep learning and multi-objective particle swarm optimization to optimize multiple ADMET endpoints, indicating that multi-objective ADMET optimization has become an important direction of computer-aided lead optimization[142]. Therefore, the value of knowledge graphs is not only to improve the accuracy of single-property prediction but also to help screen more balanced candidate molecules.
-
In translational drug discovery, target discovery should not rest on the ranking of candidate genes or proteins, but more importantly, on whether candidate targets can enter the validation cohort. Knowledge graphs can integrate disease association, genetic evidence, functional perturbation, pathway location, tissue expression, drug resistance, safety risk, and biomarker support around each target. In the case of target screening, targets with GWAS colocalization, disease-associated expression, perturbation evidence, and ligandable structures are generally more amenable to downstream research than those supported merely by literature co-occurrence or network proximity.
Previous studies have shown the possibility of the knowledge graph target hypothesis entering experimental validation. Rosalind predicts the disease-gene relationship based on heterogeneous knowledge graphs and tensor decomposition and validates candidate targets of rheumatoid arthritis in the experiment of patient-derived fibroblast-like synoviocytes, resulting in five potential genes[143]. Progeni constructed a probabilistic knowledge graph, and its top-ranked targets in melanoma and colorectal cancer were supported by knockdown experiments and pharmacological validation[104]. In idiopathic pulmonary fibrosis, Standigm ASK™ proposed AMFR, MDFIC, and NR5A2, and verified their functional relevance through siRNA knockdown experiments[144]. BenevolentAI identified baricitinib for COVID-19, and subsequent ACTT-2 and COV-BARRIER studies provided clinical evidence[145−147]. These cases show that the knowledge graph is more suitable for establishing a validation cohort, and further confirmation is still needed in the follow-up by CRISPR, RNAi, organoids, animal models, patient-derived samples, or pharmacological perturbation.
Hit discovery and DTI prediction
-
After the target ranking and preliminary validation, the next step is to find compounds or therapeutic methods that can regulate the target. DTI prediction based on a knowledge graph can sort candidate drug-target pairs, help less-studied targets find potential ligands, and suggest possible off-target interactions. DeepDTA[127], DeepAffinity[128], GraphDTA[129], DTINet[130], TriModel[131], DTKGIN[132], KGE-UNIT[133], CBKG-DTI[134], and LKE-DTA[135], which reflects that DTI prediction has gradually shifted from sequence and structure modeling to relational reasoning combined with biological background. The priority of candidate compounds should not only depend on predicted binding but also on the location of disease pathways, off-target risks, and experimental clues.
Lead optimization and molecular design
-
Lead compound optimization links computational molecular design and drug development. Real optimization can not only improve the target binding ability, but also consider the potency, selectivity, solubility, permeability, metabolic stability, clearance, hERG risk, CYP inhibition, toxicity, tissue distribution, and synthetic feasibility. Molecules that only pursue affinity may fail due to poor ADMET properties, off-target toxicity, insufficient selectivity, or difficulties in synthesis.
Knowledge graphs can connect compounds, chemical series, targets, off-target proteins, experiments, metabolic enzymes, transporters, toxicity endpoints, adverse events, pathways, and clinical risks and support multi-target scores and Pareto-frontier ranking. For molecular optimization, compounds with enhanced potency accompanied by hERG liability, CYP inhibition or hepatotoxicity demand deliberate prioritization, whereas those with moderate potency and superior druggable properties are more suitable for further optimization. The knowledge graph can also point out that the limiting factors are insufficient target affinity, off-target activity, metabolic instability, poor permeability, structural alerts, or pathway toxicity, thus providing directions for medicinal chemistry optimization.
Safety, ADMET, and risk assessment
-
The safety assessment should enter the target and compound prioritization as early as possible. Risks at the target level include target-related toxicity, critical organ expression, essential gene status, human genetic constraints, developmental functions, pathway redundancy, and disease background specificity. Compound-level risks include hERG risk, CYP inhibition, drug-induced liver injury, cardiotoxicity, genotoxicity, reactive substructure, and transporter-related risks (Fig. 4f).
Knowledge graphs can integrate FAERS[148], SIDER[149], CTD[150], DrugBank, ToxCast[151], Open Targets Safety[152], electronic health records, toxicological genomics data, and pathway annotations. Bean et al. constructed a knowledge graph containing drugs, targets, indications, and adverse reactions, and verified the high-confidence prediction with electronic health records[153]. Subsequent studies have improved ADR risk identification ability through representation learning[154] and extended it to multi-drug adverse reaction prediction[155]. MVDSA integrates multi-source knowledge, relational semantics, and multi-perspective features for joint association prediction[156]; graph representation and transformer model have also been used for multi-label ADR prediction[157].
ADMET evaluation focuses on absorption, distribution, metabolism, excretion, and toxicity. Machine learning frameworks are gradually replacing traditional empirical rules[158]. However, the pharmacovigilance database may have reporting bias, confounding indications, the impact of multiple drug combinations, and a lack of denominator information. Therefore, the associative signal is more suitable for interpretation as a risk association rather than a causal safety conclusion.
Drug repositioning and combination therapy
-
Drug repositioning aims to find new indications for approved or clinical-stage compounds (Fig. 4e). Existing drugs usually accumulate safety, pharmacokinetics, manufacturing, and clinical information, so repositioning may shorten the development cycle and reduce uncertainty[159,160]. PREDICT predicts indications based on drug-disease similarity[161]. During COVID-19, network proximity, network diffusion, and AI-based prioritization were used to screen drug candidates[162]. Rephetio integrates multiple biomedical entities and associations for systematic screening of drug-disease combinations[90]. OREGANO provides a standardized repositioning knowledge base[163]. DREAMwalk alleviates the problem of uneven gene distribution through semantically guided random walk[164]. TxGNN framed drug repositioning as a zero-shot prediction task and demonstrated the potential of ranking indications and contraindications[165]. Coronavirus-related research also used knowledge graphs to integrate literature, databases, and deep learning results to screen drug candidates[166,167].
Combination therapy needs to distinguish synergy prediction from DDI prediction. Synergy prediction focuses on whether multiple drugs can enhance therapeutic benefit; DDI prediction focuses on whether co-administration increases toxicity, changes pharmacokinetics, or produces unexpected pharmacodynamic effects. SynergyGraph combines cell-line background, knowledge graph representation, and hypergraph modeling[168]. HyperSynergyX integrates hypergraph modeling and knowledge graph-enhanced retrieval for mechanism interpretation[169]. MDDI-SCL[170], HGTSynergy[171], and Pisces[172] illustrate that synergy prediction and DDI prediction are moving towards joint modeling of shared knowledge. In DDI prediction, SumGNN[173], LaGAT[174], KG-CLDDI[175], Heterogeneous drug knowledge graph[176], predicting rich DDI[177], KA-DDI[178], and LLM-DDI[179], which reflects the fusion trend of graph structure, text information, and representation learning. Subsequent models also need to incorporate dose, administration time, tissue exposure, pharmacokinetics, pharmacodynamics, and toxicity overlap.
Biomarkers and precision medicine
-
Biomarker discovery and patient stratification aim to link molecular characteristics with diagnosis, prognosis, treatment response, and clinical decision-making. Knowledge graphs can integrate mutations, gene expression, proteins, pathways, drug responses, phenotypes, electronic health records, and clinical outcomes, and transform dispersed molecular signals into clinical hypotheses that are easier to interpret.
The Monarch Initiative integrates gene disease and phenotype information to provide the basis for cross-species biomarker discovery[180]. The Clinical Knowledge Atlas can connect proteomic changes with literature and pathway knowledge to form a more grounded biomarker hypothesis[181]. In non-small cell lung cancer, incorporating prior knowledge of knowledge graphs into machine learning can improve survival prediction and enhance candidate biomarker interpretability[182]. The patient-specific knowledge graph can integrate medical history, genetic variation, molecular characteristics, treatment exposure, and health outcomes to build an individualized representation[183]. PreMedKB integrates diseases, genes, variants, and drugs to provide a unified entry for the interpretation of evidence in precision medicine[184].
Biomarker evidence needs to be evaluated hierarchically. Retrospective association, cross-cohort validation, prospective validation, clinical utility assessment, and companion diagnostic approval represent different levels of evidence. Cohort bias, batch effects, population differences, overfitting, treatment heterogeneity, and endpoint definitions all affect the reliability of the results. Therefore, in the absence of prospective clinical validation, biomarker discovery supported by knowledge graphs is more suitable to be regarded as a patient stratification hypothesis.
Dynamic KGs and drug resistance
-
Most biomedical knowledge graphs are static, but drug responses and disease progression are dynamic processes. Under the pressure of treatment, tumors, pathogens, and diseased tissues may undergo genetic, epigenetic, transcriptional, protein, and microenvironment remodeling. This can be illustrated by drug-resistant mutations. EGFR T790M will change the sensitivity of kinase inhibitors in lung cancer. ALK G1202R is associated with ALK inhibitor resistance. BCR-ABL T315I will affect the response of a variety of tyrosine kinase inhibitors[185−187]. These mutations may change the binding pocket, reduce drug affinity, change the conformational state, activate bypass signals, or remodel downstream pathways.
The static knowledge graph can record the mutation drug-resistance association, but it is difficult to explain when the mutation appears, under what treatment pressure it is selected, and how the pathway activity changes. Dynamic knowledge graphs, temporal knowledge graphs, versioned knowledge graphs, longitudinal omics, single-cell perturbation data, and treatment response trajectories can represent changes in disease status, clonal composition, pathway activation, target dependence, and drug sensitivity. In the future, knowledge graphs need to move from static evidence integration to time-aware decision support, especially for tumors, infectious diseases, immune diseases, and chronic diseases.
-
In general, biomedical knowledge graphs can break through the barriers of multi-source heterogeneous data and build a multi-level computable knowledge network from molecular to clinical. At present, AI technology has promoted its automatic application and has shown obvious potential in target identification, drug target prediction, drug repurposing, and other tasks.
However, several key challenges remain in this area. First, the quality of multi-source data integration remains problematic. Biomedical knowledge is scattered in different fields, so it is difficult to integrate multi-scale and multi-modal data with high fidelity. In the case of GWAS data mapping, population bias may be introduced. Activity data from different platforms can also undermine edge weight confidence due to disparate experimental conditions[8]. In the future, it is necessary to establish a more stringent confidence evaluation mechanism in the data fusion stage[188], filter false positive associations by combining uncertainty modeling[189], and carry out automatic fact verification relying on domain ontology[190].
The second is that the generalization ability of the model is still insufficient. At present, reasoning modules are facing problems such as cold start and insufficient out-of-distribution generalization[191−193]. Methods such as transfer learning and zero-shot learning have been widely studied[194,195]. In the future, it is also necessary to combine the anomaly detection mechanism to improve the reliability of real-world deployment.
In addition, most current biomedical KGs are still static snapshots. This static nature is a fundamental limitation for resistance prediction and adaptive therapy design, because resistance is not a fixed attribute of a drug-target pair but an evolving process shaped by clonal selection, compensatory pathways, tumor microenvironment, drug pressure, and prior treatment history. A static KG may correctly encode that a drug inhibits a target, but it cannot represent when a resistant clone emerges, which bypass pathway becomes dominant, or how sequential therapy changes future sensitivity. As a result, predictions based only on fixed associations may lag behind real disease trajectories and may overestimate the durability of a candidate target or regimen.
Self-evolving KGs should therefore be defined more concretely than through continuous updating. They need timestamped entities and edges, versioned evidence, edge confidence that changes with new assays or clinical observations, and mechanisms for retiring contradicted relations. For adaptive treatment, this means representing patient-state transitions, therapy-induced perturbations, emerging resistance mechanisms, and feedback from longitudinal omics, imaging, electronic health records, and trial data. Temporal KG analysis during COVID-19 has shown how scientific understanding can change over time[196], and incremental embedding methods can maintain representations as graphs grow[197]. However, the key challenge is not only adding new triples but also enabling reasoning models to compare graph states before and after treatment and to recommend interventions that remain valid under evolving biology.
Finally, privacy protection and federated knowledge graphs are the key to breaking the institutional data silos. Privacy and compliance constraints keep a large amount of data inside the organization, while federated KG can support inter-agency collaboration without disclosing the original data. Existing studies have verified its effectiveness[198−200]. In the future, it is still necessary to optimize the model architecture, improve the performance of decentralized reasoning, and support secure cross-agency sharing.
With the continuous breakthroughs in these directions, biomedical knowledge graphs are expected to further promote the implementation of AI drug discovery and provide more powerful support for drug discovery and development, and precision medicine.
-
Not applicable.
-
The authors confirm their contributions to the work as follows: conceptualization: Li S, Li H; manuscript writing: Dou P, Yang X; manuscript revision: Jiang Y, Xie X, Geng X, Huang H. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing is not applicable to this review as no datasets were generated or analyzed.
-
This work was supported in part by the National Natural Science Foundation of China (82425104 to Honglin Li), the National Key R&D Program of China (2022YFC3400504). Shiliang Li is also sponsored by the Shanghai Rising-Star Program (23QA1402800).
-
The authors declare that there is no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/targetome-0026-0025.
-
#Authors contributed equally: Pan Dou, Xiaobo Yang
- Supplementary Table S1 Common data sources for biomedical KG construction.
- Supplementary Table S2 Representative biomedical knowledge graphs.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Dou P, Yang X, Jiang Y, Xie X, Geng X, et al. 2026. Biomedical knowledge graphs driving new target identification and novel drug discovery. Targetome 2(3): e026 doi: 10.48130/targetome-0026-0025 

Biomedical knowledge graphs driving new target identification and novel drug discovery
- Received: 27 April 2026
- Revised: 31 May 2026
- Accepted: 03 June 2026
- Published online: 25 June 2026
Abstract: Biomedical knowledge graphs (KGs) provide a structured and traceable framework for target-oriented, AI-assisted drug discovery. By linking genes, proteins, diseases, phenotypes, compounds, pathways, assays, structures, adverse events, and clinical evidence, biomedical KGs can partially mitigate evidence fragmentation and support hypothesis generation across molecular, cellular, and clinical levels. This review summarizes how biomedical KGs are constructed, reasoned over, evaluated, and translated into practical drug-discovery tasks. We first outline major data resources, entity and relation extraction, knowledge fusion, quality control, and large language model-assisted KG construction, with emphasis on ontology grounding, evidence tracing and hallucination control. We then compare key KG reasoning paradigms, including symbolic rule-based reasoning, representation learning, path- and subgraph-aware reasoning, graph Transformer models, and KG-LLM hybrid reasoning. Their strengths and limitations are discussed in relation to interpretability, scalability, leakage risk, benchmark bias, and prospective validation. We further highlight the need to incorporate three-dimensional structural evidence, including protein structures, binding pockets, variants, conformational states, and ligand compatibility, so that graph-based relevance can be connected with chemical tractability. Finally, we discuss applications of biomedical KGs in target prioritization, druggability assessment, mechanism interpretation, drug-target interaction prediction, molecular generation, lead optimization, ADMET and safety assessment, drug repurposing, combination therapy, biomarker discovery, precision medicine, and drug-resistance modeling. Current limitations include heterogeneous data quality, incomplete causal evidence, static graph representations, privacy constraints, and insufficient experimental or clinical validation. Overall, biomedical KGs should be viewed as evidence-integration and prioritization engines rather than automatic proof-generating systems for drug discovery, guiding rigorous, iterative, and experimentally grounded translational decisions.
-
Key words:
- Biomedical knowledge graph /
- Drug discovery /
- Target identification /
- Knowledge reasoning