








-
As the most abundant form of carbohydrates in nature[1], polysaccharides consist of more than 10 monosaccharide units that are linked by glycosidic bonds either in the form of sugar residues or covalently bonded to other structures like peptides, amino acids, and lipids[2,3]. Polysaccharides, which belong to the third major class of biopolymers, play critical roles in various biological and physiological activities, including antioxidant, anti-inflammatory, antitumor, and immunomodulatory activities[4,5]. Consequently, a variety of polysaccharide-based drugs have been developed, as summarized in Table 1. In addition, polysaccharides have been extensively used in diverse other applications, particularly in drug delivery systems as drug carriers[6,7], building blocks and functional excipients[8−10], as well as in tissue engineering[11], cosmetics[12], and wound healing[13]. The functional diversity of polysaccharides is rooted in their highly complex molecular structures, which also present significant research challenges.
Table 1. Approved natural product-derived polysaccharide drugs.
Category Generic name Source Indications Year Country Adjuvant therapy Lentinan Lentinus edodes mycelium/fruiting body Gastric cancer 1985 Japan Polysaccharide K Trametes versicolor (CM-101 strain) mycelium Gastric and colorectal cancer 1998 Japan Sizofiran Schizophyllum commune mycelium Cervical cancer 1986 Japan Poria cocos polysaccharide Poria cocos sclerotium/mycelium/fruiting body Cancers and Hepatitis B 2005 China Astragalus polysaccharide Astragalus membranaceus root Leukopenia/cancer-related fatigue 2001 China Polysaccharopeptide Trametes versicolor (COV-1 strain) mycelium Gastric and lung cancer 1970s China; Japan Fucoidan Brown seaweed (Phaeophyceae) thallus Cancers and immunodeficiency 2003 China Ganoderma lucidum polysaccharide Ganoderma lucidum fruiting body/mycelium/spore Cancers/immunodeficiency 2000 China Polyporus umbellatus polysaccharide Polyporus umbellatus sclerotium/mycelium/fruiting body Lung cancer/Hepatitis B ~1990 China Panax ginseng polysaccharide Panax ginseng root Leukopenia and immunodeficiency 2006 China Phellinus linteus polysaccharide Phellinus linteus fruiting body/mycelium Gastrointestinal cancer 1993 South Korea Tremella fuciformis polysaccharide Tremella fuciformis fruiting body/mycelium/spore Chemotherapy-induced leukopenia 2002 China Plasma volume expander Dextran 40/70 Leuconostoc mesenteroides (NRRL B-512F strain) fermentation broth Hypovolemia 1953;1962 USA Hydroxyethyl starch Modified waxy maize starch/potato starch Hypovolemia 2000 Germany Chondroitin sulfate Bovine, porcine, chicken, and shark cartilage Osteoarthritis 1983 Switzerland Symptomatic management Heparin sodium Porcine intestinal mucosa/bovine lung Anticoagulant 1939 USA Sodium alginate Brown seaweed (Phaeophyceae) thallus/bacterial fermentation broth Gastroesophageal reflux disease 1961 Japan Sodium hyaluronate Streptococcus equi fermentation broth/Avian (rooster combs) Osteoarthritis and dry eye 1987 Japan Compared with the remarkable progress in protein and nucleic acid research, polysaccharide research has long lagged behind due to the intrinsic chemical complexity, physical entanglement within natural matrices, and unclear interactions with biological targets. Purified polysaccharides are not chemically uniform entities, but rather complex mixtures of structurally similar yet non-identical homologs, which exhibit microheterogeneity in their monosaccharide composition, linkage patterns, and molecular weight distributions[14,15]. In contrast to the uniform linkage between two amino acids, the linkage between two identical hexoses can theoretically generate dozens of distinct disaccharides due to their multiple reactive hydroxyl groups and variable anomeric configurations (α/β). Such a combinatorial explosion is further magnified by increasing degrees of polymerization, intricate branching, and extensive post-synthetic modifications such as sulfation, phosphorylation, and acetylation[16−18]. Additionally, the polar glycan chains of polysaccharides exist as highly dynamic and heterogeneous conformational states in solution[19,20], which limits the applicability of traditional high-resolution structural determination methods such as X-ray crystallography and nuclear magnetic resonance (NMR)[21,22]. Meanwhile, polysaccharides are typically in the form of complex supramolecular networks, associated with lignin, lipids, proteins and nucleic acids within plant cell walls[23], the extracellular matrix of animal cells[24], and fungal capsules[25]. Despite their effectiveness in releasing polysaccharides from biological matrices, harsh extraction conditions, including treatment with strong alkali, prolonged heating, and high-power ultrasound, risk disrupting native conformations, randomly cleaving glycosidic bonds, and irreversibly removing labile bioactive side-chain modifications such as sulfate groups[26]. Conversely, mild conditions fail to break the crosslinked matrix, resulting in ineffective extraction of polysaccharides. Distinct from nucleic acids and proteins, polysaccharides are biosynthesized in a template-independent manner and consequently lack any mature in vitro enzymatic amplification technology for the precise replication and large-scale production of a desired polysaccharide[27]. Further structure-activity relationship (SAR) analysis is therefore directly limited by low-yield and heterogeneous polysaccharides extracted from natural biomass[28].
Traditional research methods are severely limited by the inner structural heterogeneity of polysaccharides, usually generating extensive and multidimensional data space characterized by the nonlinear interactions of multiple extraction variables, whether in extraction optimization or structural analysis. Therefore, conventional strategies have greatly relied on empirical and stepwise experimentation, frequently leading to time-consuming, labor-intensive but still unsatisfactory results[29,30]. In addition, the structural information derived from these methods is mainly encoded in machine-unreadable formats, including static images or human-readable but non-standardized textual notations, making computational integration and large-scale analysis difficult. While decades of research have indeed generated a wealth of experimental data on polysaccharides, the majority of these valuable findings are unfortunately trapped in machine-unreadable formats, leaving the SARs poorly understood and the biomedical potential of polysaccharides largely unrealized[31,32].
AI has been emerging as a transformative force, offering a paradigm shift from conventional experimental approaches to data-driven extraction, structural elucidation, bioactivity prediction, and even intelligent design of polysaccharides[33]. AI development can be divided into three historical stages. It began with symbolic AI, featuring rule-based expert systems and logic programming. The second stage marked the emergence of statistical methods and machine learning (ML), including support vector machines (SVM), random forests (RF), and Bayesian methods. We are currently in the deep learning (DL) era, characterized by neural network expansion through big data and Graphics Processing Unit (GPU) acceleration[34,35]. While symbolic AI achieved early success in protein and nucleic acid research with rule-based approaches such as the Chou–Fasman method and software platforms like IntelliGenetics[36−39], its application to polysaccharides remained remarkably limited given the mismatch between the need for explicit, predefined knowledge and the unpredictable structural features of polysaccharides. By contrast, ML and DL are capable of learning implicit patterns directly from data, enabling precise modeling of nonlinear, multifactorial relationships, interpretable analysis of variable importance, and integration of high-dimensional information from diverse spectroscopic and chromatographic data. AI therefore facilitates the entire workflow of polysaccharide research, spanning extraction optimization, structural characterization, and functional prediction. As the Nobel Prize-winning achievement in AI, AlphaFold 2 has achieved a breakthrough in predicting protein three-dimensional structures directly from amino acid sequences[40]. AlphaFold 3 has been recently extended to glycan modeling, demonstrating its ability to generate stereochemically valid glycan structures and predict their interactions[41]. However, it should also be noted that these models are basically static snapshots requiring careful interpretation. Confidence metrics remain blind to stereochemical errors, and complementary methods such as molecular dynamics simulations are still required to capture glycan dynamics.
This review provides a comprehensive analysis of AI applications in polysaccharide research. The field of polysaccharide research is undergoing a major transformation driven by AI, from repetitive manual benchwork to data-driven intelligence. Through examining AI's role in distinct stages of polysaccharide research from extraction to functional discovery, this work aims to provide valuable insights for researchers and practitioners in related fields, supporting the development of polysaccharide-based products for biomedical and pharmaceutical applications (Fig. 1).
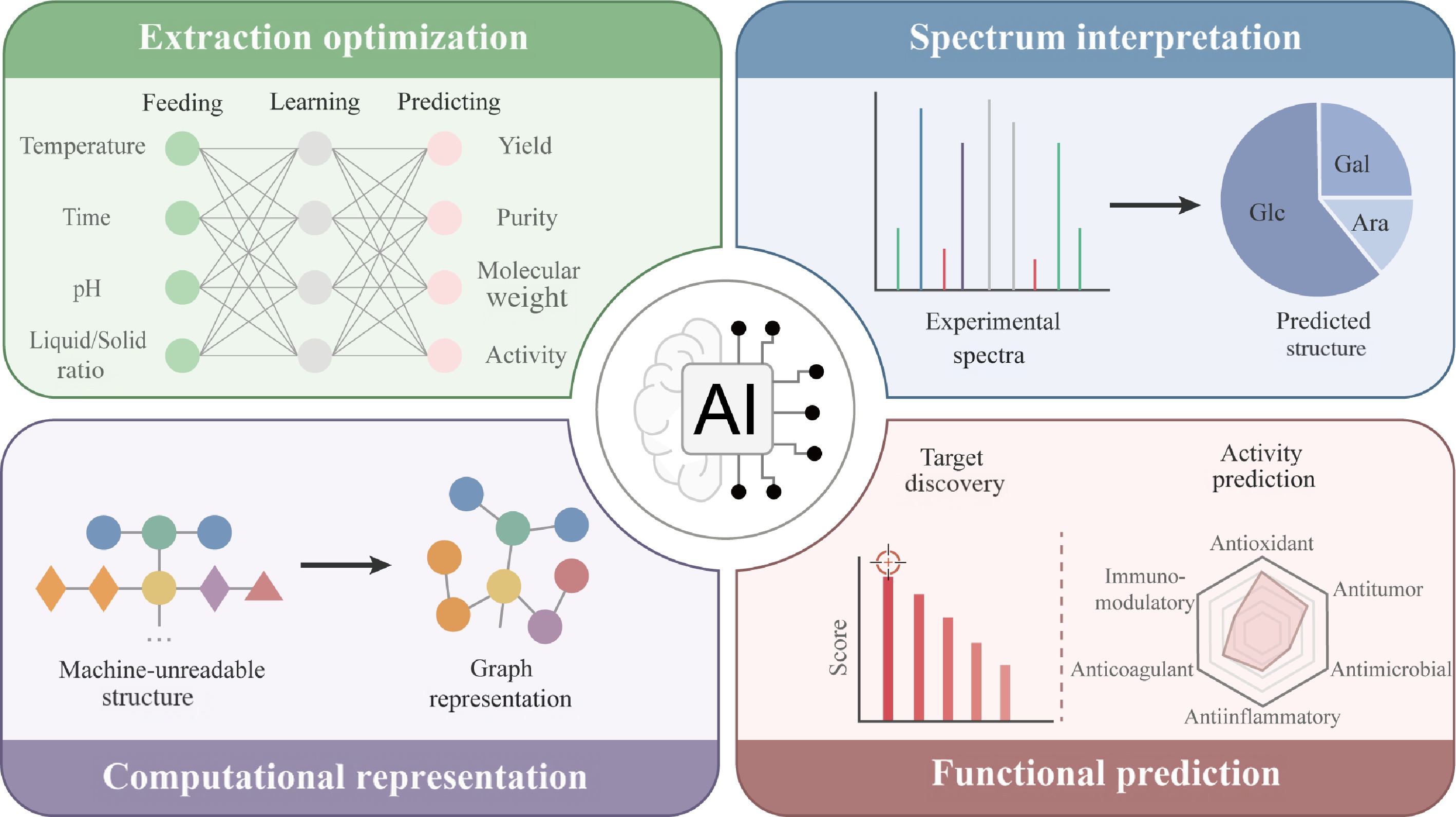
Figure 1.
AI powered stages in polysaccharide drug development. The diagram summarizes the integration of AI methodologies across the pipeline of polysaccharide-based drug discovery and development, highlighting how data-driven approaches accelerate each stage from raw material processing to bioactivity prediction.
-
Given that almost all methods for polysaccharide extraction fundamentally rely on the solvent to dissolve and release target polysaccharides from solid biomass, the solvent selection is critical as the first step for obtaining bioactive polysaccharides with higher yield and purity[42]. Deep eutectic solvents (DESs) have emerged as a highly promising green alternative. Typically formed by combining a hydrogen-bond acceptor (HBA) with a donor (HBD) at a specific molar ratio, DESs greatly enhance extraction by establishing intermolecular hydrogen bonds to solubilize polysaccharides and competitively disrupting the hydrogen-bond network within cell walls[43,44]. The choline chloride-malic acid (ChCl-MA) system achieved a yield more than three times higher than that of conventional hot-water extraction for Auricularia auricula-judae polysaccharides[45]. In order to increase mass transfer efficiency, ternary deep eutectic solvents (TDESs) have been recently introduced to address the high viscosity of binary DESs[46]. DESs offer tunable polarity, making it easier to match the hydrophilicity of target polysaccharides. However, with hundreds of commonly used HBAs and HBDs, each adjustable in molar ratio and water content, the number of potential DESs can reach tens of millions[47]. Even restricting the choice to four components from a library of 100 yields over 4 million distinct combinations, a scale at which conventional experimental screening becomes effectively impractical. Additionally, such conventional methods are mainly focused on macroscale parameters like extraction yield and crude polysaccharide properties, thus failing to reveal underlying microscopic mechanisms[48].
The integration of quantum-mechanical computational tools with machine learning (ML) has shifted solvent optimization from repetitive manual experimentation toward predictive, rational design. For lentinan extraction, a theoretical screening of 372 potential natural DESs was performed using the Conductor-like Screening Model for Real Solvents (COSMO-RS), which was originally developed in 1995[49] and later refined and parametrized in 1998 by Klamt and his team[50]. This model predicts thermodynamic properties, including solubility from quantum-chemically derived surface charge distributions and chemical potentials, enabling high-throughput in silico screening. Employed by Wang et al., the model successfully predicted the relative solubility of lentinan, thereby identifying the carnitine-urea-water ternary system as the most promising solvent[51]. In contrast to single-model screening, Deng et al. developed a synergistic strategy, training the artificial neural network (ANN) with the molecular descriptors derived from COSMO-RS to predict solvent pH. Choline chloride-sorbitol was ultimately identified as the top-performing DES for extracting Camellia oleifera polysaccharides[52]. It should be noted that the optimal composition depends heavily on the specific objective function, such as prioritizing yield over purity and the thermodynamic model's assumptions, which may not fully capture the kinetic mass transfer limitations of complex cellular matrices. Similarly, Takuya Uto has established an intelligent screening platform integrating molecular dynamics (MD) simulations and ML to efficiently predict and rank the performance of over 3,000 imidazolium-based ionic liquids for cellulose and chitin. Molecular mechanisms such as hydrogen bond cleavage and ion adsorption during cellulose and chitin dissolution in ionic liquids are first extracted by MD, revealing the quantitative correlation between intermolecular hydrogen bond count and the experimentally measured solubility, which is further used as fundamental training data for an ML model to predict the solvation capability of entirely new ionic liquid candidates based solely on chemical structures[53]. However, reliable prediction requires that new candidates stay within the chemical space of the training set and that the MD force fields used to generate the training data are accurately parameterized for the functional groups present in those candidates.
Global optimization of extraction conditions
-
Polysaccharide extraction is often the defining step for yield and quality[54]. Conventional optimization mainly relies on single-factor experiments, orthogonal designs, or response surface methodology (RSM). While RSM can model interactions between variables, its reliance on local second-order polynomial approximations often leads to poor generalization and a typically low R2 between 0.7 and 0.8, especially when applied to multi-variable dynamic processes such as polysaccharide extraction[55]. Such methods are also limited to local optimization within predetermined experimental points, possibly missing the global optimum. Meanwhile, traditional strategies are mostly focused on single-objective optimization such as maximizing yield, making it difficult to effectively balance various performance parameters like bioactivity, energy consumption, and cycle time. The initial design space is also heavily dependent on experience, which inevitably results in a lack of data-driven learning and thus severely limits the capability of performing multi-objective and high-throughput optimization. AI can offer a solution to these bottlenecks with adaptive learning, global search capabilities, and superior nonlinear fitting performance[56]. However, typical data sources for such AI models are matrices derived from conventional experimental design, which frequently produce small datasets with inherent measurement noise. High-dimensional process-monitoring data, such as time-resolved Raman spectroscopy, are also being explored to enhance model transferability across dynamic extraction conditions. Given the typically small sample sizes, rigorous resampling strategies are essential to prevent overfitting. K-fold cross-validation coupled with grid search is widely adopted to stabilize generalization error estimates, while leave-one-out cross-validation (LOOCV) is preferentially used when the dataset is extremely scarce, as it yields an almost unbiased evaluation of predictive capability. Beyond conventional R2 and mean absolute error (MAE), the root mean square error (RMSE) is emphasized because its quadratic penalty on large deviations serves as a critical indicator for detecting potential catastrophic failures in industrial batch processing. Meanwhile, the mean absolute percentage error (MAPE) provides a scale-independent assessment, enabling fair comparisons of model robustness across diverse biological matrices and extraction systems. Collectively, these validation strategies and metrics ensure that AI-driven optimization is not merely memorizing the training data but faithfully learning the underlying extraction principles required for industrial translation.
Meanwhile, despite the remarkable success of AI-driven models in optimizing polysaccharide extraction at the laboratory scale, their direct application to industrial production remains challenging. When scaled up from a small laboratory reactor to a multi-ton system, physical constraints that are negligible in the lab become decisive. For example, the limited penetration depth of microwave irradiation inevitably creates severe thermal gradients inside large reactors, overheating the periphery while leaving the core under-extracted. Ultrasonic energy also decays exponentially with distance, and suspended solids further scatter acoustic waves, so the homogeneous cavitation field assumed by lab-trained models cannot be maintained. Variability among different batches of natural feedstocks arising from genetic, geographic, seasonal, and post-harvest processing differences further adds unpredictability. Static machine learning models such as artificial neural networks and random forests, which excel at interpolation within their training domain, lack the physical awareness required for robust extrapolation across scales and thus cannot automatically accommodate such variability. Bridging this gap between laboratory and industrial scales therefore calls for a shift from purely data-driven modelling to hybrid approaches that embed physical laws like heat and momentum conservation into the learning framework, for example, physics-informed neural networks, combined with real-time process analytical technologies that enable adaptive control based on the actual state of the extraction system.
Artificial neural network (ANN) and genetic algorithm (GA)
-
ANNs are the most widely used AI technology for parameter optimization in polysaccharide extraction, dealing with the complicated interactions among multiple factors by establishing complex nonlinear mappings. Among these, the Multilayer Perceptron (MLP) trained with the backpropagation (BP) algorithm is currently the most common architecture, which typically consists of an input layer, one or more hidden layers, and an output layer[57]. Each neuron in a hidden layer computes a weighted sum of its inputs, then applies a nonlinear transformation typically via a Sigmoid or ReLU activation function. The BP algorithm iteratively adjusts the weights across layers based on the error between predicted and actual outputs, which allows the network to capture complex, nonlinear relationships in polysaccharide extraction. For example, Dos Santos et al. built a feedforward neural network with a single hidden layer (four neurons) to predict hemicellulosic sugar yields from spent coffee grounds under dilute acid hydrolysis[58]. Inputs were temperature, acid concentration, solid-to-liquid ratio, and reaction time. Trained with the Levenberg–Marquardt algorithm, the MLP achieved high accuracy with an R2 of 0.99 across training, validation, and test sets, with an average error of 9.20%, effectively modeling the complex hydrolysis process.
However, the MLP-based ANN remains a static prediction model. It forecasts yield accurately for any given set of parameters but does not actively search for the optimal set. Manual effort is thus still required to locate the optimal condition through a passive screening across the entire parameter space, risking identifying a local optimum. In practice, ANN is typically paired with an optimization algorithm. Inspired by the principles of natural selection and survival of the fittest, GA is a powerful population-based heuristic for global optimization. GA encodes key parameters like temperature, time, and the solvent-to-material ratio into chromosomes within a population. A fitness function, defined to reflect extraction performance, drives iterative refinement through selection, crossover, and mutation, converging efficiently on optimal conditions. In the ultrasonic-assisted extraction of polysaccharides from Polygonum perfoliatum L., a GA-BP model was constructed with liquid-to-solid ratio, extraction time, extraction temperature, and ultrasonic power as input variables, and the comprehensive score of polysaccharide extraction based on transfer rate and purity was set as the output target. The GA model was configured with an initial population size of 30, a maximum of 50 iterations, a crossover probability of 0.8, and a mutation probability of 0.2. Through iterative evolutionary optimization, the optimal conditions were determined as a liquid-to-solid ratio of 35 mL·g−1, an extraction time of 29 min, an extraction temperature of 72 °C, and an ultrasonic power of 415 W. The experimental comprehensive score reached (96.96% ± 6.23%) using these parameters, significantly surpassing the result obtained from traditional RSM optimization (85.27% ± 4.38%)[59]. It must be noted that the superiority of GA-BP neural networks depends on rigorous hyperparameter tuning and sufficient data volume to prevent overfitting. In systems with limited data or lower non-linearity, RSM may still offer more robust generalization.
Beyond the MLP-BP architecture, the Radial Basis Function Neural Network (RBFNN) is also frequently integrated with GA. In contrast to the global nonlinear transformations of MLP-BP, RBFNN uses localized radial basis functions, primarily including Gaussian, allowing for a rapid two-phase training of unsupervised clustering followed by linear regression. Consequently, for low-dimensional extraction processes, RBFNN offers faster training and higher fidelity in local approximation compared with MLP-BP. But the advantage is highly dependent on the optimal selection of Gaussian spread parameters, and its performance may degrade in high-dimensional feature spaces due to the curse of dimensionality, where MLP-BP models typically scale more effectively. For instance, in a study focused on the extraction of polysaccharides from a traditional Chinese medicine formula, researchers trained an RBFNN to accurately characterize the effects of critical process parameters such as ethanol concentration, extraction time, and solid-to-liquid ratio on multiple performance indicators. Considering the competing objectives, including polysaccharide yield, molecular weight distribution, and the content of bioactive compounds like schisandrin, the Non-dominated Sorting Genetic Algorithm II (NSGA-II) was introduced as a multi-objective optimizer with the RBFNN model embedded in its evaluation loop to guide the evolutionary search. The optimal process conditions were an ethanol concentration of 60%, an extraction time of 1 h, and a solid-to-liquid ratio of 1:10 (w/v). Under these conditions, total sugar content increased by 14.13% relative to the conventional orthogonal design, hesperidin content by 22.03%, and extraction time was reduced by one-third[60].
While the ANN-GA strategy exhibits powerful fitting capabilities for highly nonlinear, multi-objective optimization cases, particularly when the extraction parameters are governed by complex dynamic interactions that place the process well beyond the reach of conventional polynomial models, it is critically dependent on the availability of large-scale, high-quality training data. Without sufficient training data, the network tends to overfit experimental noise because its vast parameter space, combined with empirical risk minimization, offers no inherent mechanism to distinguish random error from genuine patterns. In contrast, support vector regression, with its structural risk minimization and reliance on a sparse set of support vectors, can often maintain more stable predictions when applied to limited datasets. Furthermore, the weight matrices of an ANN carry no direct physicochemical meaning, depriving the model of mechanistic interpretability. The highly non-convex nature of its loss landscape also makes the final solution sensitive to weight initialization, with different training runs potentially converging to substantially different predictions and thus complicating reproducibility. Hence, ANN-GA should be preferred when an ample corpus of high-quality data is available and maximizing predictive accuracy stands as the central objective. In early-stage laboratory explorations where data are usually limited, the potential instability and lack of interpretability call for careful deliberation.
Support vector regression (SVR) and particle swarm optimization (PSO)
-
SVR, a supervised learning method designed for regression tasks, handles nonlinear relationships across multiple inputs against a continuous output, even when experimental data are limited. Unlike ordinary linear regression, which minimizes pointwise errors across all data points, SVR seeks a function that balances structural complexity with accuracy within a predefined ε-insensitive margin. The result is a sparse model defined only by critical data points, known as the support vectors. This sparsity improves robustness against overfitting and experimental noise. Furthermore, kernel functions, typically the radial basis function (RBF), implicitly map inputs into a higher-dimensional feature space, allowing a linear model to capture the nonlinearities inherent in extraction processes. However, its performance is highly reliant on the selection of hyperparameters, chiefly including the regularization parameter C, the kernel parameter γ, and the margin ε. Efficient optimization algorithms such as Particle Swarm Optimization are therefore required to identify the optimal configuration for a given dataset. PSO simulates the social foraging behavior of bird flocks or fish schools through a population-based search. Each particle adjusts its position by balancing its own best solution (pbest) and the swarm's global best solution, efficiently locating the optimal configuration of C, γ, and ε.
In the ultrasound-assisted extraction of Rosa laevigata polysaccharides (RLMP), a PSO-SVR model was trained to optimize critical parameters including ultrasound time, liquid-to-solid ratio, ultrasonic power, temperature, and particle size. The PSO algorithm was configured with the following hyperparameters: a population size of 50 particles, a maximum of 50 iterations, an inertia weight of 0.8, and both cognitive and social learning factors (c1, c2) set to 2. After the PSO search for tuned SVR hyperparameters, the globally optimal process combination identified by the refined SVR model was an ultrasound time of 34 min, a liquid-to-solid ratio of 19 mL·g−1, an ultrasonic power of 180 W, a temperature of 41 °C and a particle size of 0.355 mm, leading to an improved experimental extraction yield of 11.07% which closely matched the predicted value and was significantly higher than the RSM-optimized yield[61]. While this specific configuration outperformed the RSM-optimized yield within this dataset, the model's performance and its identified optimal conditions are strictly dependent on the predefined data ranges and experimental settings and, therefore, may shift if the initial parameter boundaries are altered.
The SVR-PSO model is defined solely by a subset of critical support vectors rather than by all data points, making it naturally insensitive to outliers and experimental deviations. It thus tends to yield predictions that are more reliable and reproducible than those attainable with an over-parameterized ANN when the dataset is limited. Such robustness, however, comes at the cost of acute sensitivity to hyperparameter configuration. The penalty coefficient C governs the trade-off between empirical risk and model complexity; the kernel parameter γ shapes the distribution of data in the high-dimensional feature space, and the insensitive margin ε directly controls the sparsity of the support vector set. Their joint influence determines the model's ultimate performance. Although PSO can automate the search for suitable hyperparameters, the population-based iterative process itself introduces considerable computational overhead. Moreover, the computational complexity of SVR grows super-linearly with the number of samples, making it far less efficient than tree-based ensemble methods that exploit divide-and-conquer strategies when confronted with large-scale industrial datasets, and the choice of kernel function, in the absence of sufficient prior knowledge, presents another practical hurdle.
Random forest (RF) and XGBoost
-
Ensemble learning methods based on decision trees like RF and eXtreme Gradient Boosting (XGBoost) are characterized by their proficiency in modeling structured tabular experimental data, making them particularly suited for AI-driven parameter optimization in polysaccharide extraction. Combining bootstrap aggregating with random feature selection, the RF algorithm achieves higher predictive accuracy and improved generalization by controlling model overfitting. RF builds a collection of decorrelated decision trees, each trained on a distinct bootstrap sample, with a random subset of process variables considered at each node. This dual-randomness strategy diversifies the individual trees, and aggregating their outputs yields stable, robust predictions. XGBoost, by contrast, is a sequential ensemble method based on gradient boosting. It constructs trees in a stagewise additive manner: the first tree predicts the target variable (e.g., yield), and each subsequent tree fits the negative gradient of the loss function relative to the current ensemble's predictions. XGBoost also incorporates regularization terms directly into its objective function to penalize model complexity, which improves generalization and often delivers higher predictive accuracy than RF[62]. Both RF and XGBoost can quantify variable importance by measuring how much each input contributes to error reduction across trees, allowing researchers to identify not only optimal parameters but also influential interactions among variables.
In a study on optimizing selenium-enriched Yacon-apple juice fermentation, key process parameters were identified as 34.8 °C, a 1:2.2 apple-to-yacon ratio, and 0.65 g·L−1 enzyme addition by integrating RSM and ML modeling, including RF and XGBoost. Meanwhile, XGBoost demonstrated superior predictive accuracy, achieving an R2 of 0.953, which outperformed the traditional RSM (R2 = 0.872), SVR (R2 = 0.901), and RF (R2 = 0.928) in capturing the complex process dynamics, resulting in high bioactive yields including 1.4942 mg·mL−1 polysaccharides[63]. However, tree-based algorithms are mathematically incapable of extrapolation due to their reliance on piecewise constant functions. Their superiority is strictly limited to interpolative predictions, whereas continuous function models like RSM remain necessary for trend projection outside the training domain.
RF and XGBoost both offer strong predictive performance on structured tabular data, but they excel in distinct tasks. RF, with its dual randomness of bootstrap aggregation and random subspace selection, is suited for exploratory data analysis and assisting mechanistic interpretation. The feature importance rankings generated by RF provide a quantification of the relative contributions of temperature, time, solid-to-liquid ratio, and other process variables, supplying direct evidence for identifying dominant factors. XGBoost, however, incorporates regularization terms directly into its objective function and adopts sequential gradient boosting, aiming to approach the predictive accuracy limit within the observed data distribution. Therefore, it frequently achieves the highest R2 values in complex fermentation kinetics or multi-stage extractions. Such performance advantage, however, is accompanied by a high-dimensional hyperparameter space including learning rate, tree depth, column subsampling, minimum leaf weight, and numerous other parameters that interact in intricate ways, requiring both computational resources and domain-specific machine learning expertise. Meanwhile, limited by the hard-partitioning mechanism of decision trees, both methods struggle to generalize beyond the training data range, in notable contrast to the continuous function mapping of ANN, which can at least offer trend-based inferences that carry indicative value. XGBoost and RF are therefore the preferred choices when the goal is precise optimization within a known process space or identification of key influencing factors. When the task shifts to extrapolation, these tree-based methods are best complemented by approaches with continuous mapping capabilities, each serving as a cross-check against the other.
-
The biological activity of natural polysaccharides depends on their complex, multi-layered structure (primary, secondary, and higher-order structures). Polysaccharide structures are usually characterized by NMR, gas chromatography-mass spectrometry (GC-MS), and Fourier transform infrared spectroscopy (FT-IR)[64]. Traditional characterization methods face challenges such as severe signal overlap, time-consuming data processing, and excessive reliance on expert experience[65,66]. AI can effectively process multiple structural pieces of information at the same time, rapidly extract features from complex raw data, reduce the dependence on human experience, improve data processing efficiency, and reveal the structure-activity relationship by analyzing the structure of polysaccharides, providing strong technical support for the design and optimization of polysaccharide structure.
Application of AI in Fourier transform infrared spectroscopy (FT-IR) for polysaccharide structure analysis
-
Fourier transform infrared spectroscopy is the key technology of polysaccharide structure analysis. It reveals the absorption peaks of characteristic functional groups and chemical bonds in polysaccharide molecules at specific wave numbers, thus inferring the backbone characteristics of sugar chains and substituents. The infrared spectrum of polysaccharides is usually divided into two regions: the functional group region (4,000–1,000 cm–1) and fingerprint region (1,000–400 cm–1). The functional group region reflects the basic structural characteristics of polysaccharides, while the fingerprint region reflects the core information of polysaccharide glycosidic bond configuration. However, due to the dense and highly overlapping original signals in the infrared spectra of polysaccharides and their sensitivity to baseline drift, traditional manual analysis is very difficult to capture subtle changes in the structure of functional groups[67,68]. Compared with the traditional manual interpretation of infrared spectra, applying AI and DL models to infrared spectra can analyze the peak patterns in the whole spectrum, rather than focusing only on specific regions. Researchers from the Anhui Institute of Optical Fine Mechanics and Physics proposed a DL model based on information density adaptive band selection (ID-ABS). The model can dynamically evaluate the information density distribution in the whole spectrum, optimize the inversion parameters of each component, determine the optimal inversion band, and finally update the parameters through nonlinear multiple regression iteration until convergence. This model provides the possibility of using FT-IR to analyze the full band structure of polysaccharides[69]. In the study of chitosan in two kinds of red mushrooms, the FT-IR spectrum of chitosan was processed by combining the second derivative spectrum and Fourier self-deconvolution spectrum (FSD spectrum). This processing improves the resolution of the spectrum and can determine the wave position of the superimposed peak. By further combining this with curve fitting analysis, researchers successfully separated the characteristic absorption peaks involved in the superposition[70]. In the study of Polygonatum polysaccharides, ATR-FTIR spectroscopy combined with convolutional neural network (CNN), partial least squares discriminant analysis (PLS-DA), and support vector machine (SVM) was used to extract information from the characteristic band screening and data fusion. Adaptive feature learning was completed through CNN, thus establishing a correlation model between spectrum and polysaccharide content, enabling both variety identification and polysaccharide content prediction[71].
Application of AI in the analysis of polysaccharide structures using nuclear magnetic resonance (NMR) technology
-
In polysaccharide structure analysis, chemical shift and coupling data obtained by nuclear magnetic resonance spectroscopy are important for determining glycosidic bond linkages, isomer sites, and stereoconfigurations of terminal carbons[72,73]. However, NMR technology has always suffered from severe signal overlap, complex spectra due to conformational flexibility, and high cost of manual analysis, which has prevented NMR technology from being fully applied in structural analysis[74]. CASPER (Computer Assisted Spectrum Evaluation of Regular polysaccharides) is a computer program specifically designed for the automatic interpretation of NMR spectra of oligosaccharides and polysaccharides. It can process experimental signals that are not fully identified in NMR spectra and compare them with simulated spectra generated by the program. When combined with the Pure Shift NMR method, this program has achieved a leap from 'human experience-based attribution' to 'computer-aided automatic resolution', successfully resolving typical HMOs such as 2'-FL (2'-fucosylated lactose), 6'-SL (6'-sialylated lactose), and LNnT (lactose-N-neotetrasaccharide), solving the problem of signal attribution for glycan isomers with severe overlap[75,76]. The successful application of the CASPER program in oligosaccharide structure analysis has laid a solid foundation for its application in the more complex polysaccharide research field.
Application of AI in mass spectrometry (MS) for polysaccharide structure determination
-
Tandem mass spectrometry induces the breakage of chemical bonds in polysaccharide molecules through physical collisions, producing fragments. The formation pattern of the fragments reflects the breakage mode of the polysaccharide chain and can then be used to deduce information such as the sugar chain composition, branching, linkage type, and end modification of the monosaccharides[77−80]. However, due to the complexity of polysaccharide structures, such as the presence of isomers and diverse branching patterns, labor-intensive interpretation has been a major challenge. The interpretation process has encountered problems such as difficulty in distinguishing between isomers and ambiguous fragment ion assignment[81,82]. After AI technology was introduced into this field, the above problems were largely addressed. Urban et al. created the CandyCrunch DL model, which predicts glycan structures using LC-MS/MS data. The database used to train the model contains nearly 500,000 MS/MS spectra. After training, the model achieves an accuracy of up to 90%, accurately predicts glycan structures, and can complete the analysis of the entire dataset in a few seconds, significantly reducing manual labor costs[83]. In practice, application of such models to macromolecular polysaccharides typically requires prior depolymerization into oligosaccharide fragments, further highlighting the gap between current capabilities and the ultimate goal of predicting intact polysaccharide–protein interactions. Data scarcity remains a significant challenge due to the immense chemical diversity of glycans, including numerous isomers and the high cost of accurate annotation. CandyCrunch addresses data scarcity by systematically curating and re-annotating heterogeneous LC-MS/MS data from decades of published glycomics studies, ultimately constructing a large-scale dataset of nearly 500,000 high-quality, structurally annotated spectra. It introduced a deep residual network for end-to-end supervised learning without transfer learning, presumably because the raw spectral features differ markedly from existing pre-training domains such as protein sequences, and the spectrum-to-glycan mapping is highly specific, making supervised learning a more straightforward and effective choice here. In addition, researchers have developed two glycan prediction models based on Transformers: GlycoBERT (Bidirectional Encoder Representations from Transformers) and GlycoBART (Bidirectional and Auto-Regressive Transformers). Unlike CandyCrunch, GlycoBERT and GlycoBART adopt a strategy of self-supervised pre-training followed by transfer learning to alleviate labeled data scarcity. Both models are pre-trained on the same MS/MS spectral database curated by CandyCrunch to learn the implicit mapping between spectra and glycan structures. GlycoBERT is trained as a sequence classifier that can accurately classify mass spectra into specific glycan structures. After pre-training, it fine-tunes the learned representations on specific glycan structure recognition tasks, achieving > 95% accuracy with small amounts of labeled data. However, the model can only predict structures that already exist in the training data. GlycoBART is a generative model that can infer glycan structures from scratch, thereby predicting novel glycans outside the scope of the existing database[84]. The combined paradigm of pre-training, transfer, and generation provides a generalizable solution for high-precision structural elucidation under data-scarce conditions.
-
The translation of polysaccharide structures into computer-processable formats remains a significant challenge in structural bioinformatics. In contrast to linear and template-driven biopolymers such as DNA and proteins synthesized from limited and conserved building blocks, polysaccharides are secondary gene products without a direct genetic template[85]. Their assembly involves the coordinated action of glycosyltransferases, generating macromolecules with non-linear, highly branched, tree-like topologies constructed from a pool of over 100 distinct monosaccharides. Many plant-derived bioactive polysaccharides are primarily cell wall-derived[86,87], which typically contain rare monosaccharides and even non-glycosidic bonds, supporting both mechanical strength and resistance to degradation[88]. For instance, as a pectic polysaccharide with significant immunomodulatory activity, rhamnogalacturonan-II (RG-II) not only contains rare monosaccharides such as apiose, Kdo, and Dha but also forms unique borate diester cross-links, falling outside the predefined vocabularies of formats like GlycoCT and WURCS and consequently triggering parsing errors[89]. Furthermore, polysaccharides are frequently characterized by non-stoichiometric substitutions and microheterogeneity with side chains and chemical modifications like acetylation, sulfation, and feruloylation distributed randomly instead of in a predetermined manner along the polymer backbone[90,91]. Such probabilistic distribution conflicts directly with the deterministic requirements of standard encoding schemas, which expect exact sequences and linkage positions. Meanwhile, fine structural details of polysaccharides are closely linked to the therapeutic activity, necessitating the discrimination of subtle structural features in computational representation. For example, the immunomodulatory potency of licorice polysaccharide GPS-1, which is a homogalacturonan-type pectin, highly depends on its branching pattern[92]; the antioxidant effects of Codonopsis polysaccharides correlate with particular 1→4 and 1→6 glycosidic linkages[93,94]. Likewise, for animal-derived polysaccharides like heparin, the non-uniform sulfation patterns are associated with their anticoagulant activity[95,96].
However, precise structural elucidation of polysaccharides remains a challenge that complicates their computational representation. Mass spectrometry, for instance, cannot distinguish isobaric stereoisomers like glucose from galactose without derivatization. Meanwhile, NMR suffers from severe signal overlap in highly branched polysaccharides, limiting resolution and making it difficult to resolve side-chain arrangements or non-stoichiometric modifications[97−99]. Therefore, databases become populated with underdetermined structures with records lacking the structural precision required for molecular docking and dynamics simulations[100]. AI-augmented spectroscopic methods introduced previously generally yield either fully resolved sequences for glycans of limited structural complexity or, more commonly, coarse-grained structural features for heterogeneous plant polysaccharides. Accordingly, the encoding and representation learning strategies discussed here are primarily applicable to glycans whose structures have been sufficiently characterized, particularly certain polysaccharides with well-defined repeating units such as hyaluronic acid. For the majority of structurally complex and incompletely resolved plant polysaccharides, applying such representation learning frameworks remains challenging. Downstream functional prediction in such cases still relies heavily on feature vectors such as molecular weight, monosaccharide molar ratios, and estimated linkage-type frequencies derived from chemical analyses and partial structural characterization.
Linear strings, connection tables, and feature vectors
-
Prior to the application of AI, constructing machine-readable encoding standards for polysaccharides was undoubtedly essential for glycoinformatics. As a well-established human-readable system effective for describing molecular entities, IUPAC nomenclature also provides systematic naming rules for polysaccharides. However, the flexibility of IUPAC rules for branched polysaccharides frequently results in multiple valid names for a single structure, posing significant challenges for computational handling[101].
For defined oligosaccharide sequences, IUPAC Condensed was separately released to offer a compact and human-readable linear syntax[102], forming the basis for several computational linear notations such as LINUCS[103]. GlyLES was designed by Joeres et al., allowing conversion between IUPAC-condensed notations of glycans and SMILES strings[104]. While providing a unique machine-interpretable name for a given structure, linear string representations are incapable of handling complex carbohydrates with repeating units, cyclic topologies, non-stoichiometric substitutions, or ambiguous linkage positions. GlycoCT, developed by Herget et al., overcomes these limitations based on a connection table approach instead of a linear encoding scheme, guaranteeing a canonical identifier regardless of structural complexity[105]. Different databases have introduced GlycoCT as a standard encoding system. GlycoCT has been adopted as a standard encoding format within UniCarbKB, which focuses on glycoprotein glycan structures[106]. Animal-derived glycosaminoglycans, including chondroitin sulfate, are also available in GlycoCT format in MatrixDB and GlyTouCan[107,108]. Nevertheless, GlycoCT is, in general, nonlinear, but can be made linear by appending each line to a single string. To this end, Tanaka et al. developed Web3 Unique Representation of Carbohydrate Structures (WURCS), facilitating a unique and URI-compatible linear notation for any glycan structure through canonical sorting of backbones, modifications, and linkages[109]. Dedicated symbols were also introduced to encode uncertain structural features, providing explicit representation of ambiguity frequently observed in real glycan data.
Translating glycan structures into linear codes and connection tables further makes computational processing possible. Based on their in-house KCF format, which is a connection-table representation for glycan structures, Aoki et al. developed a BLAST for glycans called KEGG Carbohydrate Matcher (KCaM), enabling the structure-based similarity search of carbohydrate sugar chains. Technically, KCaM treats glycans as tree structures and performs alignment through dynamic programming adapted from the Smith–Waterman algorithm. Each tree is traversed from root to leaves to establish order, and scores are computed bottom-up by comparing monosaccharide names, linkage information, and optimal child subtree alignments[110].
Conventional supervised learning algorithms such as SVMs and RFs are mathematically restricted to processing fixed-dimensional numerical feature vectors. However, glycan structures encoded in connection tables and linear notations vary in length and are thus incompatible with this input format. In order to input a glycan structure into the neural network, Carpenter et al. adapted the q-gram fingerprinting approach, encoding each glycan as a feature vector comprising counts of how often each feature occurs. The major features included were contiguous 1-, 2-, and 3-monosaccharide subgraphs of the glycan structure involving the connecting anomeric linkages, terminal monosaccharide frequencies, as well as site-specific modifications. The resulting 272-element feature vector containing counts of every substructure feature observed in the training set was then suitable for neural network input[111]. Despite its effectiveness in terms of mammalian glycan array data, the q-gram fingerprinting method is generally based on a predefined lexicon of monosaccharide fragments and completely ignores the location and connectivity of these substructures, simplifying each glycan structure to a frequency vector[112,113].
Graph-based representations
-
Distinct from the structurally resolved and branching-restricted mammalian N- and O-glycans, plant polysaccharides are characterized by diverse and sometimes incompletely determined glycosidic linkages, monosaccharide composition that cannot be exhaustively predefined, and intrinsic microheterogeneity, making their structures difficult to represent in feature vectors. Rather than relying on a list of fixed monosaccharides, Bojar and colleagues developed SweetTalk to automatically learn to represent glycans by introducing a natural language processing (NLP) framework[114]. Each glycan was deconstructed into 'glycowords', which consist of three monosaccharides connected by two glycosidic bonds, the largest substructures with strict linearity by definition. Every monosaccharide or linkage was then defined as a 'glycoletter'. A character-level language model implemented as a bidirectional recurrent neural network was pretrained to predict the next character in a glycoletter's name, thereby acquiring distributed vector representations that capture distributional similarities between glycoletters. For instance, sulfated and unmodified galactose would be positioned close to each other in the embedding space. Glycoword embeddings were subsequently derived from averaging the vectors of their constituent glycoletters, and the glycan representations were further computed as the arithmetic mean of all glycoword embeddings within a given glycan (Fig. 2). The SweetTalk model is thus capable of predicting human immunogenicity directly from a raw glycan sequence, achieving approximately 92% accuracy on a validation set and successfully recognizing established immunogenic motifs such as the α-gal epitope. A microbiome-derived inflammatory glycan absent from the training data was also identified, demonstrating the model's generalization ability.
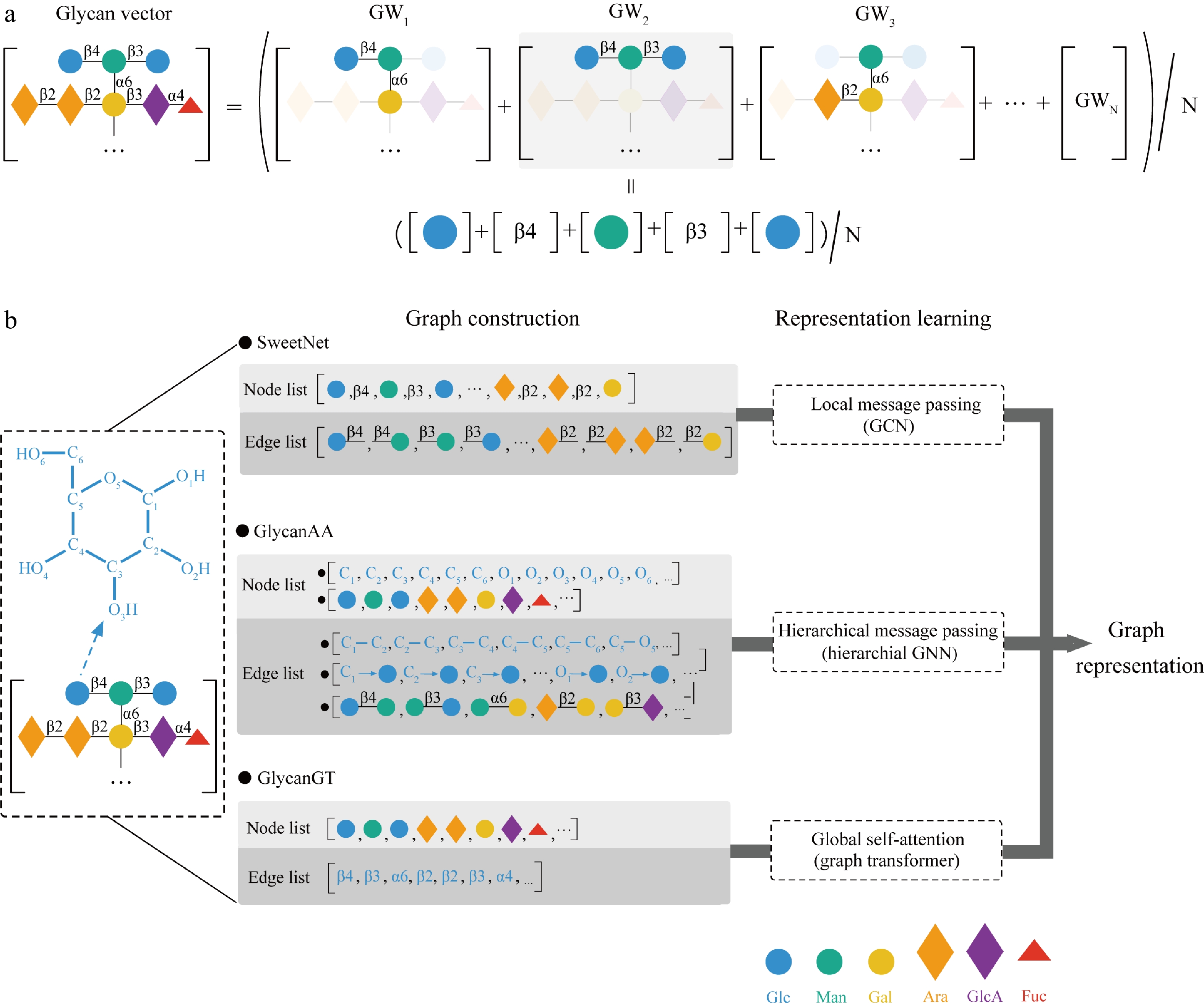
Figure 2.
Schematic illustration of glycan representation learning. (a) SweetTalk: a recurrent neural network (RNN)-based language model. GW: glycoword (three monosaccharides and two bonds). Glycans are featurized by extracting GWs, whose embeddings are obtained by averaging their constituent glycoletter embeddings. The corresponding embeddings for whole glycans are then constructed by averaging GW embeddings for downstream tasks. (b) SweetNet, GlycanAA, and GlycanGT: three graph-based glycan encoders with distinct node/edge definitions and information propagation strategies. SweetNet uses monosaccharides and linkages as nodes with connections between them as edges and performs local message passing via graph convolutional layers. GlycanAA constructs a heterogeneous graph with atom and monosaccharide nodes, connected by atom-atom, atom-monosaccharide, and monosaccharide-monosaccharide edges, enabling hierarchical message passing from the atomic to the monosaccharide level. GlycanGT treats monosaccharides and glycosidic bonds as tokens and captures global context through full self-attention.
However, the SweetTalk model cannot fully capture the branched or tree-like architecture that is seen in most glycans. Bojar and his team introduced graph convolutional networks (GCNNs) named SweetNet to treat glycan sequences as molecular graphs, viewing monosaccharides and linkages as nodes and the connections that define the glycan's topology as edges[115]. By converting the linearized IUPAC condensed bracket notation of a glycan into a node list and an edge index list, researchers constructed the graph-based representation and further fed it into multiple graph convolutional layers that learn to aggregate feature information from neighboring nodes, thereby capturing both local structural contexts like disaccharide motifs and global topological patterns including overall branching complexity (Fig. 2). A dendrogram that grouped plants according to shared phenotypic and environmental traits was successfully generated through averaging the SweetNet-generated representations of all Fabales glycans from distinct species and performing hierarchical clustering, revealing that biologically meaningful information can be captured by graph-based representations. Additionally, a GCNN framework was also adopted by Mohapatra to model glycans with nodes and edges, respectively, representing monosaccharides and glycosidic bonds[116]. Comprehensive biochemical information was directly encoded into each monosaccharide node feature through extended connectivity fingerprints, capturing atomic-level details such as stereochemistry, substituent modifications, and local structural environments. Similarly, based on graph-based representations, GlycanAA, developed by Xu et al., achieves atomic-level resolution in glycan modeling by forming a heterogeneous all-atom graph where each heavy atom within a monosaccharide is treated as an individual node[117]. However, monosaccharides were still preserved as higher-level nodes, enabling the integration of three distinct edge types, including atom-atom covalent bonds, atom-monosaccharide connections, and monosaccharide-monosaccharide glycosidic bonds. A hierarchical message-passing scheme was therefore introduced to process such rich structure, starting with propagating information among atoms within a monosaccharide, followed by updating both atom and monosaccharide representations through atom-monosaccharide message passing and ultimately exchanging information between monosaccharides via glycosidic linkages (Fig. 2). Furthermore, the model is enhanced through self-supervised pre-training on a curated dataset of over 40,000 high-quality glycans using a multi-scale mask prediction task. On the GlycanML benchmark, GlycanAA surpasses existing glycan encoders, with its pre-trained counterpart PreGlycanAA achieving top performance on nearly all tasks. It should also be noted that such an all-atom representation may lead to prohibitive computational costs due to the exponential growth of graph nodes and edges, especially when applied to plant polysaccharides, possibly containing even thousands of monosaccharide units.
Unlike previous GNN-based approaches relying on local message passing, GlycanGT introduced a graph transformer pretrained on a large-scale corpus of over 80,000 curated glycans, using global self-attention to capture both local and global structural relationships (Fig. 2)[118]. GlycanGT can also infer missing structural elements by predicting masked monosaccharides and linkages from their surrounding context through masked language modeling (MLM) pretraining, enabling automated reconstruction of incompletely annotated glycans. Meanwhile, GlycanGT learns to generate [Graph] token embeddings that can be directly applied as feature vectors for diverse downstream tasks without fine-tuning, bridging structural, functional and omics-level analyses. While GlycanAA achieved better performance in tasks emphasizing local structural motifs, GlycanGT excelled in tasks requiring long-range contextual dependencies across entire glycan structures, such as taxonomic classification at several hierarchical levels.
-
Current research on the structure–activity relationship of polysaccharides mainly focuses on the effects of structural features such as molecular weight, monosaccharide composition, glycosidic bond type, and side-chain branching pattern on the biological activities of polysaccharides, including immunomodulation, anti-inflammation, anti-oxidation, and anti-tumor[119,120]. Based on the conversion of spectral analyses into machine-readable embedded representations, artificial intelligence is now increasingly applied to associate these defined structural characteristics with their corresponding bioactivities[121]. Unlike traditional methods that focus on isolated structural pharmacological effects, AI models can directly receive encoded representations derived from spectral data such as graph embedding or eigenvectors[122,123]. This can not only effectively and accurately observe the structure–activity correlation but also predict the pharmacological effect only from the structural coding, and even de novo design polysaccharides for specific indications in the case of sufficient data.
Application of AI in polysaccharide structure–activity relationship research
-
Polysaccharides, as one of the most abundant biological macromolecules in nature, have a variety of biological activities such as immunomodulation, anti-tumor, and antioxidant effects, and are widely used in the food and biomedical fields. However, the complexity and diversity of polysaccharide structures, as well as the singularity of traditional research models, have caused their functional research progress to lag far behind that of biological macromolecules such as proteins, lipids, and nucleic acids. AI has enabled the effective modeling of correlations between polysaccharide structural features and biological activities by constructing nonlinear models with multiple inputs and outputs. In AI-driven polysaccharide structure–activity relationship research, various ML algorithms have been applied to predict polysaccharide activity. In a study on the relationship between the structure and immune activity of raspberry polysaccharides, an ANN was used to predict this relationship, and the gradient-weighted class activation mapping (Grad-CAM) algorithm was used to interpret it. In the ANN prediction model, the molecular weight, monosaccharide composition, and glycosidic bond linkage of raspberry polysaccharides were used as inputs, and cell viability, TNF-α, IL-6, and other immune activity data were used as outputs to construct a quantitative structure–activity relationship (QSAR) model. Through model training, key structural features affecting the immune activity of raspberries were identified, including the molecular weight of raspberry polysaccharides, the content of arabinose, and galactose. The mean squared errors of the training set and the test set were stable at around 0.003 and 0.013, respectively, and the mean absolute percentage errors were 0.21% and 0.98%, respectively. This shows that even with a small sample size and low dataset dimensionality, ML can effectively identify key structural features[124] (Fig. 3).
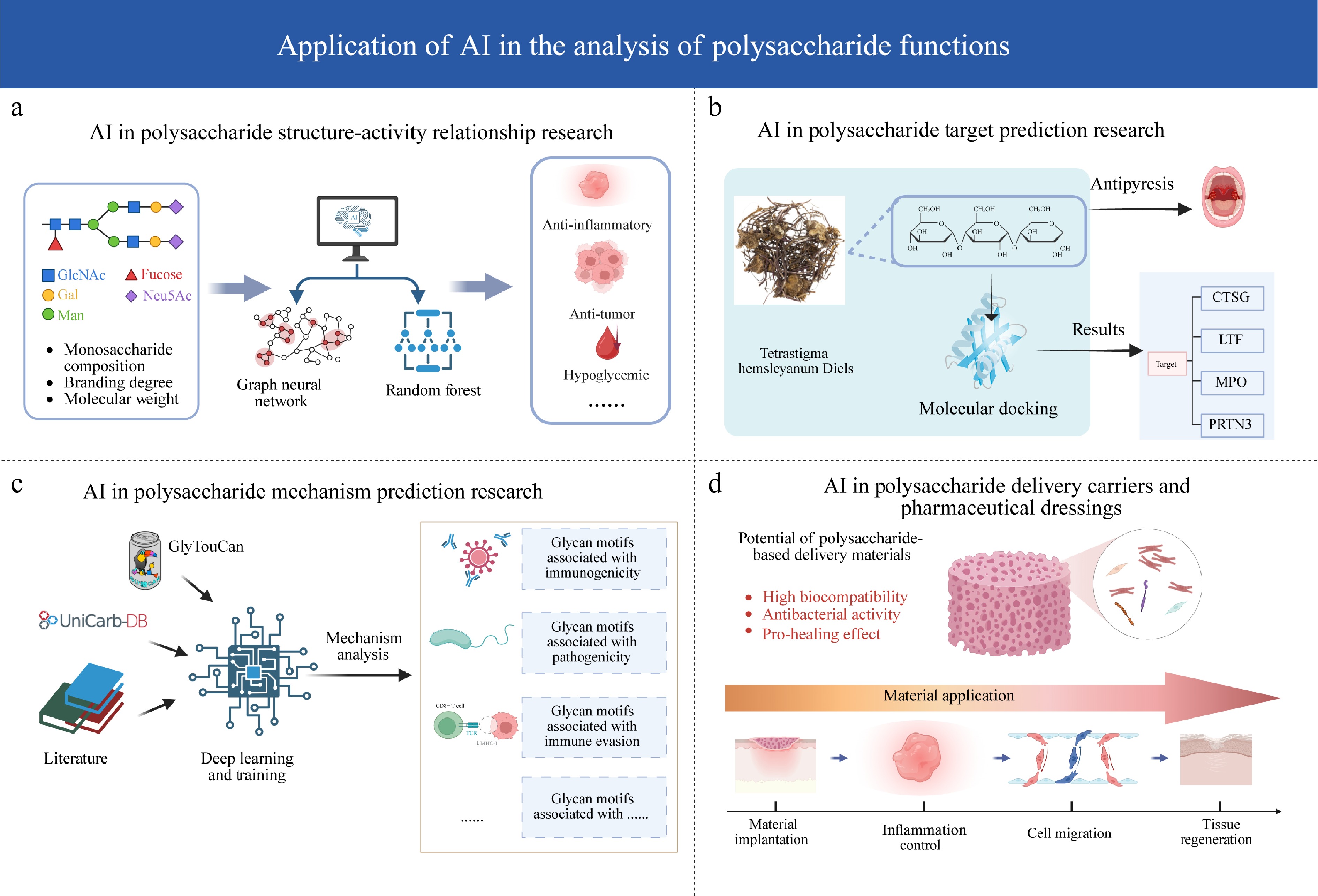
Figure 3.
Application of AI in the analysis of polysaccharide functions. (a) AI in polysaccharide structure–activity relationship research: structural features (composition, branching, molecular weight) are processed by machine learning models (graph neural networks, random forests) to predict biological activities (anti-inflammatory, anti-tumor, hypoglycemic). (b) AI in polysaccharide target prediction research: integration of chemical structures with molecular docking to identify protein targets (e.g., CTSG, LTF, MPO, PRTN3) linked to specific therapeutic results like antipyresis. (c) AI in polysaccharide mechanism prediction research: data from literature and databases (GlyTouCan, UniCarb-DB) are utilized via deep learning to predict functional mechanisms, identifying glycan motifs associated with immunogenicity, pathogenicity, and immune evasion. (d) AI in polysaccharide delivery carriers and pharmaceutical dressings: evaluation of polysaccharide-based biomaterials with key properties (biocompatibility, antibacterial, pro-healing) across consecutive stages of material application, from implantation to tissue regeneration.
The clinical translation of polysaccharide-based drugs heavily depends on mechanistic clarity; thus, the 'black-box' nature of conventional AI models poses a significant challenge. Explainable artificial intelligence (XAI) offers considerable potential to address this issue by making model decisions transparent. For polysaccharide SAR studies, XAI can not only identify which structural features (e.g., specific monosaccharide composition or glycosidic linkages) drive biological activity but also reveal how these features interact nonlinearly to modulate activity. This interpretability facilitates rational polysaccharide design and optimization, thereby accelerating clinical translation. However, several obstacles remain. The inherent complexity and heterogeneity of polysaccharides, such as branching patterns, molecular weight distribution, and conformational flexibility, make it difficult for XAI methods to generate stable and chemically meaningful explanations. Meanwhile, most XAI techniques, including Grad-CAM, were originally designed for image or sequence data and may not fully capture the hierarchical and multivariate nature of polysaccharide features. The scarcity of high-quality, annotated polysaccharide datasets also limits the robustness of both predictive models and their interpretability. Overcoming these barriers will require domain-adapted XAI algorithms, integration of chemical knowledge into explanation generation, and collaborative efforts to build standardized polysaccharide bioactivity databases.
Application of AI in polysaccharide target prediction research
-
Polysaccharide target prediction is one of the most cutting-edge directions in glycoinformatics, aiming to answer which sites polysaccharides bind to, to exert their functions. PeSTo is a Geometric Transformer model used to predict protein-carbohydrate binding interfaces. This model uses only geometric coordinates and elemental information to describe atomic structure without any artificial preprocessing of protein structure. This shows that the atomic arrangement (atomic geometry) information of the protein surface itself is sufficient to describe the characteristics of the protein-sugar binding pocket. Using this model to predict the binding interface between cyclodextrin and protein resulted in extremely high accuracy[125]. The PeSTo-Carbs model has established a stable and comprehensive carbohydrate-binding site predictor, which has made a significant contribution to a deeper understanding of carbohydrate–protein interactions and their biological significance. A cross-domain transfer learning strategy is employed to overcome the scarcity of carbohydrate-protein complex structures. PeSTo, as the base model, is first pre-trained on abundant protein–protein interaction interfaces to learn general geometric features including van der Waals contacts and hydrogen-bonding geometry, and is subsequently transferred to carbohydrate-binding interface prediction, requiring only a small set of glycan-protein complexes for fine-tuning. Such a strategy enables the model to maintain an AUC above 0.85 even for data-scarce systems such as cyclodextrins, significantly outperforming models trained from scratch. DeepGlycanSite is a deep equivariant neural network (EGNN) learning model integrated with the geometric and evolutionary features of proteins based on the Transformer architecture. It can accurately predict carbohydrate-binding sites on proteins based on the structure of the target protein. In addition, by introducing the Uni-Mol pre-trained model into polysaccharide binding prediction, DeepGlycanSite can also provide predictions of specific binding sites[126]. Uni-Mol is pre-trained on over 200 million small-molecule 3D conformations through masked atom prediction and coordinate recovery, learning atomic quantum chemical environments and spatial topology. By transferring these generic geometric representations to the task of carbohydrate-binding site prediction, DeepGlycanSite achieves a high MCC of 0.625 with only a limited number of glycan-protein co-crystal structures. The transfer of geometric knowledge from small molecules to polysaccharides effectively alleviates data scarcity. MCNet is an ML model used to predict the interaction between proteins and polysaccharides. The model effectively incorporates atomic composition and atomic chirality information and can predict the binding probability of glycans to protein based solely on the atomic-level structure of the glycan. Atomic-level chirality encoding is independent of predefined monosaccharide vocabularies, enabling transfer learning of general binding principles across different stereochemical configurations to alleviate the scarcity of labeled data for novel glycan types. In addition, the model can identify and infer the structure of L-type polysaccharide enantiomers. After standardizing polysaccharide microarray data and affinity measurement data using a 'binding score' to expand usable training data by integrating heterogeneous sources, the data were used to train the model. MCNet can predict the quantitative interaction results between proteins and common polysaccharide enantiomers without considering the monosaccharide composition[127] (Fig. 3).
At present, many deep learning models have made progress in this field, but they have differences in task definition, data sources, and evaluation strategies, which directly affect their generalization ability in practical applications. PeSTo, DeepGlycanSite, MCNet, and other models undertake different prediction tasks. PeSTo is a geometric transformer model. The input is the atomic coordinates and element types of the protein without any structural pretreatment, and the output is the probability that each residue or atom on the protein surface belongs to the carbohydrate-binding interface, so as to identify the binding interface. This model is suitable for scanning the protein surface rapidly and unbiased, but it is not sensitive to the internal conformational changes of polysaccharides and cannot distinguish different monosaccharide types. DeepGlycanSite is based on the equivariant neural network and transformer architecture. The input is the three-dimensional structure of the target protein, which can combine the sequence evolution characteristics; the output is the carbohydrate binding probability of each residue on the protein, and it can further predict the specific binding site. The model has high accuracy, but it relies on a high-quality protein structure, and the conformational diversity of the polysaccharide itself is not considered enough. MCNet focuses on predicting the interaction strength between the protein and the polysaccharide: the input is the atomic-level structure of the polysaccharide, including atomic composition and chiral information, and the output is the binding probability or quantitative binding fraction. Its unique advantage is that it can process L-type polysaccharide enantiomers, but it requires pre-standardized polysaccharide microarray data as a training set, and does not directly predict the binding interface, but outputs the global binding possibility. Overall, these models can be divided into two categories according to the task: binding interface localization (PeSTo, DeepGlycanSite) and binding affinity prediction (MCNet). The former is suitable for defining the binding pocket on the protein, while the latter is suitable for screening polysaccharide molecules with potential binding ability, but both of them have difficulty dealing with the flexible conformation and multi-target non-specific binding of polysaccharides.
The training and evaluation of the above models mainly rely on the structural databases of protein glycocomplexes (such as carbohydrate protein complexes resolved in PDB) and the affinity data of sugar microarray experiments. However, there is a lack of standardized evaluation protocols for the processing of these data. PeSTo and DeepGlycanSite usually adopt training/test set partitioning based on time or sequence similarity to avoid data leakage. MCNet divides the training set and validation set after normalizing the microarray data through a custom 'binding score'. In addition to the common R2 and MAE, the indicators that can better reflect the generalization ability of the model include the area under the precision-recall curve, F1 score, Matthews correlation coefficient, and cross-validation performance on different protein families or polysaccharide types. At present, there are few systematic reports of these indicators in the literature, making it still difficult to judge the true generalization ability of the model on unknown proteins or polysaccharides.
Application of AI in polysaccharide mechanism prediction research
-
Although polysaccharides have a wide range of biological activities, due to their large molecular weight, significant differences in monosaccharide composition, diverse glycosidic bond linkages, and complex higher conformations, the biological effects are often multi-targeted and non-specific[128−130]. Therefore, it is difficult to systematically elucidate the mechanism of action of polysaccharides in vivo by relying solely on experiments to evaluate their biological activity. In a study on the antipyretic mechanism of ADHP1 polysaccharide, researchers combined network pharmacology with ML. They first evaluated the antipyretic effect of ADHP1 in a mouse fever model and then performed molecular docking on key proteins identified by GEO enrichment and protein–protein interaction (PPI) analysis. They predicted that ADHP1 may exert its antipyretic effect by activating CTSG, LTF, MPO, and PRTN3 proteins, providing a specific research direction for explaining the antipyretic mechanism of ADHP1[131]. Bojar et al. developed a DL model for glycan research and functional prediction using natural language processing techniques. This model consists of three modules: SweetTalk (a glycan language model), SweetOrigins (a language model-based classifier), and SugarBase (a database containing annotations for 12,674 glycans and their species). The model was trained using 19,299 carefully selected glycan datasets. Combined with glycan comparison, this model can be used to predict the immunogenicity of glycans and the pathogenicity of E. coli strains. In addition, by incorporating glycan evolution information provided by SweetOrigins, this model can also predict glycan-mediated molecular mimicry and immune escape mechanisms. The model helps to reveal the similarities between different sugar chains at the evolutionary and functional levels, such as showing how bacteria can achieve immune escape by mimicking the host glycan pattern, and establishes a powerful and universal platform for further understanding the immune regulation mechanism of glycans[132] (Fig. 3).
Research and application of AI in polysaccharide delivery carriers and pharmaceutical dressings
-
In addition to possessing a variety of biological activities, polysaccharides also exhibit unique physicochemical properties, such as biocompatibility, low immunogenicity, renewability, biodegradability, modifiability, non-toxicity, and the ability to assemble with other bioactive molecules to treat diseases. This makes polysaccharides both drugs for treating diseases and carriers for targeted drug delivery, making them highly valuable biomolecules for research[133,134] (Fig. 3). Unlike the research pipeline for polysaccharides as drug entities covered in previous sections, which usually involves complex extraction from highly heterogeneous natural biomass, laborious purification, and precise elucidation of fine structural features such as specific glycosidic linkages and active modifications, delivery carriers and pharmaceutical dressings typically start from stable and industrially mass-produced polysaccharides, focusing primarily on semi-synthetic modifications or other formulation strategies (e.g., crosslinking, grafting, ionic gelation) to optimize macroscopic physicochemical properties. Consequently, the specific AI tasks in functional prediction are fundamentally different. AI tasks for drug entities are heavily oriented toward pharmacological activity mapping and target discovery, including SAR modeling, predicting geometric protein-carbohydrate binding interfaces, and inferring molecular interaction networks. In contrast, AI tasks for polysaccharide-based functional materials mainly highlight pharmaceutical formulation engineering and release kinetics. In this context, AI is usually tasked with mapping material and processing parameters to critical quality attributes (CQAs) like particle size and encapsulation efficiency, as well as providing time-series predictions for drug release dynamics under varying physiological conditions. To accurately assess these distinct objectives, the evaluation metrics employed by AI models also differ markedly. Drug entity models are judged by biological metrics like binding free energy, EC50/IC50, molecular docking scores, or root-mean-square deviation (RMSD) for conformational alignment, whereas formulation models are assessed against pharmaceutical standards including the similarity factor (f2) and difference factor (f1) for dissolution profiles, along with cumulative release errors at discrete time points.
Application of AI in the design and performance prediction of polysaccharide-targeted delivery systems
-
AI, especially ML, can improve the performance and efficiency of polysaccharide nanocarrier drug delivery by building high-precision predictive models. Abdalla et al. innovatively used Raman spectroscopy to characterize polysaccharides and employed ML models (LightGBM, XGBoost, RF, KNN, and SVM) to predict the release of 5-aminosalicylic acid from 13 polysaccharide coatings (maltodextrin, inulin, maltitol, okra extract, coix seed extract, raffinose, pregelatinized starch, cooked corn starch, xylan, rice starch, resistant corn starch, wolfberry extract, and isomaltulose) in simulated human, rat, and canine colonic environments. Raman spectral data were collected at 2, 8, and 24 h as input data to predict drug release curves. The results showed that the RF model had high prediction accuracy (R2 = 0.81, MAE = 0.08)[135]. In a study predicting the performance of polysaccharide-coated colonic drug delivery, researchers modeled drug release as the sole target variable by combining formulation parameters (coating type, medium, and release time) with Raman spectroscopy data as input parameters. Partial least squares (PLS) was used to reduce the dimensionality of the dataset (155 samples and over 1,500 spectral variables). Multiple regression models, including AdaBoost linear regression, MLP, and Theil-Sen, were applied. After optimization using the PSO algorithm, AdaBoost-MLP performed best (R2 = 0.994, MSE = 0.000368), making it the optimal choice for detecting nonlinear correlations in the data. This study combines spectral features with component factors, providing a modeling foundation for evaluating colon-targeted drug delivery kinetics[136]. Ac(e)Dex is a highly promising nanopolysaccharide delivery material with advantages such as hydrophobicity, pH sensitivity, and biodegradability. In our standardization research, 36 Ac(e)Dex derivatives with different molar masses, types, and functions were synthesized. High-throughput screening (> 1,000) was performed using a liquid handling robot to optimize polymer concentration, solvent, and additives. The selected superior formulations were then scaled up for production, and their stability was evaluated. To understand the structure-property relationship, an ML model was developed to predict the degradation of Ac(e)Dex nanocarriers using formulation data, providing support for their future clinical development[137].
Research and application of AI in polysaccharide tissue repair
-
Polysaccharide hydrogel polymers are three-dimensional network structures formed by cross-linking of polymer chains. Due to their good biocompatibility and biodegradability, they are widely used in drug delivery, tissue engineering, and wound dressings[138,139]. In wound dressing development, AI technology can predict the biocompatibility, antibacterial properties, and healing-promoting effects of polysaccharide hydrogels based on their physicochemical properties and biological data. In the research of Garcia-Del Rio et al., an AI tool, atomic-induced topological theory (AIT), was used to develop and characterize thermosensitive and mucosal-adhesive hydrogels for oral mucosal vaccination. The physicochemical properties, mucosal adhesion, and antigen-like microsphere release of the hydrogel polymer were characterized using AIT technology. The biocompatibility of the polymer and its immunostimulatory activity in human macrophages were also evaluated. Finally, a ternary hydrogel with immunostimulatory properties, strong mucosal adhesion, and controllable microsphere release was prepared[140]. In their research, Deng et al. constructed a light- and pH-triggered smart hydrogel dressing for early-stage diabetes with acidic wound pH. They used GelMA/CMCSMAP-GACo hydrogel for diabetes treatment. This gel exhibits rapid and reversible color changes within a pH range of 5.0–9.0, accurately indicating wound pH. By capturing color images of the wound with a smartphone and using ML to intelligently analyze the wound color, researchers could predict the condition of diabetic wounds. This provides visualized pH detection and treatment capabilities, aiding in diabetic wound management and demonstrating significant application potential[141].
Naturally derived polysaccharides are biological macromolecules that combine the functions of food and medicine, as well as drug and adjuvant. Due to their excellent biocompatibility, low toxicity, and multi-target properties, they have become a highly regarded area for drug research and pharmaceutical development. Polysaccharides exert immunomodulatory effects at multiple levels. At the organ and cellular level, they can promote the development of immune organs and the proliferation of immune cells and effectively activate key effector cells such as macrophages, dendritic cells, natural killer (NK) cells, and T-/B-lymphocytes. At the molecular level, they can induce the secretion of interleukins, interferons, and tumor necrosis factor-α, thereby regulating the immune system and antibody production and achieving systemic immune enhancement. In terms of anti-tumor activity, polysaccharide-based anti-tumor research is currently focused on several directions, including liver cancer, lung cancer, cervical cancer, and gastric cancer. Their antitumor mechanism research includes enhancing immune regulation function, inhibiting tumor cell growth, migration, and invasion, etc. Studies on their structure–activity relationship in antitumor activity focus on the relationship between antitumor activity and molecular weight, monosaccharide composition, glycosidic bond type, and side chain composition. In anti-inflammatory activity studies, various polysaccharides, such as astragalus polysaccharide, fucoidan, and chondroitin sulfate, exert anti-inflammatory activity by acting on the NF-κB and MAPK inflammatory pathways, reducing the levels of pro-inflammatory cytokines, and regulating immune cell polarization. In addition to the three bioactivities mentioned above that have been extensively studied, polysaccharides also exhibit good bioactivities in anticoagulation, antiviral activity, gut microbiota regulation, and hypoglycemic effects, making them highly valuable for research. Due to the complexity of polysaccharide structures, there are currently few drug-grade polysaccharides, and further research is needed in various aspects to enhance the prospects of developing polysaccharide-based drugs.
Currently, polysaccharides are mainly obtained by extraction from plants, animals, and microorganisms. At the molecular level, these naturally derived polysaccharides are usually mixtures rather than compounds with well-defined and identical structures, thus exhibiting characteristics of structural heterogeneity and non-uniformity. The biosynthesis of polysaccharides is extremely complex. Their structure is not directly determined by genes, and they often have complex modifications, making the traditional method of predicting the mechanism of action of polysaccharides from genes unsuitable for glycobiology. More importantly, the mechanism of action of polysaccharides is not a single 'key and lock' mechanism. Instead, they interact with multiple targets (lectins, receptors, enzymes) through a unique glycan sequence. The complex chemical structure leads to a 'multi-target' effect in their mechanism of action, and they may even act directly on target cell populations, exhibiting higher-dimensional functional regulation[142−144].
-
AI has been integrated into the entire process of polysaccharide research, including extraction, processing, structural analysis, functional analysis, and structure–activity relationship analysis, enabling polysaccharide research to enter a stage of high-quality, efficient, and rapid development. The rise of DL models has accelerated the development of polysaccharide phenotype screening towards phenotype prediction. On the one hand, the application of high-throughput technologies such as lectin microarrays to polysaccharide phenotype screening has made it possible to quickly resolve the interaction mechanism between polysaccharides and proteins based on polysaccharide structure. The experiment identified the cell wall glycans related to bacterial gene function by analyzing the aggregation ability of gene mutants with lectins[145]. On the other hand, the application of DL models has greatly improved the ability of structure-based phenotypic prediction. The LectinOracle DL model combines a Transformer (processing lectin information) with a graph neural network (processing glycan information) to predict the interaction between lectins and glycans without experiments[146].
In recent years, AI has been breaking through the bottlenecks in polysaccharide–protein interaction research from three levels: structural modeling, site identification, and affinity prediction. In terms of structure prediction, AlphaFold 3 achieves end-to-end modeling of protein-glycan chains using a diffusion module, and the prediction accuracy for ST6GAL1 substrates shows moderate agreement with the experimental structures[147]. Evaluations based on the BCAPIN benchmark show that top-1 predictions of mainstream models (e.g., AlphaFold 3, Boltz-1, Chai-1, RFAA) can achieve at least 80% docking success (DockQC ≥ 0.25) for protein–carbohydrate complexes, but performance decreases with increasing glycan polymerization degree[148]. A 2026 systematic evaluation also revealed that AlphaFold 3 produces stereochemical errors in 85.8% of carbohydrate ligand models, including chirality violations, planar ring distortions, and artificial double bonds. Using the BondedAtomPairs (BAP) syntax hybridized with CCD building blocks can eliminate these errors, but systematically removes the reducing-end oxygen, introducing a new artifact. Direct application of AlphaFold 3 to long-chain or branched polysaccharides therefore remains unreliable, and manual inspection of predicted glycan structures is still essential[149]. Many of the current deep learning advances in site recognition and affinity prediction have been demonstrated primarily on protein-protein or protein-small molecule systems. At the site recognition level, DL has developed a 'zero-shot' prediction paradigm. Models like MaSIF and InDeepNet can predict the interaction interface based on the protein structure[150]; MotifGen uses an SE(3)-equivariant graph neural network to predict ligand groups based on the receptor surface, thereby improving the accuracy of binding site recognition[151]. For affinity prediction, ProtAttBA predicts binding free energy changes solely from the sequence using a protein language model, which is more robust when antibody structure is uncertain[152]. To bridge this domain gap, emerging glycan-specific frameworks (e.g., PeSTo-Carbs, DeepGlycanSite, and MCNet) have successfully adapted geometric learning and equivariant networks to capture the unique atomic and chiral features of carbohydrates. However, these cutting-edge models are primarily trained on and validated against simple oligosaccharides or well-defined glycan motifs. Scaling them to predict the interactions of highly branched, macro-molecular polysaccharides is still fundamentally hindered by the severe scarcity of fully resolved target datasets and the intrinsic microheterogeneity of polysaccharide chains. With the optimization of general models, the improvement of glycan-specific benchmarks, and the application of multimodal frameworks, it is expected to achieve high-confidence analysis of complex polysaccharide–protein interaction groups, laying a theoretical foundation for the rational design of carbohydrate drugs.
Although AI has made breakthrough progress in many areas of polysaccharide research and development, its application in areas such as de novo polysaccharide design, phenotypic function prediction, metabolic prediction, and clinical evaluation of polysaccharide drugs is still in its early stages and requires further research efforts. In terms of de novo drug design, current AI drug design focuses mainly on proteins and short peptides. However, due to the complexity of polysaccharide structures, there have been few breakthroughs, and progress has been relatively slow. There is an urgent need to develop adversarial networks or diffusion models specifically for glycan generation, integrating the topological features of the sugar library, so as to achieve scientific and evidence-based de novo design of polysaccharide molecules. In terms of phenotypic function prediction, there are few polysaccharide molecular structures that can be accurately and completely resolved. This results in insufficient training data for existing models, making it difficult to introduce DL models to analyze and classify polysaccharide structures, and the prediction of polysaccharide structure–activity relationships lacks depth. The current challenges in predicting polysaccharide metabolism lie in the scarcity of high-quality glycomic data, the uninterpretability of DL models, and the low precision and dimensionality of the observational data input to the models, which makes it impossible to accurately and comprehensively describe the dynamic changes in metabolic processes. As a result, it is difficult to use predictive models to reflect the true process of polysaccharide metabolism in vivo. The clinical evaluation of polysaccharide drugs also faces the challenge of insufficient high-quality data. Pharmacokinetic and pharmacodynamic evaluation data are heterogeneous and difficult to use in training DL models. It is necessary to establish an AI-assisted clinical trial platform to conduct clinical safety and efficacy evaluation based on multi-omics data in order to improve the drug-likeness of polysaccharide drugs.
In summary, the ability of AI to design molecules has been extended to higher dimensions, such as multi-parameter collaborative optimization and precise control of post-translational modifications. These successful practices provide an important theoretical basis and knowledge for the de novo design of polysaccharide molecules. The structural complexity of polysaccharide molecules far exceeds that of peptides and proteins, which is expected to promote the discovery of carbohydrate drugs in the programmable era (Fig. 4).
Figure 4.
Challenges and future directions of AI in polysaccharide research. (a) De novo polysaccharide design: Illustration of using AI to generate completely novel polysaccharide molecular structures, highlighting the challenge and goal of achieving an 'Optimized structure'. (b) Phenotypic function prediction: schematic of the workflow mapping Cell phenotype input' through an AI model to hit a specific 'Target', resulting in a 'Predicted function' for biological activity. (c) Polysaccharide metabolism prediction: representation of human metabolic tracking, highlighting current bottlenecks such as 'Insufficient data' and a 'Model inexplicable', (lack of interpretability) within the predictive pipeline. (d) Clinical evaluation of polysaccharide drugs: demonstration of AI acting as an 'Auxiliary clinical trial' tool to simulate patient group cohorts, aimed at ensuring 'Effective' and 'Safe' outcomes for polysaccharide-based therapeutics.
-
Not applicable.
-
The authors confirm contributions to the review as follows: conception and design: Xu H, Luo C; draft manuscript preparation: Yu J, Liu N. All authors reviewed and approved the final version of the manuscript.
-
Data sharing is not applicable to this article as no datasets were generated or analyzed.
-
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0830300 to Cheng Luo), Science and Technology Department of Guizhou Province (Grant No. [2024]015), the Science and Technology Commission of Shanghai Municipality (YDZX20233100004032 to Cheng Luo, 24JS2830200, 25JS2830300, 25ZR1402556 to Heng Xu), the Applied Basic Research Foundation of Yunnan Province (202501BC070005), and the project of National Multidisciplinary Innovation Team of Traditional Chinese Medicine (ZYYCXTD-D-202004 to Cheng Luo).
-
The authors declare that they have no conflict of interest.
-
#Authors contributed equally: Jiayi Yu, Ningyun Liu
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of China Pharmaceutical University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yu J, Liu N, Xu H, Luo C. 2026. Artificial intelligence empowering polysaccharide research in drug development. Targetome 2(3): e027 doi: 10.48130/targetome-0026-0026 

Artificial intelligence empowering polysaccharide research in drug development
- Received: 28 March 2026
- Revised: 29 May 2026
- Accepted: 09 June 2026
- Published online: 26 June 2026
Abstract: Polysaccharides, together with proteins and nucleic acids, are typically considered the three fundamental macromolecules essential for life. Unlike well-studied proteins and nucleic acids, polysaccharides remain poorly characterized. Their inherent structural heterogeneity makes them particularly challenging to study with conventional techniques. Artificial intelligence (AI) has emerged as a transformative technology in driving the paradigm shift of polysaccharide research to data-driven intelligence, thereby enabling efficient analysis of extensive data. Herein, we systematically review AI applications in polysaccharide research, mainly focusing on various stages in polysaccharide drug development. The limitations and outlooks are discussed as well, following the review of the advantages of AI in this field.