









-
Jaboticaba (Myrciaria cauliflora (Mart.) O. Berg.) also termed Plinia, is a grape-shaped fruit native to Brazil with many health benefits[1,2]. Jaboticaba has garnered international attention in recent years due to its notable phytochemical composition, health benefits, and potential for use in the development of derived food products[2,3]. The pericarp color of Jaboticaba fruits ranges from green to red-purple during development, and these fruits are full of phenolic constituents such as anthocyanins and ellagitannins. Anthocyanins were found as the most abundant phenolics of this fruit[4,5].
Anthocyanins have received much attention for their antioxidant capacity, which has health benefits associated with biological effects such as antioxidant action, antiviral, antimicrobial, and antitumor activities[6]. The anthocyanin biosynthetic pathway has been extensively studied in numerous plants at the biochemical, genetic, and molecular levels[7]. Several genes encoding biosynthetic enzymes and transcription factors (TFs) of the anthocyanin pathway have been extensively studied in maize, Arabidopsis, petunia, tobacco, and fruit crops such as grape, apple, and strawberry[7]. The shikimic and phenylpropanoid biosynthetic pathways are upstream of the flavonoid biosynthetic pathway, and anthocyanins, as decorated flavonoid compounds, are synthesized downstream of the flavonoid biosynthetic pathway[8]. Phenylalanine, an initial precursor of anthocyanins and other flavonoids, is successively catalyzed by phenylalanine ammonialyase (PAL), cinnamate 4-hydroxylase (C4H), 4-coumarate: CoA ligase (4CL), chalcone synthase (CHS), chalcone isomerase (CHI), flavanone 3-hydroxylase (F3H), and dihydroflavonol 4-reductase (DFR) to form unstable colorless anthocyanins[9]. Finally, colorless anthocyanins form stable and colored anthocyanins under the action of anthocyanin synthase (ANS) and UDP-glucose: flavonoid 3-O-glucosyltransferase (UFGT)[10]. In addition to structural genes, TFs serve as central regulatory components in regulating the global activity of flavonoid biosynthesis. Basic helix-loop-helix (bHLH) TFs, R2R3-MYB TFs, and WD40 proteins are the three core transcriptional regulators that directly orchestrate the precisely coordinated transactivation of structural genes encoding anthocyanin biosynthesis enzymes. They modulate structural gene expression mainly by forming MYB/bHLH/WD40 (MBW) complexes, and MYB TFs are the main determinants in these complexes[11]. Although the anthocyanin biosynthetic pathway has been studied in numerous model plants, it has not been reported in Jaboticaba. The characterization of the Jaboticaba transcriptome is urgently needed to provide insights into its gene functions and regulatory mechanisms.
Illumina second-generation sequencing is a powerful tool for evaluating gene expression levels[12]. However, even in organisms with a reference genome, comprehensively understanding all spliced RNAs within a transcriptome using second-generation sequencing is difficult. Single-molecule real-time (SMRT) sequencing developed by PacBio, also called third-generation sequencing, is an improvement over current second-generation sequencing technologies based on read length, and it avoids the transcriptome assembly required in second-generation sequencing, thereby strengthening the understanding of a complex transcriptome[13]. Recently, SMRT technology has been used to characterize the complex transcriptomes of Paulownia catalpifolia[14], dragon fruit, Oryza rufipogon[15], and other plants.
In this study, SMRT and Illumina high-throughput deep sequencing technology, transcriptome sequencing, and metabolite profiling were performed to identify the anthocyanin biosynthetic pathway in Jaboticaba. Furthermore, both long non-coding RNA (lncRNA) prediction and expressed sequence tag-simple sequence repeat (EST-SSR) marker analysis were performed to acquire an in-depth understanding of anthocyanin biosynthesis. Our results show a regulatory network and a basic genetic information for anthocyanin biosynthesis and other metabolites in Jaboticaba.
-
Jaboticaba (M. cauliflora) were grown in an orchard in Yuanjiang City, Yunnan Province, China. For anthocyanin measurements, green and red fruit peel samples from a single Jaboticaba tree were collected. The samples were snap-frozen in liquid nitrogen and preserved under standardized cryogenic storage for downstream experimental analyses. For anthocyanin analysis, anthocyanin content was quantified as described by Zambrano-Moreno[16]. All experiments were repeated three times.
RNA extraction, library construction, and PacBio and Illumina sequencing
-
Total mRNA was extracted using the Plant RNA Kit (Qiagen, Valencia, CA, USA). RNA purity and concentration were assessed with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Rockland, DE, USA) using an OD260/280 reading. RNA integrity was evaluated by agarose gel electrophoresis with Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). High-quality mRNA from each tissue was then used to construct the mRNA-seq library. Equal amounts of mRNA from the three samples were pooled for library construction. cDNA was size-selected for fragments, < 4 kb and > 4 kb. The isoform sequencing (Iso-Seq) library was prepared following the Iso-Seq protocol using the Clontech SMARTer PCR cDNA Synthesis Kit and BluePippin Size-Selection System (Pacific Biosciences, PN 100-092-800-03). The Illumina sequencing library was constructed using the Illumina TruSeq RNA Sample Preparation Kit. The transcriptome is publicly available in the National Center for Biotechnology Information (NCBI) database.
Functional annotation
-
Unigenes were annotated based on the following seven protein and nucleic acid databases: NR, NT, SwissProt, Gene Ontology (GO), Clusters of Orthologous Groups of proteins (COG), Karyotic Ortholog Groups (KOG), Pfam, and Kyoto Encyclopedia of Genes and Genomes (KEGG). We selected the best alignment from the matches with an E-value ≤ 10–10. We assigned GO terms to the assembled unigene sequences using the Blast2GO platform. The KEGG pathways were assigned to the assembled unigene sequences using the online KAAS-KEGG automatic annotation server (
www.genome.jp/kegg/kaas ).lncRNA prediction
-
The coding potential of transcripts was performed by predictor of long non-coding RNAs and messenger RNAs based on an improved k-mer scheme (PLEK)[17] and Coding–Non-Coding Index (CNCI)[18]. These predicted transcript sequences were then blasted with protein databases using the Coding Potential Calculator (CPC) software[19]. To obtain lncRNA sequences, HMMER hmmscan homologous search using the Pfam protein families and domains database was performed for the transcript sequences predicted by PLEK, CNCI, and CPC software.
EST-SSR detection
-
The MicroSatellite identification tool (MISA;
http://pgrc.ipk-gatersleben.de/misa/ ) was used for EST-SSR mining of the whole transcriptome, and the characteristics of repeat motif types were further analyzed statistically. In this study, the SSR loci were identified according to the criteria: the number of mononucleotide repeat motifs ≥ 10, and that of di-, tri-, tetra-, penta-, and hexanucleotide repeat motifs ≥ 6, 5, 5, 5, and 5, respectively.Metabolite identification and quantification
-
The high-performance liquid chromatography column effluent was connected to a electrospray ionization (ESI)–triple quadrupole linear ion trap tandem mass spectrometry system (Applied Biosystems 4500 Q TRAP). The ESI source was set to the positive ionization mode, with a temperature of 550 °C and a capillary voltage of 5.5 kV. The mode was set to multiple-reaction monitoring (MRM). Metabolites were identified by measuring secondary spectral information using a compiled metabolite database MWDB (Metware Biotechnology Co., Ltd., Wuhan, China). Partial least squares–discriminant analysis was performed with the identified metabolites. Metabolite quantification was performed using MRM, and the obtained spectrometry data was processed using Analyst 1.6.3 software. Metabolites with significant differences in content were set with thresholds of variable importance in projection ≥ 1, and fold change ≥ 1.5 or ≤ 0.67.
Differentially expressed gene (DEG) analysis
-
Data from RNA-seq were mapped to the non-redundant SMRT reference by RSEM software. The unigene expression levels were represented as fragments per kilobase of transcript per million mapped reads (FPKM) values, and genes were considered not expressed for FPKM < 1. Differentially expressed genes (false discovery rate < 0.01, and fold change ≥ 1.5 or ≤ 0.67) were obtained using EBSeq software.
qRT-PCR analysis
-
To validate the bioinformatics analysis results of gene expression in the anthocyanin biosynthetic pathway during fruit ripening, qRT-PCR assays were performed using green fruit (GF) and ripening fruit (RF) of Jaboticaba. A total of 31 DEGs were selected and analyzed by qRT-PCR to verify the RNA-seq output of genes. The total RNA was extracted from selected samples using the RNAprep Pure Plant Kit (Tiangen Biotech Co., Beijing, China). qRT-PCR was performed with a Fast Real-Time PCR System using TB Green® Premix Ex Taq™ II (Takara, Japan). PCR was performed as follows: 95 °C for 30 s for initial denaturation, then 35 cycles at 95 °C for 5 s, and 60 °C for 34 s. Each experiment was performed in triplicate. The primer sequences used for qRT-PCR are listed in Supplementary Table S1.
-
To obtain the whole transcriptome profile of Jaboticaba, SMRT sequencing was performed using a mixture of leaves, green fruits (GF), and ripening fruits (RF). A total of 6,772,478 subreads (19.17 Gb) were obtained by removing all adapter sequences and low-quality reads from the raw data (Supplementary Table S2). A total of 417,506 circular consensus sequences (Fig. 1a, Supplementary Table S2) and 360,262 full-length non-chimeric sequences (mean length 3,298 bp) were generated (Fig. 1b, Supplementary Table S2). The iterative isoform-clustering algorithm and Quiver software were employed to polish the consensus isoforms, and 169,212 high-quality polished consensuses with N50 of 3,987 bp were generated (Fig. 1c, Supplementary Table S2). In addition, high-quality reads totaling 281,189,080 clean reads (Supplementary Table S3) were obtained after removal of sequencing adapters and primer sequences, and we obtained 86,758 unigene sequences (non-redundant sequences) with an N50 length of 3,987 bp and mean length of 3,239 bp (Fig. 1d, Supplementary Table S2). High-quality RNA sequencing reads were submitted to the NCBI Short Read Archive for subsequent annotation analysis. Furthermore, our dataset had a high amount of data (nucleotide) and good assembly results (average length and N50; Supplementary Table S4). The results supplied useful data for analysis of secondary metabolites in Jaboticaba, such as flavone, flavonol, and anthoxanthin, etc.
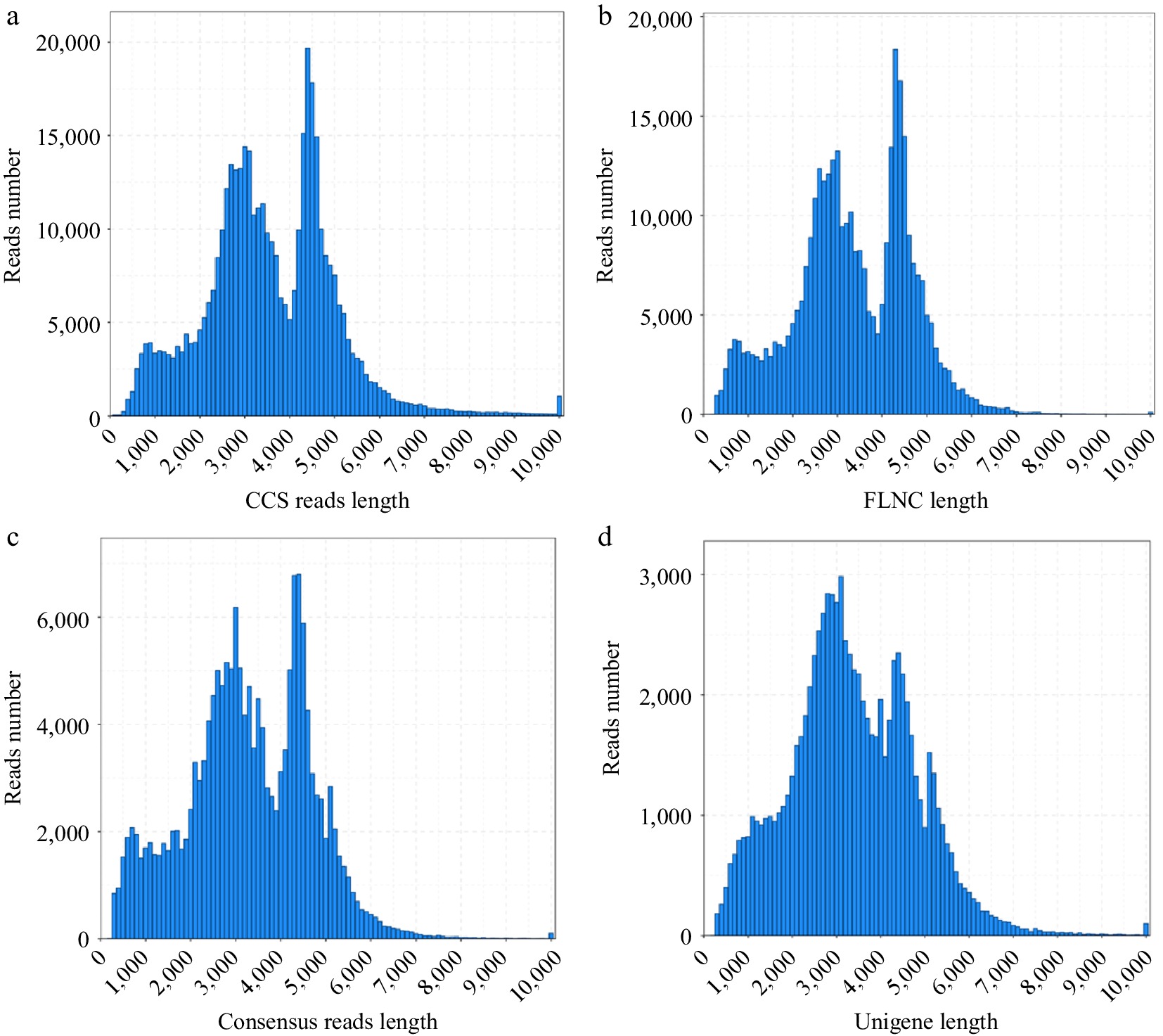
Figure 1.
Length distributions of PacBio SMRT sequences. (a) Number and length distributions of circular consensus sequences. (b) Number and length distributions of full-length non-chimeric sequences. (c) Number and length distributions of consensus isoforms. (d) Number and length distributions of unigenes.
Functional annotation and classification of unigenes
-
Similarity analysis of the unigenes functional annotation was performed using BLASTX and compared against the NR database. Among the amino acid sequences, the Jaboticaba unigenes had the highest number of hits against Eucalyptus grandis at 72,015 hits (86.82%; Fig. 2a), indicating that both the transcriptome and genome of E. grandis could be used as references for further research.
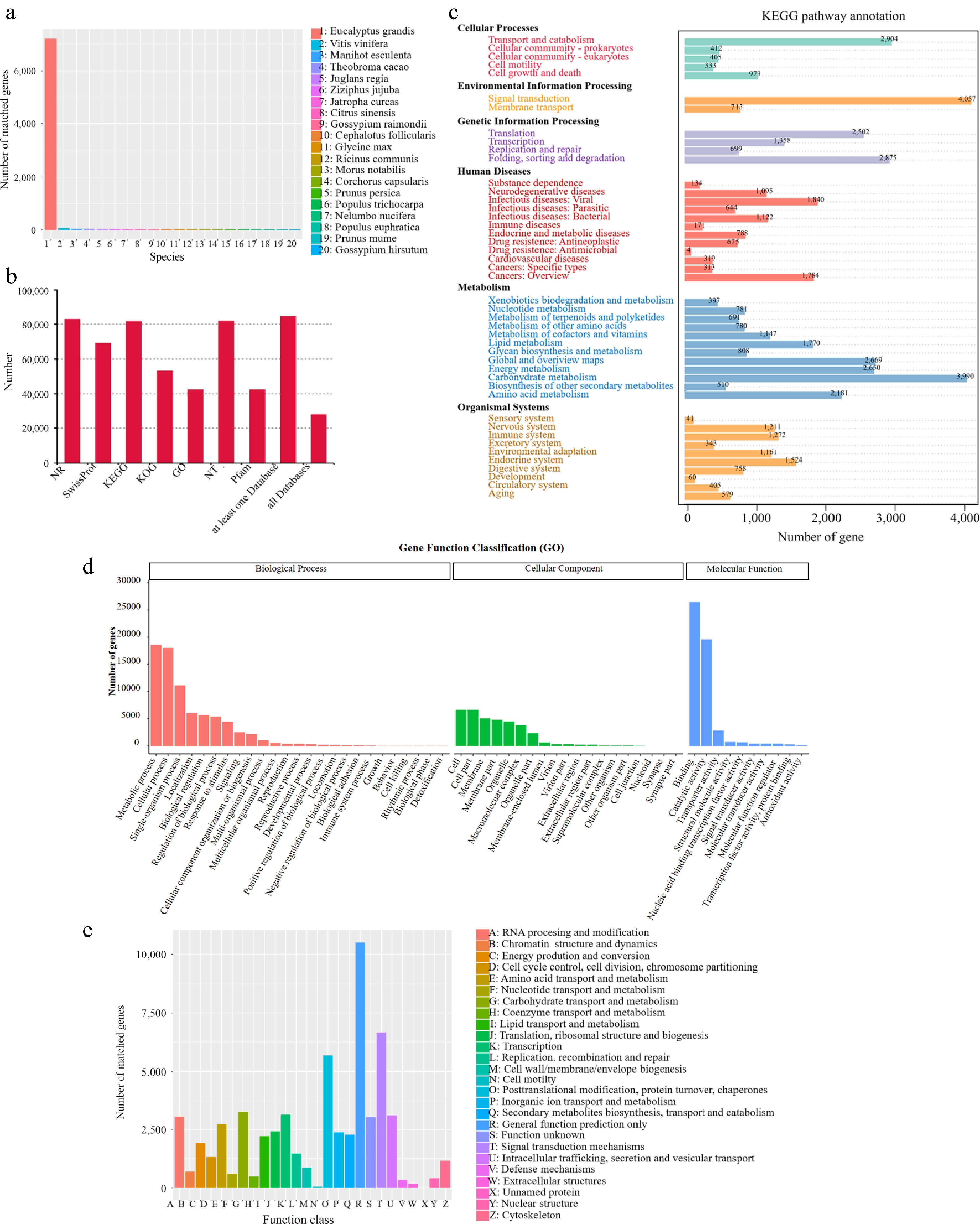
Figure 2.
Annotation of Jaboticaba unigenes. (a) NR homologous species distribution of unigenes. (b) Function annotation of unigenes in all databases. (c) KEGG pathways enriched for unigenes. (d) Distribution of GO terms for all annotated unigenes. (e) Searching of unigenes against the KOG database.
To determine highly homologous sequences, the 86,758 unigenes were blasted against the NR, Swissprot, KEGG, KOG, GO, NT, and Pfam databases. The results showed that 82,952 (95.61%), 69,182 (79.74%), 81,719 (94.19%), 53,217 (61.34%), 42,334 (48.80%), 81,849 (94.34%), and 42,334 (48.80%) unigenes were similar to accessions in NR, Swissprot, KEGG, KOG, GO, NT, and Pfam databases, respectively (Fig. 2b). Moreover, 28,578 unigenes were annotated in all databases.
A total of 81,719 unigenes, annotated by the KEGG database, were grouped into 38 KEGG pathways associated with 94.2% of the whole annotated dataset (Fig. 2c). They were divided into six broad classes: cellular processes (5,027 unigenes, 6.15%), environmental information processing (4,770 unigenes, 5.84%), genetic information processing (7,434 unigenes, 9.10%), human disease (8,889 unigenes, 10.88%), metabolism (18,374 unigenes, 22.48%), and organismal systems (7,354 unigenes, 9.00%). The GO database was searched against unigenes to analyze their functions. In GO annotation, biological process (77,603, 89.5%) was more abundant than molecular function (51,933, 59.9%) and cellular components (35,871, 41.35%; Fig. 2d). Within these functional groups, the high number of sequences was annotated with molecular function (GO: 0005488-binding; 26,427, 30.46%), molecular function (GO: 0003824-catalytic activity; 19,588, 22.58%), and biological process (GO: 0008152-metabolic process; 18,553, 21.4%).
In addition, the assembled unigenes were searched against the KOG database. A total of 53,217 unigenes were similar to accessions in the KOG database (Fig. 2e). The highest term was general function prediction only (10,502, 19.7%), followed by signal transduction mechanisms (6,669, 12.5%), translation, post-translational modification, protein turnover, and chaperones (5,674, 10.7%), carbohydrate transport and metabolism (3,252, 6.11%), and transcription (3,132, 5.89%).
Prediction and discovery of lncRNA and EST-SSR markers
-
A total of 1,127 lncRNA sequences were present during all methods, and these sequences were considered candidate lncRNAs in the target dataset (Supplementary Fig. S1). Additionally, 9,732 EST-SSRs, including 1,465 complex SSRs and 8,267 complete SSRs, were identified in Jaboticaba. There were 14,824 sequences containing SSRs, of which 8,882 sequences contained one SSR marker and 2,000 sequences contained two or more SSRs. SSRs occurred at a frequency of 10.03% (100% × Total number of sequences containing SSRs/Total number of examined sequences), and the SSR appearance frequency was 28.4% (100% × Total number of SSRs identified/Total number of examined sequences) (Supplementary Table S5). The number of complete SSRs was 8,267 and accounted for 85% of the total SSRs, including 1,833 mononucleotides (21.3%), 2,517 dinucleotides (29.3%), 3,824 trinucleotides (44.5%), 38 tetranucleotides (0.44%), six pentanucleotides (0.06%), and 49 hexanucleotides (0.57%). SSRs with five repeat motifs were the most frequent and accounted for 17.4% (2,583) of all SSRs, followed by SSRs with 10 repeat motifs (897, 13.6%), three repeat motifs (834, 12.63%), and six repeat motifs (2,089, 14.1%), respectively, whereas 5,400 SSRs had repeat motifs > 12, accounting for 36.4% of all SSR loci identified. A total of 48 repeat motifs were identified among the complete SSRs; these were two mononucleotides, four dinucleotides, 10 trinucleotides, 13 tetranucleotides, five pentanucleotides, and 24 hexanucleotides, respectively. Statistical analysis of all SSR loci showed that the five most frequent repeat motif types were in the order A/T (5,527, 37.3%), AG/CT (3,114, 21%), C/G (1,281, 8.64%), CCG/CGG (1115, 7.52%), and AGG/CTT (1026, 6.92%). The large set of EST-SSR markers identified in this study can provide a novel genomic toolkit for evolutionary genetics and precision breeding applications in Jaboticaba.
Anthocyanins accumulated during fruit ripening in Jaboticaba
-
To identify the varieties and regulatory pathways of anthocyanins in Jaboticaba, ripening fruits were collected for anthocyanin measurement. The results showed that ripening fruits accumulated 920 mg/100 g fresh weight of anthocyanins, whereas no anthocyanins were detected in both leaves and skin of green fruit (Fig. 3). To identify anthocyanins in ripening fruits, a targeted secondary metabolite assay was performed. A total of 72 metabolites with significant content differences were identified from green and ripening fruits, containing six anthocyanins, five chalcones, five dihydroflavones, five dihydroflavonols, four flavanols, 14 flavonoids, 33 flavonols, and one isoflavone. Compared with green fruits, six anthocyanins, pelargonidin-3-O-glucoside, peonidin-3-O-glucoside cyanidin-3-O-glucoside (kuromanin), peonidin-3-O-glucoside, delphinidin-3-O-glucoside (mirtillin), cyanidin-3-O-(6''-O-caffeoyl) glucoside, and delphinidin-3-O-(6''-O-caffeoyl) glucoside were found with 499.7-, 189.0-, 3,471,074.1-, 70.4-, 71.7-, and 3.2-fold increments in ripening fruits, respectively, thereby explaining the accumulation of purple pigment in ripening fruit peels (Supplementary Table S6).
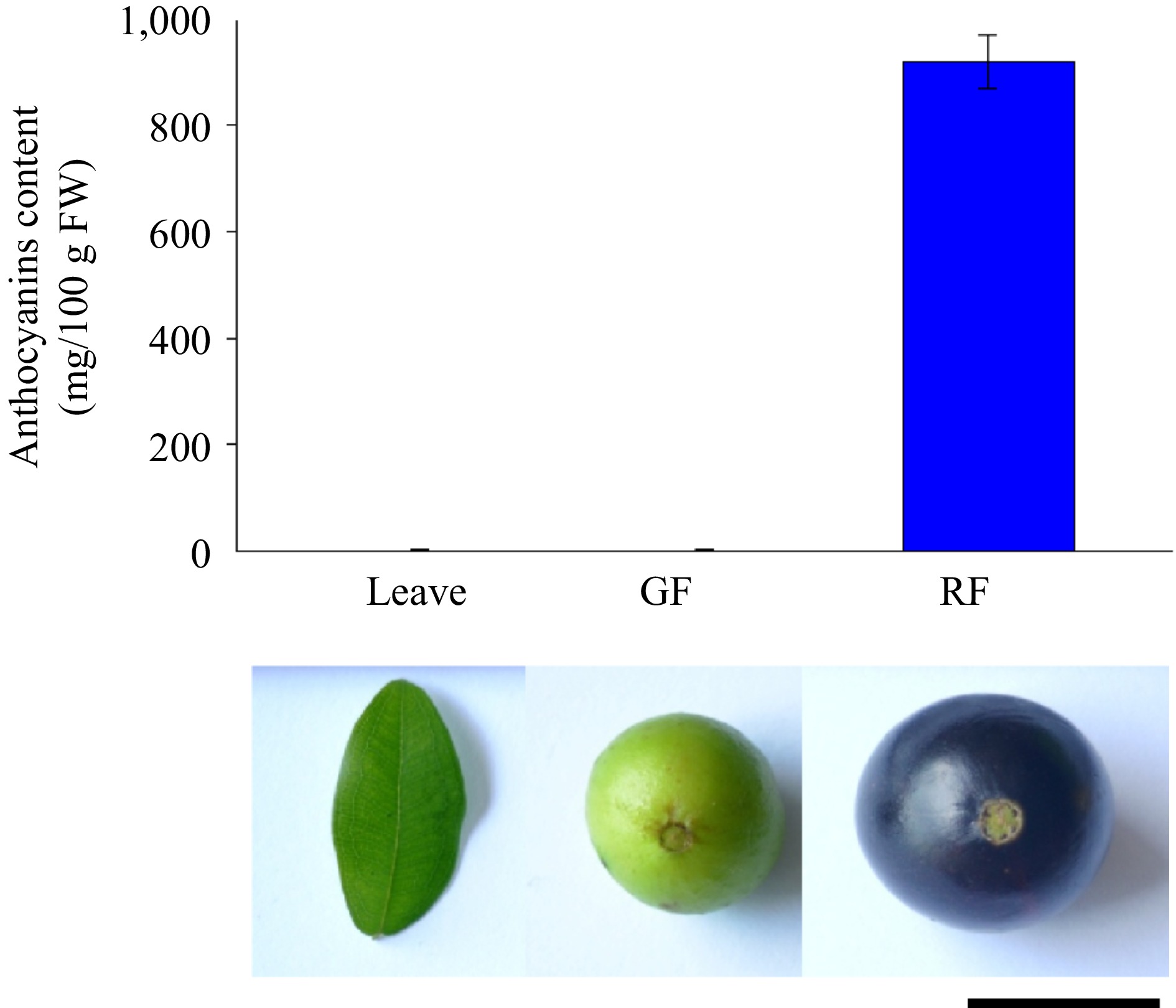
Figure 3.
Anthocyanin content in different tissues of Jaboticaba.
Modulation of anthocyanin biosynthetic genes during fruit ripening in Jaboticaba
-
A total of 79 unigenes were identified to be associated with 12 enzymes of the anthocyanin biosynthetic pathway in Jaboticaba (Supplementary Table S7). Interestingly, more than one unigene was annotated as encoding the same enzyme, especially the final step involving 3-glycoside formation by UFGT, indicating that some functional redundancy genes might encode anthocyanin biosynthesis enzymes. Among the 79 potential anthocyanin biosynthesis genes, 12 DEGs that might be involved in phenylpropanoid, flavonoid, and anthocyanin biosynthetic pathways were upregulated during fruit ripening (Supplementary Table S8).
In ripening fruits, the phenylpropanoid pathway was strengthened by upregulating gene expression. Compared with green fruits, three PAL genes (McPAL1, McPAL2, and McPAL3), catalyzing the formation of cinnamic acid and p-coumaroyl-CoA, were upregulated (7.85-, 4.67-, and 6.30-fold, respectively) at the beginning of the phenylpropanoid pathway in line with the increased cinnamic acid content in ripening fruits, and three 4CL genes (Mc4CL1–Mc4CL3) were upregulated (3.56-, 3.56-, and 1.87-, respectively; Fig. 4). Eight phenylpropanoid biosynthetic-related genes were highly expressed inducing the biosynthetic pathway to enter the flavonoid biosynthetic pathway, of which the end-products were anthocyanins. Simultaneously, large-scale upregulation of DEGs related to flavonoid and anthocyanin biosynthetic genes found in ripening fruits, including F3'H, LAR, ANS, UFGT (one DEG each), and DFR (two DEGs), dominated secondary metabolite synthesis modulation, enhancing the flux in the flavonoid and anthocyanidin biosynthetic pathways. The transcript level of McF3'H, which catalyzes eriodictyol synthesis also increased, resulting in higher eriodictyol accumulation in ripening fruits. Besides, the 3.38- and 4.27-fold increments of McF3H and McDFR1 led to high dihydroquercetin and leucocyanidin content, respectively. In addition, leucodelphinidin was transformed into catechin and gallocatechin, which were regulated by McLAR (3.83-fold upregulation). McANS, synthesizing the conversion from colorless anthocyanins to unstable colored anthocyanins, showed 5.00-fold upregulation. Finally, the unstable colored anthocyanins transformed into blue-violet, brick-red, or magenta glycosides by McUFGT (2.60-fold upregulation), which favors the high accumulation of the four anthocyanins (Fig. 4a). From our transcriptomic and metabolomic data, F3H seemed to favor eriodictyol, rather than dihydromyricetin and dihydrokaempferol, to produce dihydroquercetin. DFR and LAR favor dihydroquercetin to produce leucocyanidin and catechin, indicating cyanidin glycosides were the dominant anthocyanins. However, high delphinidin-3-O-glucoside and pelargonidin-3-O-glucoside content in ripening Jaboticaba fruits suggested that other mechanisms are involved in anthocyanin biosynthesis.
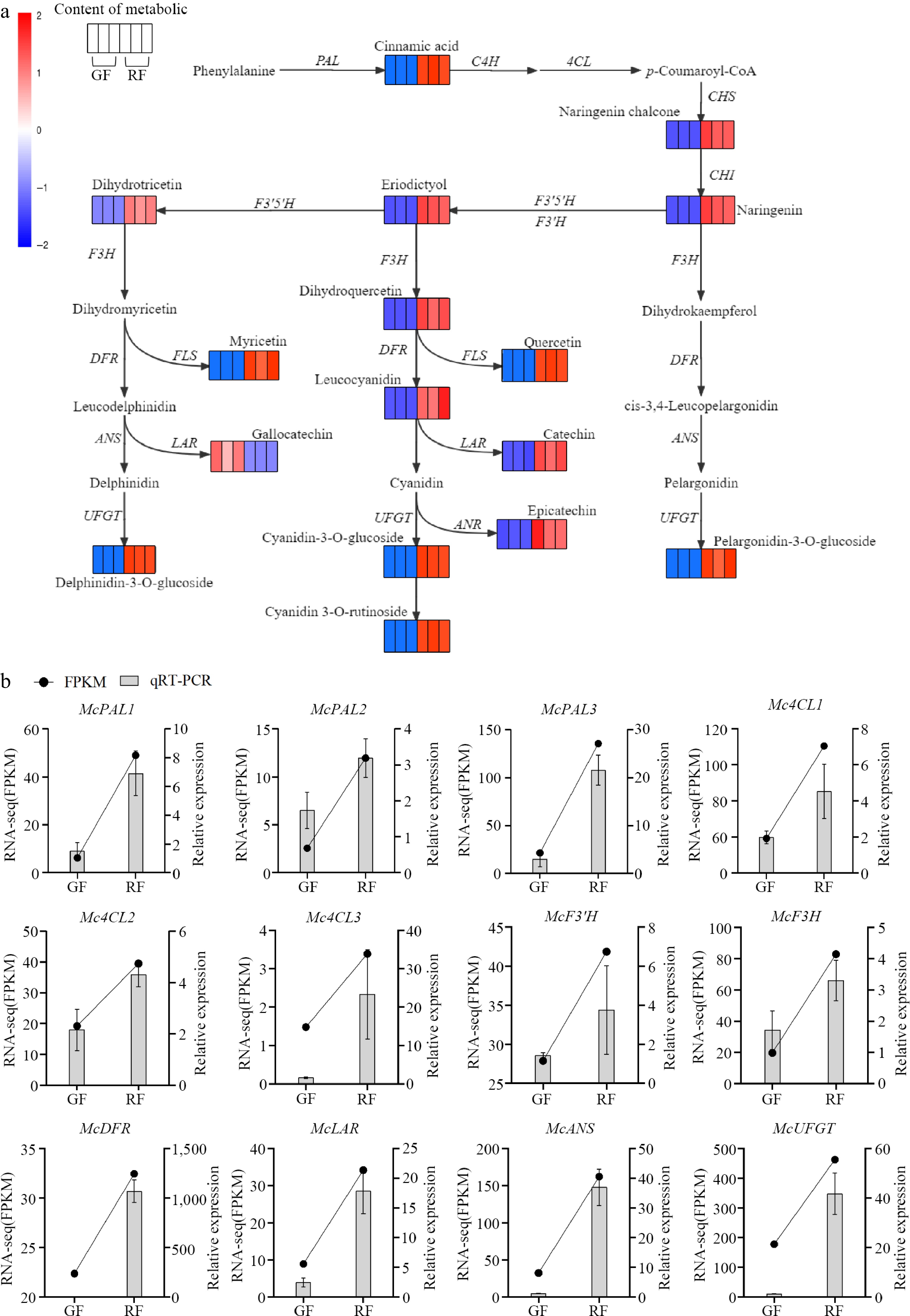
Figure 4.
Anthocyanin biosynthesis pathway in Jaboticaba. (a) Transcript and metabolic profiling of genes and metabolites in the phenylpropanoid, flavonoid and anthocyanin biosynthetic pathways in green fruits (GF) and ripening fruits (RF). Each colored cell represents the log2(FPKM) and log10(Content) value of each pathway gene and metabolite according to the color scale, respectively. PAL, phenylalanine ammonia-lyase; C4H, cinnamic acid 4-hydroxylase; 4CL, 4-coumarate CoA ligase; CHS, chalcone synthase; CHI, chalcone isomerase; F3H, flavanone 3-hydroxylase; F3'H, flavanoid 3'-hydroxylase; DFR, dihydroflavonol 4-reductase; ANS, anthocyanidin synthase; UFGT, UDP glucose-flavonoid 3-O-glcosyl-transferase; FLS, flavonol synthesis; LAR, leucocyanidin reductase; ANR, anthocyanin reductase. (b) Expression levels of the color-related genes in green fruits (GF) and ripening fruits (RF) validated by RNA-seq and qRT-PCR.
qRT-PCR assays were performed to validate the bioinformatics analysis results of the 12 DEGs (six phenylpropanoid biosynthetic-related genes, five flavonoid biosynthetic-related genes, and one anthocyanin biosynthetic-related gene). When the fruit matured, the relative expression of phenylpropanoid pathway genes (Mc4CL1, Mc4CL2, Mc4CL3, and McPAL1), flavonoid biosynthetic pathway genes (McF3H, McF3'H, McDFR2, and McANS), and McUFGT were upregulated significantly (Fig. 4b). These results were consistent with those of the transcriptomic analysis, indicating that the 12 DEGs might be the most significant genes enhancing anthocyanin accumulation in ripening Jaboticaba fruits.
Identification and analysis of TFs involved in the anthocyanin biosynthetic pathway
-
In addition, TFs that regulated the activation of anthocyanin biosynthetic genes were also analyzed based on the sequence and annotation information from NR, NT, SwissProt, GO, KOG, Pfam, and KEGG databases. There were 3,562 proteins were identified as possible TFs belonging to different TF families (Fig. 5a), and 67 MYB TFs and 107 bHLH TFs were identified (Supplementary Table S9). Three MYB and 13 bHLH TFs of them were highly expressed in GF as compared to that in RF (Supplementary Table S10).
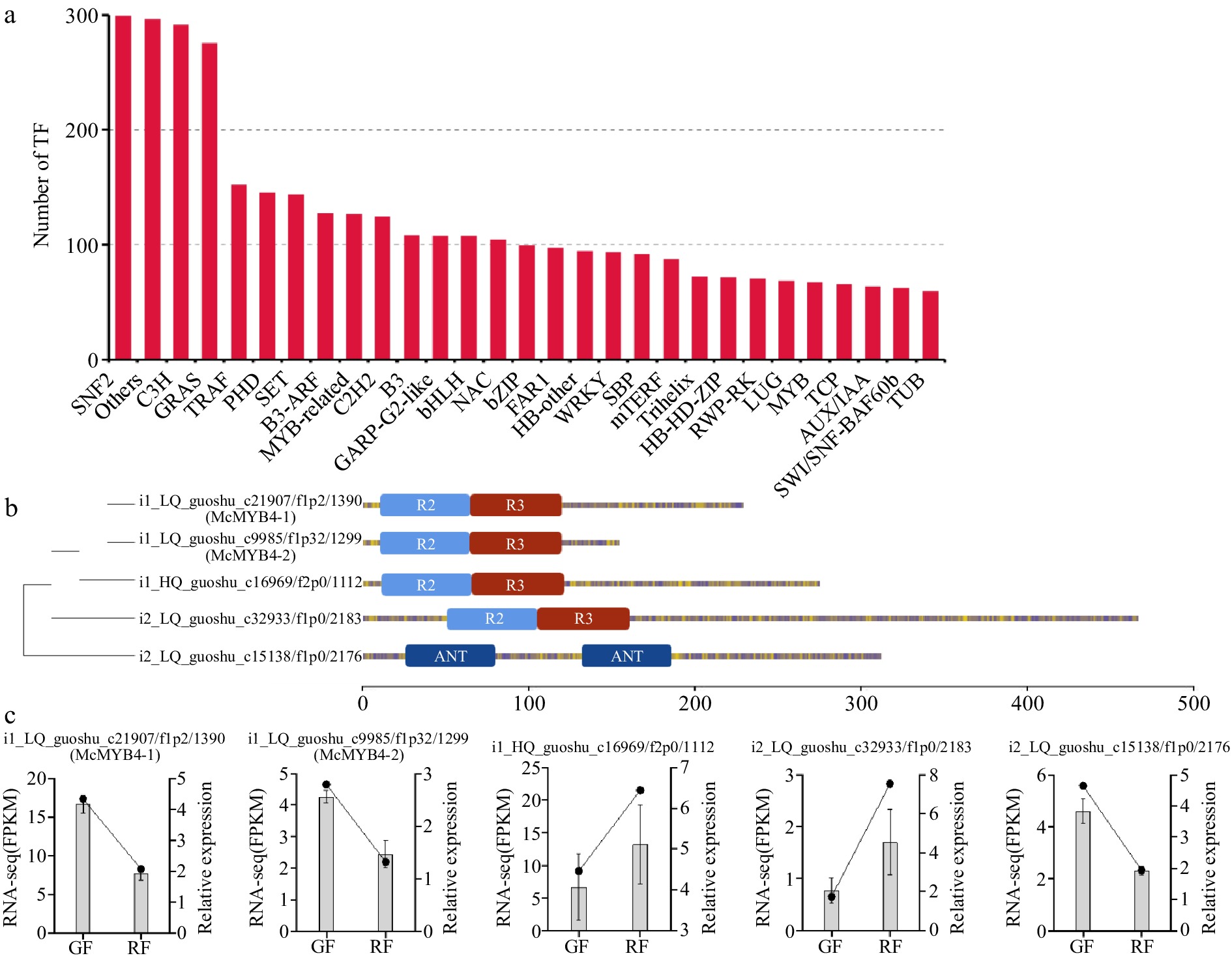
Figure 5.
McMYBs analysis in Jaboticaba. (a) The number of TFs belonging to different TF families. (b) Phylogenetic relationships and gene structure analysis of five McMYBs. (c) Expression levels of the five McMYBs in green fruits (GF), and ripening fruits (RF) validated by RNA-seq and qRT-PCR.
Among five MYBs differentially expressed, four were R2R3-MYBs, while one MYB TF contained two ANT domains, functionally divergent from the canonical MYB DNA-binding domain (Fig. 5b). Two R2R3-MYBs were upregulated in RF, and the other two were downregulated (Fig. 5c). The R2R3-MYBs were often considered to be closely related with accumulation of flavonoids and anthocyanins. To analyze the phylogenetic relationships between the four R2R3-MYBs in Jaboticaba and 126 R2R3-MYBs in Arabidopsis, a phylogenetic tree was constructed. The functions of AtMYBs are listed in Supplementary Table S11. The results showed that two McMYBs and three SG7 (Subgroup seven) AtMYBs, AtMYB11, AtMYB12, and AtMYB111, formed a clade, indicating that two MYBs in Jaboticaba were SG7 MYB proteins (Fig. 6a). The two SG7 McMYBs downregulated in ripening fruits most closely related to the RaMYB4 in Rhodamnia argentea were named with McMYB4-1 and McMYB4-2 (Supplementary Fig. S2). The R2R3-MYB TF McMYB4-1 contained conserved SG7 and EAR motif at the C-terminus, while McMYB4-2 had a conserved R2R3-MYB domain at the N-terminus but lacked both the SG7(-2) motif and EAR motif at the C-terminus (Fig. 6b). Phylogenetic analysis grouped McMYB4-1 and McMYB4-2 within the R2R3-MYB subfamily associated with anthocyanin repression (Fig. 6c). Furthermore, conserved SG7(-2) motifs were not identified in their protein sequences. These findings, combined with their declining expression during anthocyanin accumulation, suggest that McMYB4-1 and McMYB4-2 may act as potential negative regulators of anthocyanin biosynthesis in Jaboticaba. Furthermore, GtMYBP4 with the same EAR motif in McMYB4-1 inhibited the expression of GtF3H, indicating that McMYB4-1 may act as a repressive transcriptional activation of flavonoid structase genes at the same time. Above all, the results showed that McMYB4-1 and McMYB4-2 were typical SG7 R2R3-MYBs and played a role in decreasing anthocyanin contents in Jaboticaba fruits.
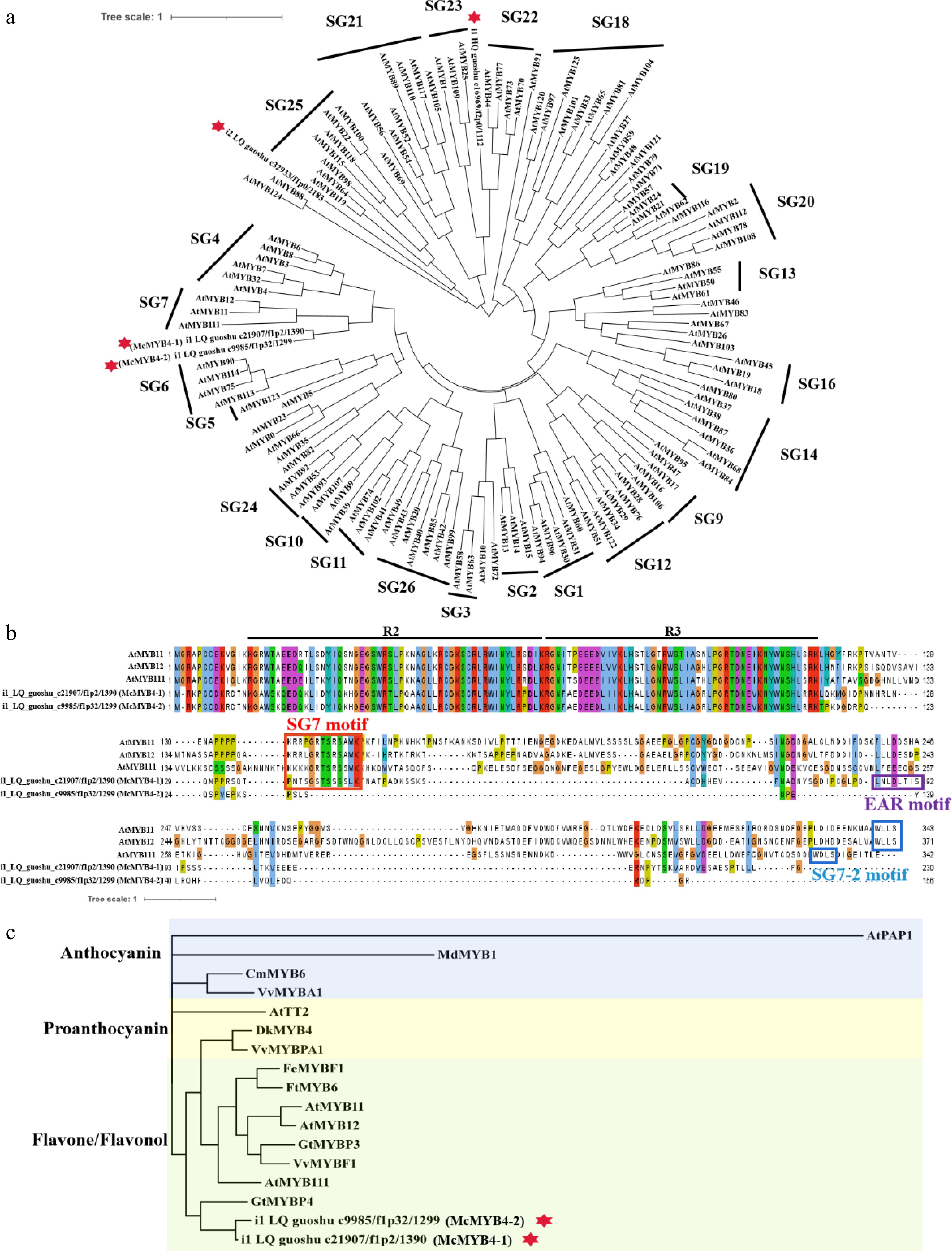
Figure 6.
Subgroup and phylogenetic relationships of McMYBs. (a) Phylogenetic relationships of four McMYBs and R2R3 MYBs from Arabidopsis. A total of 126 protein sequences of the R2R3 MYBs in Arabidopsis were obtained from The Arabidopsis Information Resource (TAIR) database. (b) Multiple alignment of SG7 R2R3-MYB proteins. The R2, R3 MYB domains, SG7, SG7-2 and EAR motif are marked. (c) Phylogenetic relationships of McMYB4-1, McMYB4-2 and R2R3 MYBs from other species. At, Arabidopsis thaliana; Cm, Chrysanthemum morifolium; Dk, Diospyros kaki; Fe, Fagopyrum esculentum; Ft, Fagopyrum tataricum; Gt, Gentiana trifloral; Md, Malus domestica; Vv, Vitis vinifera.
The expression of 14 bHLH TFs was measured via qRT-PCR. The 13 McbHLHs showed higher expression in GF than that in RF (Fig. 7a). To investigate the evolutionary relationships of McbHLHs in Jaboticaba, a maximum-likelihood method was constructed with amino acid sequences of 14 McbHLHs from Myrciaria cauliflora and 45 AtbHLHs from Arabidopsis (Fig. 7b). The functions of AtbHLHs are listed in Supplementary Table S12. In phylogenetic analysis, all the bHLH proteins were divided into 15 subgroups, and the regulatory roles of McbHLHs were predicted based on AtbHLH classification. But none of the McbHLHs were assigned to SG IIIf, in which the majority of bHLHs might regulate the biosynthesis of anthocyanin or proanthocyanin, suggesting that none of the 14 McbHLHs are involved in the anthocyanin biosynthetic pathway.
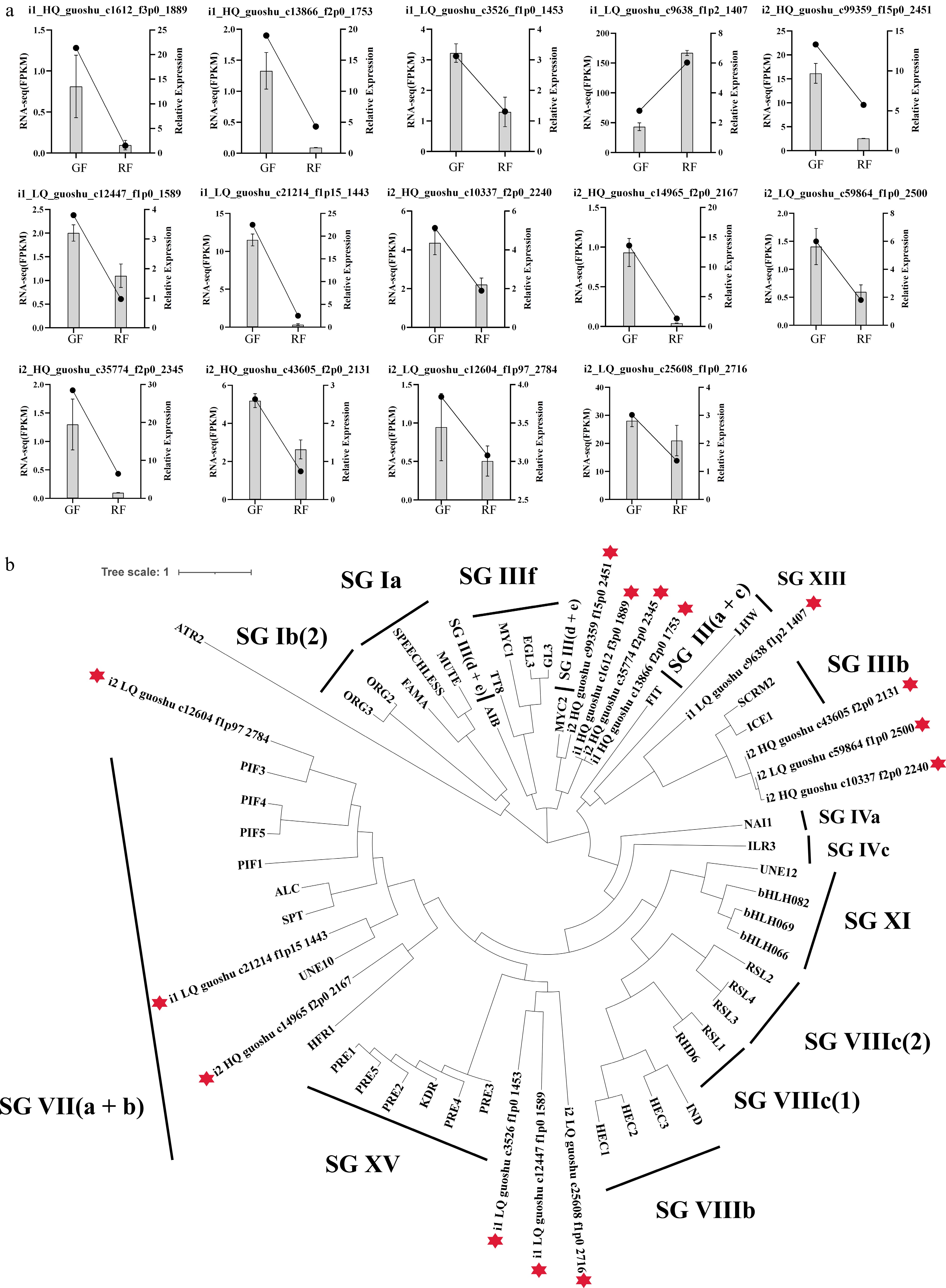
Figure 7.
Expression and phylogenetic analysis of McbHLH transcription factors. (a) Expression levels of the 14 McbHLHs in green fruits (GF), and ripening fruits (RF) validated by RNA-seq and qRT-PCR. (b) Phylogenetic relationships of 14 McbHLHs and bHLHs from Arabidopsis. A total of 45 protein sequences of the bHLHs in Arabidopsis were obtained from the TAIR database.
-
Jaboticaba is a native fruit of South America that has been gaining attention due to its high anthocyanin content and potential benefits. While previous studies have reported various phenolic compounds in Jaboticaba, including anthocyanins, flavonoids, and phenolic acids[4,6], a comprehensive understanding of its molecular mechanisms, particularly regarding anthocyanin biosynthesis, has been limited. Our study presents a significant advancement in this area, providing the first full transcriptomic characterization of Jaboticaba, which led to the identification of 86,758 unigenes, 9,732 EST-SSRs, and 1,127 long non-coding RNA sequences. These findings contribute to our understanding of the molecular basis of pigment accumulation and will facilitate future genetic research, especially in the areas of genetic structure characterization and breeding for improved fruit traits.
The six anthocyanins identified in Jaboticaba—including cyanidin-3-O-glucoside, delphinidin-3-O-glucoside, and peonidin-3-O-glucoside—are consistent with anthocyanins found in other fruits like grapes (Vitis vinifera), apples (Malus domestica), and strawberries (Fragaria vesca)[20−27]. However, our study uniquely identifies the key regulatory genes involved in anthocyanin biosyntheses, such as McPAL1-3, Mc4CL1-3, McF3H, McF3'H, McDFR, McANS, McUFGT, and McLAR. The expression of these genes during fruit ripening suggests a significant role in regulating the flavonoid and anthocyanin biosynthetic pathways in Jaboticaba, particularly through the upregulation of enzymes involved in the late stages of anthocyanin synthesis.
In comparison to other plants, Jaboticaba shows distinct transcriptional regulation of anthocyanin biosynthesis. For example, the MYB-bHLH-WD40 (MBW) complex is well-characterized in plants like Arabidopsis thaliana and Malus domestica as a key regulator of anthocyanin biosynthesis[28]. However, the role of MYB transcription factors in Jaboticaba presents some novel findings. Specifically, two R2R3-MYB transcription factors, McMYB4-1 and McMYB4-2, were identified as potential repressors of anthocyanin biosynthesis, which contrasts with the positive regulation observed in other species. This finding suggests a unique regulatory mechanism in Jaboticaba that could offer insights into the modulation of flavonoid and anthocyanin accumulation.
Our study also highlights the potential role of long non-coding RNAs (lncRNAs) in regulating anthocyanin biosynthesis in Jaboticaba, a novel area of research that has not been extensively explored in fruit crops. The identification of 1,127 lncRNA sequences opens up new avenues for understanding the epigenetic regulation of pigment production and other metabolic processes in this species. Given that lncRNAs are involved in critical biological processes such as cell differentiation and stress responses[29], their potential regulatory role in secondary metabolite production warrants further investigation.
Moving forward, future studies should focus on the functional validation of the identified genes and transcription factors using advanced genetic tools such as CRISPR/Cas9. Additionally, a more comprehensive study of lncRNA interactions and their impact on anthocyanin biosynthesis would enhance our understanding of the epigenetic regulation of flavonoid production. These efforts could pave the way for breeding programs aimed at enhancing the nutritional and aesthetic value of Jaboticaba.
In conclusion, while Jaboticaba shares common features with other anthocyanin-rich fruits, our study identifies unique genetic and regulatory mechanisms that contribute to its distinct pigmentation. This research provides a valuable foundation for future studies aimed at optimizing pigment production and improving the genetic resources for Jaboticaba breeding.
-
This study provides the first comprehensive SMRT transcriptome sequencing of Jaboticaba (Myrciaria cauliflora), focusing on the leaves, flowers, and fruits at different developmental stages. Our findings uncover a wealth of genetic information, including 86,758 unigenes and 9,732 EST-SSRs, which lay the groundwork for future genetic research in this valuable fruit. More importantly, we identify key metabolites responsible for the distinctive purple coloration of Jaboticaba fruits, including six anthocyanins, and reveal a significant accumulation of leucocyanidin, dihydroquercetin, and eriodictyol during fruit ripening. By integrating transcriptomic and metabolomic data, we uncover a set of 15 differentially expressed genes (DEGs) involved in the phenylpropanoid, flavonoid, and anthocyanin biosynthetic pathways, contributing to anthocyanin accumulation. Notably, two R2R3-MYB transcription factors, McMYB4-1 and McMYB4-2, were found to negatively regulate anthocyanin biosynthesis. Our findings not only provide valuable insights into the molecular mechanisms regulating anthocyanin biosynthesis but also offer new perspectives on the nutritional and functional potential of Jaboticaba as a source of bioactive compounds.
This work was financially supported by grants from the Major Special Projects of Yunnan Provincial Science and Technology Plan (202302AE090005), and Yunnan Province Key Projects of Applied Basic Research (202401AS070145). We would like to thank TopEdit (
www.topeditsci.com ) for linguistic assistance during the preparation of this manuscript.-
The authors confirm contribution to the paper as follows: study conception and design: Xu W, Liu DD; data collection: Fan SC, Yang XY; analysis and interpretation of results: Fan SC, Yang XY; draft manuscript preparation: Liu CN, Chen LY. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
-
The authors declare that they have no conflict of interest.
-
accompanies this paper at (https://www.maxapress.com/article/doi/10.48130/tp-0025-0016)
-
Received 17 December 2024; Accepted 10 March 2025; Published online 28 July 2025
-
This study conducted the first comprehensive transcriptomic analysis of Jaboticaba, identifying 86,758 unigenes, 9,732 EST-SSRs, and 1,127 long non-coding RNA (lncRNA) sequences, providing critical genomic resources for understanding the molecular mechanisms of Jaboticaba.
The study identified key regulatory genes involved in anthocyanin biosynthesis in Jaboticaba, such as McDFR, McANS, and McUFGT, revealing the critical enzyme genes that regulate anthocyanin biosynthesis in fruits.
Unlike the positive regulatory role of MYB transcription factors in traditional plants such as Arabidopsis thaliana and Malus domestica, this study found that two R2R3-MYB transcription factors (McMYB4-1 and McMYB4-2) in Jaboticaba may act as repressors of anthocyanin biosynthesis, highlighting a unique transcriptional regulatory mechanism in Jaboticaba.
This research provides a valuable foundation for the genetic characterization and improvement of fruit traits in Jaboticaba, with significant potential applications in optimizing anthocyanin accumulation and enhancing the nutritional and aesthetic value of the fruit.
-
# Authors contributed equally: Shu-Chen Fan, Xuan-Yu Yang
- Supplementary Table S1 Primers used for qRT-PCR assays.
- Supplementary Table S2 Summary of SMRT transcriptome sequencing for Jaboticaba.
- Supplementary Table S3 Summary of Illumina transcriptome sequencing for Jaboticaba.
- Supplementary Table S4 Transcriptome dataset compared with the previously published dataset.
- Supplementary Table S5 Six types of SSR repeat motifs and their frequency in Jaboticaba.
- Supplementary Table S6 Differentially accumulated phenolic compounds in the peel of green and ripening fruits.
- Supplementary Table S7 List of unigenes that cover anthocyanin biosynthesis pathway 9 selected genes.
- Supplementary Table S8 List of differentially expressed genes (DEGs) involved in the phenylpropanoid, flavonoid, and anthocyanin biosynthetic pathways.
- Supplementary Table S9 List of unigenes that cover MYB and bHLH TFs.
- Supplementary Table S10 Screening for MYB and bHLH candidate target genes by transcriptome analysis.
- Supplementary Table S11 Functions of plant R2R3-MYBs mentioned in this review.
- Supplementary Table S12 Functions of bHLHs in Arabidopsis mentioned in this review.
- Supplementary Fig. S1 Venn diagram of the number of lncRNAs predicted by Coding-Non-Coding Index (CNCI), Pfam, PLEK, and Coding Potential Calculator (CPC).
- Supplementary Fig. S2 Multiple alignments of RaMYB4 and two McMYBs.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Fan SC, Yang XY, Chen LY, Liu CN, Xu W, et al. 2025. Long-read transcriptome sequencing for the in-depth understanding of anthocyanin biosynthesis in Jaboticaba. Tropical Plants 4: e026 doi: 10.48130/tp-0025-0016 

Long-read transcriptome sequencing for the in-depth understanding of anthocyanin biosynthesis in Jaboticaba
- Received: 17 December 2024
- Revised: 20 February 2025
- Accepted: 10 March 2025
- Published online: 28 July 2025
Abstract: Jaboticaba (Myrciaria cauliflora) is a grape-shaped fruit characterized by high concentrations of anthocyanins concentrated within its peel, representing a valuable natural source of functional pigments. Here, we report long-read transcriptome sequencing in Jaboticaba, and 86,758 unigene sequences, 9,732 expressed sequence tag-simple sequence repeats, and 1,127 long non-coding RNA sequences were uncovered. In addition, integrated transcriptomic and metabolomic analysis of pigment accumulation during fruit ripening showed that six anthocyanins accumulated in Jaboticaba fruit, with 47 flavonoid synthesis-related genes, and 12 differentially expressed genes. Compared with green fruits, these candidate genes encoded eight upregulated enzymes particularly expressed in ripening fruits—4CL, PAL, F3H, F3'H, DFR, ANS, LAR, and UFGT—and related to the regulation of phenylpropanoid, flavonoid, and anthocyanin biosynthetic pathways. Furthermore, two R2R3-MYB transcription factors McMYB4-1 and McMYB4-2 might negatively regulate anthocyanin accumulation. The results reveal key regulatory mechanisms governing flavonoid and anthocyanin biosynthetic pathways, thereby offering critical perspectives on plant secondary metabolite production in Jaboticaba.