









-
Dendrobium is a genus of perennial herbaceous plants in the orchid family (Orchidaceae). These plants are widely distributed across tropical and subtropical regions of Asia, particularly in China, India, and Southeast Asia[1]. Dendrobium plants are edible, medicinal, and ornamental, with effects such as nourishing yin energy, moistening the lungs, promoting the production of bodily fluids, and quenching thirst[2,3]. Modern pharmacological research has shown that Dendrobium contains large numbers of polysaccharides, alkaloids, phenanthrenes, bibenzyls, sesquiterpenoids, and other chemical constituents with pharmacological effects, including enhancing immunity, lowering blood sugar, and exhibiting antitumor, antioxidant, and anti-inflammatory effects[4,5].
The phenotypes, compositions, and contents of these components vary greatly among Dendrobium species, and the pharmacological activities and medicinal effects of these species are not the same[6]. For example, the stems of the common Dendrobium species D. officinale, D. huoshanense, D. nobile, and D. chrysotoxum are of different sizes and shapes, and their polysaccharide, alkaloid, and bibenzyl contents also differ. D. officinale stems are long, slender, and cylindrical, and their main active ingredients include polysaccharides and flavonoids[7]. D. nobile stems are erect, fleshy and thick, slightly flattened, and cylindrical. Their major effective chemical components include alkaloids and sesquiterpenes[8]. D. chrysotoxum stems are partially fleshy and fusiform, with more rounded and blunt strips of ribs, and their primary active compounds are in the bibenzyl and phenanthrene classes[9]. D. huoshanense has shorter stems, which are thicker above the base and thinner in the upper part; the predominant compounds in its stems are polysaccharides and flavonoids[10]. In-depth research has revealed that the polysaccharide and flavonoid contents are higher in D. officinale and D. huoshanense than in other Dendrobium species, making these two species expensive and of excellent quality. In recent years, increasing numbers of studies have investigated Dendrobium polysaccharides, with the majority of studies focusing on D. officinale. However, while significant attention has been devoted to developing quality evaluation methodologies for D. officinale, few studies have explored the genetic mechanisms underlying polysaccharide biosynthesis in this medicinal plant.
The declining cost of sequencing and the development of bioinformatics technologies have made it possible to obtain the genome sequences of many plant species and to carry out evolutionary studies at the genome-wide level. At the same time, the large amount of sequencing data has enabled comparative analyses of genomes across species[11]. Through comparative genomics, researchers can identify conserved coding/non-coding regulatory elements and lineage-specific sequences, elucidate both conserved and divergent genomic features through orthologous sequence alignment, trace phylogenetic relationships and ancestral origins via homology analysis, facilitate structural annotation and functional prediction of protein-coding genes, and reconstruct phylogenetic relationships while deciphering molecular adaptation mechanisms underlying species evolution[12,13]. Comparative genomics has been widely used to study plants such as Arabidopsis thaliana and rice (Oryza sativa)[14].
Sugar serves as the primary energy source for plants and plays critical roles in plant growth, reproduction, development, and adaptation to stress[15]. In plants, carbon flux is coordinated by sugar transporters[16], including SUT (sucrose transporter), MST (monosaccharide transporter), and SWEET (sugar will eventually be exported transporter) proteins[17]. SWEET proteins are a conserved class of bidirectional sugar transporters that function in fundamental physiological processes by mediating the transmembrane translocation and intercellular allocation of saccharides. These membrane-spanning proteins function as bidirectional facilitators of sucrose efflux, orchestrating source-to-sink carbohydrate partitioning to ensure proper growth, developmental transition, and stress adaptation in plants[18]. SWEET proteins play major roles in fructose transport within vesicles[19] and affect sugar transport in the phloem, leaves, and seeds, while also regulating sugar accumulation in fruits[20]. Furthermore, SWEET proteins transport monosaccharides or disaccharides across the plasma membrane or intracellular membranes[21], thereby affecting sugar accumulation and metabolism in plants. SWEET genes play multiple roles in plant growth and development and resistance to biotic and abiotic stresses[22]. Plants under biological stress usually regulate sugar levels in vivo by upregulating or downregulating the expression of SWEET genes. Plants reduce osmotic pressure under stress by increasing sugar content in the plant body or by regulating sugar transfer and redistribution to maintain the balance of osmotic pressure and thus ensure their survival[23].
During the domestication of Dendrobium species from wild to cultivated, drastic changes in their growth environment led to variations in physiological phenotypes and genotypes and the formation of a rich variety of Dendrobium species, such as D. officinale and D. huoshanense (with high polysaccharide contents) and D. nobile (with high alkaloid contents)[24]. However, little systematic research has focused on the underlying gene expression patterns and the basis of genome evolution in this genus owing to the lack of coverage and functional gene mining of the whole-genome sequences of Dendrobium species.
In this study, comparative genomics was used to analyze four Dendrobium species with high-quality genome information (D. officinale, D. huoshanense, D. nobile, and D. chrysotoxum) and to elucidate the mechanism underlying the high polysaccharide contents of D. officinale from an evolutionary perspective with a focus on the SWEET gene family. These findings shed light on the evolution of this important genus and the roles of key DoSWEET proteins in polysaccharide accumulation. In addition, they lay the foundation for further exploring the molecular mechanism of quality formation in Dendrobium.
-
Soil-grown Dendrobium plants were used in this study. The seedlings were grown under a 12/12 h light/dark cycle at 22–25 °C. The plants were maintained under normal growth light (GL) conditions using white LEDs (100 μmol·m−2·s−1).
Genomic comparison of four Dendrobium species
-
The homologous chromosomes of D. officinale from D. nobile, D. huoshanense, and D. chrysotoxum were identified using Minimap2 (
https://github.com/lh3/minimap2/releases?after=v2.21 ), and genome phasing and analysis of the specific enrichment of long terminal repeat (LTR) retrotransposons (LTR-RTs) among chromosomes were performed using SubPhaser. OrthoVenn3 was employed to identify orthologous gene clusters, followed by Gene Ontology (GO) enrichment analysis to investigate the functional relevance of these clusters. The analysis focused on identifying genes associated with biological processes related to carbohydrate metabolism and transport.Identification of the SWEET gene family in Dendrobium
-
The PFAM profile hidden Markov model (HMM) for SWEET domain MtN3_slv (PF03083) was downloaded from the PFAM database (
http://pfam.xfam.org/ ), and the SWEET protein sequences of D. officinale, D. nobile, D. huoshanense, and D. chrysotoxum were searched with a threshold value of 1e−5. The protein sequence files of the SWEET gene family of A. thaliana were compared with those of D. officinale, D. nobile, D. huoshanense, and D. chrysotoxum by BLAST analysis with a threshold of 1e−10. After merging the HMM search results with the results of this comparison, the National Center for Biotechnology Information (NCBI;www.ncbi.nlm.nih.gov ) CD-Search tool was used to conduct domain searches and to screen candidate SWEET gene family members.Phylogenetic analysis of SWEET gene family members
-
OrthoVenn3 (
https://orthovenn3.bioinfotoolkits.net/ ) was used to compare and annotate the orthologous gene clusters among D. nobile, D. chrysotoxum, D. huoshanense, and D. officinale. Using the gene sequences and intergenic regions of all samples, gene/region-specific alignments were performed using MAFFT (v7.299b). The sequences of low quality were automatically removed using trimAl v1.4 software with default values. The approximate maximum likelihood tree was generated by FastTree v2.1.7 software (www.microbesonline.org/fasttree ). Support values were computed by the Shimodaira-Hasegawa test with 1,000 resamples[25−27].Analysis of conserved structures and cis-regulatory elements
-
The deduced amino acid sequences of Dendrobium SWEET proteins were uploaded to the MEME Suite (
https://meme-suite.org/meme/doc/meme.html ) and to the NCBI Conserved Domain Database (www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi ) to search and obtain the conserved motif model and conserved structural domains; the 2,000 bp upstream sequences of the coding regions of the genes were submitted to PlantCARE (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/ ) to predict cis-regulatory elements in the promoter regions.RNA-Seq data acquisition and analysis
-
The original transcriptome data for different D. officinale organs (PRJNA715099), D. officinale hormone treatment (PRJNA763165), D. officinale salt stress treatment (PRJNA715099), and D. officinale cold stress (PRJNA949802) were downloaded from NCBI SRA. The transcriptional data of D. officinale varieties under high-light treatment were acquired by the research team. Fastp software was used for the quality control of the original data, and hisat2 software was used for genome comparison. The feature counts were used to calculate the count of reads aligned to each gene. Transcripts per million (TPM) were calculated by DESeq2, and log-transformed TPM [log2(TPM + 1)], referred to as log-TPM, was used for the downstream analysis. The TPM values were used the log2(TPM + 1) to construct the heatmap by TBtools.
Expression analysis of DoSWEET genes in different tissues and under abiotic stress treatment
-
RNA-seq data for SWEET genes in D. officinale in different tissues and under abiotic stress were downloaded from NCBI (
www.ncbi.nlm.nih.gov ) under the following accession numbers: cold stress (PRJNA949802), hormone treatment (PRJNA763165), salt stress (PRJNA715099), and different tissues (PRJNA715099). The data for SWEET genes under high-light treatment included in this study are available upon request from the corresponding author. All fragment per kilobase million mapped reads values were log2 transformed and used to construct gene expression heatmaps with TBtools software.Reverse-transcription quantitative PCR (RT-qPCR)
-
Total RNA was extracted from the samples with RNA Plant Plus Reagent (Magen). RNA concentrations were measured with a NanoDrop 2000 spectrophotometer, and an Evo M-MLV Tracking Kit (AG11734; Accurate Biotechnology Co., Ltd, China) was used to produce complementary DNA (cDNA). qPCR was conducted using the LightCycler 480 Real-Time PCR System (Roche, Basel, Switzerland). All reactions were performed with SYBR Green Premix Pro Taq HS qPCR according to the manufacturer's protocol (AG11735; Accurate Biotechnology Co., Ltd, China). The Actin genes from D. officinale, D. nobile, D. huoshanense, and D. chrysotoxum were used as internal controls, and relative transcript levels were determined using the ΔΔCt method and normalized. All transcript-level data were obtained by RT-qPCR; three biological samples and three replicates were performed per sample (Supplementary Table S1).
Vector construction for virus-induced gene silencing
-
Specific primers targeting the DoSWEET genes were designed using
https://crm.vazyme.com/cetool/singlefragment.html (Supplementary Table S1). The DoSWEET fragments were PCR amplified and cloned into pTRV2, which was digested with KpnI and EcoRI. The recombinant plasmids were introduced into Agrobacterium tumefaciens strain GV3101. The volume of TRV1 culture inoculated was equal to the sum of all pTRV2 cultures, and the Agrobacterium cells were grown at 28 °C in LB medium with 50 µg·mL−1 kanamycin and 25 µg·mL−1 rifampicin for 24 h. Cultures were pelleted at 3,000× g for 15 min and resuspended by gentle vortexing in one volume of infiltration buffer (1 mM MES, 1 mM MgCl2, 0.1 µM acetosyringone, and 0.002% Triton). The pelleting was repeated, the cells were resuspended again in one-half volume of infiltration buffer, and the OD600 was measured. Suspensions were diluted to an OD600 = 1.0 with infiltration buffer. Equal volumes of TRV1 and each separate TRV2 suspension were mixed by gentle inversion and incubated in the dark for 3 h at 22 °C to induce viral gene expression. Forty-day-old seedlings of D. officinale were used to conduct the virus-induced gene silencing (VIGS) experiment. To facilitate the vacuum infiltration, when the vacuum (generated by a vacuum pump) reached roughly 0.02 mbar, the vacuum pressure was maintained for 60 s, the vacuum pump was turned off, the system was left alone for 3 min, and the pressure was slowly released. After that, the plants were transplanted to soil and grown under a 12 h light/12 h dark cycle at 22 °C with a relative humidity of 65% for 30 d (Supplementary Fig. S1).Measurement of polysaccharide contents
-
Total polysaccharide contents in the samples were measured using the phenol-sulfuric acid method with a Plant Polysaccharide Test Kit (Sangon Biotech, Shanghai, China) following the manufacturer's protocol.
-
Dendrobium has attracted much attention owing to its important medicinal and ornamental value. To date, high-quality genomes of D. officinale, D. nobile, D. huoshanense, and D. chrysotoxum with different polysaccharide contents, alkaloid contents, and phenotypes (Fig. 1a) have been published. However, no studies have explored the different Dendrobium species in depth. MCscan was used to search for collinearity among the D. nobile, D. chrysotoxum, D. huoshanense, and D. officinale genomes. Ultraviolet (UV) spectrophotometry was then employed to determine the polysaccharide contents in the stems of these four species. D. huoshanense exhibited the highest polysaccharide contents, followed by D. officinale, while D. chrysotoxum showed the lowest polysaccharide contents (Fig. 1b).
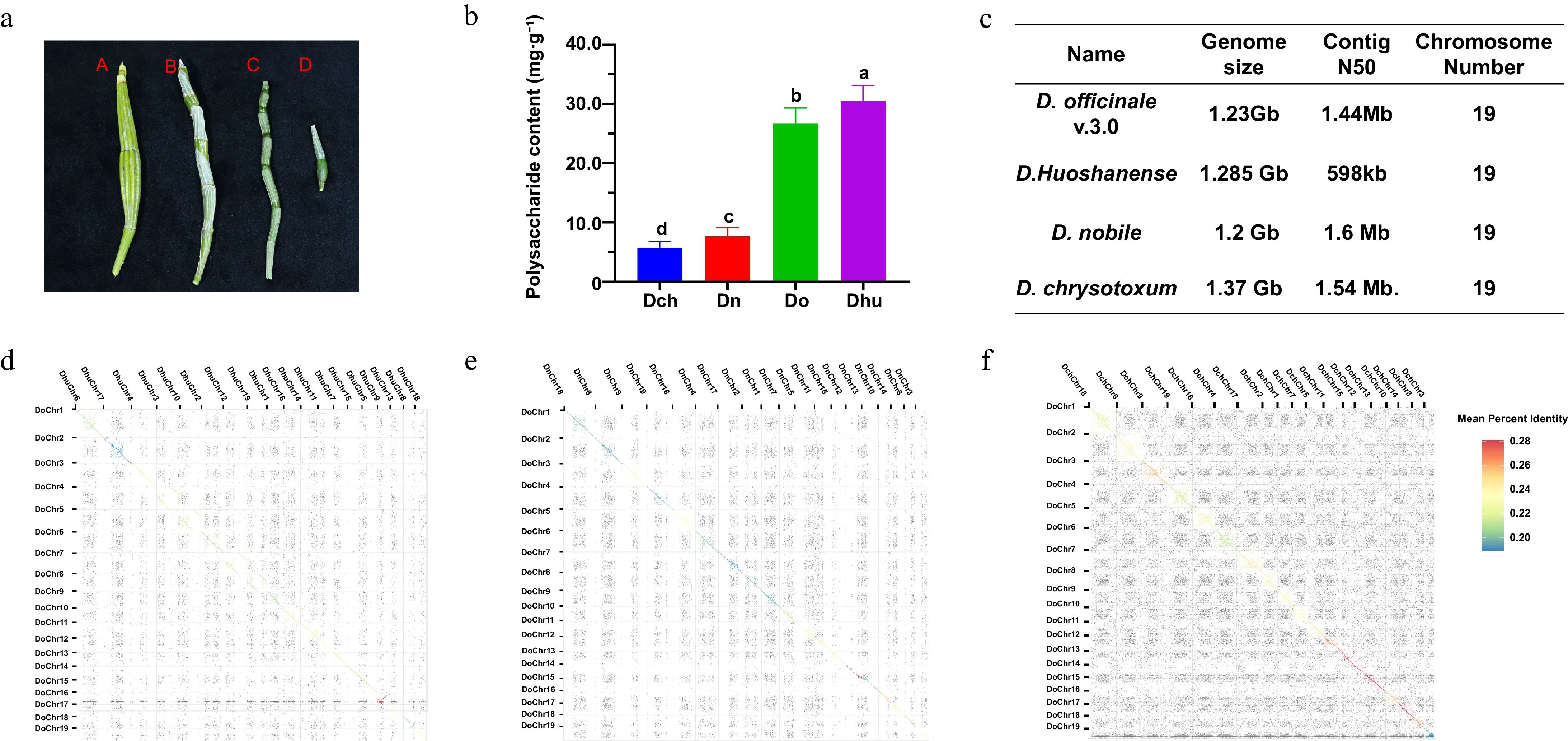
Figure 1.
Characterization of four Dendrobium species. (a) Phenotypes of stems of the four Dendrobium species (A D. chrysotoxum [Dch], B D. nobile [Dn], C D. officinale [Do], D D. huoshanense [Dhu]. Scale bar is 1 cm). (b) Polysaccharide contents of the four Dendrobium species. Different letters indicate significant differences among groups, as determined by one-way ANOVA with Tukey's multiple-comparisons test (p < 0.05). (c) Summary of sequencing data for the four Dendrobium genome assemblies. (d) Syntenic relationship of D. officinale and D. huoshanense. (e) Syntenic relationship of D. officinale and D. nobile. (f) Syntenic relationship of D. officinale and D. chrysotoxum.
To investigate whether the polysaccharide contents in these four Dendrobium species (D. officinale, D. nobile, D. huoshanense, and D. chrysotoxum) are associated with their genomic characteristics (Fig. 1c), a comprehensive analysis of their genome sizes and assembly status was conducted. All four species exhibited genome sizes exceeding 1 Gb, with a diploid chromosome number of 2n = 38 (19 chromosome pairs). The focus was on D. officinale, a widely used medicinal species, as the reference genome. Minimap2 (v2.24) was employed for comparative genomic analysis to detect homologous regions across D. officinale, D. nobile, D. chrysotoxum, and D. huoshanense and visualized the alignment results with dotPlotly to assess macro-synteny and evolutionary conservation.
The D. officinale and D. huoshanense genomes share a substantial number of syntenic genes, indicating a close evolutionary relationship between them (Fig. 1d). Chromosomes DoChr3, DoChr13, DoChr14, DoChr15, DoChr16, DoChr17, and DoChr18 of D. officinale share strong homology with those of D. nobile (Fig. 1e), and chromosome DoChr14 is highly homologous to that of D. chrysotoxum (Fig. 1f). The chromosomal homology between the genomes of D. officinale and D. huoshanense is low, but homology occurs at the top and bottom positions of chromosomes DoChr4 and DhuChr4, DoChr5 and DhuChr3, DoChr7 and DhuChr2, and DoChr8 and DhuChr12, thus preserving a large proportion of syntenic blocks. The variation between D. officinale and D. huoshanense during the evolutionary process is thought to be mainly due to the changes in chromosome positions, leading to species differentiation. The plant phenotypes and polysaccharide contents of D. officinale and D. huoshanense were similar, indicating that they did not change significantly after species differentiation. These results suggest that the genes controlling the polysaccharide contents and phenotypes of Dendrobium are primarily distributed in the top and bottom ends of chromosomes DoChr4 and DhuChr4, DoChr5 and DhuChr3, DoChr7 and DhuChr2, and DoChr8 and DhuChr12.
Genome characteristics and analysis of LTR-RT insertion times in four Dendrobium species
-
To gain an in-depth understanding of the genomic characteristics of the four Dendrobium plants, SubPhaser was used to phase their genomes (Fig. 2a, b). kmer heatmap clustering and principal component analysis revealed similarities among the homologous chromosomes in these species (Fig. 2c, d), suggesting that each genome shares specific features, as expected, and that inter-genome-specific features are present.
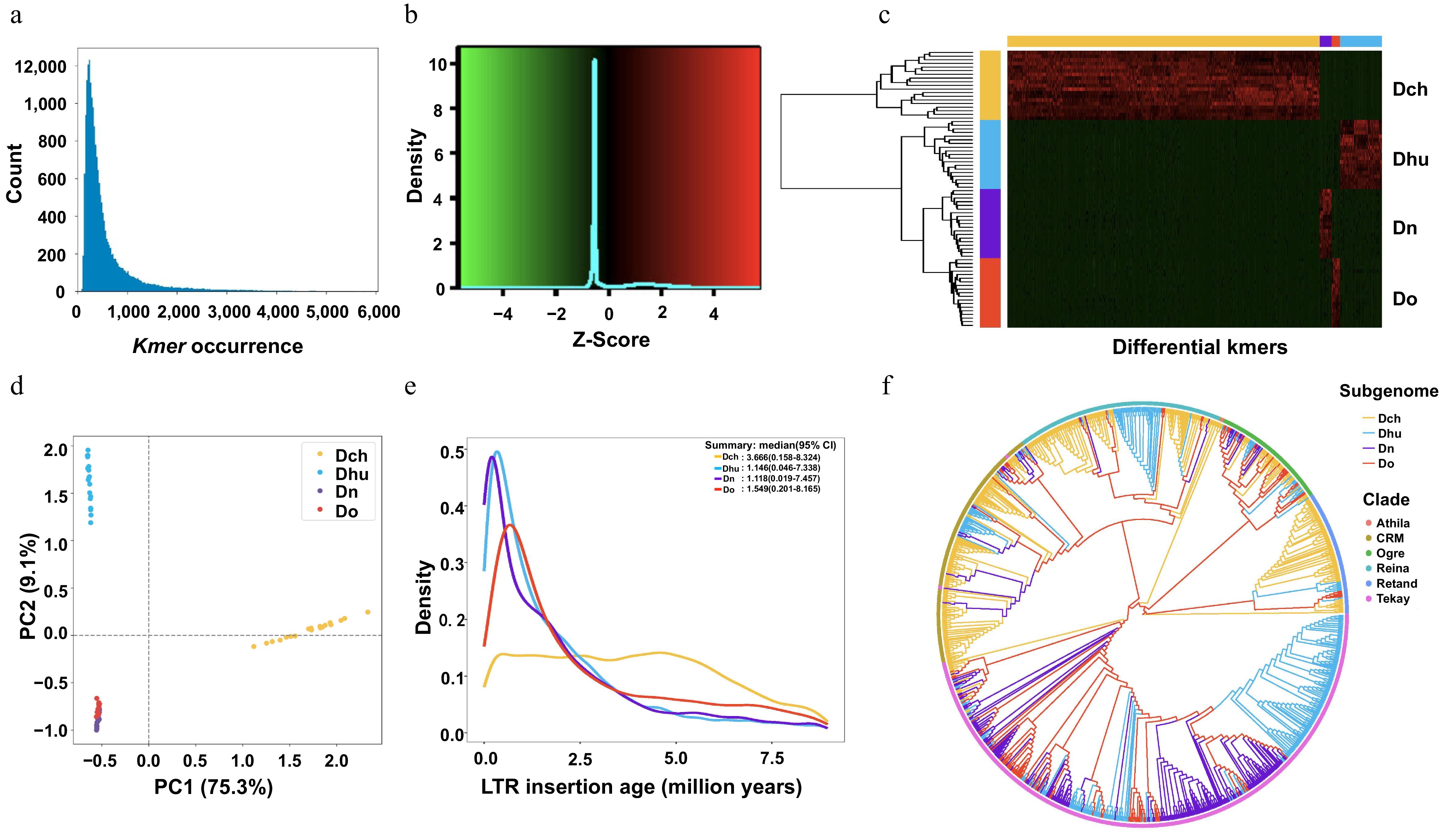
Figure 2.
Genome phasing of four Dendrobium species (Dch, Dn, Do, and Dhu) using SubPhaser. (a) Distribution frequency of different 15-mers in homologous chromosomes of the four species. (b) Heatmap showing the Z-scale relative abundance of kmers. (c) Unsupervised hierarchical clustering heatmap; horizontal color bar at the top indicates kmer specificity to the genome, and vertical color bar on the left indicates chromosomes. (d) Principal component analysis of different 15-mers. (e) Genome-specific long terminal repeat (LTR) retrotransposon (LTR-RT) insertion times (95% confidence interval). (f) Phylogenetic tree of 1,000 Gypsy LTR-RTs from random data resampling.
By analyzing the insertion times of long terminal repeat (LTR) retrotransposons (LTR-RTs), it was determined that, with the exception of D. nobile, the LTR-RT insertion events in Dendrobium species were predominantly concentrated between 1.395 and 1.863 million years ago (MYA). By contrast, the LTR-RT insertion events in D. nobile occurred approximately 5.222 MYA. This distinctive timing may be associated with a significant increase in global carbon dioxide concentrations during that period (Fig. 2e). D. officinale is thought to have originated in the Yunnan-Guizhou Plateau, adjacent to the Xizang Plateau, where continuous plateau uplift occurred between 0 and 8 MYA[24]. Approximately 2.4 MYA, LTR-RT insertion events also appeared in D. officinale, suggesting that these events during this period might be an important cause of intraspecific differentiation within D. officinale. Many LTR-RTs in the genomes of the four Dendrobium species occurred during the 1–2.5 MYA period, far from the time when species differentiation occurred (22 MYA), but the global climate and environment changed greatly during this time. The insertion events during this period might have been caused by natural selection during plant responses to environmental changes and might have been an important factor leading to changes in plant gene function. For this study, 1,000 Gypsy LTR sequences were randomly sampled for phylogenetic analysis. The LTR sequences of the Athila-Gypsy family were highly prevalent, dominating the genomes of D. nobile, D. huoshanense, and D. officinale (Fig. 2f), indicating that the LTR sequences of this family underwent a large-scale expansion during evolution that promoted the differentiation of these three species.
Whole-genome comparisons reveal evolutionary divergence in four Dendrobium species
-
Climate change directly or indirectly affects the adaptation of plants to the environment, plant diversity, and species migration. According to predictions about the four Dendrobium species using the TimeTree database, the light intensity gradually increased approximately 22 MYA and the four Dendrobium species differentiated, suggesting that light intensity was an important factor causing the species differentiation of Dendrobium (Fig. 3a). To investigate potential gene-exchange events between homologous chromosomal segments across distinct Dendrobium genomes, molecular typing and cluster analysis of chromosomal sequences was systematically conducted using an integrative approach combining whole-chromosome sequencing with kmer frequency profiling (Fig. 3b). Comparative Circos mapping of the four Dendrobium genomes revealed distinct patterns of intergenomic chromosomal exchanges. Notably, chromosomes DoChr4 and DoChr6 in D. officinale exhibited pronounced kmer enrichment patterns corresponding to DnChr11 and DnChr17 in D. nobile, suggesting substantial chromosomal rearrangements between these species. By contrast, the exchange patterns observed between DoChr16 of D. officinale and DnChr16/DnChr6 of D. nobile demonstrated a singular genomic interaction event exclusively with D. huoshanense. However, DoChr16 of D. officinale and DnChr16 and DnChr6 of D. nobile only experienced one genome exchange with D. huoshanense, showing different exchange patterns, suggesting that D. nobile and D. officinale might come from the same ancestor.
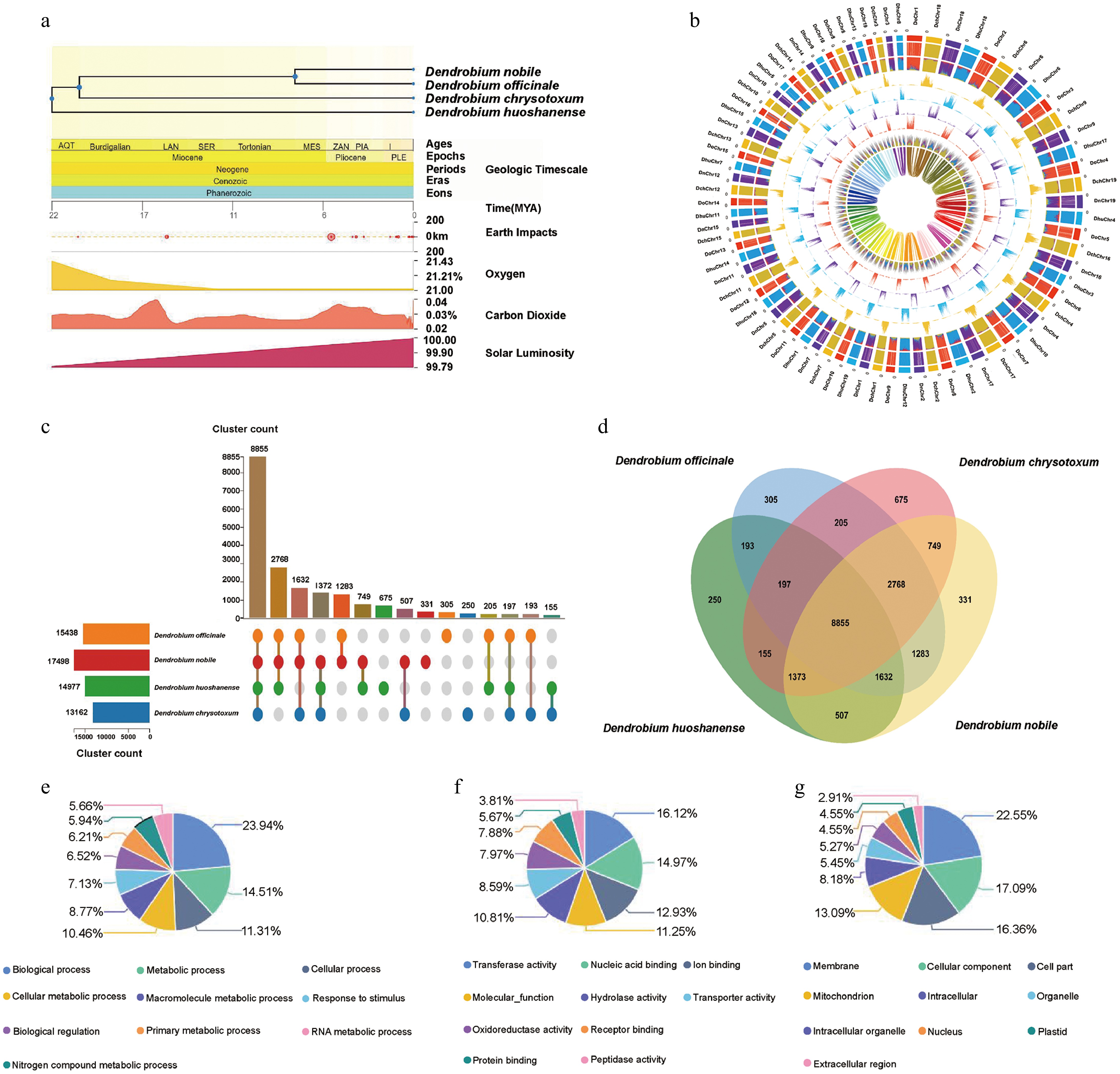
Figure 3.
Genomic comparisons and evolutionary divergence among four Dendrobium species. (a) Speciation of the four Dendrobium species and the timing of geo-environmental change events. (b) Chromosome characterization of the four Dendrobium species. From the inner to outer circles (1–8): (1) genome types assigned based on the K-means algorithm; (2) enriched genome-specific kmers; (3) genome-specific kmers normalized against each other; (3–6) absolute values of the pooled counts of each genome-specific kmer; and (8) the density of long terminal repeat (LTR) retrotransposons (LTR-RTs); a color consistent with the genome class indicates that LTR-RTs are significantly enriched in these genome-specific kmers, and gray indicates non-specific LTR-RTs. (c) The number of orthologous clusters in each species. (d) Venn diagram of intersecting direct homologous gene clusters. (e) GO enrichment analysis of biological processes. (f) GO enrichment analysis of molecular functions. (g) GO enrichment analysis of cellular components.
To explore the functions of the genes in the four genomes in detail, OrthoVenn3 was used to identify the orthologous genes between the genomes and to cluster the genes (Fig. 3c). The four Dendrobium species shared 8,855 common gene clusters. D. nobile contained the most gene clusters (17,498), followed by D. officinale (15,438), and D. huoshanense contained the fewest gene clusters (13,162; Fig. 3c, d). Gene Ontology (GO) analysis revealed that 23.94% of the genes in the ten most abundant gene clusters were enriched in biological process-related GO terms, and 14.51% were enriched in metabolic process–related terms. Moreover, genes responsible for transporter activity and membrane components accounted for 8.59% of molecular function-related terms and 22.55% of cell component–related terms, respectively (Fig. 3e–g). Among the enriched GO terms associated with biological activity, 283 genes were involved in the metabolism and transport of carbohydrates. A significant number of SWEET genes are annotated in the AmiGO2 database as key genes responsible for intercellular carbohydrate-directed transport. SWEET genes might have undergone subfunctionalization (e.g., specializing in transporting different types of sugars) or neofunctionalization (e.g., participating in stress responses) during evolution.
Identification and distribution of cis-promoter elements in SWEET genes in Dendrobium
-
Sugars account for over 50% of the total organic matter in plants and play critical roles in plant growth and development. SWEET genes play key roles in sugar transport, participating in fructose transport in vacuoles; sugar transport in the phloem, seeds, and leaves; and the regulation of sugar accumulation in fruits. Numerous SWEET genes that were annotated as key genes in the targeted transport of carbohydrates were identified between cells using the AmiGO2 database. To explore the evolutionary relationships and classification of the Dendrobium SWEET genes, OrthoVenn3 (
https://orthovenn3.bioinfotoolkits.net/ ) was used to compare and annotate the orthologous gene clusters among D. nobile, D. chrysotoxum, D. huoshanense, and D. officinale. Using the gene sequences and intergenic regions of all samples, gene/region-specific alignments were performed using MAFFT (v7.299b). The protein sequence files of the SWEET gene family in A. thaliana were then compared with those of D. officinale, D. nobile, D. chrysotoxum, and D. huoshanense by BLAST analysis with a threshold of 1e−10 (Supplementary Table S2). The sequences of low quality were automatically removed using trimAl v1.4 software with default values. The approximate maximum likelihood tree was generated by FastTree v2.1.7 software (www.microbesonline.org/fasttree ). Support values were computed by the Shimodaira-Hasegawa test with 1,000 resamples. Following this, 105 SWEET gene family members were identified, including 24 in D. officinale (DoSWEETs), 21 in D. huoshanense (DhuSWEETs), 25 in D. chrysotoxum (DchSWEETs), and 35 in D. nobile (DnSWEETs) (Supplementary Table S3). A phylogenetic tree containing these 105 Dendrobium SWEET genes and A. thaliana SWEET genes was constructed using FastTree software (Fig. 4a). The SWEET genes in the five species were divided into four clades (I, II, III, and IV) according to evolutionary distance. Clade II is the largest, containing 54 SWEET genes, including seven DhuSWEET, 16 DnSWEET, 13 DoSWEET, 13 DchSWEET, and five AtSWEET genes. The second largest clade (Clade III) contains six DhuSWEET, nine DnSWEET, eight DoSWEET, six DchSWEET, and seven AtSWEET genes. Clade I contains five DhuSWEET, five DnSWEET, two DoSWEET, three DchSWEET, and three AtSWEET genes, while Clade IV includes two DhuSWEET, one DnSWEET, one DoSWEET, two DchSWEET, and two AtSWEET genes. A previous study showed that Clade II SWEET proteins have a preference for hexose transport[28]. Hexoses are monosaccharides containing six carbon atoms. Common hexoses include glucose, mannose, fructose, and galactose, and the major polysaccharides in Dendrobium are composed mainly of mannose and glucose. The different polysaccharide contents and monosaccharide compositions of Dendrobium stems might be caused by the differences in the activity of sugar transporters.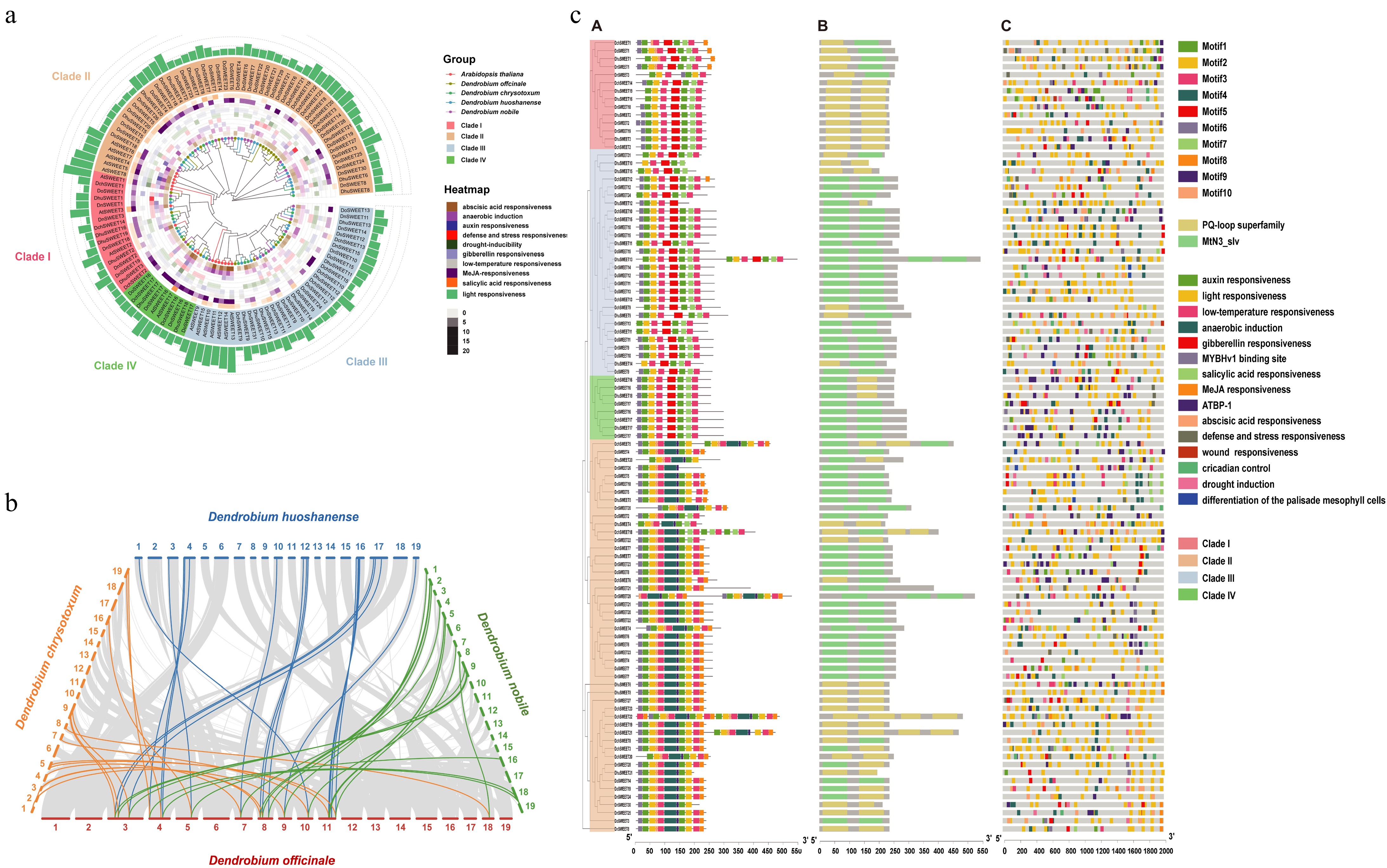
Figure 4.
Genome-wide identification and functional analysis of the SWEET gene family in Dendrobium. (a) Phylogenetic relationships of SWEET proteins in Arabidopsis thaliana (At), D. officinale (Do), D. chrysotoxum (Dch), D. huoshanense (Dhu), and D. nobile (Dn). The trees were constructed based on protein sequence alignment using the maximum likelihood method. The four clades and different cis-elements in the heatmap are marked with different colors. (b) Syntenic relationships of D. officinale, D. chrysotoxum, D. huoshanense, and D. nobile SWEET are genes shown on the chromosome maps. (c) Analysis of conserved motifs and conserved domains in SWEETs. ([A] Conserved motifs and gene exon-intron structures in the SWEETs. [B] Conserved domains in SWEET proteins identified by a search using NCBI (
www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi ). [C] Predicted cis-acting elements in the upstream regions of SWEET genes).To further investigate the syntenic relationships of SWEET genes in the genomes of the four Dendrobium species, collinearity analysis was conducted using MCScanX. Limited chromosomal homology was detected between D. officinale and D. huoshanense. However, synteny was detected in four chromosome pairs, DoChr4–DhuChr4, DoChr5–DhuChr3, DoChr7–DhuChr2, and DoChr8–DhuChr12, which have retained a significant proportion of homologous sequences (Fig. 4b). Phylogenetic analysis revealed that chromosomal rearrangements, rather than whole-genome duplication, were the dominant drivers of speciation between these two medicinal Dendrobium species. The plant phenotypes and polysaccharide contents of D. officinale and D. huoshanense were similar, indicating that the phenotypes and compound contents did not change significantly following species differentiation, suggesting that the genes controlling the polysaccharide contents and phenotypes of Dendrobium might be mainly distributed in the top and bottom ends of chromosomes DoChr4 and DhuChr4, DoChr5 and DhuChr3, DoChr7 and DhuChr2, and DoChr8 and DhuChr12. These findings suggest that the SWEET gene family might be responsible for the differences in polysaccharide contents in stems among Dendrobium species.
To further explore the structural characteristics and conservation of the SWEET gene family in Dendrobium, MEME Suite was used to identify conserved motifs in SWEET proteins (Fig. 4c). Most of these proteins contain two MTN3 motifs. However, DhuSWEET13 and DchSWEET5, 18, 21, and 22 contain four such motifs, perhaps resulting from gene duplication and replicative transposition. The distribution patterns of ten conserved motifs were also analyzed, finding that Motif1, Motif3, and Motif4 were the most widely distributed and highly conserved in the MtN3_slv domain. Notably, Motif4 and Motif9 are exclusively found in Clade II. This restricted distribution may reflect gene replacement or insertion events that occurred during the evolution of the SWEET gene family.
To study the response of SWEET genes in Dendrobium to various abiotic stresses, their promoters (2 kb upstream of the transcription start site) were submitted to PlantCARE to predict the cis-acting elements in each promoter. Fifteen cis-acting elements related to plant hormones, stress, and development, were identified, including five plant hormone response elements (auxin response, MeJA response, abscisic acid response, gibberellin response, and salicylic acid response) and six stress response elements (light induction, abscission induction, drought induction, low-temperature induction, defense and stress induction, and trauma induction). The distribution of the 15 major cis-acting elements in the Dendrobium SWEET gene family is shown in Fig. 4c, with a large number of MeJA- and salicylic acid-responsive elements widely distributed in each species in Clade II. Numerous abscisic acid response elements are present in each species of Clade III. The variable number of light-responsive binding elements in the promoters of SWEET genes of various Dendrobium species is notable, suggesting that light might be one of the main factors driving the evolutionary differentiation of Dendrobium species. These results suggest that the SWEET gene family in each Dendrobium species is functionally differentiated and is involved in multiple biological processes, as suggested by the variation in transcriptional levels and responsiveness to environmental stress of the SWEET genes.
Expression patterns of SWEET genes in different organs under abiotic stress
Expression patterns of SWEET genes in different organs of D. officinale
-
To examine the expression patterns of the SWEET genes, the commonly studied species D. officinale was chosen. The expression levels of SWEET genes in different organs of D. officinale were analyzed (Fig. 5a). Of the 24 DoSWEET genes, 22 exhibited significant differential expression across the organs. Six DoSWEET genes (DoSWEET1, 2, 9, 15, 16, and 24) were highly expressed in leaves and flowers, and ten (DoSWEET3, DoSWEET5, DoSWEET6, DoSWEET13, DoSWEET14, DoSWEET15, DoSWEET17, DoSWEET19, DoSWEET20, and DoSWEET24) were highly expressed in flowers. Four DoSWEET genes (DoSWEET4, 12, 21, and 22) were highly expressed in roots, and six (DoSWEET2, 7, 8, 9, 11, and 18) were highly expressed in stems. DoSWEET7, 8, 11, and 18 were more highly expressed in stems than in leaves, flowers, and roots, suggesting that the higher polysaccharide contents in stems than in roots, leaves, and flowers are mainly due to the differential activities of these DoSWEET genes in different organs.
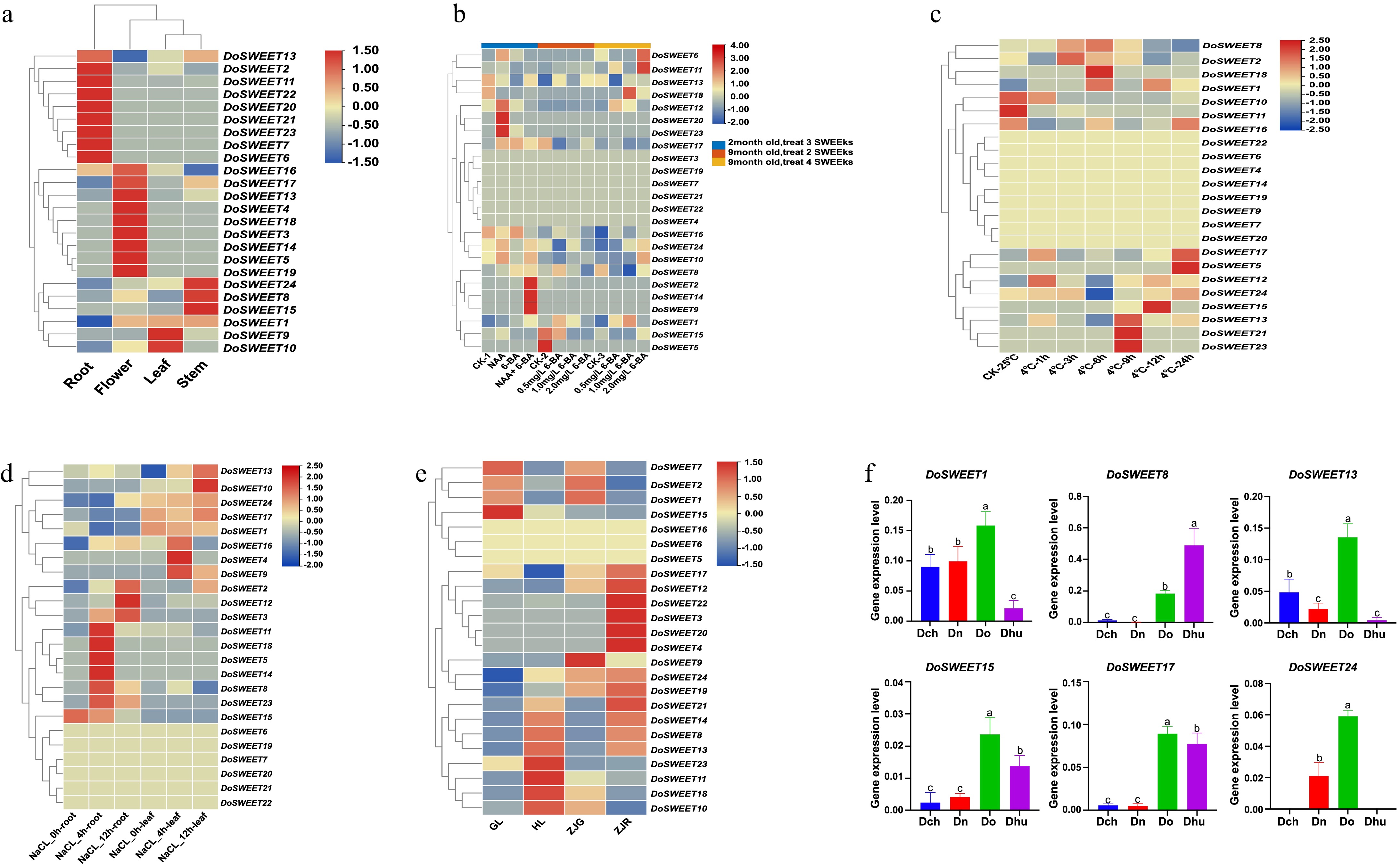
Figure 5.
Expression patterns of SWEET genes in D. officinale. (a) Heatmap of the differential expression of SWEET genes in four D. officinale organs. (b)–(d) Heatmaps of the differential expression of DoSWEET genes in response to different (b) phytohormones, (c) Cold, (d) salt, and (e) differences in light conditions and ecotype. ZJG and ZJR indicate ecotypes with green stems and red stems, respectively, from Zhejiang Province, China, that were maintained under normal growth light. GL and HL indicate normal growth light and high-light conditions, respectively, applied to the YNG ecotype. (a)–(e) The expression levels of SWEET genes are represented by log2-transformed transcripts per million (TPM) values acquired from RNA-seq data. (f) Expression analysis of SWEET genes in the four Dendrobium species (Do, Dhu, Dn, and Dch), as determined by RT-qPCR. The data are representative of three independent experiments. The error bars indicate SD, and different lowercase letters indicate significant differences in expression levels at the 95% confidence level.
Expression patterns of SWEET genes in D. officinale treated with plant hormones
-
Analysis of cis-elements in the SWEET gene promoters revealed many phytohormone-responsive elements. To explore the potential roles of DoSWEETs in plant hormonal regulation, the SWEET gene expression profiles of D. officinale treated with different concentrations of 6-BA and NAA were analyzed (Fig. 5b). The expression levels of DoSWEETs in D. officinale were generally not high in the 2-month-old control (CK) group, but some SWEET genes were significantly induced after three weeks of phytohormone treatment. DoSWEET12, 20, and 23 were highly induced by NAA treatment, while DoSWEET2, 9, and 14 were highly induced by the combination of NAA and 6-BA. In 9-month-old plants, DoSWEET5 and 15 were highly expressed in the CK group, but their expression decreased in response to 6-BA treatment. However, after four weeks of treatment, DoSWEET6 and 11 were significantly induced by 2.0 mg·L−1 6-BA treatment, and DoSWEET18 was highly induced by 1.0 mg·L−1 6-BA treatment. These results suggest that the expression levels of DoSWEETs are related to the phytohormones NAA and 6-BA.
Expression patterns of SWEET genes in D. officinale under cold stress
-
Analysis of cis-elements in the promoters of SWEET genes revealed the presence of many low-temperature-responsive elements. To explore the potential functions of DoSWEETs in the plant response to cold stress, the gene expression profiles of D. officinale seedlings were analyzed at different temperatures (Fig. 5c). DoSWEET 10, 11, and 16 were highly expressed under normal conditions, while DoSWEET1, 17, and 12 were highly inhibited by this treatment. DoSWEET10, 12, and 17 were highly expressed under 4 °C-1 h; DoSWEET8 and 12 were highly expressed under 4 °C-3 h; DoSWEET1, 8, and 18 were highly expressed under 4 °C-6 h; DoSWEET13, 21, and 23 were highly expressed under 4 °C-9 h; DoSWEET1 and 15 were highly expressed under 4 °C-12 h. DoSWEET5, 16, and 17 were highly induced by cold treatment (4 °C-24 h). These results suggest that the expression patterns of DoSWEETs were affected by cold temperature treatment.
Expression patterns of SWEET genes in D. officinale under salt stress
-
Analysis of cis-elements in the SWEET gene promoters revealed numerous stress-responsive elements. To explore the potential functions of DoSWEET genes in plant responses to salt stress, their expression profiles were analyzed in leaves and roots after 4 and 12 h of salt stress (Fig. 5d). DoSWEET15 was highly expressed in roots under normal conditions, but its expression decreased gradually with increasing salt stress treatment, while DoSWEET11, 18, 5, 14, 8, and 23 expression was induced by 4 h of salt stress treatment. After 12 h of salt stress treatment, DoSWEET2, 12, and 3 were highly induced in roots. DoSWEET13 was expressed at low levels in leaves under normal conditions but was highly upregulated in response to salt stress. After 4 h of salt stress, DoSWEET16, 4, and 9 were highly expressed; this effect decreased after 12 h of salt stress treatment, while DoSWEET8 expression was significantly reduced by this treatment. Therefore, the expression patterns of DoSWEETs under salt stress were affected by the duration of salt stress and the plant organ.
Expression patterns of SWEET genes in D. officinale under high-light stress
-
Climate change directly or indirectly affects plant adaptation to the environment, plant diversity, and species migration. According to the predictions by the TimeTree database, the light intensity gradually increased before 22 MYA, and the four Dendrobium plants differentiated, suggesting that light intensity is an important factor causing the species differentiation. Analysis of the cis-elements in the SWEET gene promoters revealed numerous light-responsive elements, providing additional evidence that light might be a driver of Dendrobium evolution. This is in agreement with the hypothesis stated earlier (Fig. 3a). In a previous study in which D. officinale was divided into green-stem, purple-stem, and red-stem ecotypes, anthocyanin content increased with the deepening of stem color. The depth of stem color in D. officinale is mainly determined by the anthocyanin content, and the formation of anthocyanin is dependent on light[29]. Therefore, in this study, the effects of normal growth light (GL; control) treatment were examined on D. officinale ecotypes ZJR (with red stems) and ZJG (with green stems), both from Zhejiang Province, China. A third ecotype, D. officinale YNG (green-stemmed D. officinale varieties from Yunnan), was exposed to both GL and high-light (HL) treatment. Under GL conditions, the expression levels of DoSWEET4, 20, 3, 22, and 12 were higher in ZJR than in ZJG, while the expression levels of DoSWEET9, 2, and 1 were higher in ZJG than in ZJR, as revealed by transcriptome analysis. Notably, the expression levels of DoSWEET11 in ZJG and DoSWEET8, 13, and 14 in ZJR under GL conditions were close to those of the HL group. However, in D. officinale YNG, DoSWEET14, 8, 13, 23, 11, 18, and 10 were highly induced under HL treatment. Finally, in D. officinale YNG, DoSWEET7, 2, 1, and 15 were highly expressed under GL conditions and were significantly inhibited under HL conditions. These results suggest that DoSWEETs are critical for the HL response and that DoSWEET8, 13, and 14 might be key genes in the response of D. officinale to high light (Fig. 5e).
RT-qPCR analysis of candidate genes associated with polysaccharide accumulation
-
Among the tissues of Dendrobium, the stems contain the highest polysaccharide levels, followed by the flowers and leaves, while the roots contain the lowest polysaccharide levels. To validate the correlation between differentially expressed genes identified from the RNA-seq data and polysaccharide accumulation, six genes that are highly expressed specifically in stems—SWEET1, 8, 13, 15, 17, and 24—were selcted and their expression patterns were analyzed across the four Dendrobium species using RT-qPCR. SWEET8, 15, and 17 were expressed at significantly higher levels in D. officinale and D. huoshanense than in D. nobile and D. chrysotoxum. SWEET1 and 13 were more highly expressed in D. nobile, D. chrysotoxum, and D. officinale than in D. huoshanense, whereas SWEET24 was more highly expressed in D. nobile and D. officinale than in D. huoshanense and D. chrysotoxum (Fig. 5f). D. officinale and D. huoshanense are the two species with the highest polysaccharide contents among the four species studied, suggesting that SWEET8, 15, and 17 are closely associated with polysaccharide accumulation and serve as key genes determining polysaccharide levels in Dendrobium species. The expression patterns of these genes align strongly with the distribution of polysaccharides, further supporting their potential roles in polysaccharide metabolism.
Functional validation of SWEET genes in D. officinale
-
Finally, to investigate the roles of DoSWEET1, 8, 13, 15, 17, and 24 in polysaccharide biosynthesis during D. officinale growth and metabolism, virus-induced gene silencing (VIGS) was performed. Prior to initiating formal VIGS experiments, D. officinale seedlings were pre-infiltrated with the TRV2-GFP vector to confirm the successful establishment of the VIGS system. Observation using in vivo imaging on day 3 post-infiltration revealed the following results: Plants infiltrated with pTRV2-GFP emitted strong green fluorescence (Supplementary Fig. S2a); specific GFP fluorescence signals were also clearly visible under UV light illumination (Supplementary Fig. S2b). Further examination using the THUNDER wide-field high-definition imaging system showed that GFP fluorescence intensity in the vascular bundles and surrounding parenchyma cells of the stems was significantly higher in the pTRV2-GFP-infiltrated group compared to the wild-type(WT) control (Supplementary Fig. S2c). Collectively, these results successfully tracked the dynamic changes of the virus using the fluorescence encoded by EGFP tagged on the TRV2 vector, further confirming that TRV can infect D. officinale. This preliminarily demonstrates the successful construction of the VIGS system. Building upon this successfully established VIGS system, the DoSWEET1, 8, 13, 15, 17, and 24 genes were targeted for silencing. At 30 d post-infiltration, relative gene expression levels were measured in the plants using a UV-VIS spectrophotometer (Thermo Fisher Scientific Inc., USA), and RT-qPCR, and polysaccharide contents were examined via a phenol-sulfuric acid assay. Polysaccharide accumulation was markedly reduced across all silenced lines (14.3%–22.2% reduction) compared to the pTRV2-GFP control, with DoSWEET1, 8, 13, 17, and 24 showing the most pronounced effects (> 10% reduction; Fig. 6a). As shown in Fig. 6b, targeted silencing of DoSWEET1, 8, 13, 15, 17, and 24 resulted in their significant downregulation compared to the pTRV2-GFP control (Original data are in Supplementary Table S4). These results suggest that SWEET-mediated monosaccharide transport is critical for polysaccharide biosynthesis in D. officinale, with specific SWEET proteins (DoSWEET1, 8, 13, 17, and 24) serving as key regulators of carbohydrate partitioning and storage.
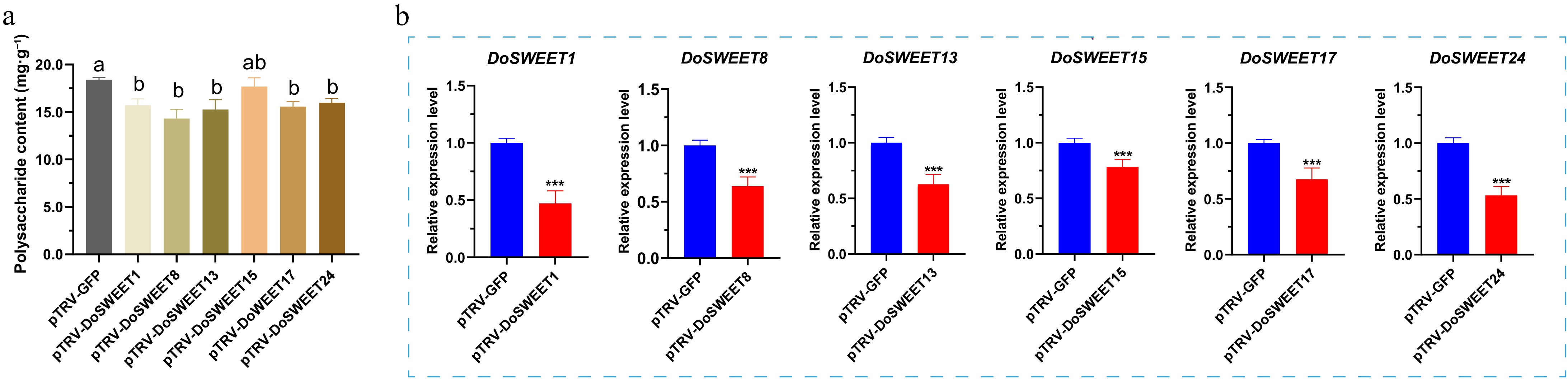
Figure 6.
The SWEET gene family plays a critical role in polysaccharide accumulation in D. officinale. (a) Total polysaccharide contents in D. officinale after virus-induced gene silencing (VIGS). Different lowercase letters indicate significant differences at the 95% confidence level. (b) Expression patterns of DoSWEET1, 8, 13, 15, 17, and 24 in D. officinale stems after VIGS. Error bars indicate the standard deviation of three biologically independent repeats. Student's t-test: *** p < 0.001.
-
The SWEET protein family in plants represents a novel type of transporter capable of transporting sugars, sugar alcohols, and hormones[30]. By regulating sugar translocation and distribution, this protein family plays crucial roles in plant growth and development[20]. In recent years, SWEET genes have been shown to affect polysaccharide accumulation in other species. In Polygonatum cyrtonema Hua, polysaccharide accumulation is highly correlated with the expression levels of SUS, INV, SWEET, and PLST genes, and SWEETs affect polysaccharide biosynthesis via the transport of large amounts of sucrose and several monosaccharides[31]. From a chromosome-scale assembly of the D. chrysotoxum genome, SWEET genes were identified, and phylogenetic analysis showed that 17 SWEET genes might be associated with fleshy stems that are abundant in polysaccharides and other medicinal compounds[32]. In a similar study in apple, analysis of sugar accumulation and the underlying mechanisms in the F2 progenies of a hybridization between the high-sugar apple (Malus × domestica) variety 'Gala' and the high-flavonoid apple germplasm 'CSR6R6', MdSWEET9b was shown to help mediate sugar accumulation in fruits. Moreover, MdWRKY9 binds to the MdSWEET9b promoter and regulates its activity in response to abscisic acid signaling, which enhances its regulation of MdSWEET9b expression[33]. Here, multiple SWEET genes were identified in four Dendrobium species, with numbers comparable to those in model species such as Arabidopsis, rice, and soybean. Phylogenetic analysis classified these genes into four distinct clades (Fig. 4a), consistent with previous studies. Notably, all of these SWEET proteins contained the conserved mtN3/saliva domain. Similar features were observed in studies of the medicinal plant Bletilla striata and Euphorbiaceae plants, indicating that SWEET gene family members are highly conserved.
The main bioactive substances of Dendrobium contain polysaccharides, dendrobine, phenanthrenes, amino acids, and trace mineral elements[34,35]. Polysaccharides, which make up a substantial proportion of D. officinale plant biomass, are the predominant bioactive compounds in Dendrobium and are used as a quality index for this plant in the Chinese pharmacopoeia. In this study, it was found that the content of Dendrobium polysaccharides was correlated with the expression of SWEET genes. Specifically, in the varipolysaccharide contents of D. officinale and D. huoshanense with higher (Fig. 1b), DoSWEET8, DoSWEET15, and DoSWEET17 exhibit a significantly higher expression (Fig. 5f). It is hypothesized that SWEET-mediated sugar transport promotes polysaccharide accumulation. Among the three DoSWEET genes in D. officinale, DoSWEET8 belongs to Clade II, similar to Arabidopsis SWEET13, which transports sugars and supports pollen grain development[36]. In Citrus sinensis, CsSWEET15 plays a crucial role in sucrose accumulation in the juice sac[37]. DoSWEET17, on the other hand, is assigned to Clade IV, similarly to Arabidopsis SWEET17, which is involved in sugar transport in vacuoles[38]. In the tuber of the medicinal plant Bletilla striata, as polysaccharides continuously accumulate, BsSWEET1 participates in the transport and accumulation of sugars. Similarly, in the medicinal plant Potentilla anserina, PaSWEET7, PaSWEET9, and PaSWEET12 promote the formation and development of taproots. In this study, the DoSWEET genes in D. officinale were analyzed and it was found that the expression levels of DoSWEET1, DoSWEET8, DoSWEET13, DoSWEET15, DoSWEET17, and DoSWEET24 in the stem are higher than those of other DoSWEETs (Fig. 5a). Given that the stem has the highest polysaccharide contents among different tissues of D. officinale, DoSWEET1, DoSWEET8, DoSWEET13, DoSWEET15, DoSWEET17, and DoSWEET24 may play important roles in the transport and accumulation of polysaccharides in the stem of D. officinale, thereby influencing its medicinal value and quality formation.
Environmental factors strongly influence polysaccharide dynamics in Dendrobium. Temperature-dependent polysaccharide degradation patterns were previously observed in this genus, with low-temperature storage effectively preserving carbohydrate content, likely through enhanced osmotic adjustment via polysaccharides and compatible solutes during cold-induced declines in water potential[39]. Sugars play multifaceted roles in plant physiology, functioning not only as energy sources and structural components for biopolymers such as starch and cellulose but also as osmoregulators that mitigate abiotic stress-induced cellular damage. The SWEET genes in plants widely respond to abiotic stresses, such as low temperature, drought, and salt stress[40−42]. In this study, the expression levels of many of the SWEET genes (such as DoSWEET1, DoSWEET8, DoSWEET13, DoSWEET15, DoSWEET17, and DoSWEET24) showed significant differences during the cold, NaCl, and high-light treatments (Fig. 5d, e). This result is consistent with many studies on the SWEET gene family; SWEET1 has been recently reported to play an important role in cold resistance in longan[41], and homologous genes of CsSWEET13 show increased expression levels under salt stress[43].
This study on D. chrysotoxum, D. nobile, D. officinale, and D. huoshanense revealed a noteworthy phenomenon: no significant positive correlation was observed between stem robustness and polysaccharide contents. Specifically, compared to D. officinale and D. huoshanense, which have relatively slender stems, the polysaccharide contents in D. chrysotoxum and D. nobile, which have stouter stems, were significantly lower (Fig. 1a, b). This finding suggests that the accumulation mechanism of polysaccharides—a key active component in Dendrobium medicinal materials—may operate independently of intuitive morphological traits (such as stem robustness). It appears that the biosynthesis and accumulation of polysaccharides are primarily regulated by intrinsic genetic programs, rather than being driven by morphogenesis. Sugar transport efficiency is a crucial link connecting primary photosynthetic products with the synthesis of secondary metabolites[44,45]. The analysis of the SWEET gene family in these four Dendrobium species demonstrates that the expression and regulation of genes in this family play key roles in polysaccharide accumulation. Therefore, targeting SWEET genes represents a promising avenue for enhancing the medicinal quality of authentic Dendrobium materials through molecular-assisted breeding strategies.
-
In summary, this study provides comprehensive insights into the genomic similarity of D. nobile, D. chrysotoxum, D. huoshanense, and D. officinale. Through comparative genomic analysis, it was determined that genes regulating polysaccharide contents and phenotypic traits in Dendrobium species are predominantly localized at the terminal regions of chromosomes DoChr4 and DhuChr4, DoChr5 and DhuChr3, DoChr7 and DhuChr2, and DoChr8 and DhuChr12. Additionally, an analysis of LTR-RT insertion times revealed that the amplification of the Athila-Gypsy family LTR-RT sequences contributed substantially to the divergence of these species. Analysis of chromosomal characteristics suggested that D. officinale and D. nobile may share a common ancestor. Moreover, comparative genomics combined with functional annotation using the AmiGO2 database highlighted SWEET family proteins as critical regulators of carbohydrate transport between cells.
A total of 105 SWEET genes were identified across the four Dendrobium species, including 24 DoSWEETs, 21 DhuSWEETs, 25 DchSWEETs, and 35 DnSWEETs. The distribution of SWEET gene domains and cis-promoter elements revealed functional divergence within this family, indicating that they function in diverse biological processes. Expression profiling of DoSWEETs demonstrated their differential expression across various organs, environmental conditions (e.g., high light and low temperature), developmental stages (seedling age), abiotic stress (salt stress), and phytohormone treatments. Notably, VIGS identified SWEET1, SWEET8, SWEET13, SWEET17, and SWEET24 as key contributors to polysaccharide accumulation in Dendrobium.
Overall, the evolutionary dynamics of the SWEET gene family were elucidated in D. nobile, D. chrysotoxum, D. huoshanense, and D. officinale, as well as its role in plant responses to abiotic stress. Collectively, the evolutionary dynamics of the SWEET gene family (such as duplication, selection, and expression regulation) and the evolutionary history of Dendrobium species (such as divergence times and habitat adaptation) have shaped the diversity of polysaccharide biosynthesis capacity in this genus. These findings provide a foundation for further exploring the molecular mechanisms underlying polysaccharide biosynthesis in D. officinale and hold significant implications for its functional genomics and biotechnological applications.
The authors acknowledge the financial support provided by the National Natural Science Foundation of China (Grant Nos U22A20446, 32322007, and 82404991); the Guangdong Major Project of Basic Research (Grant No. 2023B0303000022), and the 2024 Annual Science and Technology Research and Development Incubation Project by the Guangdong Provincial Laboratory of Traditional Chinese Medicine (Grant No. HQL2024PZ024). The Innovation and Entrepreneurship Training Program for College Students (Grant No. 202310572001), and the Special Fund for the Cultivation of Guangdong College Students' Scientific and Technological Innovation (Grant No. pdjh2024a099).
-
The authors confirm contributions to the paper as follows: study conception and design: Jin HL, Wang HB; data collection: Li D, Lu P, Li F, Qi Q, Ren Z, Lin Y, Zeng J, Mai H; analysis and interpretation of results: Lai Z, Li J, Wang Z, Hong Z, Tian J, Lu P; draft manuscript preparation: Qi Q, Li D; revision of the manuscript: Li D, Lu P, Jin H. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Dongxiao Li, Peijun Lu
- Supplementary Table S1 List of primers.
- Supplementary Table S2 Gene data.
- Supplementary Table S3 SWEET protein family data.
- Supplementary Table S4 qRT-PCR original Ct value.
- Supplementary Fig. S1 VIGS experiment original picture.
- Supplementary Fig. S2 Expression of the GFP reporter gene in D. officinale.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Li D, Lu P, Li F, Qi Q, Lai Z, et al. 2025. SWEET transporters in Dendrobium species: molecular insights into the regulation of polysaccharide biosynthesis. Medicinal Plant Biology 4: e030 doi: 10.48130/mpb-0025-0028 

SWEET transporters in Dendrobium species: molecular insights into the regulation of polysaccharide biosynthesis
- Received: 29 April 2025
- Revised: 25 July 2025
- Accepted: 29 July 2025
- Published online: 28 September 2025
Abstract: Dendrobium, a large genus in the orchid family, is globally valued for its medicinal and ornamental properties. However, Dendrobium species exhibit significant variations in their phenotypic traits, chemical compositions, and contents of bioactive compounds, particularly polysaccharides, which are a key determinant of quality. This study employed comparative genomics to investigate the evolutionary trajectories of four Dendrobium species with distinct polysaccharide profiles and phenotypic characteristics to identify putative genomic loci controlling polysaccharide biosynthesis. The analysis revealed differential evolutionary patterns potentially associated with polysaccharide accumulation. The SWEET gene family was systematically characterized, encoding sugar transporters implicated in polysaccharide metabolism, across Dendrobium species. Promoter analysis demonstrated that SWEET genes predominantly contain abiotic stress-responsive cis-acting elements. Using D. officinale as a model, SWEET gene expression dynamics were investigated through transcriptomic profiling and RT-qPCR validation. Heatmap analysis revealed tissue-specific expression patterns and differential responses to environmental stress factors (high light, cold, and salinity) and phytohormones. Integrated expression profiling identified six candidate genes (SWEET1, 8, 13, 15, 17, and 24) as potential key regulators of the quality formation mechanism of D. officinale. These six genes were silenced via virus-induced gene silencing. Polysaccharide accumulation was significantly reduced in all silenced lines, with a 14%–22% decrease observed across lines. Silencing DoSWEET1, 8, 13, 17, and 24 had the most pronounced effects on polysaccharide contents. This study enhances the understanding of the mechanisms underlying quality formation in medicinal plants and provides critical genetic resources for the improvement and breeding of Dendrobium species.